
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmonic nanoparticles in chemical analysis
Jan Krajczewski
,
Karol Kołątaj
and
Andrzej Kudelski
 *
*
Department of Chemistry, Faculty of Chemistry, University of Warsaw, Pasteur 1, 02-093 Warsaw, Poland. E-mail: akudel@chem.uw.edu.pl; Fax: +48-225526434
First published on 20th March 2017
Abstract
Many very sensitive analytical methods utilising specific properties of plasmonic metal nanoparticles have been developed. Some of these techniques are so sensitive that observation of the reliable signal even from a single molecule of the analyte is possible. In this review article we present the most important analytical techniques based on the plasmonic properties of selected metal nanoparticles and the basic theoretical background of these analytical techniques, including the mechanism of the interaction of the electromagnetic radiation with the plasmonic nanoparticles. The analytical techniques presented in this article include methods based on the change of the optical properties of plasmonic nanoparticles caused by analyte-induced aggregation, etching or the change of the growth of plasmonic nanoparticles, and techniques utilising increased efficiency of some optical processes in the proximity of the plasmonic nanoparticles, e.g. surface-enhanced Raman scattering (SERS), surface enhanced infra-red absorption (SEIRA), and metal enhanced fluorescence (MEF). Recently, an observed increase in the number of applications of techniques utilising surface plasmon resonance for the analysis of various industrial, biological, medical, and environmental samples allows us to predict a large increase of the significance of these techniques in the near future.
 Jan Krajczewski | Jan Krajczewski is pursuing his PhD at the University of Warsaw, Poland. His main research interest is surface-enhanced Raman spectroscopy (SERS) and synthesis of various silver and gold nanoresonators. |
 Karol Kołątaj | Karol Kołątaj is pursuing his PhD at the University of Warsaw, Poland. His main research interest is shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) and synthesis of electromagnetic nanoresonators for SHINERS measurements. |
 Andrzej Kudelski | Andrzej Kudelski is a professor at the University of Warsaw, Poland. His area of expertise is vibrational spectroscopy of molecules at metal surfaces, mainly SERS, SHINERS, SEROA and SEIRA spectroscopy and synthesis of electromagnetic nanoresonators for various surface-enhanced vibrational spectroscopy. |
1. Introduction
When electromagnetic radiation interacts with metal nanoparticles with a negative real and small positive imaginary dielectric constant (e.g. silver and gold nanoparticles) it induces a collective oscillation of surface conduction electrons called surface plasmons.1–5 Excitation of surface plasmons leads to an enhanced electromagnetic field at some places on the illuminated nanoparticles. Surface plasmons excited in silver and gold nanoparticles during their interaction with light are also responsible for the bright colours of Ag and Au colloids. The intensive colour of suspensions of Ag and Au nanoparticles was the reason for using these materials in ancient times. For example, in ancient Rome gold and silver nanoparticles were used (of course without an understanding of the phenomenon) for dyeing glass and ceramics. Such use of nanoparticles continued over time and many examples of stained glasses dyed using nanoparticles can be found in churches from the Middle Ages. Various colours of glass were obtained by the formation of nanoparticles with different diameters. For example, red glass often contained ca. 20 nm gold nanoparticles, pink glass contained ca. 30 nm gold nanoparticles and orange glass contained gold nanoparticles larger than 80 nm or silver nanoparticles. This method of glass dyeing is so durable that glass dyed centuries ago is still colourful.In this work some analytical applications of plasmonic metal nanoparticles are reviewed. We have focused on analytical applications based on the change of the plasmonic properties of Ag and Au nanoparticles caused by analyte-induced aggregation, etching or change of the grow of plasmonic nanoparticles, and on the analytical methods utilising increase of the efficiency of various optical processes in the proximity of gold and silver nanoparticles, such as surface-enhanced Raman scattering (SERS), surface enhanced infra-red absorption (SEIRA), and metal enhanced fluorescence (MEF).6,7 We have also briefly described the mechanism of the interaction of the electromagnetic wave with the plasmonic nanoobjects. All mentioned above plasmonic properties of gold and silver are only observed when gold and silver structures have sizes in the nanometers range (or the Au/Ag surface is nanostructured). The appearance of new functionalities of materials after decreasing the size of its individual parts into the nanometers range is a realization of the futuristic concept of Richard Feynman presented in 1959 in the speech called: “There's Plenty of Room at the Bottom”. In this lecture Feynman described some new possibilities that comes from use of systems in the nanometric scale. Futuristic nanotechnology idea described in 1959 by Feynman is today's reality and presented in this work applications of gold and silver nanoparticles is only a small part of the practical applications of nanomaterials.
2. Surface plasmon resonance (SPR) and distribution of electromagnetic field around spherical “plasmonic” nanoparticles
Optical properties of nanoparticles from IB metals are very important for many of their applications. While a gold ingot is yellow and a silver ingot is greyish, colour of Au and Ag nanoparticles strongly depends on their size and shape, and can have all colours of rainbow from red through green to violet.8 Silver and gold nanoparticles have their intensive and various colours because of plasmons excited on their surfaces. Metal can be considered as positively charged atomic nuclei surrounded by a plasma of free electrons from the conduction band. Surface plasmon resonance (SPR) is a collective oscillation of electron plasma near nanoparticle's surface when the nanoparticle is irradiated – see Fig. 1a.9,10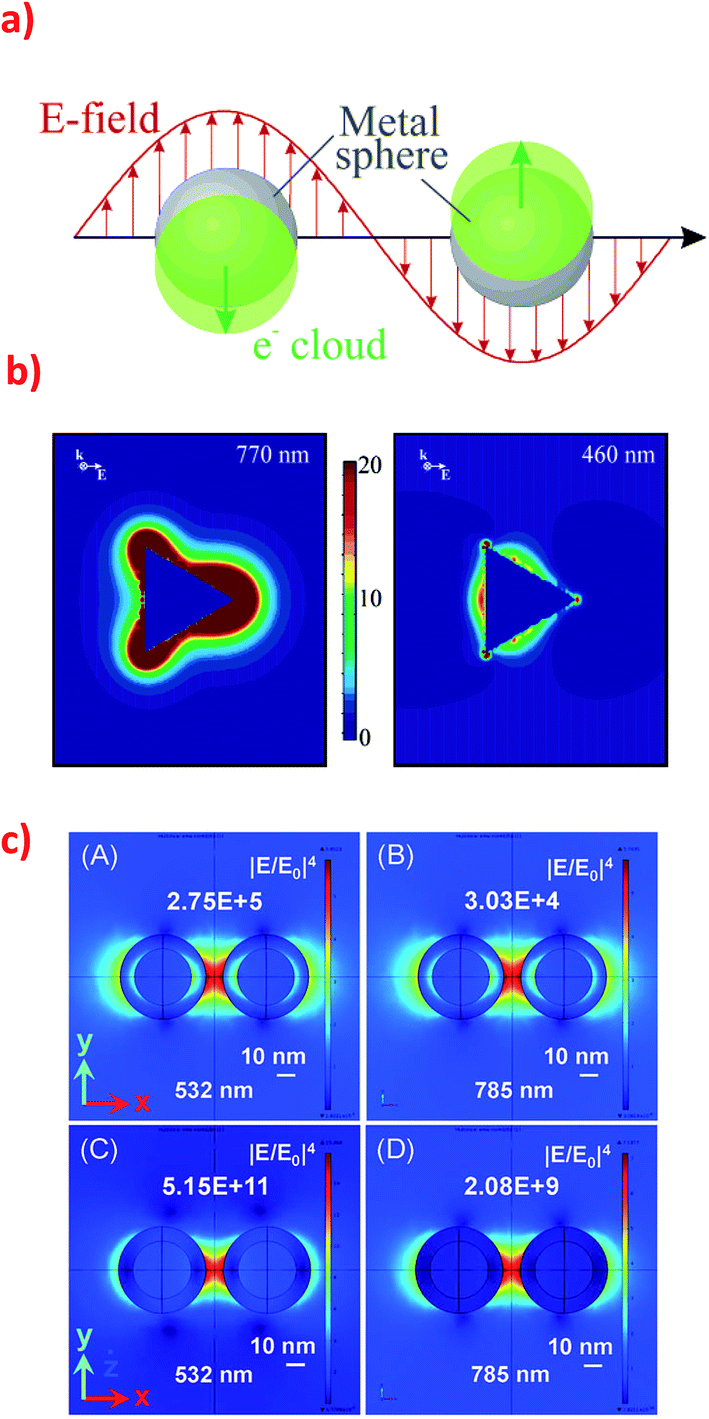 | ||
| Fig. 1 (a) Schematic of plasmon oscillation in spherical plasmonic nanoparticle showing the displacement of the conduction electron charge cloud relative to the nuclei. Reprinted with permission from ref. 10. Copyright 2003 American Chemical Society. (b) Electric field enhancement contours around the silver trigonal prism for a plane that is perpendicular to the trigonal axis and that passes midway through the prism. The prism is illuminated by the light that have k along the trigonal axis and E along the abscissa. The size of the prism: side length = 100 nm, thickness = 16 nm. The wavelength of the excitation radiation: for the left image 770 nm, and for the right image 460 nm. Reprinted with permission from ref. 10. Copyright 2003 American Chemical Society. (c) Electromagnetic field distribution in the dimers consisting of two types of nanoparticles: (A) and (B) Silver nanoparticles (30 nm) with a thin silica layer (5 nm) and (C) and (D) silica nanoparticles (30 nm) with a thin silver layer (5 nm) at two excitation wavelength (532 nm and 785 nm). Reprinted with permission from ref. 31. Copyright 2015 the Owner Societies of PCCP. | ||
Dislocation of metal electrons in the nanoparticle during irradiation leads to creation of the electric dipole and, as a consequence, to the additional electric field near the nanoparticle's surface. The combined electric field depends on the electric field of the irradiating electromagnetic wave and on the field created near nanoparticle's surface by the surface plasmon resonance. For a spherical nanoparticle the electric field outside its surface is described by the following equation:
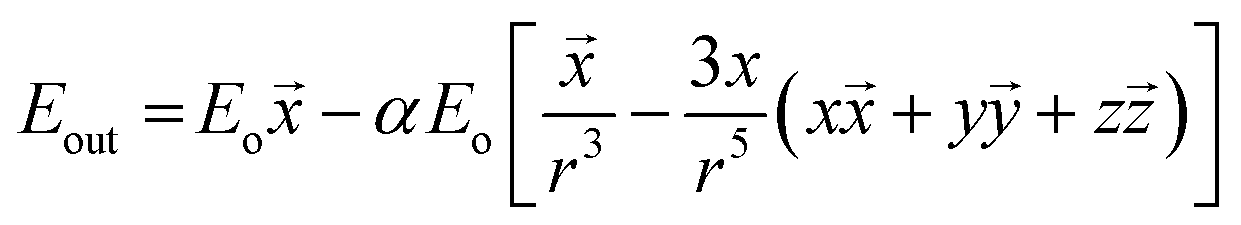
![[x with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0078_20d1.gif) ,
, ![[y with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0079_20d1.gif) ,
, ![[z with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_007a_20d1.gif) are unit vectors.10,11 As can be seen, the first part of the above equation is the electric field of irradiating light and the second part is the electric field from the induced dipole created on the nanoparticle's surface. For a nanoparticle with a spherical geometry polarizability α can be expressed as:
are unit vectors.10,11 As can be seen, the first part of the above equation is the electric field of irradiating light and the second part is the electric field from the induced dipole created on the nanoparticle's surface. For a nanoparticle with a spherical geometry polarizability α can be expressed as:| α = gda3 |
where a is a radius of specific nanoparticle,
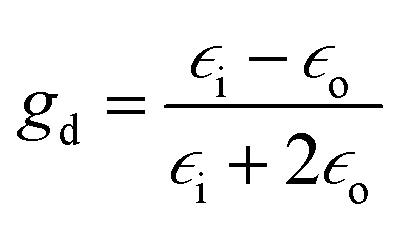
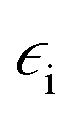 is the electric permittivity of the metal and
is the electric permittivity of the metal and 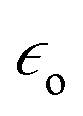 is the electric permittivity of nanoparticle's environment.10,11 As we can see from the above equations, the strongest surface plasmon resonance occurs when
is the electric permittivity of nanoparticle's environment.10,11 As we can see from the above equations, the strongest surface plasmon resonance occurs when 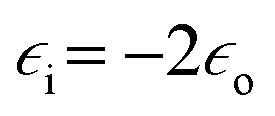 , so for nanoparticles with strong plasmonic activity we can anticipate the existence of a band on the UV-vis extinction spectrum.12 Moreover, for effective surface plasmon resonance the imaginary part of the dielectric permittivity (which is due to absorption) must be small. Such conditions are fulfilled only by nanoparticles from a few metals (for example Au, Ag, Cu, Al) and in this group silver nanoparticles has the greatest capacity to support surface plasmon resonance when using visible radiation.13
, so for nanoparticles with strong plasmonic activity we can anticipate the existence of a band on the UV-vis extinction spectrum.12 Moreover, for effective surface plasmon resonance the imaginary part of the dielectric permittivity (which is due to absorption) must be small. Such conditions are fulfilled only by nanoparticles from a few metals (for example Au, Ag, Cu, Al) and in this group silver nanoparticles has the greatest capacity to support surface plasmon resonance when using visible radiation.13
The incident electromagnetic wave irradiating nanoparticles is absorbed and scattered. Hence, the observed extinction of radiation by metal sols is the sum of these two phenomena: absorption and scattering. Absorption and scattering cross-section for spherical nanoparticles with radius a are expressed as:
