
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene quantum dots: effect of size, composition and curvature on their assembly†
Paolo Elvati a,
Elizabeth Baumeisterb and
Angela Violi*abc
a,
Elizabeth Baumeisterb and
Angela Violi*abc
aMechanical Engineering, University of Michigan, Ann Arbor, MI, USA. E-mail: avioli@umich.edu
bChemical Engineering, University of Michigan, Ann Arbor, MI, USA
cMacromolecular Science and Engineering, Biophysics Program, University of Michigan, Ann Arbor, MI, USA
First published on 21st March 2017
Abstract
Graphene Quantum Dots (GQDs) are a relatively new class of molecules that have ignited tremendous research interest due to their extraordinary and tunable optical, electrical, chemical and structural properties. In this work, we report a molecular-level elucidation of the key mechanisms and physical–chemical factors controlling the assembly and stability of nanostructures formed by GQDs in an aqueous environment, using molecular dynamics simulations. We observe the general tendency to form small aggregates and three recurring configurations, one of them with a single layer of water separating two GQDs. The type and characteristics of the structure are mostly determined by the hydrophobicity of the GQDs as well as the steric hindrance of the dangling groups. The composition of the terminal groups plays a key role in determining the configuration of the GQDs, which is also markedly affected by the formation of clusters. Notably, the aggregated GQDs assume strongly correlated shapes and, in some cases, display a radically different conformation distribution compared to single molecules. This cooperative effect prolongs the lifetime of the GQD configurations and can explain the observed persistence of chiral conformations that are only marginally more stable than their specular images.
Introduction
Graphene quantum dots (GQDs), defined as graphene sheets with lateral dimensions less than 100 nm in single-, double- and few- (less than 10) layers,1–4 hold great promise as a new class of material for a wide range of technologies including energy applications, biomedical treatments, photovoltaic devices and catalysis.1,5 The unique combination of a number of key merits, including biocompatibility, excellent photostability, small size, highly tunable photoluminescence, electrochemiluminescence, exceptional multiphoton excitation, ease of functionalization, and chemical inertness provide an ongoing enthusiasm for these compounds.To date, a variety of techniques have been developed to synthesize nanosized graphene by physical and/or chemical methods, including nanopatterning of graphene by electron beam lithography,6 cage opening of fullerene,7 breakage of graphene oxide from micro- into nanosize by hydrothermal and electrochemical methods,8,9 strong acid assisted cleavage of graphite nanomaterials (nanographite, carbon fibers),10,11 organic synthesis using polyphenylene dendritic precursors,12 and self-assembly of hexa-peri-hexabenzocoronene.13 One of the unavoidable consequences of this variety of procedures is the large and uncontrolled heterogeneity of GQDs synthesized by the current approaches. Even considering only the statistically most significant products of each methodology, the range of GQD structures that is available is still quite wide.1,14 This variability is extremely relevant to the solubility, biocompatibility, and optical properties of GQDs. As an example, GQDs, composed of C and H, fluoresce from green to near infrared on varying their size. Also, the synthesized GQDs often bear functional groups, such as OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and COOH, which can red-shift the emission peaks because of band gap reduction. For example, attaching OH groups to edge carbon atoms (varying from 0–100% coverage) continuously tunes emissions from green (572.4 nm) to red (732.3 nm).15 In addition, as we have recently reported, size and chemical moieties influence ostensibly the chirality of GQDs16 and therefore their potential applications in chemistry, biology, and medicine.17
O, and COOH, which can red-shift the emission peaks because of band gap reduction. For example, attaching OH groups to edge carbon atoms (varying from 0–100% coverage) continuously tunes emissions from green (572.4 nm) to red (732.3 nm).15 In addition, as we have recently reported, size and chemical moieties influence ostensibly the chirality of GQDs16 and therefore their potential applications in chemistry, biology, and medicine.17
In this paper, we investigated the effects of the morphology and hydrophobicity of GQDs on their self-assembly. Driven by experimental evidence, we examined how the morphology of aggregates depends on the size and chemistry of the building blocks. Currently this task cannot be accomplished experimentally, as depending on the preparation method, the measured values (e.g. the size distribution, percent composition, and number of layers) are often rough estimates or broad distributions, and exact structural contributions cannot be identified.
The most striking feature of the simulation results presented below is the type of recurring sub-structure generated inside the aggregates. Notably, even minimal aggregation can induce a strong correlation in the assumed conformations, a property that can potentially be leveraged to prolong the lifetime of specific configurations for medical and optical applications.
Methods
Molecular dynamics simulations were performed using the NAMD18 code, employing the CHARMM general force field19–21 (version 3.0.1) for the GQDs and the TIP3P model for water. To use a timestep of 2 fs, all the hydrogen bonds were constrained with the SHAKE algorithm.22 After 2 ns of equilibration, 20 ns simulations were run in the NPT ensemble (310 K, 1.0325 bar, thermostat time constant of 100 fs, and a piston period and decay of 100 fs and 50 fs, respectively). To calculate the interplanar distances and puckering coordinates,23 we computed the best fitting plane (with respect to the distance of all the atoms of each GQD’s carbon matrix) that contained the GQD’s center of mass (COM), using the atomic coordinates saved every picosecond. The GQD–GQD planar distance was then obtained by projecting the COM–COM distance on the normal to the planes of both GQDs and calculating the average.To compute the puckering coordinates, we first obtained the distances from the GQD’s plane (zi) of the six vertices of the hexagonal aromatic part of the molecule and then computed Q, qx, and qy:
| Q2 = q22 + q32 |
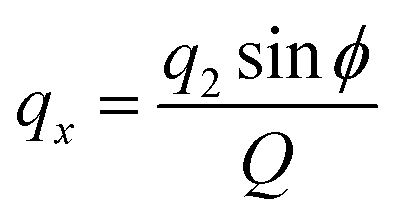
where q2, q3, and ϕ are defined by the following equations:
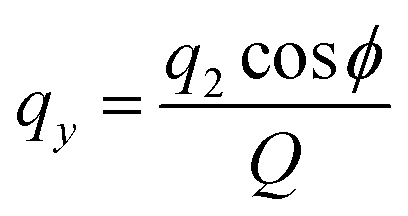
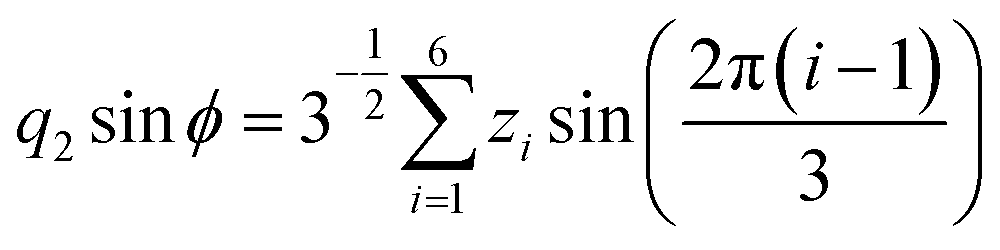
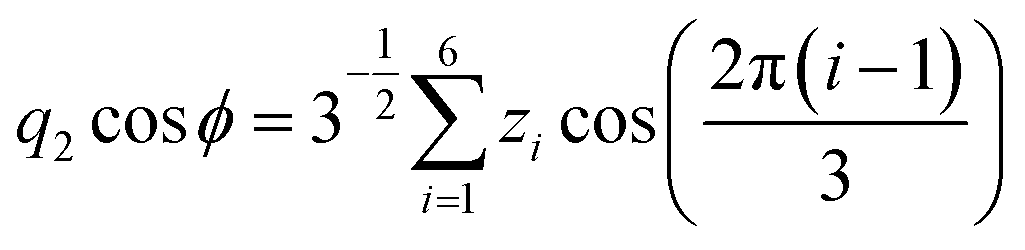
Q is a measure of the total deviation of the six centers from a planar configuration, while qx and qy identify the shape of the conformations assumed by the GQDs as shown in Fig. 1.

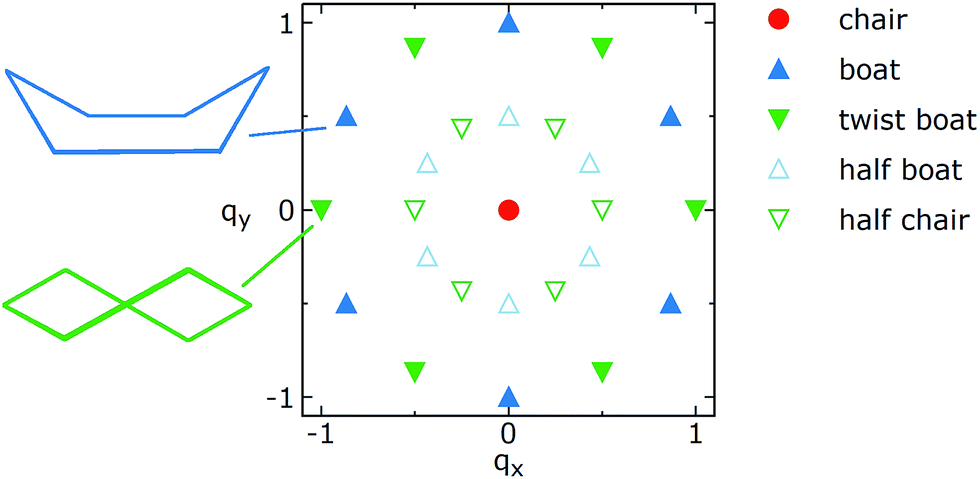 | ||
| Fig. 1 Schematic representation of the locations of the most common conformations of a six-membered ring on a qx, qy plot. See text for the definitions of qx and qy. | ||
Borrowing the nomenclature commonly used for cyclohexane, the chair conformation is located at qx = 0, qy = 0, while there are six boat conformations, one every 60° on the perimeter of a circle of radius 1, starting from qx = 0, qy = 1 (see Fig. 1). The twist boat conformations are also on the perimeter of the circle but rotated by 30° with respect to the boat. The half-boat and half-chair conformations are located midway between the center (qx = 0, qy = 0) and any boat or twist boat, respectively.