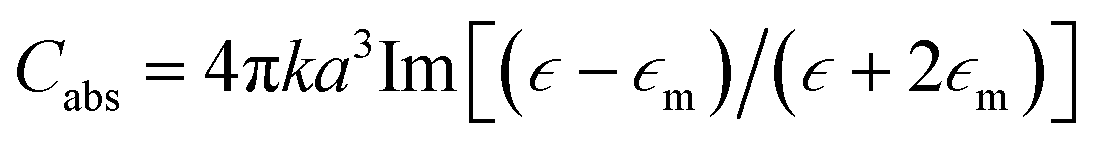
where k = 2π/λ and λ is wavelength. As can be seen from equations above Cabs scales with a3 while Csca scales with a6.14 It means that absorption is more important for smaller nanoparticles (for such nanoparticles scattering can be neglected) while for larger nanoparticles light scattering becomes dominant.14
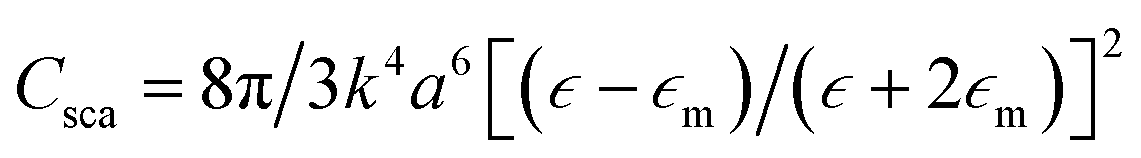
Surface plasmon resonance depends on many parameters of the nanoparticle, such as: its size and shape (Fig. 2 shows various nanoparticles practically used for SERS measurements in our group, see also ref. 15 and 16 for other examples), dielectric properties of the metal from which the nanoparticle is created, and the dielectric permittivity of the environment.17,18 Nanoparticles may have different plasmon modes (for more detailed description of the excitation of plasmons, the reader is referred to ref. 1 and 2). For small spherical nanoparticles only dipole plasmons are excited whereas for anisotropic nanoparticles also higher order plasmon modes can be excited.19 For example, UV-vis absorption spectrum of silver nanoprisms obtained by the plasmon-driven transformation revealed three peaks: at 340 nm (out of plane quadrupole resonance), 470 nm (in-plane quadrupole resonance), and 640 nm (in-plane dipole resonance).20 In the case of nanorods there are usually two separated absorption bands, one from the longitudinal excitation, and the second from the transverse excitation. Let us assume that the width of obtained nanorods is constant, and the length is changing. In this case the wavelength of the transverse mode is constant (for example 400 nm) and the wavelength of the longitudinal excitation is blue shifted when the length of the nanorod is decreasing. When both dimensions are the same (nanoparticles are spherical) only one peak in spectrum is present. In general nanorods may reveal many multipole modes: dipoles, tripoles, quadrupoles, pentapoles, hexapoles, and even heptapoles, the position of these modes strongly depends on the size and the aspect ratios (length/width) of the nanorod and is usually red shifted when the size of the nanorod is increasing.21
 | ||
| Fig. 2 TEM micrographs presenting Ag and Au nanoparticles with different geometry and shapes practically used as electromagnetic nanoresonators for SERS measurements: (a) quasi-spherical silver nanoparticles, (b) hollow spherical silver nanoparticles, (c) cubic silver nanoparticles, (d) gold–silver nanostars, (e) silver decahedral nanoparticles, (f) gold bipyramid. | ||
From the practical point of view it is important that in the case of spherical silver and gold nanoparticles it is very difficult to shift the position of the SPR band. For example, spherical Ag nanoparticles with diameter of 60 nm absorb at ca. 400 nm, while 5 nm nanoparticles have absorption band at 390 nm.22 Analogously, spherical gold nanoparticles with average diameter of 16 nm exhibit SPR band at 520 nm, when bigger 59 nm nanoparticles exhibit SPR at 534.5 nm.23
Interesting systems with tunable plasmonic properties are spherical hollow nanoparticles.24 For example, for spherical hollow gold nanoparticles it is possible to change the position of the plasmon band by more than 300 nm by changing the external and internal radiuses.24 Analysis of spectra of hollow gold structures showed that increasing of the outer diameter of nanoparticles and keeping constant thickness of the shell causes shift of the SPR band into longer wavelength (red shift).24 Increasing the thickness of the shell and keeping constant outer diameter induces shift of the SPR band into shorter wavelength (blue shift) – see Fig. 3.24 Also for silver nanoparticles there are significant differences between plasmonic properties of full (“solid”) and hollow nanoparticles with the same outer diameter. For example, UV-vis extinction spectrum of sol containing solid spherical silver nanoparticles with the diameter of about 60 nm reveals strong extinction peak at 400 nm, whereas in the case of spherical hollow nanoparticles (with the ratio of external and internal radiuses equal to 1.55) the plasmon peak is shifted to about 550 nm.25
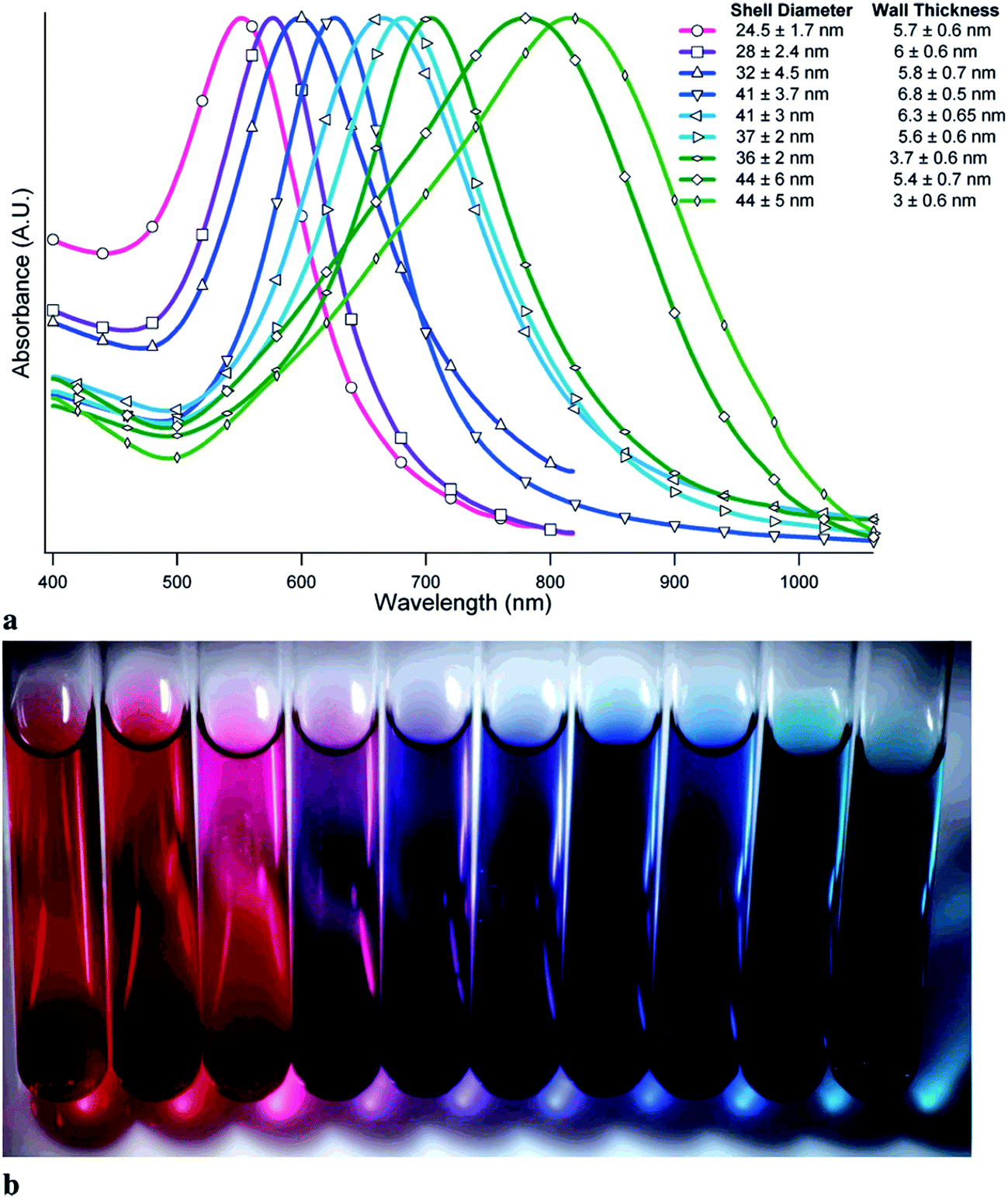 | ||
| Fig. 3 (a) UV-visible absorption spectra of samples of gold hollow nanospheres with varying diameters and wall thicknesses. (b) Visual appearance of various samples of gold hollow nanospheres. Reprinted with permission from ref. 24. Copyright 2006 American Chemical Society. | ||
3. Distribution of electromagnetic field around anisotropic “plasmonic” systems
When surface plasmons are excited in metal nanoparticles the intensity of the electromagnetic field at some places may be significantly larger than the intensity of the incident radiation. In the literature there are numerous publications about the distribution of the electromagnetic field around single “plasmonic” nanoparticles with different shape and around agglomerates of two or more “plasmonic” nanoparticles. In the case of an isolated nanoparticle the strongest field enhancement usually occurs on its sharp edges (see Fig. 1b).10,26 In the case of dimers and other agglomerates very large enhancement is observed when nanoparticles are relative close to each other – the strongest field is in the slit/slits between nanoparticles (see Fig. 1c).27–29 Places with especially high field enhancements are called “hot-spots”.30 In some cases it is possible to obtain enhancement of the intensity of the electromagnetic field even above 102 times.27 This property of some nanoparticles (especially Au or Ag ones) allows to carry out many surface-specific spectroscopic measurements such as: surface-enhanced Raman scattering (SERS), surface-enhanced hyper-Raman scattering (SEHRS), surface-enhanced infrared absorption (SEIRA) or metal-enhanced fluorescence (MEF).Distribution of electromagnetic field around nanoparticles strongly depends on the nanoparticle shape. As mentioned above the strongest enhancement of the electromagnetic field is usually observed on sharp edges of nanoparticles, however, in some cases, the strongest field enhancement appears in other places. For example, in 2003 Schatz's group analyzed the enhancement of the electric field outside silver nanoprism under various irradiation – see Fig. 1b.10 Simulations have been carried out for 16 nm thick silver right nanoprism with 100 nm edges of the bases. Irradiating light was polarized along the bisector of the base of the nanoprism. Schatz found that while irradiating nanoprism using radiation with the wavelength of 770 nm the highest field enhancement around nanoprism (over 22 times) is observed at the tips of the nanoprism. While irradiating using radiation with the wavelength of 460 nm the highest enhancement occurs at the middle of triangle's sides opposite to the triangle's edge, but this enhancement is much smaller than the enhancement at tips when using 770 nm radiation – see Fig. 1b.10 This behaviour is due to the possibility of the excitation in nanoprisms of various plasmonic modes. Nanoprisms of all kinds has four plasmon resonance peaks: out-of-plane quadrupole, in-plane quadrupole, in-plane dipole, and weak out-of-plane dipole. When 770 nm radiation is used for excitation of surface plasmons the distribution of electric field outside the nanoprism is similar to this obtained from dipole excitation in spherical nanoparticles while for excitation with 460 nm radiation the distribution of the electromagnetic field outside the nanoprism is similar to the distribution of the electromagnetic field due to the quadrupole excitation in spherical nanoparticle.10
It is worth to note that the maximum achievable field enhancement factor for anisotropic nanoparticles is significantly larger than for isotropic spherical nanoparticles. For example, the maximum enhancement factor for spherical Ag nanoparticles (with the diameter of 20 nm) irradiated using the light with the wavelength of 700 nm is calculated to be 14 times the applied field, whereas the maximal field enhancement factors for nanoprisms, nanorods, and nanospheroids irradiated using radiation with the same wavelength are estimated as equal to 59.1, 67, and 68.5, respectively.27
Very interesting plasmonic properties can be observed for dimers and larger agglomerates of metal nanoparticles. For example, for a dimer of spherical nanoparticles irradiated with the light polarized parallel to the interparticle axis two plasmon peaks are observed: one at 520 nm that is due to a dipole resonance, and the second (weaker) at 430 nm that is due to a quadrupole resonance. For these systems very large enhancement of the electric field may be also obtained in narrow slits between nanoparticles – even ca. 103 for dimers of spherical nanoobjects (see Fig. 1c).31 For dimer of triangular nanoprisms positioned on a common plane with tips directed toward each other and irradiated with light polarized parallel to the interparticle axis the theoretically calculated field enhancements are very large even when considering only dipole or only quadrupole resonance. Considering only the dipole resonance the field enhancement was calculated to be equal to 2.3 × 102, whereas considering only the quadrupole resonance the field enhancement was calculated to be equal 75.27 Such big enhancement factor for some places between nanoparticles (so called “hot spots”) enable, for example, single molecule measurements using SERS spectroscopy.32
Because of a large increase of the efficiency of many optical processes occurring for molecules being present in the “hot spots”, some groups developed techniques which allow on more efficient trapping of molecules of an analyte in such places.33 For example, Kumacheva et al. presented a method for trapping molecules of dyes at the “hot spots” formed between ends of the self-assembled gold nanorods.34 First, gold nanorods were modified with thiol-terminated polystyrene and then nanorods were self-assembled in the presence of analyte, which facilitates trapping of molecules of analyte (SERS dyes) at the “hot spots” formed between the ends of the gold nanorods.34 Another approach has been applied by Suh et al.35 In the first stage they formed dimers from gold nanoparticles surface-modified with DNA (some of DNA were dye-labelled).35 In the second stage, the interparticle gap between gold nanoparticles forming dimer was decreased by coating a silver shell on them, which significantly increase efficiency of electromagnetic hot spots between plasmonic metal nanoparticles.35
4. Selected analytical applications of gold and silver nanoparticles
4.1. Methods based on measurement of UV-vis spectra
Plasmonic optical properties of metal nanostructures depend on their geometrical parameters (shape and size) and dielectric properties of the metal and the surroundings (on the presence of other plasmonic object in the close proximity and on the dielectric properties of the environment). The frequency of the plasmon resonance is particularly strongly dependent on the shape of metal nanostructures and on the possible electromagnetic coupling with the localized surface plasmons in nearby other plasmonic objects. Therefore, for sols of plasmonic nanoparticles (like Ag and Au) even small change of the degree of agglomeration may be often easily detected by the observation of the change of their UV-vis spectrum. In some cases aggregation of nanoparticles may cause visible change of the colour of the solution. Very easy detection of agglomeration of the plasmonic nanoparticles induced by some compounds (analytes) resulted in the development of many analytical procedures based on the observation of the change of the extinction of the plasmonic sols. For example, Hupp et al. developed analytical method for determination of the concentrations of lead, cadmium, and mercury cations (in the concentration range between 50–200 μM) based on aggregation of gold nanoparticles.36 Addition of ions of heavy metals causes change of the colour of the solution from blue to red. Red to blue change in colour of the solution could be reversed by the addition of complexing agent like EDTA. For these measurements gold nanoparticles are functionalized by heavy-metal ion receptor, 11-mercaptoundecanoic acid (MUA). The reversible aggregation of nanoparticles are induced by coordination of cations of heavy atoms by MUA. This leads to decrease of negative charge on the nanoparticle surfaces causing the nanoparticles to repel each other much more weaker. Similar method based on the agglomeration of gold nanoparticles can be effectively used to measure concentrations of lithium ions in the 10–100 mM range and copper cations in the range 50–500 μM.37,38 Obare et al. showed that using smaller nanoparticles for this kind of analysis allows for achieving lower detection limit of the analyte.37 An analogous aggregation method has been developed by Fan et al. for detection of platinum ions.39 In this case gold nanoparticles have been modified by streptavidin aptamer. Using this method Fan et al. were able to determine colorimetrically concentration of Pt(II) ions in the range from 0.6 μM to 12.5 μM.39 Silver nanoparticles stabilized by starch can be used for colorimetric detection of copper(II) ions.40 This method has low detection limit, high selectivity, and could be used for real environmental water samples. In some cases it is even possible to use unmodified (bare) metal nanoparticles for such colorimetric measurements. For example, Shellaiah et al. observed aggregation of gold nanoparticles after addition of Cr3+ ions, which allows for using bare gold nanoparticles for rapid colorimetric detection of Cr3+ ions in aqueous media.41 The detection limit of chromium ions was determined as equal to ca. 13 nM and it was also found that the colorimetric detection of Cr3+ ions may be carried out in the presence of many other metal ions.41Very interesting and novel method for detection of Hg2+ ions has been recently described by Chen et al.42 This method is based not on agglomeration of plasmonic nanoparticles but on the change of their plasmonic properties during their growth. Chen et al. added analysed samples containing mercury ions into gold colloids prepared by the standard citrate reduction.42 Next, they added hydroxylamine (NH2OH) and chloroauric acid (HAuCl4). In this conditions hydroxylamine is able to reduce HAuCl4 into metallic gold. However, mercury ions are also reduced on the surface of gold nanoparticles. Depending on the amount of added mercury ions the growth of nanoparticles can be different. Gold nanoparticles with low mercury cover grow into spherical objects with slow growth rate which results in red-coloured solutions, while gold nanoparticles with high mercury coverage grow very quickly, nanoparticles with larger diameters are formed and colour is changing from red to blue. What is important other cations do not disturb this analysis. The detection limit of this method was estimated as equal to 0.26 nM.42
Determination of metal ions may be also based on de-aggregation of plasmonic metal nanoparticles. For example, He and Zhang developed sensitive method of colorimetric detection of Mn2+ ions based on de-aggregation of modified silver nanoparticles.43 He and Zhang prepared Ag nanoparticles modified with L-arginine. Addition of L-arginine causes efficient aggregation of Ag nanoparticles, which leads to fading of the colour of the Ag sol. However, addition of manganese ions can reverse aggregation of arginine-modified silver nanoparticles and induces appearance of intensive yellow colour. Therefore, by the measurement of the absorbance of the solutions at 390 nm one can determine the amount of manganese ions added (the higher adsorption, the higher concentration of the Mn2+ ions). This colorimetric method allows for determination of Mn2+ ions in the 300 nM to 60 μM concentration range.43 Moreover, such colorimetric detection of Mn2+ ions exhibits very high selectivity because addition of different metal ions like: K+, Ca2+, Na+, Mg2+, Ba2+, Al3+, Fe2+, Fe3+, Cu2+, Sr2+, Cs+, Pb2+, Ni2+, Co2+, Cr3+, and Hg2+ does not cause observable de-aggregation of arginine-modified silver nanoparticles.41 Similar method for detection of manganese ions has been proposed by Wu et al.44 In this case silver nanoparticles have been modified with tripolyphosphate (P3O105−). Due to high selectivity and good linearity (from 0.05 μM to 20 μM) this method can be used for detection of Mn2+ ions in real environmental water samples like lake and tap water.44
The other interesting colorimetric method utilising plasmonic nanoparticles is detection based on etching of the existing plasmonic nanoparticles that generates a colour change. For example, Gao et al. and Guo et al. demonstrated the possibility of using of the process of etching of triangular silver nanoprisms for the detection of various compounds.45–47 In the simplest approach Guo et al. directly detected the etching agent (hydrogen sulphide) by the analysis of the SPR band of silver nanoparticles – increase in the concentration of hydrogen sulphide (etching agent) results in a decrease in absorbance and red shift of the SPR peak of the silver nanoprisms as they are etched.47 Using non direct approach (the actual etching agent was H2O2, which was produced in a catalytic reaction from the dissolved molecular oxygen) Gao et al. was even able to detect target DNA at concentrations as low as 6.0 fM.46
In addition to mentioned above colorimetric detection of DNA, methods utilising plasmonic nanoparticles and based on various processes influencing plasmon resonance (usually aggregation of plasmonic nanoparticles or changing of the electric permittivity of the environment) are often used for detection of organic compounds, even such as proteins and nucleic acids.48–52 For example, Feng et al. described sensitive method for phosgene detection based on aggregation of gold nanoparticles.52 In this case gold nanoparticles are capped by L-cysteine due to the strong affinity of the –SH group to gold surface. L-Cysteine molecule is attached to the gold surface via S–Au bond. Gold nanoparticles exhibit strong absorption peak at 520 nm, but after addition of phosgene their colour changes from wine-red to purple-blue (see Fig. 4).52 It is caused by the formation of new absorption peak at 640 nm, which is ascribed to the agglomerates of gold nanoparticles (see Fig. 4).52 Additionally, non-modified gold nanoparticles have been also tested for phosgene detection. Modified and non-modified gold nanoparticles exhibits strong absorption peak at 520 nm, which shows that cysteine molecule hardly affects the spectroscopic properties of Au nanoparticles. Subsequent addition of phosgene in a case of non-modified nanoparticles did not cause any obvious colour change. It means that the detection of phosgene molecules is only possible due to the modification of the surface of gold nanoparticles by chemisorbed cysteine. This method provides a linear response in the concentration range of phosgene between 5 and 50 μM.52
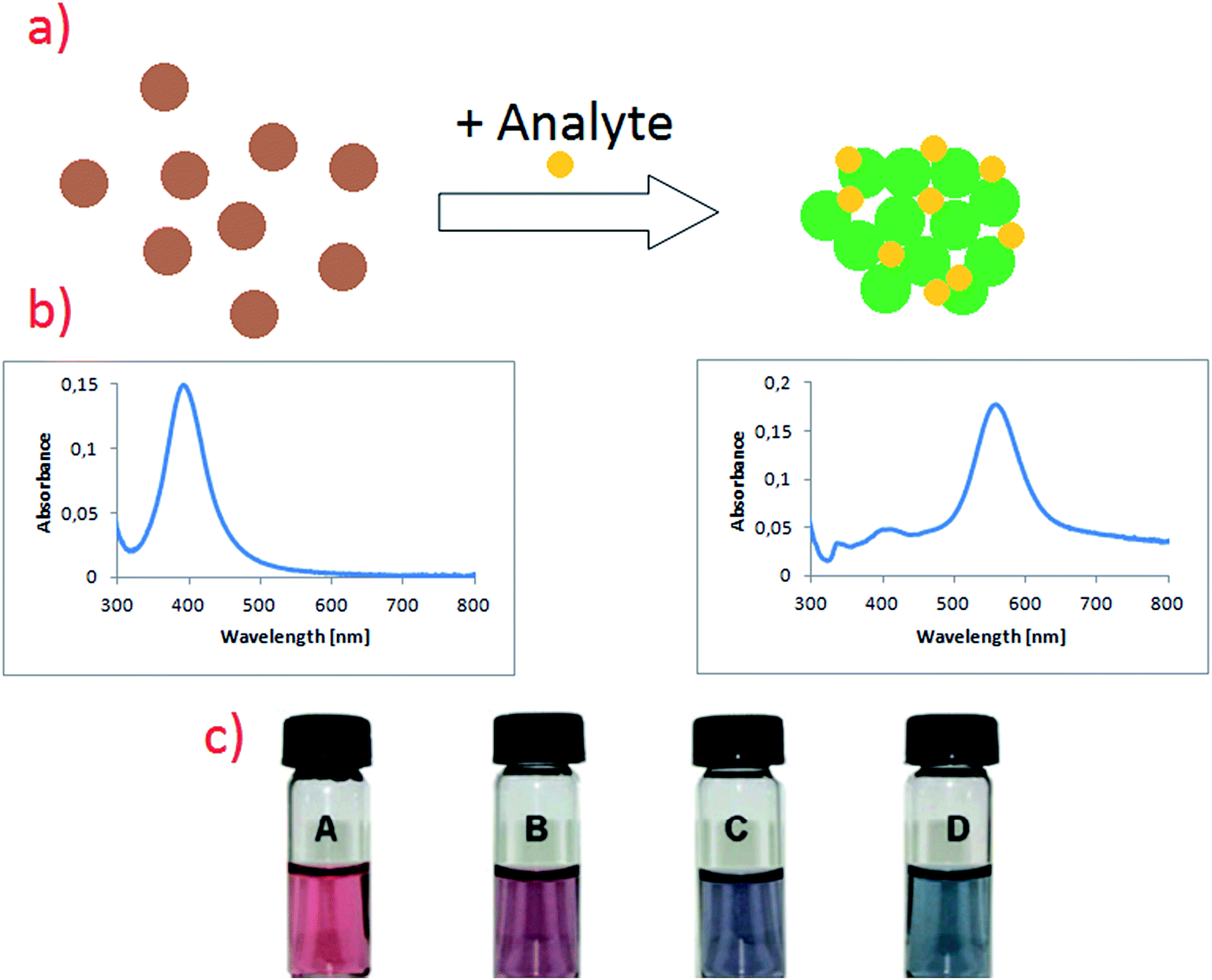 | ||
| Fig. 4 (a) Schematic illustration showing the change in noble metal nanoparticles solutions after addition of analyte. (b) Respectively UV-vis spectra of sol of noble metal nanoparticles before and after addition of analyte. (c) Colour change in L-cysteine modified gold nanoparticles after exposure to different concentrations of phosgene in air: (A) 0, (B) 1.4, (C) 1.9, (D) 2.5 mg ml−1. Reprinted with permission from ref. 52. Copyright 2010 The Royal Society of Chemistry. | ||
Interesting aggregation technique has been also applied by Guo et al. for the detection of target DNA (see Fig. 5).47 In this strategy, unlike the conventional colorimetric methods based on formation of large nanoparticle aggregates, dimers of plasmonic nanoparticles are selectively formed upon target binding, which results in significantly improved long-term stability and a more than 2 orders of magnitude wider dynamic range of detection than that of the conventional colorimetric sensors.47 Moreover, significant decreasing of the interparticle gap through the formation of a Y-shaped DNA duplex (see Fig. 5) enables to increase the limit of detection by 104 times.47
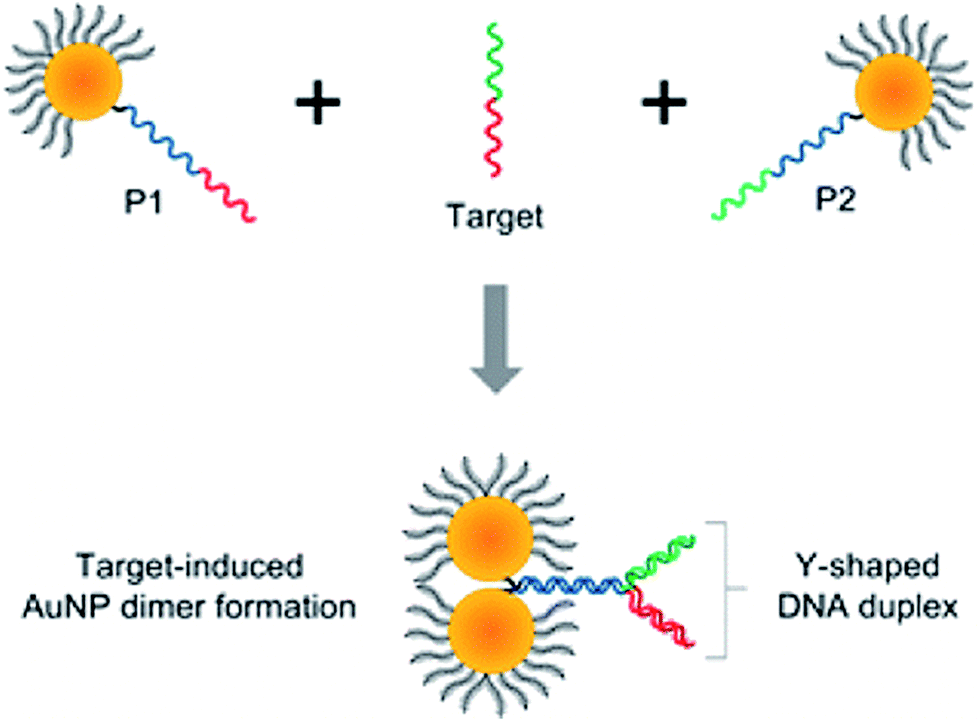 | ||
| Fig. 5 Schematic representation of the formation of nanoparticle dimers upon target DNA binding using the asymmetrically modified gold nanoparticles. Reprinted with permission from ref. 47. Copyright 2013 American Chemical Society. | ||
Plasmonic sensors for detection of various species may also utilise the influence on the plasmon resonance of the electric permittivity of the environment of the plasmonic structures. For example, Ding et al. fabricated a triangular silver nanoparticle array, to which protein p53 antibody has been chemically attached.53 Then, such modified nanoparticle arrays have been incubated with various serums containing protein p53.53 In healthy humans the level of protein p53 is ca. 16 pg ml−1 whereas in a case of patients with some cancers diseases concentration of p53 is elevated to ca. 60 pg ml−1. Ding et al. found that difference in the position of the plasmon peak (due to the adsorption of the molecules of analyte on the plasmonic structures) for sensor soaked in serum from a patient and a control serum is equal to ca. 18 nm, which is significantly larger than differences observed for the control group (<5 nm).53