Results
Using molecular dynamics simulations, we investigated the assembly of nine GQDs, whose molecular structures are reported in Fig. 2. Five types of side chains (all H, all OH, all CHO, all COOH, and alternating OH and COOH) were selected to reproduce the variety of experimental values of O/C atomic ratios.1,4,6,14,24–27 Beside these groups, other fragments are sometimes found on GQDs, for example cyclic anhydrides, lactones, lactols, aldehydes, and quinone as well as pyrone-like structures.28 However, these groups are relatively uncommon or reactive in an aqueous environment. In our study, we have only considered edge substitutes, as the GQDs’ edge sites are more reactive than the carbon matrix, especially considering the small size of the GQDs investigated here. As the medium for our analysis, we used water due to its relevance in synthesis methods as well as biological systems.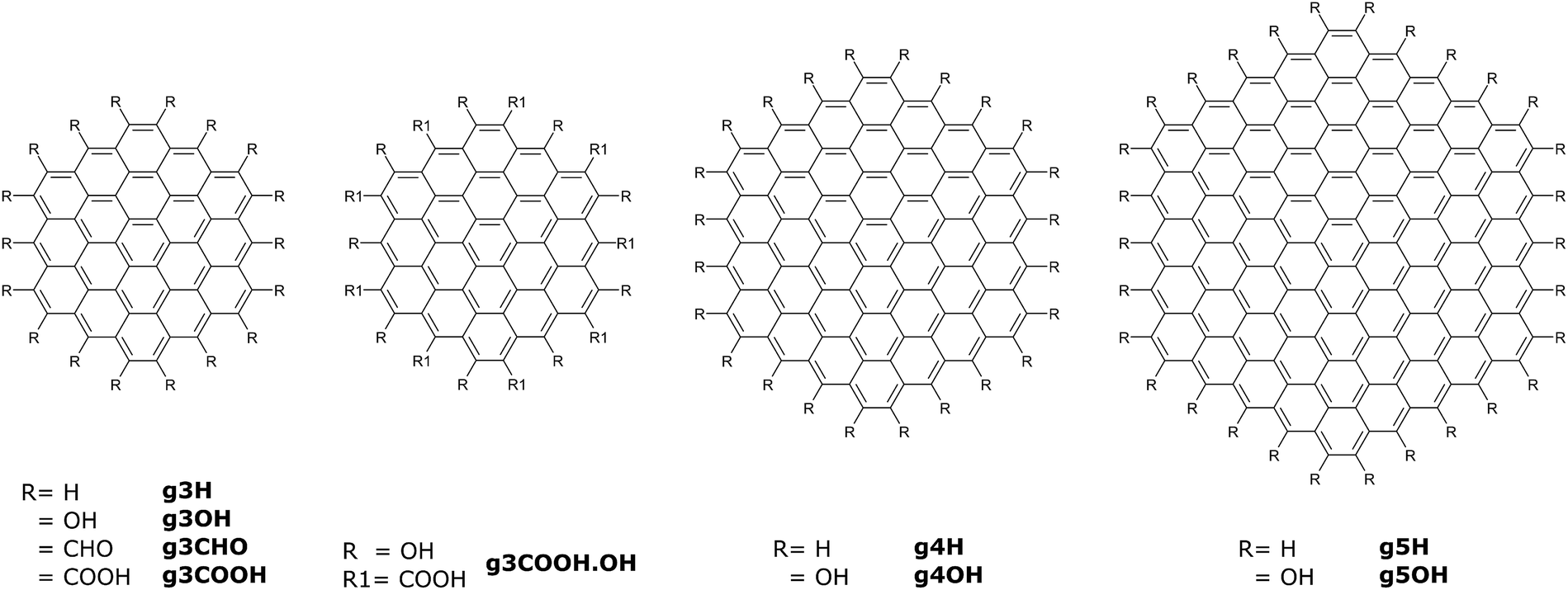 | ||
| Fig. 2 Molecular structures and corresponding labels (in bold) of GQDs simulated in this work. | ||
One of the most important morphological properties of a GQD is its curvature, as it plays an important role in its biocompatibility.16,29 Since GQDs are relatively flexible, in this context the term curvature is meant as the distribution of configurations that deviates from a perfectly planar atomic arrangement. To this end, we computed the puckering values of their hexagonal carbonaceous core, employing a general definition of ring-puckering coordinates that can be applied to any cyclic molecule given only the coordinates of the atoms in the ring.23 The geometry of the puckering, relative to this plane, is then described by the amplitude Q, and the distortion type is specified by two variables qx and qy. Different from treatments based on torsion angles, no mathematical approximations are involved. Depending on ring size, the three-dimensional shapes of the cyclic structures can vary and interconvert such that conformational isomerism is displayed. Examples of conformations include the chair, boat, twist boat, half-boat, and half chair (see Fig. 1). The intensity of the deviation from the planar conformation, Q, for different GQDs is shown in Fig. 3.