Colorimetric methods may be also applied for detection of larger bio-objects, such as cancer cells (see Fig. 6).54 This method relies on aptamer-conjugated gold nanoparticles. Aptamers are oligonucleotide strands that bind to their targets with high affinity and selectivity. For example, Medley et al. conjugated aptamer sequences to 20 nm gold nanoparticles via thiol functional groups.54 In this experiment five samples with different concentration of nanoparticles with target and control cells was prepared. Absorbance (especially at ca. 680 nm) of Au nanoparticles with target cells (due to their agglomeration) is significantly higher than in the case of the same amount of nanoparticles with control cells (see Fig. 6). These results show that Au nanoparticles are binding selectively to the target cells which causes increase in the absorption and scattering of the solution (see Fig. 6).54
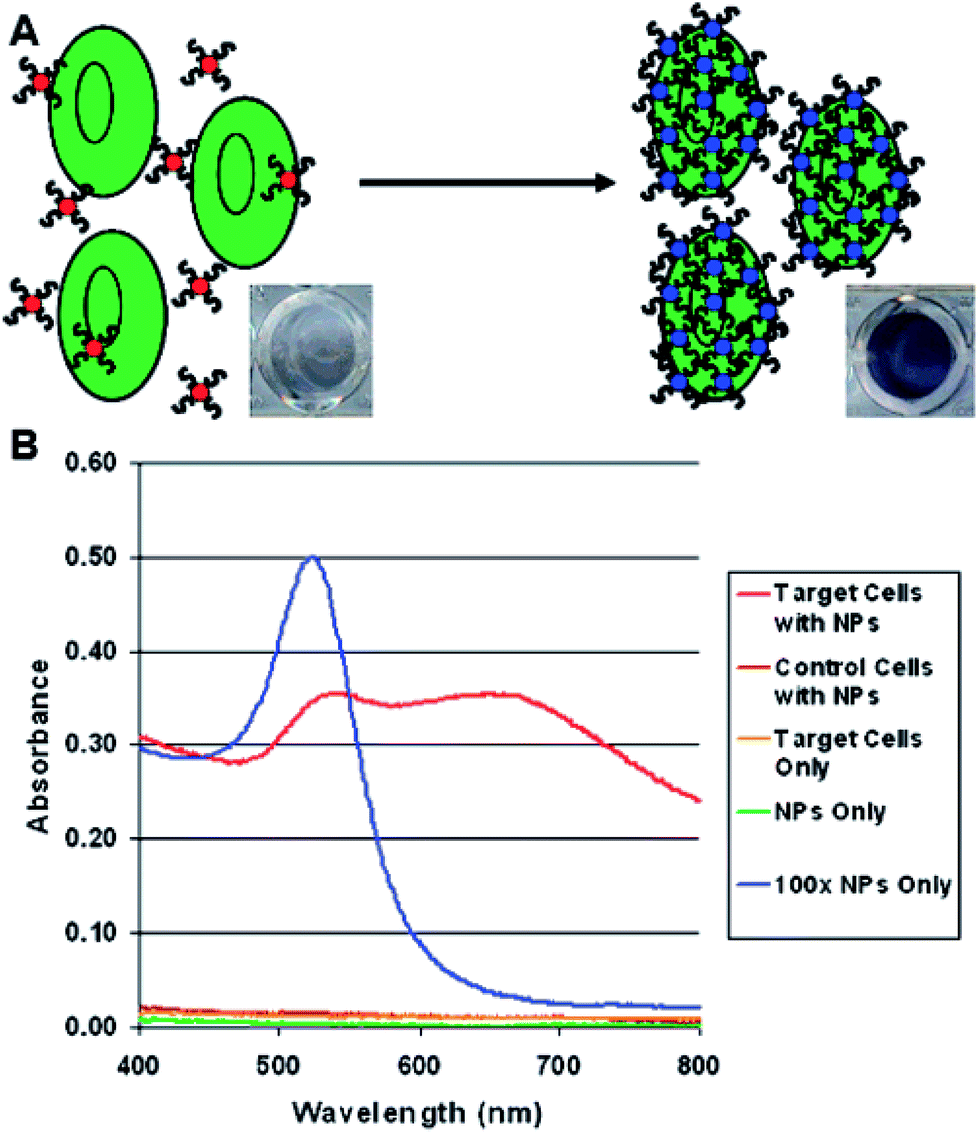 | ||
| Fig. 6 (A) Schematic representation of the colorimetric detection of cancer cells using aptamer-conjugated gold nanoparticles. (B) Plots depicting the absorption spectra obtained for various samples analyzed using aptamer-conjugated gold nanoparticles. The spectra illustrate the differences in the spectral characteristics observed after the aptamer-conjugated gold nanoparticles bind to the target cells. Reprinted with permission from ref. 54. Copyright 2008 American Chemical Society. | ||
4.2. Surface-enhanced Raman scattering
As mentioned in the Introduction the local enhancement of the intensity of the electromagnetic field in the proximity of plasmonic nanoparticles may cause significant increase of the efficiency of several optical processes, which are commonly applied in the chemical analysis, such as fluorescence, infrared absorption, and Raman scattering (which includes also Raman optical activity, hyper-Raman scattering and coherent anti-Stokes Raman scattering). From the practical point of view the most important application of plasmonic nanoparticles is increasing of the efficiency of Raman scattering, and therefore, we will start characterization of spectroscopic methods utilizing metal nanoresonators from description of the applications of plasmonic nanoparticles in this field of spectroscopy.Raman scattering relies on an inelastic photon scattering. In contrast to elastic Rayleigh scattering, inelastically scattered photons have different energy from the incident ones. For many years Raman spectroscopy has not been considered a useful analytical tool because of very low efficiency of the “normal” Raman scattering. Typical total Raman scattering cross-section is only ca. 10−29 cm2 per molecule, whereas typical cross-sections for absorption in ultraviolet and infrared are ca. 10−18 and 10−21 cm2 per molecule, respectively.55 In 1974 Fleischmann et al. reported a significant growth of the Raman signal measured from pyridine adsorbed on electrochemically roughened (nanostructured) silver substrate.56 They interpreted this effect as a result of an increase of the surface area of electrochemically roughened silver electrode and hence adsorption of a significantly larger number of pyridine molecules. Three years later two independent research groups found true reasons for the very large growth of the intensity of the Raman signal observed by Fleischmann et al.56 Jeanmaire and van Duyne, and Albrecht and Creighton connected very strong Raman signal observed for pyridine adsorbed on nanostructured Ag surface with the increase of the efficiency of Raman scattering.57,58 The observed effect was called SERS (surface-enhanced Raman scattering). As mentioned above, the increase of the efficiency of Raman scattering for molecules being in the close proximity to the plasmonic nanoparticles is mainly due to the increase of the intensity of the electromagnetic field. Kerker et al. found that for molecules at the surface of metallic spheres the electromagnetic SERS enhancement (EF) can be calculated from the following simple expression:59
| EF = 5 × |(1 + 2 × gd(ωo)) × (1 + 2 × gd(ω))|2 |
ωo and ω are the wavenumbers of the incident and Raman radiations, respectively,
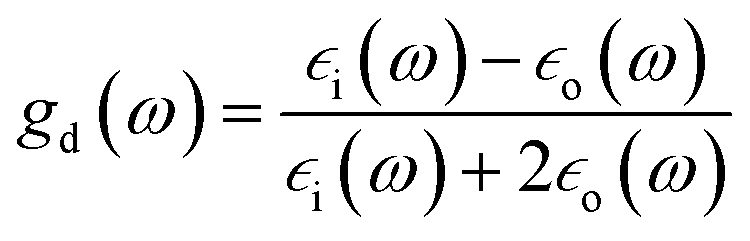
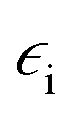 is the electric permittivity of the metal and
is the electric permittivity of the metal and 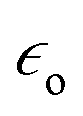 is the electric permittivity of nanoparticle's environment. It can be shown that for Raman bands with small so-called Raman shift (it means difference between ωo and ω) EF is roughly proportional to the fourth power of the field enhancement.55,60–62
is the electric permittivity of nanoparticle's environment. It can be shown that for Raman bands with small so-called Raman shift (it means difference between ωo and ω) EF is roughly proportional to the fourth power of the field enhancement.55,60–62
In addition to the increase of the efficiency of Raman scattering due to the local enhancement of the intensity of the electromagnetic field, the enhancement of the Raman signal for molecules adsorbed on metal nanoclusters is also due to the so-called charge transfer mechanism that resembles the ordinary resonance Raman process. The largest enhancement in so-called charge transfer mechanism is observed when the energy of photons in the incident beam is fitted to the difference between energies of Fermi level in the metal and an unoccupied molecular orbital of absorbed molecule or between the highest occupied molecular orbital of absorbed molecule and Fermi level in metal. By the modification of the potential in the electrochemical systems it is possible to control the energy of Fermi level in metal, and hence to influence the value of the enhancement due to the charge transfer mechanism.63–65
Utilising both SERS and resonance Raman effects, the Raman scattering cross-section can be sometimes increased even to 2 × 10−14 cm2 per molecule (i.e. by about 15 orders of magnitude in comparison to the cross-section of normal Raman scattering) making possible observation of SERS spectra even of a single molecule.66–68 Although, as far as we known, single molecule SERS spectroscopy has not been used for practical analysis due to the poor reproducibility of the measured signal (for example see Fig. 7, which shows single molecule SERS spectra of adenine measured from the solution with the concentration of 10−12 M),69 its strong fluctuation and possibility of the complete disappearance of the signal due to the photodecomposition of the investigated molecule, SERS is actually one of the most sensitive analytical tools with the limit of detection for some analytes of the order of 10−18 mol dm−3.70 The other problem connected with very strong local enhancement of the intensity of the electromagnetic field in so-called “hot spots” is appearing in the measured SERS spectra of a background from “carbon impurities” (carbon clusters are produced by the photocatalytic decomposition of various organic compounds in SERS “hot spots”). The more detailed analysis of this problem may be found in ref. 71 and 72.
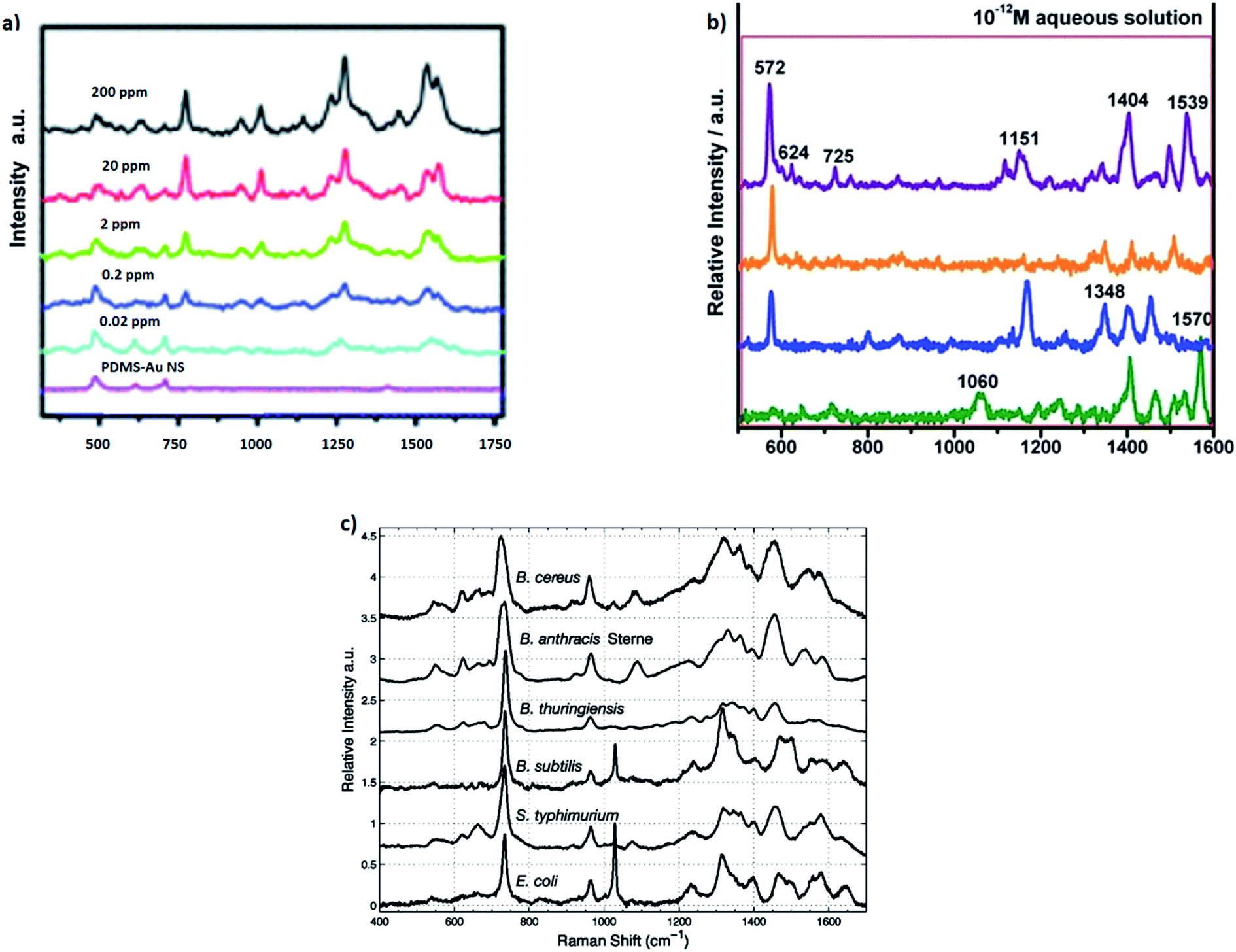 | ||
| Fig. 7 (a) SERS spectra of the different concentrations of thiabendazole deposited on apple fruit skin with gold nanostars used as optical nanoresonators. Reprinted with permission from ref. 82. Copyright 2014 Royal Society of Chemistry. (b) Single molecule SERS spectra of adenine measured from the solution with the concentration of 10−12 M. Reprinted with permission from ref. 69. Copyright 2014 Royal Society of Chemistry. (c) SERS spectra of different Gram-positive and Gram-negative bacteria. Reprinted with permission from ref. 87. Copyright 2005 American Chemical Society. | ||
The simplest SERS analytical procedures are based on the measurement of the SERS signal of the analyte. For example, some anthraquinone dyes can be detected by recording of the SERS spectrum from highly SERS-active substrate immersed in the analysed solution (SERS substrate can be prepared, for example, by evaporating a layer of silver nanoparticles on the surface of Al2O3 or by deposition of a layer of silver nanoparticles directly on pieces of the filter paper). The limit of detection of alizarin (one of the studied dyes) using this simple SERS measurement was estimated to be 7 × 10−15 g.73 In 2015 Lian et al. synthesised very efficient SERS substrates by deposition of Au nanoparticles on MoO3 nanowires.74 Using such SERS substrates Lian et al. were able to detect melamine in the solution with the concentration of 0.08 nM (0.1 ppb).74
Solid samples may be analysed using SERS spectroscopy by covering of their surfaces by the plasmonic metal nanoparticles (electromagnetic nanoresonators) and subsequent recording of the Raman spectra. Such approach has been applied, for example, to detect dyes in textiles.75 Using silver nanoresonators Leona et al. analysed fibre from a sixteenth-century Dutch tapestry. In the recorded Raman spectra of such sample Leona et al. identified bands characteristic to alizarine, and hence, proved the presence of this dye in the analysed sample.75 Recorded SERS spectra are very characteristic of scattering molecules, and therefore this technique is recommended for atypical samples, for which unexpected compounds present in the sample could seriously interfere with the result of the analysis carried out with many standard analytical methods, e.g., electrochemical analytical techniques or UV-vis absorption spectroscopy.
In some cases plasmonic metal nanoparticles for surface Raman measurements are covered by ultrathin layer of SiO2, MnO2 or Al2O3 separating metal nanoparticles from direct contact with the probed material and keeping them from agglomerating.25,76–80 This Raman technique for surface analysis is called SHINERS (shell-isolated nanoparticle-enhanced Raman spectroscopy).25,76–80 Oxide layers do not exhibit plasmonic properties, however, if they are very thin and used material is transparent, they have not damped significantly surface electromagnetic enhancement (see Fig. 8). Fig. 8a and b shows transmission electron microscopy (TEM) micrographs of example nanoresonators used for SHINERS measurement (solid and hollow silver nanoparticles covered by a SiO2 layer).
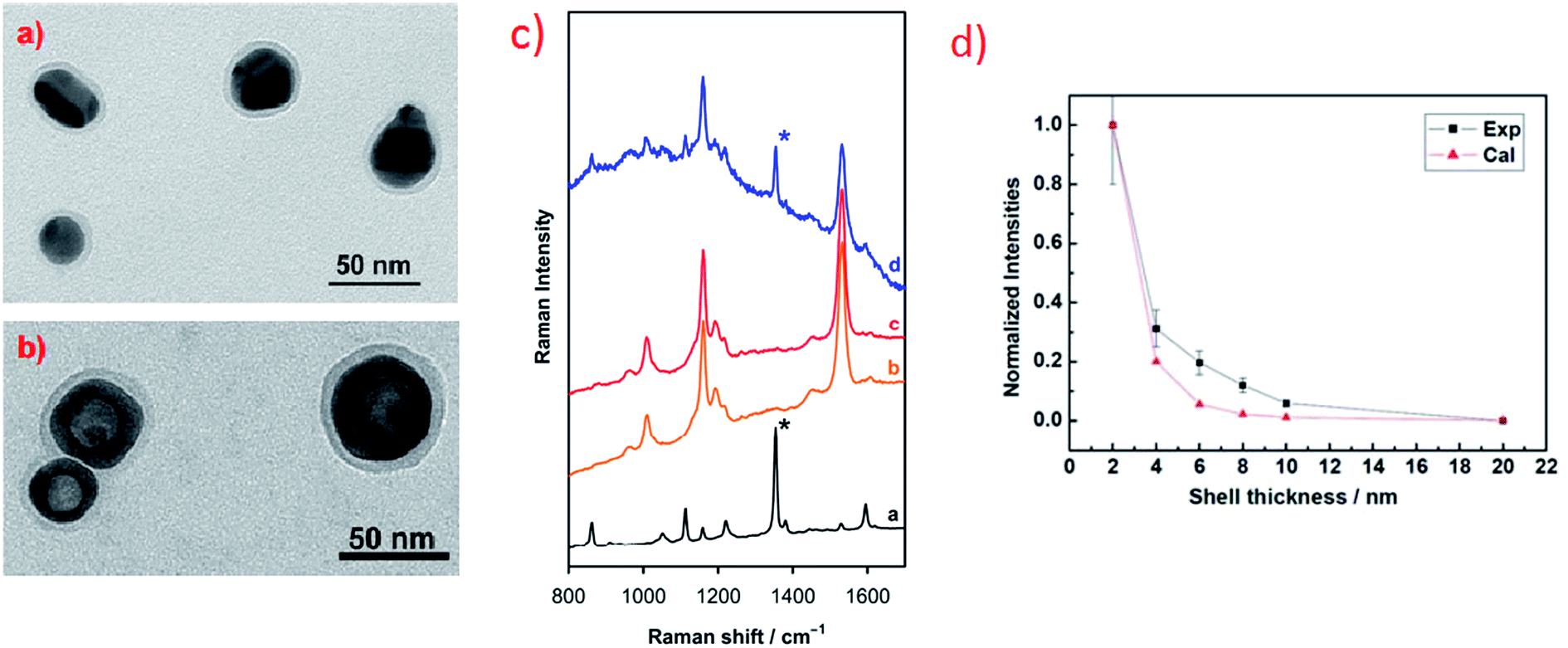 | ||
| Fig. 8 (a) and (b) TEM micrographs of example nanoresonators for SHINERS measurement: (a) Ag@SiO2 nanoparticles, (b) hollow-Ag@SiO2 nanoparticles. Reprinted with permission from ref. 25. Copyright 2015 American Chemical Society. (c) Raman spectra of: [a] methyl parathion, [b] skin of the orange fruit, [c] skin of the orange fruit contaminated by methyl parathion, [d] skin of the orange fruit contaminated by methyl parathion and covered with Ag@MnO2 nanoparticles. Reprinted from ref. 80 with permission from Elsevier. Copyright 2016 Elsevier. (d) Experimental and theoretical calculations of the influence of the width of silica layer deposited on nanoresonators on the intensity of Raman signal. Reprinted with permission from ref. 76. Copyright 2010 Nature Publishing Group. | ||
Fig. 8c shows an example result of SHINERS analysis: detection of traces of methyl parathion (which is an efficient insecticide) on the surface of an orange fruit. As can be seen in Fig. 8c there are no significant differences between Raman spectra recorded from the surface of the clean orange fruit and the surface of the orange fruit contaminated with methyl parathion.80 However, when SHINERS nanoresonators are deposited on the surface of the orange fruit contaminated with methyl parathion, the Raman band due to methyl parathion at 1350 cm−1 (marked with asterisk in Fig. 8c) can be clearly noticed in the measured spectra.80
Interesting SERS method useful also for detection of traces of pesticides on the surface of fruits has been recently developed by Singh et al.81 This group has synthesized large area flexible SERS substrates by embedding Ag nanorods into the polydimethylsiloxane polymer. These flexible SERS substrates may be used, for example, for direct extracting of trace amount (∼10−9 g cm−2) of thiram pesticide directly from fruit peels via simple “paste and peel off” method. The SERS detection of thiram pesticide is based on measurement of the intensity of the Raman band at 1386 cm−1, which is due to the vibration of methyl group in thiram molecule. This analytical method allows for detection of surface concentrations of thiram even 3 orders of magnitude lower than the level currently permissible in farming.81 In another study Liz-Marzan et al. showed utilization of gold nanostars (efficient plasmonic nanoparticles) for SERS detection of thiabendazole pesticide on the surface of apple skin.82 As can be seen in Fig. 7a, Liz-Marzan et al. were able to obtain very small detection limit of thiabendazole (0.02 ppm).82
SERS spectroscopy may be also applied for detection of relatively complex biological agents, such as viruses, fungi, bacteria, and bacterial spores. Rapid and reliable detection of some of these bioagents is very important in order to detect a terrorist attack with biological warfare. Detection of spores of Bacillus anthracis, which are dangerous pathogens for the disease anthrax, is particularly important from the practical point of view. The procedures of detection of this pathogen utilizing SERS spectroscopy have been developed by several groups.83–86 The SERS detection of Bacillus anthracis spores is usually based on the detection of calcium dipicolinate, which exists in large quantity (about 10% of the spore's dry weight) in the protective layer of the spore.86,87
Covering the surface of many microorganisms (bacteria, viruses) with plasmonic metal nanoparticles allows for measurements of so-called whole-organism SERS spectra. Due to the specific enhancement mechanism, the recorded SERS spectra are dominated by the contribution from compounds that are near the surface of the metal clusters which means that they are in the outermost parts of the microorganisms (the cell wall of a bacterium or the shell of a virus). Since bacterial cell walls differ more between various species than the composition of the bacteria's bulk, SERS spectra of bacteria exhibit usually greater differentiation for various species than their standard Raman spectra.87,88 Some examples of identification of various bacteria based on their SERS spectra can be found in ref. 87–95. For example, Premasiri et al. used SERS spectroscopy to study cell walls of Gram-positive and Gram-negative bacteria.87 This method enabled to distinguish both bacterial species and strains from SERS spectra of their nanoparticle-covered cell walls (see Fig. 7c).87 It means that in some cases SERS can be considered as a label-free whole-organism fingerprinting technique.
SERS spectroscopy may be also used for detection of some enzymes. Such analysis is usually based on SERS detection of products of the enzyme-catalyzed reactions. For example, Ruan et al. proposed detection of enzyme alkaline phosphatase by the detection of indigo dye produced in the catalytic reaction from 5-bromo-4-chloro-3-indolyl.96 The detection limit of alkaline phosphatase using this method was estimated as equal to 10−15 M.96
Important analytical application of SERS spectroscopy is measurement of pH using pH nanosensors. Typical SERS pH sensors are composed of plasmonic metallic nanostructures with molecules of so-called Raman reporters (molecules that significantly change their SERS spectrum with the change of the pH of the surrounding solution) immobilised on their surfaces. Due to their nanoscale size, such sensors can be successfully introduced into cells of living organisms and track the distribution of the pH inside the cell. In the interior of the eukaryotic cell there are various structures (organelles) having different functions. Therefore, the distribution of the concentration of H+ ions is not homogeneous, and hence, measurements of local pH at various places in the interior of the living cell is important. Interesting pH sensor was developed by Huser et al.97 As a molecule sensitive to changes in pH 4-mercaptobenzoic acid was used. Binding of these molecules to the surface of gold/silver nanoparticles is realized via their thiol groups. The determination of pH is realized by the measurement of the relative intensity of the SERS band at 1430 cm−1 due to the vibration of the carboxyl group. As one can expect the relative intensity of this band (calculated versus the intensity of the band at 1590 cm−1 due to the vibration of the aromatic ring) increases with increasing of pH. Kneipp and co-workers introduced this pH sensor to the live fibroblast NIH/3T3 cell.98 As a result, they obtained a map of the distribution of H+ ions in the living cell (see Fig. 9).98 SERS pH nanosensors may be also based on other Raman reporters such as 4-aminothiophenol or 4-ethynylpyridine.99,100
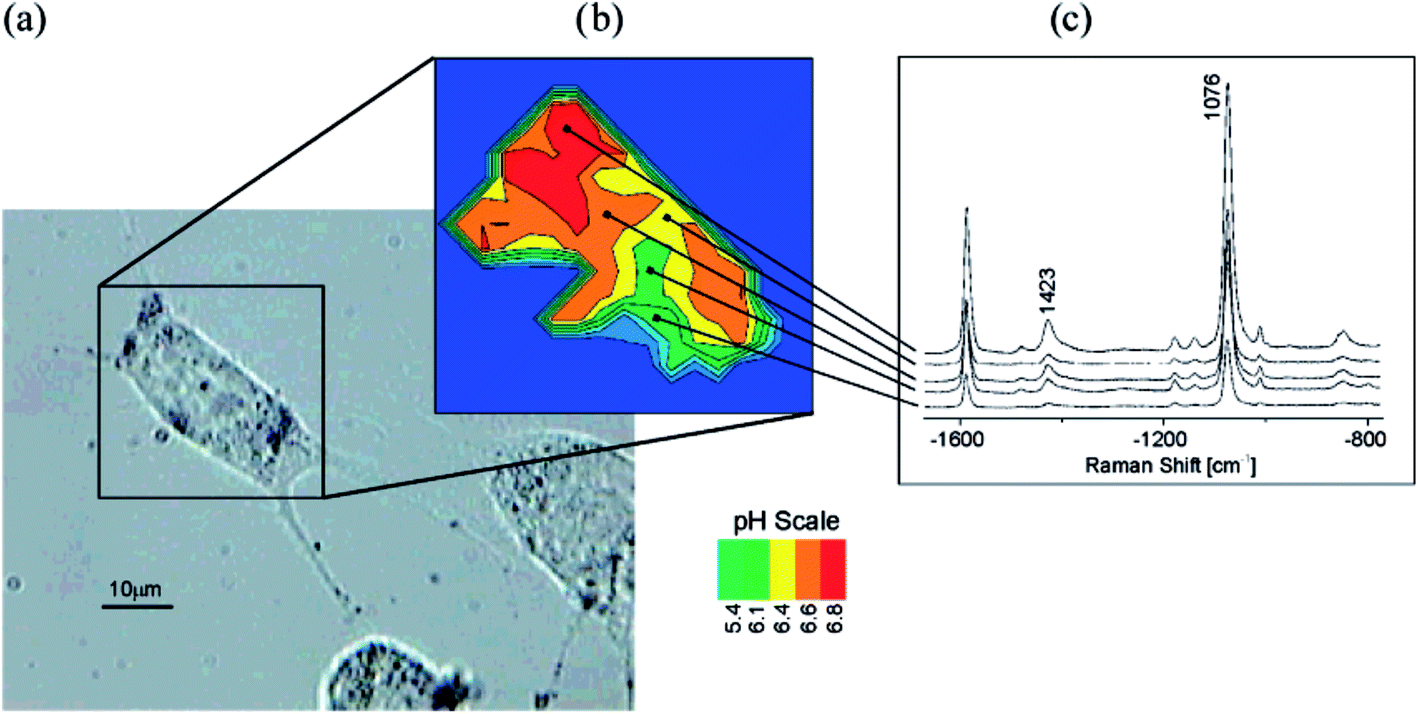 | ||
| Fig. 9 Probing and imaging pH values in individual live cells using a SERS nanosensor. (a) Photomicrograph of an NIH/3T3 cell after incubation with gold nanoparticles functionalized with 4-mercaptobenzoic acid. (b) pH map of the cell displayed as false colour plot of the ratios of the intensity of SERS bands at 1423 and 1076 cm−1. (c) Typical SERS spectra collected in the endosomal compartments with different pH. Reprinted with permission from ref. 98. Copyright 2007 American Chemical Society. | ||
Similar SERS nanosensors could be also used for detection of other cations. For example, Piotrowski and Bukowska showed that silver nanoparticles covered by the monolayer of mercaptoethanesulfonate can serve as a sensor for many metal cations.101 Sulfonate groups of mercaptoethanesulfonate may form contact-ion pairs with metal cations, and therefore νs(SO3−) band can be split into two components, one at a higher wavenumber (1065 cm−1) for sulfonic groups interacting directly with co-adsorbed cations and one at a lower wavenumber (1040 cm−1) for sulfonic groups which are not involved in the formation of direct bonds with co-adsorbed cations. Piotrowski and Bukowska demonstrated a big potential of such functionalized Ag nanoparticles for detection of a great variety of cations, including alkaline and alkaline earth metal cations, down to 10−8 M in the case of Ca2+.101 SERS sensor developed by Chen et al. for detection of As3+ cations utilises other mechanism. In this case addition of As3+ cations induces aggregation of glutathione-modified silver nanoparticles which results in increase of the intensity of the measured SERS spectra.102 Aggregation of glutathione-modified silver nanoparticles is due to the high affinity of glutathione to As3+ ions, each As3+ ion could bind by the As–O bond to three glutathione molecules (also attached to different Ag nanoparticles) and hence O–As–O bridges can bond various silver nanoparticles.102 This SERS As3+ sensor has good linear response in a wide concentration range (4–300 ppb), low detection limit (0.76 ppb), and has high selectivity – practically only As3+ ions could induce the aggregation of glutathione-modified silver nanoparticles.102 Similar aggregation-based SERS sensor has been also developed for detection of cadmium ions by Dasary et al.103 In this case Cd2+ induces aggregation of alizarin-modified gold nanoparticles (alizarin is a very efficient Raman scatterer) which were further modified with 3-mercaptopropionic acid and 2,6-pyridinedicarboxylic acid.103 As in the previous case, aggregation of nanoparticles significantly increases the intensity of measured SERS signal of alizarin (Raman reporter).103 Such SERS sensor could detect traces of Cd2+ ions even at 10 ppt level.103
Very promising plasmonic method which may be used for chemical analysis is surface enhanced Raman optical activity (SEROA). Raman optical activity (ROA) is a phenomenon of differential scattering of right and left circularly polarized light by chiral molecules (ROA is also manifested as a small circularly polarized component in the scattered light when the incident radiation is linearly polarized). Standard ROA spectrum is very weak, usually 3–5 orders of magnitude weaker than those of the parent Raman scattering.104 Therefore, increasing of the intensity of the ROA signal by the utilising of metal nanoresonators have been tried by many groups. Since ROA and SEROA spectra collected for L- and D-enantiomers are almost mirror images of each other (when at a given frequency the D-isomer exhibits positive peak the L-form should exhibit negative peak) those analytical techniques allow for distinction of various isomers.
One of the first reports about measurement of the SEROA spectra were published by Kneipp et al.105 They recorded SEROA spectra of adenine (with the concentration of 8 × 10−5 M) adsorbed on silver colloid. This report states that using SEROA method allows for considerably shortening data collection time and decreasing concentration of the target molecules.105 In 2010 Osinska et al. measured SEROA spectra for L- and D-cysteine and proved for the first time that SEROA spectra for L- and D-enantiomers are actually mirror images of each other.106 In 2011 Pour et al. showed similar effect for SEROA spectra of D- and L-ribose adsorbed on silver nanoparticles.107 SEROA spectroscopy is still not fully understood, however, as mentioned above, this is a potentially sensitive method for detection of chiral molecule like amino acids, nucleotides, proteins or chiral drugs molecules.