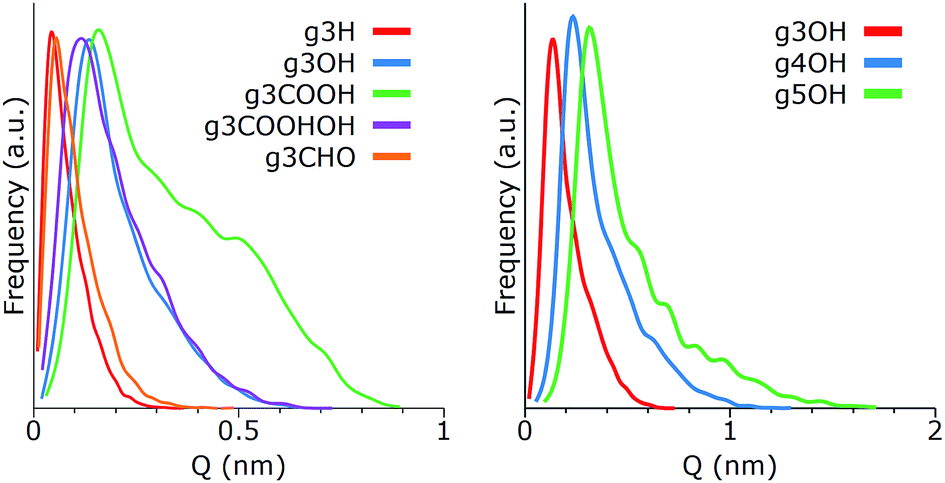 | ||
| Fig. 3 Deviation from the planar configuration for individual GQDs as a function of (left) their terminal groups and (right) their size. | ||
As expected, single GQDs tend to be more flexible as the size of the core increases, showing an approximately linear proportionality between size and Q (for additional data see Fig. X1 of the ESI†). The terminal groups play a relevant role too, with the g3H and g3CHO remaining almost flat and the g3COOH showing a strong deformation caused by the steric hindrance of the adjacent carboxylic groups. A comparison among the distributions of g3H, g3CHO, g3OH and g3COOH.OH shows that the mass of the terminal groups is not the key parameter for determining the bending of the GQD. From the analysis of the MD trajectories, we speculate that these two types of curvature behaviors (g3H and g3CHO vs. g3OH and g3COOH.OH) are due to the relevance of intramolecular hydrogen bonds. The edge groups of both g3OH and g3COOH.OH tend to have strong interactions, forming weak bonds that in turn affect the structure of the matrix. On the other hand, the GQDs in the other category tend to show minimal interactions between side groups and more (at least for g3CHO) with the solvent, exerting less strain on the GQDs.
Individual GQDs in water strongly prefer a chair conformation, with the hydroxyl terminated molecules marginally exploring all the other conformations. Fig. X2 of the ESI† contains more details about the conformation distributions of single GQDs.
The aggregation of GQDs further influences the curvature of the individual GQDs and the distribution of conformations. Experimental findings indicate that the morphology of the GQDs, including the size and number of layers, varies according to the synthesis method, but overall, most GQDs consist of no more than 5 layers.1 For this reason, we simulated the spontaneous aggregation of small GQDs and the corresponding changes in the distribution of conformations. Though a detailed study on GQD nucleation is beyond the scope of this work, we can still make some observations on recurring sub-structures generated inside the aggregates. Fig. 4 shows the three types of stable substructures observed during the simulations: a close stack (CS) where two GQDs would lie parallel to each other in close contact, a wide stack (WS) similar to the CS but with a single molecular layer of water molecules separating the GQDs, and a perpendicular stack (PS) where the main plane of a GQD lies orthogonal to the main planes of two or more GQDs. The WS and CS structures are clearly distinguished by the interlayer distance (Fig. 5), defined as the distance between the two planes that best fit the position of the matrix carbon atoms of each GQD (see the Methods section for more details). This plane-to-plane distance is for the most part only minimally affected by either the composition of the side groups and the size of the GQDs. For the CS configurations, however, bulkier groups, such as COOH, tend to stabilize the GQDs slightly further apart. Interestingly, while the interlayer distance depends on the side group composition, it is not affected by the size of the GQDs (Fig. 5) or position inside the aggregates (Fig. X3 of the ESI†).