4.3. Surface-enhanced infrared absorption
Surface enhanced infrared absorption (SEIRA) was described for the first time by Hartstein et al. in 1980.108 They found a significant enhancement of the absorption signal of an organic film covered by gold or silver nanoparticles on top. In the next contribution Hartstein reported SEIRA measurement in a configuration when the organic sample was placed as a thin film over the metal surface.108 In SEIRA measurements it is possible to obtain enhancement factor even equal to 104 (in comparison to the standard IR absorption measurements). Such large SEIRA enhancement factor is due to the combination of the electric field enhancement at the metal surface and the specific chemical interactions. A particularly important interaction is a charge transfer between analyte molecule and the metal surface, which leads to enhanced vibrational polarizability of the molecules adsorbed directly onto the metal surface.109In IR absorption spectroscopy intensities of the vibrational modes are proportional to the square of the E × μ′, where E is the electromagnetic field and μ′ is the transition dipole moment oriented vertically to the electromagnetic field.110 In SEIRA phenomenon electromagnetic field that interacts with the molecule is much stronger than the field of the incident IR beam, and as one can deduct from the mentioned above dependence the increase of the efficiency of absorption is proportional the second power of the field enhancement (in SERS experiments the increase of the intensity of measured signal is proportional to the forth power of the field enhancement). Same as in SERS measurements the field enhancement is caused by the surface plasmon resonance taking place on the metal surface (oscillating electrons create additional electromagnetic field).
The orientation of adsorbed molecules versus the metal surface plays an important role in SEIRA spectroscopy. The strongest SEIRA absorption is observed when the transition dipole moment of an adsorbed molecule is oriented vertically with the electric field vector around the metal surface. Such orientation of molecules causes much stronger IR absorption then when molecules are arranged in a chaotic manner (e.g. during physisorption or when they are in solution). It also means that the strongest SEIRA spectra are recorded for ordered monolayers formed by chemisorbed molecules.
SEIRA spectroscopy is mainly used in electrochemical measurements. For example, SEIRA measurements help to understand the mechanism of some electrocatalytic reactions, the mechanism of some processes of metal corrosion, to determine the orientation of adsorbed molecules and the structure of formed monolayers.111–114
SEIRA spectroscopy is also successfully used in chemical analysis. In some cases this kind of spectroscopy provides fast and repetitive measurement with low-level of the detection.115,116 For example, Nishikawa et al. reported SEIRA analysis of samples containing nanograms quantity of triphenyl phosphate.115 A useful SEIRA spectrum could be obtained even when the amount of deposited analyte is only equal to ca. 25 ng cm−2.115 Prati et al. analysed extract from dyed fibers and after mixing with aliquot of gold nanoparticles colloidal solution obtained by laser ablation they were able to detect acid orange on the basis of the measured SEIRS spectra.117,118
In biomedical applications surface enhanced infrared absorption spectroscopy can be used for example for detection of rat tumour cells.119 This method is based on measurement of the SEIRA spectra of nucleic acids on gold substrate. SEIRA spectra showed that secondary and tertiary structures of nucleic acids from cancer cells are significantly different than structure of nucleic acids from normal cells.120 Thus, pathologically changed cells have significantly different IR spectra than healthy (cancer-free) cells. DNA from non-pathological cells can be described as rigid structure, when structure of DNA from cancer cells seems to be flexible. As mentioned above this change in DNA structure can be monitored by SEIRA spectroscopy. Detection of the difference between the healthy and cancer cells may be also based on the analysis of SEIRA spectra of RNA. Moreover, the SEIRA spectra of RNA from tumor cells are more sensitive to the grade of tumor malignancy than the SEIRA spectra of DNA.119
It is also possible using SEIRA spectroscopy to carry out analysis based on antibody–antigen interactions and molecular and protein recognition.121,122 For example, in order to prepare SEIRA sensor for Salmonella cell Salmonella antibodies (anti-SAL) were immobilized onto a thin gold layer.121 SEIRA spectra of anti-SAL sensor with and without deposited cells of Salmonella are significantly different. SEIRA spectrum of anti-SAL deposited on gold layer exhibits two peaks at 1085 cm−1 and 990 cm−1, while in the presence of Salmonella bacteria the measured spectrum exhibits new strong band at 1045 cm−1.121 This new band is connected with P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of phospholipids in the bacteria cell wall. Presence of this band can be a clear indication of the presence of Salmonella cell in the solution. Hunter-Cevera et al. showed possibility of detection of different Gram-negative bacteria cells: Pseudomonas putida and Pseudomonas fluorescens by SEIRA measurements on the gold-coated surface of vesicular basalt.123 They described three characteristic regions in SEIRA spectra: at ca. 1740 cm−1 connected with phospholipids in the outer membrane of Gram-negative bacteria, 1650 cm−1 due to the bacterial protein amide I envelope and 1550 cm−1 connected with bacterial protein amide II envelope.
O stretching vibration of phospholipids in the bacteria cell wall. Presence of this band can be a clear indication of the presence of Salmonella cell in the solution. Hunter-Cevera et al. showed possibility of detection of different Gram-negative bacteria cells: Pseudomonas putida and Pseudomonas fluorescens by SEIRA measurements on the gold-coated surface of vesicular basalt.123 They described three characteristic regions in SEIRA spectra: at ca. 1740 cm−1 connected with phospholipids in the outer membrane of Gram-negative bacteria, 1650 cm−1 due to the bacterial protein amide I envelope and 1550 cm−1 connected with bacterial protein amide II envelope.
Interesting nanoresonators for SEIRA measurements (Fe3O4/Au nanocomposites) have been recently developed by Cai et al.124 Under strong magnetic field, the superparamagnetic Fe3O4/Au nanoparticles are highly concentrated, leading to the increase number of SEIRA “active sites” between Au nanoparticles and increase of the SEIRA enhancement factor even by one order of magnitude.
4.4. Metal-enhanced fluorescence
Fluorescence relies on emission of light by excited molecules. When a molecule in a ground singlet state (S0) absorbs a photon, the molecule goes to higher singlet (S1) quantum state. The excited molecule can relax to its ground state by two competing pathways: radiative and non-radiative. In a case of radiative relaxation there are two possibilities: transition from S1 to S0 (without changing multiplicity), which is called fluorescence. The second possibility is intersystem crossing (non-radiative process at this stage) from excited S1 state to T1 state (triplet excited state). Then molecule can come back to the ground state with emission of light. Transition with changing the multiplicity is prohibited, and therefore phosphorescence can last significantly longer than fluorescence.Noble metal nanoparticles can also enhance fluorescence.125 As in the case of other surface-enhanced spectroscopes surface plasmons excited in nanoparticles are responsible for this effect. Increase of the local electromagnetic field can accelerate both excitation and emission, and can also decrease the lifetime of the excited state. This phenomenon is called metal-enhanced fluorescence (MEF) and allows to increase the limit of detection and sensitivity in some analytical methods based on fluorescence spectroscopy. As mentioned above MEF has two main reasons.126 The first contribution is due to the enhancement of the electromagnetic field around nanoresonators. When analysed molecule is in the place when strong enhancement of the electromagnetic field occurs this leads to a significant increase of its absorption cross section which, as a result, enhances fluorescence. The second effect is due to the interactions between excited state of the molecule and surface plasmons. This interaction is responsible for the reduction of the lifetime of the excited state and also increases the quantum yield of fluorescence.
For the molecule of a fluorophore in solution, the quantum yield (Φ) and lifetime (τ) can be expressed as: Φ = Γ/(Γ + knr) and τ = 1/(Γ + knr), where Γ and knr are the radiative and nonradiative decay rates, respectively.127,128 When the structure of a fluorophore is perturbed by the interaction with a metal nanoparticle, the plasmonic coupling causes an increase in the molecular radiative decay rate by a factor of γ. Hence, the metal-enhanced quantum yield (ΦM) and lifetime (τM) become equal to: Φ = γΓ/(γΓ + knr) and τ = 1/(γΓ + knr), where γΓ represents the effective radiative decay rate.128
Very important in MEF measurements is distance between a fluorophore and a plasmonic metal structure.125,129 Fluorescence may be highly enhanced only when the distance between the plasmonic nanoparticle and fluorophore is larger than so-called quenching distance, because for very small distances the quenching of the fluorescence occurs.130 There are many methods used for controlling distance between the plasmonic structure and the fluorophore, such as the deposition of SiO2 layer or using DNA spacers.131,132 More detailed investigations of the distance-dependent quenching of fluorescence (in the range between 5 and ∼100 nm) have been carried out by Choi et al.133 They found that fluorescence quenching occurs at distances within about 15 nm from the Au surface, and MEF is observed at tens of nanometers beyond the range of quenching with the maximum enhancement at about 40–50 nm.133
One of the field where MEF is used in biochemical analysis is detection of DNA.134–136 For example, in 2010 Dragan et al. used MEF for the ultra-sensitive detection of double-stranded nucleic acids. They showed that chromophore PicoGreen creates a complex with studied DNA strands and as the result of that interaction fluorescence of chromophore is enhanced about 103 times.134 However, when this complex is adsorbed on the film of silver nanoparticles deposited on glass the enhancement of the efficiency of the fluorescence is even greater than 3 × 104. This very large enhancement factor leads to very small detection limit of double-stranded DNA using this method. The lowest DNA concentration that Dragan et al. were able to detect was ca. 1 pg ml−1.134 Dragan et al. also observed that the MEF enhancement occurred both for small and for highly polymeric DNA which makes this procedure universal method for DNA detection.134
Metal enhanced fluorescence might be also used for protein detection. As an example MEF sensor for peptide detection one can mention sensor utilizing streptavidin-functionalized silver nanoparticles with coupled fluorophore-functionalized aptamers and quencher-carrying strands hybridized in duplex.137 Such sensor may be used for detection of human platelet-derived growth factor-BB (PDGF-BB). After addition of the PDGF-BB protein, quencher-carrying strands of the duplex are displaced leading to the enhancement of the fluorescence of the fluorophore marker – see Fig. 10.137 This MEF sensor has much higher sensitivity and target specificity compared to the analogous fluorescence sensors in which plasmonic nanoparticles have not been applied.137,138
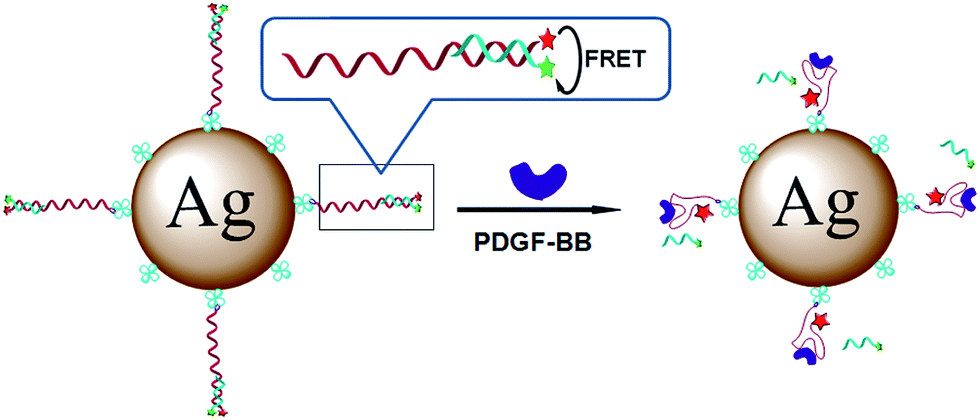 | ||
| Fig. 10 Schematic presentation of the fluorescence enhancement after addition of human platelet-derived growth factor-BB. The fluorescence enhancement is due to the displacement of the quencher-carrying strands. Reprinted with permission from ref. 136. Copyright 2013 American Chemical Society. | ||
In literature there were described silver nanoparticles modified by europium(III) as nanosensors for detection of tetracycline (Tc) – antibiotic widely used in the therapy of human and animal infection.139 It was found that fluorescence intensity of the EuTc–AgNPs sensor was 4 times higher than in case of EuTc complex without silver nanoparticles. A significant advantage of this system is a fact that addition of the amino acids (Arg, Tyr, Asp, Glu, His, Lys, Cys), ascorbic acid, and glucose does not cause any fluorescence response. The detection limit for this sensor is 20 nM and the linear relationship between the intensity of the fluorescence and the concentrations is observed in the range between 40 nM and 8 μM.139
MEF may be also used for detection of cations of heavy metals. An example MEF sensor for cations of heavy metals has been constructed by Cheng et al.140 They used both silver and gold loaded mesoporous silica materials functionalized by rhodamine as a fluorophore. The fluorescence of rhodamine at 584 nm was greatly enhanced after addition of Hg2+ ions, while there was no effect of other competitive ions – see Fig. 11.140 Both MEF sensors utilizing Ag or Au nanoparticles have high Hg2+ selectivity in the presence of interfering ions and have low detection limits (the detection limits of Hg2+ were determined as equal to 0.9 and 1.4 ppb for MEF sensors utilising Ag and Au nanoparticles, respectively).140 Different MEF sensor for detection of mercury ions uses gold nanoparticles capped by luminal.141 It was observed that fluorescent intensity decreasing with increasing concentrations of Hg2+ ions in a sample. This system could be use in the range between 10 and 600 nM, while limit of the detection was calculated as 1 nM. Geddes et al. observed that the fluorescence intensity of the complex of fluo-3 fluorophores with Ca2+ ions could be increased even 100-fold in the close proximity to the silver nanoparticles.142 Such system gives linear response (fluorescence vs. concentrations) in the range between 0.017 and 1.35 μM.142
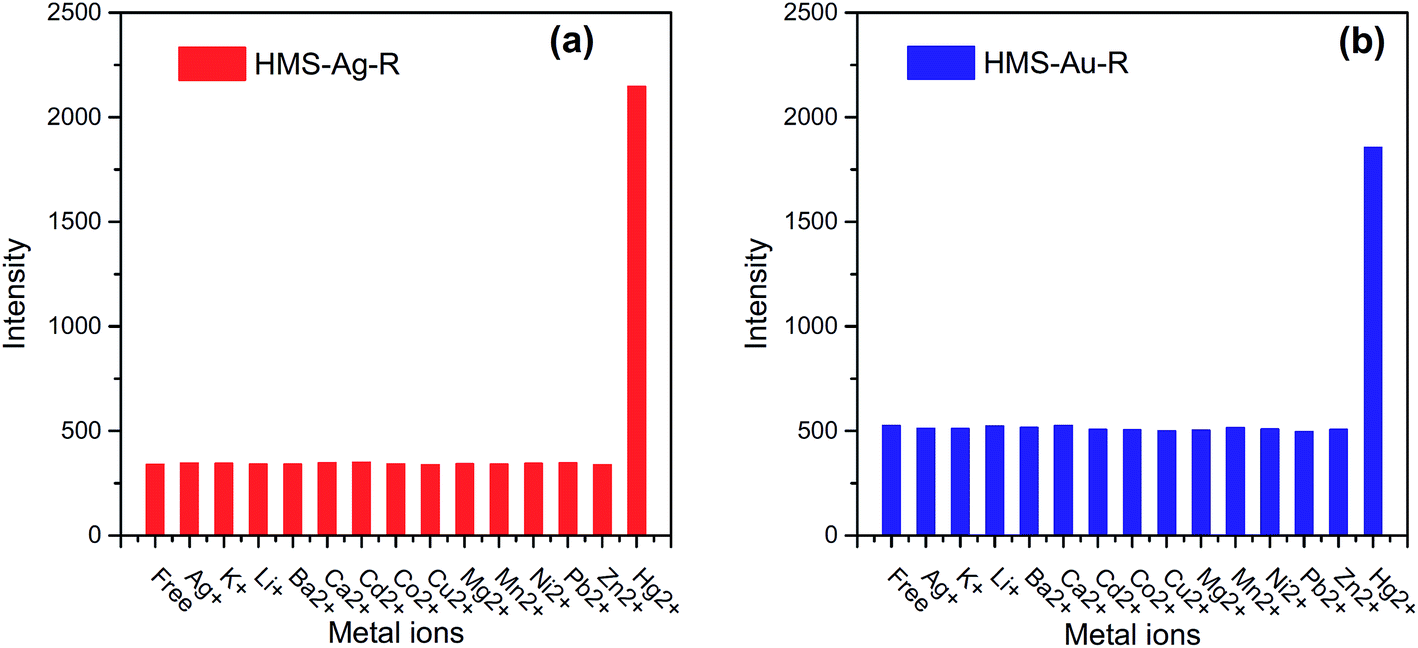 | ||
| Fig. 11 The fluorescence intensity of: (a) hexagonal mesoporous silica material loaded with Ag nanoparticles and functionalized by rhodamine (HMS-Ag-R), and (b) hexagonal mesoporous silica material loaded with Au nanoparticles and functionalized by rhodamine (HMS-Au-R) in presence of other competitive cations and Hg2+. Reprinted from ref. 140 with permission from Elsevier. Copyright 2015 Elsevier. | ||
MEF sensors can be also used for pH-sensing.143 Lakowicz et al. used dextran/carboxyseminaphthofluorescein (SNAFL-2) immobilized on silver island films.143 SNAFL-2 can be used as pH sensor because its acid form has an emission peak at 552 nm while its base form emits 665 nm light. Silver nanostructures significantly enhance intensity of the fluorophore's fluorescence (in this case about 40 times).143 The pH values were estimated from the intensity ratio of the SNAFL-2 emission at 590 and 555 nm (in base and acid form).143
Another interesting modification of MEF is MAMEF (microwave-accelerated metal-enhanced fluorescence). In this approach the effect of the fluorescence enhancement near to the metallic surface is combined with the use of the low energy microwave radiation. The used microwave does not damp the fluorescence yield increase, the MAMEF effect is due to the increased mass transport of the studied molecules to the metallic surface which results in the higher fluorescence and shorter measurement time. MAMEF has been used, for example, for protein, DNA and bacteria's cells detection.144–147 In the case of DNA detection, Geddes et al. used as DNA sensor thiol-functionalized oligonucleotides immobilized on silver nanostructures deposited on the glass surface.145 In this approach, after addition of the complementary fluorescein-labeled DNA strand, hybridization of this oligonucleotide and the one on the silver surface has been carried out. This reaction leads to the metal-enhanced fluorescein emission (MEF effect) as the probe is brought into close proximity to the silver upon hybridization. Using of the microwave irradiation induces stronger fluorescence enhancement and enables 600 times decreasing of the measurement time (less than 20 s) compared to sample without microwave irradiation.145
5. Conclusions
In this review article we have focused on analytical techniques utilising plasmonic properties of Ag and Au nanoparticles (for examples of such analysis see Table 1). Presented analytical techniques include methods based on the chance of the plasmonic properties of nanoparticles (for example caused by their aggregation) and techniques utilising increasing of the efficiency of various optical processes in the proximity of plasmonic nanoparticles, which includes surface-enhanced Raman scattering (SERS), surface enhanced infra-red absorption (SEIRA), and metal-enhanced fluorescence (MEF). The basic theory concerning interaction of the electromagnetic radiation with the plasmonic nanoparticles is also briefly presented. Analytical methods described in this article belong to the most sensitive analytical tools. Fluorescence techniques allow in some cases for reliable observation of the signal even from a single molecule. Also by utilizing plasmonic nanoresonators the Raman scattering cross-sections can be sometimes increased to the level making possible observation of the Raman spectra of a single molecule. Wide potential field of applications (especially in the medical diagnostic) of techniques utilising surface plasmon resonance suggests that the number of applications of plasmonic methods will significantly increase in the near future.| The method | Analyte | Nanoparticles | Linear range | Limit of detection (LOD) | Ref. |
|---|---|---|---|---|---|
| UV-vis | Cd2+, Hg2+, Pb2+ | Au | 50–200 μM | 2.5 nM | 36 |
| Li+ | Au | 10–100 mM | 37 | ||
| Cu2+ | Au | 50–500 μM | 20 μM | 38 | |
| Pt2+ | Au | 0.6–12.5 μM | 150 nM | 39 | |
| Hg2+ | Au | 0.5–10 nM | 0.26 nM | 42 | |
| Mn2+ | Ag | 0.3–60 μM | 20 nM | 43 | |
| Cr3+ | Au | 0.3–3 μM | 13.42 nM | 32 | |
| DNA | Ag nanoprisms | 10 fM to 100 nM | 6 fM | 45 | |
| DNA | Au | 1 pM to 10 nM | 57 | ||
| Phosgene | Au | 15–45 μM | 420 nM | 52 | |
| Dopamine | Ag | 0.2–300 μM | 0.2 μM | 48 | |
| SERS | Thiram | Ag nanorods | 10 μM to 10 mM | 10 ng cm−2 | 81 |
| Thiabendazole | Au nanostars | 0.1–100 μM | 0.1 μM | 82 | |
| Avidin | Au | 10 ng ml−1 to 1 μg ml−1 | 10 ng ml−1 | 96 | |
| Na+, K+, Ca2+, Mg2+ | Ag@SiO2 | 10 nM to 1 M | 10 nM | 101 | |
| As3+ | Ag | 4–600 ppb | 0.76 ppb | 102 | |
| Cd2+ | Au | 10–100 ppt | 10 ppt | 103 | |
| Naphthalene | Polystyrene@Au | 1–20 ppm | 1 ppm | 148 | |
| Hg2+ | Au nanostars dimers | 2 pg to 1 ng | 0.8 pg ml−1 | 149 | |
| SEIRA | p-Mercaptoaniline | Au nanoshells | 10–1000 μM | 1 μM | 150 |
| Tyrosine | Ag | 10–120 μM | 10 μM | 151 | |
| p-Nitrobenzoic acid | Ag | 17–1700 ng | 17 ng | 152 | |
| p-Nitrobenzoic acid | Ag nanorods | 10–1800 ng cm−2 | 0.08 ng cm−2 | 153 | |
| Crystal violet | Au nanostars | 4–25 mg l−1 | 4 mg l−1 | 154 | |
| MEF | DNA | Ag | 1 nM to 1 mM | 1 pg ml−1 | 134 |
| Hg2+ | Au | 10–600 nM | 1 nM | 141 | |
| Hg2+ | Ag | 10–110 ppb | 0.9 ppb | 140 | |
| Hg2+ | Au | 10–100 ppb | 1.14 ppb | 140 | |
| Hg2+ | Au | 1 nM to 1 mM | 1 nM | 155 | |
| Ca2+ | Ag | 17 nM to 135 nM | 17 nM | 142 | |
| Tetracycline | Ag | 40 nM to 8 μM | 20 nM | 139 | |
| PDGF-BB protein | Ag | 6.2–50 ng ml−1 | 0.8 ng ml−1 | 137 |
Acknowledgements
This work was financed from the funds of the National Science Centre (Poland) allocated on the basis of the decision number DEC-2013/11/B/ST5/02224. AK thanks the Faculty of Chemistry, University of Warsaw for the financial support.References
- A. Trügler, Optical Properties of Metallic Nanoparticles, Springer International Publishing, Heidelberg, 2016 Search PubMed.
- M. Pelton and G. W. Bryant, Introduction to Metal-Nanoparticle Plasmonics, John Wiley & Sons, Weinheim, 2013 Search PubMed.
- A. Kudelski, Electrochemical preparation of nanoresonators, in Handbook of Nanoelectrochemistry, ed. M. Aliofkhazraei and A. S. H. Makhlouf, Springer International Publishing, Heidelberg, 2016, pp. 47–69 Search PubMed.
- P. G. Etchegoin and E. C. Le Ru, Basic electromagnetic theory of SERS, in Surface Enhanced Raman Spectroscopy: Analytical, Biophysical and Life Science Applications, ed. S. Schlücker, Wiley-VCH, New York, 2011, pp. 1–37 Search PubMed.
- J. P. Kottmann, O. J. F. Martin, D. R. Smith and S. Schultz, Chem. Phys. Lett., 2001, 341, 1 CrossRef CAS.
- I. A. Larmour and D. Graham, Analyst, 2011, 136, 3831 RSC.
- M. Li, S. K. Cushing and N. Wu, Analyst, 2015, 140, 386 RSC.
- L. M. Luiz-Marzan, Mater. Today, 2004, 7, 26 CrossRef.
- M. G. Guzmán, J. Dille and S. Godet, Int. J. Chem. Biol. Eng., 2009, 104, 2–3 Search PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668 CrossRef CAS.
- S. Yoo and Q. H. Park, Opt. Express, 2012, 20, 16480 CrossRef.
- A. R. Sadrolhosseini, A. S. M. Noor and M. Moksin, Application of surface plasmon resonance based on a metal nanoparticle, in Plasmonics – Principles and Applications, ed. K. Y. Kim, InTech, 2012, pp. 253–282 Search PubMed.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669 CrossRef CAS PubMed.
- X. Zhang, Y. L. Chen, R. S. Liu and D. P. Tsai, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- L. Polavarapu, S. Mourdikoudis, I. Pastoriza-Santos and J. Pérez-Juste, CrystEngComm, 2015, 17, 3727 RSC.
- L. Polavarapu and L. M. Liz-Marzan, Nanoscale, 2013, 5, 4355 RSC.
- C. Xue and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 2036 CrossRef CAS PubMed.
- J. C. Scaiano and K. G. Stamplecoskie, Photochem. Photobiol., 2012, 88, 762 CrossRef PubMed.
- I. Pastoriza-Santos and L. M. Liz-Marzán, J. Mater. Chem., 2008, 18, 1724 RSC.
- R. Jin, Y. C. Cao, E. Hao, G. S. Métraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487 CrossRef CAS PubMed.
- E. K. Payne, K. L. Shuford, S. Park, G. C. Schatz and C. A. Mirkin, J. Phys. Chem. B, 2006, 110, 2150 CrossRef CAS PubMed.
- J. Krajczewski, V. Joubert and A. Kudelski, Colloids Surf., A, 2014, 456, 41 CrossRef CAS.
- D. Ghosh and N. Chattopadhyay, Opt. Photonics J., 2013, 3, 18 CrossRef CAS.
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935 CrossRef CAS PubMed.
- H. Abdulrahman, J. Krajczewski, D. Aleksandrowska and A. Kudelski, J. Phys. Chem. C, 2015, 119, 20030 CAS.
- E. Hao, G. C. Schatz and J. T. Hupp, J. Fluoresc., 2004, 14, 331 CrossRef CAS PubMed.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357 CrossRef CAS PubMed.
- K. A. Stoerzinger, W. Hasan, J. Y. Lin, A. Robles and T. W. Odom, J. Phys. Chem. Lett., 2010, 1, 1046 CrossRef CAS PubMed.
- J. Yang, F. Ren, X. Chong, D. Fan, S. Chakravarty, Z. Wang, R. T. Chen and A. X. Wang, Photonics, 2014, 1, 380 CrossRef PubMed.
- P. Pavaskar, J. Theiss and S. B. Cronin, Opt. Express, 2012, 20, 14656 CrossRef PubMed.
- D. Radziuk and H. Moehwald, Phys. Chem. Chem. Phys., 2015, 17, 21072 RSC.
- S. L. Kleinman, E. Ringe, N. Valley, K. L. Wustholz, E. Phillips, K. A. Scheidt, G. C. Schatz and R. P. van Duyne, J. Am. Chem. Soc., 2011, 133, 4115 CrossRef CAS PubMed.
- L. Polavarapu, J. Pérez-Juste, Q.-H. Xu and L. M. Liz-Marzán, J. Mater. Chem. C, 2014, 2, 7460 RSC.
- A. Lee, G. F. S. Andrade, A. Ahmed, M. L. Souza, N. Coombs, E. Tumarkin, K. Liu, R. Gordon, A. G. Brolo and E. Kumacheva, J. Am. Chem. Soc., 2011, 133, 7563 CrossRef CAS PubMed.
- D. K. Lim, K. S. Jeon, H. M. Kim, J. M. Nam and Y. D. Suh, Nat. Mater., 2010, 9, 60 CrossRef CAS PubMed.
- Y. Kim, R. C. Johnson and J. T. Hupp, Nano Lett., 2001, 1, 165 CrossRef CAS.
- S. O. Obare, R. E. Hollowell and C. J. Murphy, Langmuir, 2002, 26, 10407 CrossRef.
- Y. Zhou, S. Wang, K. Zhang and X. Jiang, Angew. Chem., Int. Ed., 2008, 47, 7454 CrossRef CAS PubMed.
- D. Fan, Q. Zhai, W. Zhou, X. Zhu, E. Wang and S. Dong, Biosens. Bioelectron., 2016, 85, 771 CrossRef CAS PubMed.
- L. J. Miao, J. W. Xin, Z. Y. Shen, Y. J. Zhang, H. Y. Wang and A. G. Wu, Sens. Actuators, B, 2013, 176, 906 CrossRef CAS.
- M. Shellaiah, T. Simon, K. W. Sun and F. H. Ko, Sens. Actuators, B, 2016, 226, 44 CrossRef CAS.
- Y. Zhao, L. Gui and Z. Chen, Sens. Actuators, B, 2017, 241, 262 CrossRef CAS.
- Y. He and X. Zhang, Sens. Actuators, B, 2016, 222, 320 CrossRef CAS.
- Y. X. Gao, J. W. Xin, Z. Y. Shen, W. Pan, X. Li and A. G. Wu, Sens. Actuators, B, 2013, 181, 288 CrossRef CAS.
- X. J. Yang, Y. B. Yu and Z. Q. Gao, ACS Nano, 2014, 8, 4902 CrossRef CAS PubMed.
- X. J. Yang, Y. Q. Ren and Z. Q. Gao, Chem.–Eur. J., 2015, 21, 988 CrossRef CAS PubMed.
- L. Guo, Y. Xu, A. R. Ferhan, G. Chen and D. H. Kim, J. Am. Chem. Soc., 2013, 135, 12338 CrossRef CAS PubMed.
- D. R. Raj, S. Prasanth, T. V. Vineeshkumar and C. Sudarsanakumar, Sens. Actuators, B, 2016, 224, 600 CrossRef.
- B. Li, H. Wei and S. Dong, Chem. Commun., 2007, 50, 73 RSC.
- R. Elghanian, J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078 CrossRef CAS PubMed.
- L. Li and L. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036 CrossRef PubMed.
- D. Feng, Y. Zhang, W. Shi, X. Lia and H. Ma, Chem. Commun., 2010, 46, 9203 RSC.
- W. Zhou, Y. Ma, H. Yang, Y. Ding and X. Luo, Int. J. Nanomed., 2011, 6, 381 CrossRef CAS PubMed.
- C. D. Medley, J. E. Smith, Z. W. Tang, Y. R. Wu, S. Bamrungsap and W. H. Tan, Anal. Chem., 2008, 80, 1067 CrossRef CAS PubMed.
- R. Aroca, Surface-enhanced vibrational spectroscopy, John Wiley & Sons Ltd, Chichester, 2006 Search PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163 CrossRef CAS.