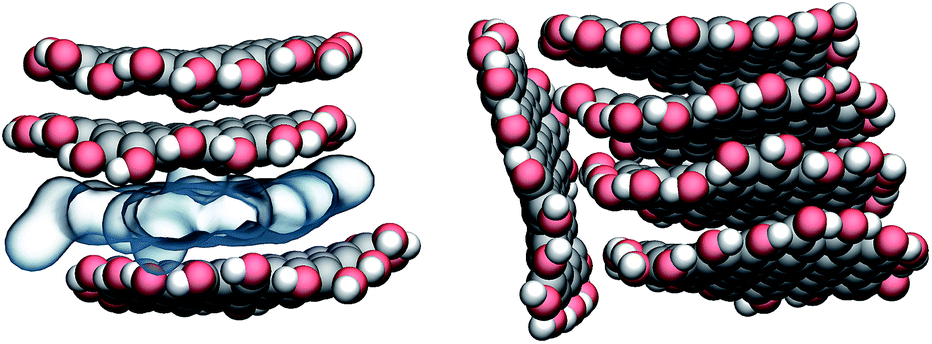 | ||
| Fig. 4 Examples of recurring substructures observed in GQD clusters. Close stack (CS, upper part on the left) and wide stack (WS, lower part on the left) are distinguished by the presence of a layer of water (shown as a blue translucent layer). The perpendicular stack (PS, on the right) is observed only when one or more CSs or WSs are already present. Carbon, oxygen, and hydrogen atoms are shown in gray, red, and white, respectively. Image prepared with VMD.30 | ||
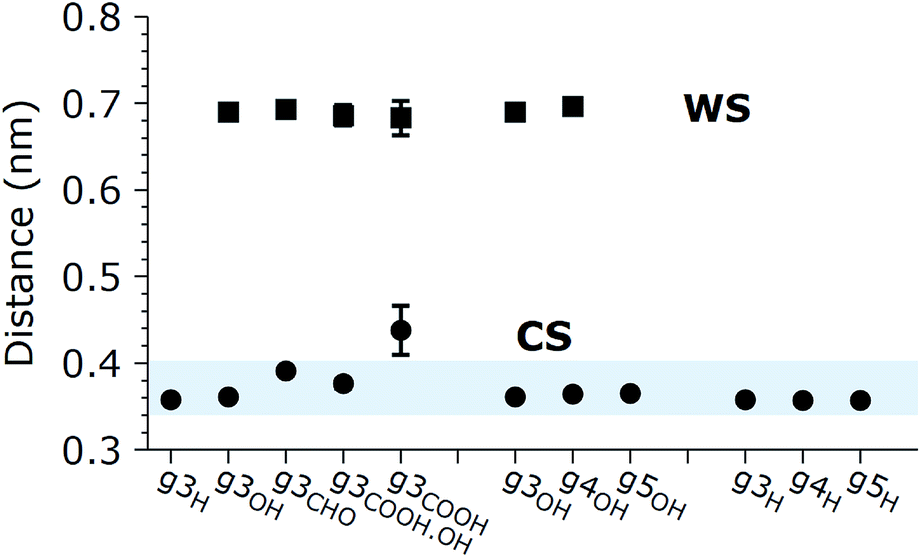 | ||
| Fig. 5 Interplanar distances between GQDs in CS and WS. The blue band indicates the interval of the experimental observations. Where not shown, statistical error (95% confidence interval) is within ±0.01 nm. | ||
The lifetime of each structure depends on both the size and side groups of the GQDs. In all cases, the various aggregates were stable for at least 0.5 ns with the CS being in general the most long lived type of stacking. The WS was not detected for the most hydrophobic systems, such as GQDs terminated by H, while the PS structure was observed even when the GQDs were terminated by hydrophilic groups, like g3OH (shown in Fig. 4), likely due to the strong intramolecular interactions that partially offset the reduced access to the solvent. The WS conformation, while relatively stable (in one simulation a WS g4OH dimer survived the whole 20 ns simulation), is in general an intermediate state to reach the CS configuration. Although not all the WS structures eventually become CS structures, once the latter are formed, the reverse process was never observed, although occasionally the aggregate would dissolve.
The only marginal exception to these observations is g3COOH for which we noticed an increase of about 20% in the layer-to-layer distance for the CS configuration. This result can be understood considering that the steric hindrance of the contiguous carboxylic groups causes marked deviations from the planar structure of the GQDs, even when they are not in a cluster. In addition, the rearrangement of the carboxylic groups is associated with a high free energy barrier, which means that better stacking (and therefore shorter interplanar distances) is potentially possible on a timescale several orders of magnitude longer than the ones considered here.
These results agree with the experimental evidence reported in the literature. The interlayer distances observed experimentally vary according to the GQD preparation methods, from 0.34 nm reported for GQDs obtained through electrochemical cutting from a graphene film and citric acid carbonization,1 to 0.403 nm for GQDs derived from carbon fibers.14 The latter method is also more likely to add a higher quantity of oxygen atoms to the carbonaceous core during the exfoliation and oxidation processes, compared to the other methods. The computational results reported in this paper agree with the experimental data in terms of values as well as trends: 0.357 for H-terminated GQDs, 0.364 for OH-terminated GQDs, and up to 0.438 for g3COOH, showing a greater interlayer distance for structures with increased oxidation of the terminal groups. In addition, experimental evidence reports a spacing of 0.852 nm when GQDs are prepared via thermal treatment, and the authors explained this result inferring the presence of oxygen-containing groups on the graphene sheets.24 In our simulations, we observed a similar, but slightly smaller distance between the GQDs when they are in the WS configuration and a layer of water is present between them. This small discrepancy could be caused by different phenomena, most likely a permanent curvature induced by the sp3 hybridization of the carbons on which out-of-plane groups are attached. Moreover, we expect that due to the directionality of hydrogen bonds, the presence of oxygenated out-of-plane groups will result in a slightly thicker solvent layer.
The clustering of GQDs strongly affects their distribution of conformations. Fig. 6 shows the correlation between Q and the stacking size, as well as the variations for the single GQDs inside an aggregate of four (all in CS).
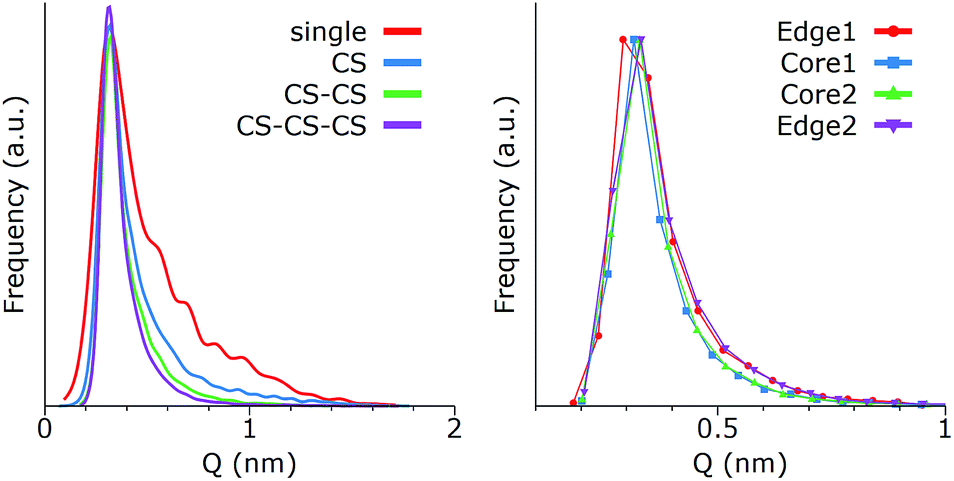 | ||
| Fig. 6 Deviations from the planar configuration for g5OH GQDs: (left) in different clusters and (right) with different positions inside an aggregate of 3 CS. Edge GQDs have a face in contact with water, while core GQDs interact with water only through their terminal groups. | ||
As reported in Fig. 6 (left panel), the peak at the maximum value of Q is only slightly shifted by the formation of one or more CSs, but the tail of the distribution at higher values progressively decreases. In other words, the preferred conformations do not change, but become statistically more dominant. This effect can be caused by at least two different phenomena: (1) a limitation of the effective volume available to GQDs, due to the presence of neighboring molecules, and/or (2) a cooperative effect, driven by the solubility, where GQDs show correlated fluctuations to minimize their interfacial areas. The similarity between the distributions of a single CS, which is completely exposed to water, and multiple CS configurations (Fig. 6 (left panel)), which also average the GQDs that are surrounded by other GQDs, suggests that the cooperative effect is the most important. To confirm this observation, we analyzed the distributions of Q for each GQD in a stack of 4 (CS–CS–CS) g5OH (Fig. 6 (right panel)). The curves are identical within error, and the external GQDs (1 and 4 in the plot) have similar deformations compared to the internal ones (labeled 2 and 3), validating the hypothesis that the GQDs assume correlated conformations. Finally, while the stability of the CS allowed us to perform a detailed analysis of this type of structure, the relatively lower stabilities of the WS and PS did not allow a similar type of analysis, although we observed that the WSs are much less affected by the presence of other GQDs, most likely because the water layer acts as a buffer between GQDs. However, due to the lower stability of the WS, and the corresponding short lifetime, we expect this type of structure to have minimal experimental relevance.
Even though the effect of clustering on the Q distribution described above is fundamentally the same for all the systems that were analyzed, the change to the preferred conformation is less uniform. When clustering, the smaller GQDs (g3) as well as g4H and g5H, maintain mostly the same chair-like conformation observed for the individual GQDs, with g3OH showing the biggest deviations (see Fig. X4 of the ESI†). The g4OH and g5OH systems, however, display an unusual behavior (Fig. 7): the GQD in the clusters assumes an almost complementary distribution of conformations with respect to the single molecule, avoiding chair structures and favoring boat and twisted boat conformations, and in smaller measure, half-chair and half-boat conformations. The reason for such behavior is still not clear, but inspection of the trajectories allowed us to exclude that these phenomena are due to the formation of intermolecular hydrogen bonds that involve the terminal groups.
 | ||
| Fig. 7 Stoddart representation of the conformations’ distributions for (left) an individual g5OH and (right) a cluster of 4 g5OH (all in CS). | ||
Discussion
The previous results show a glimpse of the complexity of the aggregation and organization behaviors of GQDs. However, for the sake of simplicity, we restricted our investigation by making several assumptions that nevertheless should be kept in mind when comparing with experiments. The most important of these aspects is the effect of side group deprotonation on aggregation, especially for GQDs with strongly acidic or basic groups. In general, the net charge accumulated on the GQDs, which will be of the same sign for similarly synthesized GQDs, will reduce the aggregation propensity, although the presence of cations in solution can effectively neutralize the repulsion. On the other hand, mixing GQDs that have opposite charge at the same pH would greatly enhance the stability of aggregates, although this would strongly affect the conformation distributions. Estimating the dissociation behavior of the side groups in the GQDs is beyond the goals of this paper, and is a complex task due to the interplay of inductive and resonance effects, which are associated with the exact position of all the GQDs’ groups. Therefore, generic predictions, like the one shown in Fig. 8, should be taken with a grain of salt.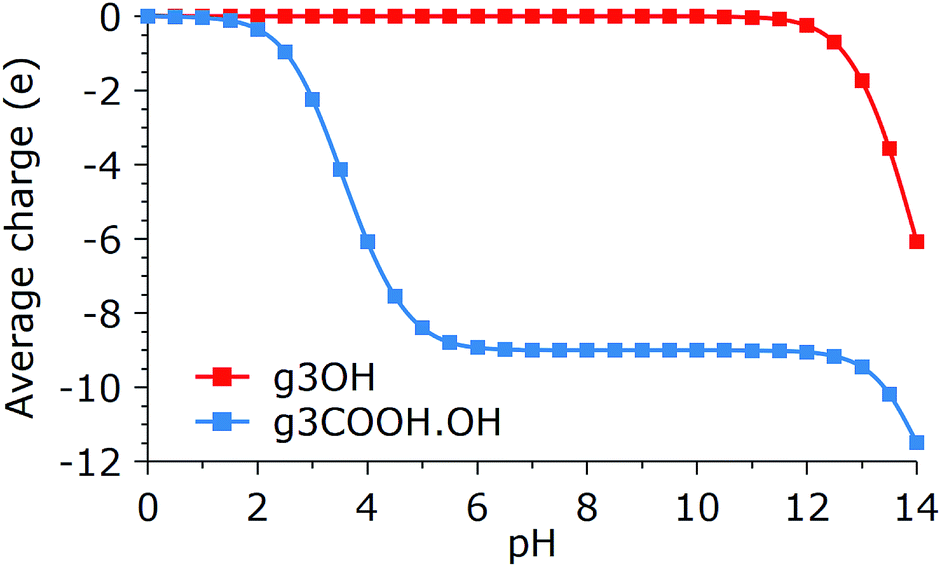 | ||
| Fig. 8 Average charge per molecule as a function of pH for g3COOH.OH and g3OH. Calculator plugins were used for structure property prediction and calculation.33 | ||
On a more general scale, the solvent also plays a crucial role, well beyond the simple change in electrostatic interactions due to variations in the dielectric constant. Our preliminary tests with implicit solvent models show that hydrogen bonding, as well as energy exchange with the solvent, is critical in reproducing the correct aggregation and conformation dynamics.
The effect of aggregation can potentially have important implications beyond the properties of GQDs in solution and biological environments, where clusters have generally different behaviors to single molecules, specifically in terms of their optical and electronic properties. We expect aggregation to have a noticeable effect on the Raman spectra, with red-shifting of the 2D peak as well as an increase in the intensity of most peaks,31,32 as the change in conformation affects the vibrational states of the GQDs. On the other hand, as aggregation does not significantly affect the electronic states, especially for GQDs that have strong intramolecular interactions, we expect a minimal effect on the aggregate photoluminescence mostly due to the changes in the vibrational states of the electronic ground state. For the same reasons, we predict negligible variations in the absorption spectra, as we don’t expect the vibrational states of the excited electronic states to be influenced by clustering. The cooperative behavior that we observe in the aggregates, however, can have important ramifications on circular dichroism spectroscopy. As GQDs can be chiral depending on the position of the edge groups, the organized conformations observed in the clusters, as well as the changes in the relative stability of different conformations and the slower rotational relaxation of the clusters, make the aggregates more likely to show a stronger dichroic signal when chiral GQDs are involved.
Conclusions
In this work, we used molecular dynamics simulations to investigate the effects of size and chemical functionalization on the agglomeration of GQDs. GQDs form small aggregates in water via the stacking of parallel molecules with an interlayer spacing of less than 1 nm. Depending on the size and terminal groups of GQDs, we observed three types of recurring substructures inside the aggregates: a close stacking with no water between the GQDs, a wide stacking with a single layer of water molecules in between, and a perpendicular stacking where the GQDs are in direct contact but assume an orthogonal configuration. Even though the relative stability of each of these structures mostly depends on the type of terminal groups (their steric hindrance and hydrophobicity), and only marginally on the GQD size, the close stacking configuration is the most stable of the three and the wide stacking appears to be an intermediate between the unbound molecules and the close stacking conformation.The distribution of GQD conformations was found to be dependent on the size, the type of terminal groups present, and clustering. Notably, even minimal aggregation can induce a strong correlation in the assumed conformations, a property that can potentially be leveraged to prolong the lifetime of specific conformations (e.g. chiral). The results reported in this study provide insightful guidance on the physical–chemical factors controlling the degree of interactions, and the resulting stability of GQDs in an aqueous environment. Aggregation can play a role not only by reducing the availability of GQDs in solution (by precipitation), but also by affecting the mechanisms, kinetics, and effects of interactions with cells. Insight into the relationships between size, morphology, chemical composition, clustering tendency, and assembly of GQDs in a geometrically well-defined fashion opens up an opportunity to control optical and electronic coupling between the individual quantum dot units and form structures with collective properties.
Acknowledgements
This research has been partially funded by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences (BES) under the Single Investigator Small Group Research (SISGR), Grant no. DE-SC0002619. This research was supported in part through computational resources and services provided by Advanced Research Computing at the University of Michigan, Ann Arbor. The authors thank Yichun Wang for the helpful discussions.References
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J.-J. Zhu, Nanoscale, 2013, 5, 4015–4039 RSC.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS PubMed.
- J. Shen, Y. Zhu, X. Yang, J. Zong, J. Zhang and C. Li, New J. Chem., 2012, 36, 97–101 RSC.
- X. Zhou, Y. Zhang, C. Wang, X. Wu, Y. Yang, B. Zheng, H. Wu, S. Guo and J. Zhang, ACS Nano, 2012, 6, 6592–6599 CrossRef CAS PubMed.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng, Y. Hou and L. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef CAS PubMed.
- L. Wang, H.-Y. Wang, Y. Wang, S.-J. Zhu, Y.-L. Zhang, J.-H. Zhang, Q.-D. Chen, W. Han, H.-L. Xu, B. Yang and H.-B. Sun, Adv. Mater., 2013, 25, 6539–6545 CrossRef CAS PubMed.
- F. Liu, M.-H. Jang, H. D. Ha, J.-H. Kim, Y.-H. Cho and T. S. Seo, Adv. Mater., 2013, 25, 3657–3662 CrossRef CAS PubMed.
- J. Lu, P. S. E. Yeo, C. K. Gan, P. Wu and K. P. Loh, Nat. Nanotechnol., 2011, 6, 247–252 CrossRef CAS PubMed.
- J. Luo, L. J. Cote, V. C. Tung, A. T. L. Tan, P. E. Goins, J. Wu and J. Huang, J. Am. Chem. Soc., 2010, 132, 17667–17669 CrossRef CAS PubMed.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- Y. Dong, C. Chen, X. Zheng, L. Gao, Z. Cui, H. Yang, C. Guo, Y. Chi and C. M. Li, J. Mater. Chem., 2012, 22, 8764–8766 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J.-J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed.
- M. A. Sk, A. Ananthanarayanan, L. Huang, K. H. Lim and P. Chen, J. Mater. Chem. C, 2014, 2, 6954–6960 RSC.
- N. Suzuki, Y. Wang, P. Elvati, Z.-B. Qu, K. Kim, S. Jiang, E. Baumeister, J. Lee, B. Yeom, J. H. Bahng, J. Lee, A. Violi and N. A. Kotov, ACS Nano, 2016, 10, 1744–1755 CrossRef CAS PubMed.
- M. Vàzquez-Nakagawa, L. Rodríguez-Péerez, M. A. Herranz and N. Martín, Chem. Commun., 2015, 52, 665–668 RSC.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell Jr, J. Comput. Chem., 2010, 31, 671–690 CAS.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- C. Hu, Y. Liu, Y. Yang, J. Cui, Z. Huang, Y. Wang, L. Yang, H. Wang, Y. Xiao and J. Rong, J. Mater. Chem. B, 2013, 1, 39–42 RSC.
- D. B. Shinde and V. K. Pillai, Chem.–Eur. J., 2012, 18, 12522–12528 CrossRef CAS PubMed.
- M. Zhang, L. Bai, W. Shang, W. Xie, H. Ma, Y. Fu, D. Fang, H. Sun, L. Fan, M. Han, C. Liu and S. Yang, J. Mater. Chem., 2012, 22, 7461–7467 RSC.
- S. Zhu, J. Zhang, C. Qiao, S. Tang, Y. Li, W. Yuan, B. Li, L. Tian, F. Liu, R. Hu, H. Gao, H. Wei, H. Zhang, H. Sun and B. Yang, Chem. Commun., 2011, 47, 6858–6860 RSC.
- H. P. Boehm, Carbon, 2002, 40, 145–149 CrossRef CAS.
- W. Shang, X. Zhang, M. Zhang, Z. Fan, Y. Sun, M. Han and L. Fan, Nanoscale, 2014, 6, 5799–5806 RSC.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- H. Cheng, Y. Zhao, Y. Fan, X. Xie, L. Qu and G. Shi, ACS Nano, 2012, 6, 2237–2244 CrossRef CAS PubMed.
- Marvin, ChemAxon 16.12.5, 2016, http://www.chemaxon.com Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01029j |
| This journal is © The Royal Society of Chemistry 2017 |