- D. L. Jeanmaire and R. P. van Duyne, J. Electroanal. Chem., 1977, 84, 1 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1974, 99, 5215 CrossRef.
- M. Kerker, D. S. Wang and H. Chew, Appl. Opt., 1980, 19, 4159 CrossRef CAS PubMed.
- R. C. Maher, L. F. Cohen and P. Etchegoin, Chem. Phys. Lett., 2002, 352, 378 CrossRef CAS.
- S. Schlucker, Angew. Chem., Int. Ed., 2014, 53, 4756 CrossRef PubMed.
- M. Procházka, Surface-Enhanced Raman Spectroscopy, Springer International Publishing, 2016 Search PubMed.
- J. R. Lombardi, R. L. Birke, T. Lu and J. Xu, J. Chem. Phys., 1986, 84, 4174 CrossRef CAS.
- M. Osawa, N. Matsudam, K. Yoshii and I. Uchida, J. Phys. Chem., 1994, 98, 12702 CrossRef CAS.
- J. F. Arenas, I. López-Tocón, M. S. Woolley, J. C. Otero and J. I. Marcos, J. Raman Spectrosc., 1998, 29, 673 CrossRef CAS.
- A. B. Zrimsek, N. L. Wong and R. P. van Duyne, J. Phys. Chem. C, 2016, 120, 5133 CAS.
- L. Li, T. Hutter, U. Steinerc and S. Mahajan, Analyst, 2013, 138, 4574 RSC.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102 CrossRef CAS PubMed.
- L. Zhang, H. Liu, L. Chen, P. Guan, B. Chen, T. Fujita, Y. Yamaguchi, H. Iwasaki, Q.-K. Xue and M. Chen, RSC Adv., 2016, 6, 2882 RSC.
- P. Etchegoin, R. C. Maher, L. F. Cohen, H. Hartigan, R. J. C. Brown, M. J. T. Milton and J. C. Gallop, Chem. Phys. Lett., 2003, 375, 84 CrossRef CAS.
- A. Kudelski, Chem. Phys. Lett., 2006, 427, 206 CrossRef CAS.
- A. Kudelski, J. Raman Spectrosc., 2007, 38, 1494 CrossRef CAS.
- K. Chen, M. Leona, K. C. Vo-Dinh, F. Yan, M. B. Wabuyele and T. Vo-Dinh, J. Raman Spectrosc., 2006, 37, 520 CrossRef CAS.
- X. Lian, X. J. Zhan, T. T. You, G. S. Wang, P. G. Yin and L. Guo, CrystEngComm, 2016, 18, 7805 RSC.
- M. Leona, J. Stenger and E. Ferloni, J. Raman Spectrosc., 2006, 37, 981 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392 CrossRef CAS PubMed.
- A. Kudelski and S. Wojtysiak, J. Phys. Chem. C, 2012, 116, 16167 CAS.
- X. D. Lin, V. Uzayisenga, J. F. Li, P. P. Fang, D. Y. Wu, B. Ren and Z. Q. Tian, J. Raman Spectrosc., 2012, 43, 40 CrossRef CAS.
- V. Uzayisenga, X. D. Lin, L. M. Li, J. R. Anema, Z. L. Yang, Y. F. Huang, H. X. Lin, S. B. Li, J. F. Li and Z. Q. Tian, Langmuir, 2012, 28, 9140 CrossRef CAS PubMed.
- H. B. Abdulrahman, K. Kołątaj, P. Lenczewski, J. Krajczewski and A. Kudelski, Appl. Surf. Sci., 2016, 388, 704 CrossRef CAS.
- S. Kumar, P. Goel and J. P. Singh, Sens. Actuators, B, 2017, 241, 577 CrossRef CAS.
- A. Shiohara, J. Langer, L. Palavarapu and L. Liz-Marzan, Nanoscale, 2014, 6, 9817 RSC.
- X. Zhang, M. A. Young, O. Lyandres and R. P. van Duyne, J. Am. Chem. Soc., 2005, 127, 4484 CrossRef CAS PubMed.
- X. Zhang, J. Zhao, A. V. Whitney, J. E. Elam and R. P. van Duyne, J. Am. Chem. Soc., 2006, 128, 10304 CrossRef CAS PubMed.
- S. E. J. Bell, J. N. Mackle and N. M. S. Sirimuthu, Analyst, 2005, 130, 545 RSC.
- D. P. Cowcher, Y. Xu and R. Goodacre, Anal. Chem., 2013, 85, 3297 CrossRef CAS PubMed.
- W. R. Premasiri, D. T. Moir, M. S. Klempner, N. Krieger, G. Jones and L. D. Ziegler, J. Phys. Chem. B, 2005, 109, 312 CrossRef CAS PubMed.
- W. Q. Wang, V. Hynninen, L. Qiu, A. W. Zhang, T. Lemmad, N. N. Zhang, H. H. Ge, J. Toppari, V. P. Hytönen and J. Wang, Sens. Actuators, B, 2017, 239, 515 CrossRef CAS.
- R. Jarvis, S. Clarke and R. Goodacre, Top. Appl. Phys., 2006, 103, 397 CrossRef CAS.
- R. M. Jarvis and R. Goodacre, Anal. Chem., 2004, 76, 40 CrossRef CAS PubMed.
- M. Harz, P. Rosch, K. D. Peschke, O. Ronneberger, H. Burkhardt and J. Popp, Analyst, 2005, 130, 1543 RSC.
- R. M. Jarvis, A. Brooker and R. Goodacre, Anal. Chem., 2004, 76, 5198 CrossRef CAS PubMed.
- P. Rosch, M. Harz, M. Schmitt, K. Peschke, O. Ronneberger, H. Burkhardt, H. Motzkus, M. Lankers, S. Hofer, H. Thiele and J. Popp, Appl. Environ. Microbiol., 2005, 71, 1626 CrossRef PubMed.
- R. M. Jarvis, A. Brooker and R. Goodacre, Faraday Discuss., 2006, 132, 281 RSC.
- T. Lemma, A. Saliniemi, V. Hynninen, V. P. Hytönen and J. J. Toppari, Vib. Spectrosc., 2016, 83, 36 CrossRef CAS.
- C. Ruan, W. Wang and B. Gu, Anal. Chem., 2006, 78, 3379 CrossRef CAS PubMed.
- C. E. Talley, L. Jusinski, C. W. Hollars, S. M. Lane and T. Huser, Anal. Chem., 2004, 76, 7064 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nano Lett., 2007, 7, 2819 CrossRef CAS PubMed.
- S. Zong, Z. Wang, J. Yang and Y. Cui, Anal. Chem., 2011, 83, 4178 CrossRef CAS PubMed.
- J. K. Lim and S. W. Joo, Appl. Spectrosc., 2006, 60, 847 CrossRef CAS PubMed.
- P. Piotrowski and J. Bukowska, Sens. Actuators, B, 2015, 221, 700 CrossRef CAS.
- J. Li, L. Chen, T. Lou and Y. Wang, ACS Appl. Mater. Interfaces, 2011, 3, 3936 CAS.
- S. S. R. Dasary, Y. K. Jones, S. L. Barnes, P. C. Ray and A. K. Singh, Sens. Actuators, B, 2016, 224, 65 CrossRef CAS PubMed.
- S. Abdali and E. W. Blanch, Chem. Soc. Rev., 2008, 37, 980 RSC.
- H. Kneipp, J. Kneipp and K. Kneipp, Anal. Chem., 2006, 78, 1363 CrossRef CAS PubMed.
- K. Osinska, M. Pecul and A. Kudelski, Chem. Phys. Lett., 2010, 496, 86 CrossRef CAS.
- S. O. Pour, S. E. J. Bellb and E. W. Blanch, Chem. Commun., 2011, 47, 4754 RSC.
- A. Hartstein, J. R. Kirtley and J. C. Tsang, Phys. Rev. Lett., 1980, 45, 201 CrossRef CAS.
- M. Osawa and M. Ikeda, J. Phys. Chem., 1991, 95, 9914 CrossRef CAS.
- R. F. Aroca, D. J. Ross and C. Domingo, Appl. Spectrosc., 2004, 58, 324A CrossRef CAS PubMed.
- G. P. C. Rao and J. Yang, Appl. Spectrosc., 2015, 69, 37 CrossRef CAS PubMed.
- K. P. Ishida and P. R. Griffiths, Anal. Chem., 1994, 66, 522 CrossRef CAS.
- Z. Zhang, T. Imae, H. Sato, A. Watanabe and Y. Ozaki, Langmuir, 2001, 17, 4564 CrossRef CAS.
- A. Hatta, T. Ohshima and W. Suëtaka, Appl. Phys. A, 1982, 29, 71 CrossRef.
- Y. Nishikawa, K. Fujiwara and T. Shima, Appl. Spectrosc., 1990, 44, 691 CrossRef CAS.
- M. Osawa, K. Ataka, M. Ikeda, H. Uchihara and R. Namba, Anal. Sci., 1991, 7, 503 CrossRef CAS.
- S. Prati, M. Quaranta, G. Sciutto, I. Bonacini, L. Litti, M. Meneghetti and R. Mazzeo, Heritage Sci., 2014, 2, 28 CrossRef.
- S. Prati, S. Sciutto, I. Bonacini and R. Mazzeo, Top. Curr. Chem., 2016, 374, 129 CrossRef PubMed.
- V. F. Chekhun, G. I. Solyanik, G. I. Kulik, V. P. Tryndiak, I. N. Todor, G. I. Dovbeshko and O. P. Repnytska, J. Exp. Clin. Cancer Res., 2002, 21, 599 CAS.
- G. I. Dobbeshko, V. I. Chegel, N. Y. Gridina, O. P. Repnytska, Y. M. Shirshob, V. P. Tryndiak, I. M. Todor and G. I. Solyanik, Biopolymers, 2002, 67, 470 CrossRef PubMed.
- C. Brown, Y. Li, J. Seelenbinder, P. Pivarnik, A. Rand, S. Letcher, M. Platek and O. Gregory, Anal. Chem., 1998, 70, 2991 CrossRef CAS PubMed.
- K. Ataka, F. Giess, W. Knoll, R. Naumann, S. Haber-Pohlmeier, B. Richter and J. Heberle, J. Am. Chem. Soc., 2004, 49, 16199 CrossRef PubMed.
- H. Y. N. Holman, D. L. Perry and J. C. Hunter-Cevera, J. Microbiol. Methods, 1998, 34, 59 CrossRef CAS.
- Q. Cai, F. Hu, S.-T. Lee, F. Liao, Y. Li and M. Shao, Appl. Phys. Lett., 2015, 106, 023107 CrossRef.
- W. Q. Lim and Z. Gao, Nano Today, 2016, 11, 168 CrossRef CAS.
- C. Geddes and J. Lakowicz, J. Fluoresc., 2012, 12, 121 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Publishing Corporation, New York, NY, USA, 2nd edn, 1999 Search PubMed.
- J. Asselin, M. L. Viger and D. Boudreau, Adv. Chem., 2014, 2014, 812313 Search PubMed.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171 CrossRef CAS PubMed.
- H. Mishra, B. L. Mali, J. Karolin, A. I. Dragan and C. D. Geddes, Phys. Chem. Chem. Phys., 2013, 15, 19538 RSC.
- A. V. Sorokin, A. A. Zabolotskii, N. V. Pereverzev, I. I. Bespalova, S. L. Yefimova, Y. V. Malyukin and A. I. Plekhanov, J. Phys. Chem. C, 2015, 119, 2743 CAS.
- Z. Zhou, H. Huang, Y. Chen, F. Liu, C. Z. Huang and N. Li, Biosens. Bioelectron., 2014, 52, 367 CrossRef CAS PubMed.
- Y. S. Chi, H. R. Byon, B. S. Lee, B. Kong, H. C. Choi and I. S. Choi, Adv. Funct. Mater., 2008, 18, 3395 CrossRef CAS.
- A. I. Dragan, E. S. Bishop, J. R. Casas-Finet, R. J. Strouse, M. A. Schenerman and C. D. Geddes, J. Immunol. Methods, 2010, 362, 95 CrossRef CAS PubMed.
- J. Lakowicz, J. Malicka and I. Gryczyński, BioTechniques, 2003, 34, 62 CAS.
- J. Lakowicz and C. Sabanayagam, Nucleic Acids Res., 2007, 35, 1 CrossRef.
- H. Li, M. Wang, C. Wang, W. Li, W. Qiang and D. Xu, Anal. Chem., 2013, 85, 4492 CrossRef CAS PubMed.
- H. Takayama, Y. Miyake, K. Nouso, F. Ikeda, H. Shiraha, A. Takaki, H. Kobashi, K. Yamamoto and K. Yanamoto, J. Gastroenterol. Hepatol., 2011, 26, 116 CrossRef CAS PubMed.
- H. Tan and Y. Chen, Sens. Actuators, B, 2012, 173, 262 CrossRef CAS.
- Z. Cheng, G. Li and M. Liu, Sens. Actuators, B, 2015, 212, 495 CrossRef CAS.
- Q. Li, J. Wang and Y. He, Sens. Actuators, B, 2016, 231, 64 CrossRef CAS.
- N. Bondre, Y. Zhang and C. D. Geddes, Sens. Actuators, B, 2011, 152, 82 CrossRef CAS.
- K. Aslan, J. R. Lakowicz, H. Szmacinski and C. D. Geddes, J. Fluoresc., 2005, 15, 37 CrossRef CAS PubMed.
- K. Aslan and C. D. Geddes, Anal. Chem., 2005, 77, 8057 CrossRef CAS PubMed.
- K. Aslan, S. Malyn, G. Bector and C. D. Geddes, Analyst, 2007, 132, 1122 RSC.
- Y. Zhang, P. Agreda, S. Kelley, C. Gaydos and C. D. Geddes, IEEE Trans. Biomed. Eng., 2011, 58, 781 CrossRef PubMed.
- S. M. Tennant, Y. Zhang, J. E. Galen, C. D. Geddes and M. M. Levine, PLoS One, 2011, 6, e18700 CAS.
- O. Peron, E. Rinnert, T. Toury, M. L. de la Chapelle and C. Compere, Analyst, 2011, 136, 1018 RSC.
- W. Ma, M. Sun, L. Xu, L. Wang, H. Kuang and C. Xu, Chem. Commun., 2013, 49, 4989 RSC.
- J. Kundub, F. Lec, P. Nordlanderc and N. J. Halas, Chem. Phys. Lett., 2008, 452, 115 CrossRef.
- G. P. C. Rao and J. Yang, Anal. Bioanal. Chem., 2011, 401, 2935 CrossRef CAS PubMed.
- H. D. Wanzenböck, B. Mizaikoff, N. Weissenbacher and R. Kellner, J. Mol. Struct., 1997, 410, 535 CrossRef.
- C. L. Leverette, S. A. Jacobs, S. Shanmukh, S. B. Chaney, R. A. Dluhy and Y. P. Zhao, Appl. Spectrosc., 2006, 60, 906 CrossRef CAS PubMed.
- O. Bibikova, J. Haas, A. I. López-Lorente, A. Popov, M. Kinnunen, I. Meglinski and B. Mizaikoff, Analyst, 2017, 142, 951 RSC.
- B. C. Ye and B. C. Yin, Angew. Chem., Int. Ed., 2008, 47, 8386 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |