
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix isolation study of the early intermediates in the ozonolysis of selected vinyl ethers†
Chen Lv,
Lin Du *,
Shanshan Tang,
Narcisse T. Tsona,
Shijie Liu,
Hailiang Zhao and
Wenxing Wang
*,
Shanshan Tang,
Narcisse T. Tsona,
Shijie Liu,
Hailiang Zhao and
Wenxing Wang
Environment Research Institute, Shandong University, Shanda South Road 27, 250100 Shandong, China. E-mail: lindu@sdu.edu.cn
First published on 31st March 2017
Abstract
The matrix isolation technique combined with infrared spectroscopy has been used to characterize Criegee Intermediates (CI) and other products formed during the ozonolysis reactions of ethyl vinyl ether and n-butyl vinyl ether. Twin jet deposition at 14 K led to a number of new bands indicating the formation of products, with an intensity increase of ∼150% to 400% when annealing to 30 K. All the infrared absorptions could be assigned to different bands, which provided direct evidence for the formation of primary ozonides, CI and secondary ozonides in the systems investigated. Theoretical calculations at the B3LYP-D3/aug-cc-pVTZ level were carried out to complement the experimental observations. Experimental and theoretical results demonstrate that the studied ozonolysis reactions predominantly follow the Criegee mechanism. The current results will allow a better assessment of the potential environmental impacts of vinyl ethers.
1. Introduction
Oxygenated volatile organic compounds (OVOCs) are important components of the tropospheric photochemistry.1,2 They are emitted into the troposphere both from natural and anthropogenic sources, as well as formed from the oxidation of hydrocarbons present in the atmosphere. Natural sources include biological and biogenic emissions, while anthropogenic sources include emissions from biomass burning, and various industrial and other commercial activities where they are used as oxygenated solvents, fuels, and fuel additives.3–5 Several studies have shown that OVOCs play an important role in the formation of ozone from photooxidation. It has also been proven that OVOCs contribute substantially to the formation of secondary organic aerosols (SOA) and hence affect the radiative budget of the atmosphere.6Vinyl ethers (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHOR, where R is an alkyl or aryl group) are one group of OVOCs used as new solvents to replace traditional ones in order to reduce damaging health effects associated with their use.7 These ethers are used in different industries, particularly as solvents, motor oil additives, and as intermediates for the synthesis of flavors, fragrances and pharmaceuticals.8 Upon their release into the atmosphere, vinyl ethers undergo either photolysis or photochemical oxidation by OH radicals and ozone during daytime and by nitrate radicals (NO3) and ozone during the nighttime.5,9–12 Reactions with Cl atoms may also be important in certain locations during certain times of the year.13
CHOR, where R is an alkyl or aryl group) are one group of OVOCs used as new solvents to replace traditional ones in order to reduce damaging health effects associated with their use.7 These ethers are used in different industries, particularly as solvents, motor oil additives, and as intermediates for the synthesis of flavors, fragrances and pharmaceuticals.8 Upon their release into the atmosphere, vinyl ethers undergo either photolysis or photochemical oxidation by OH radicals and ozone during daytime and by nitrate radicals (NO3) and ozone during the nighttime.5,9–12 Reactions with Cl atoms may also be important in certain locations during certain times of the year.13
Some experimental and theoretical studies on the reaction of ozone with selected vinyl ethers have been performed. The rate coefficient of the ozonolysis of ethyl vinyl ether (EVE) measured at ambient temperatures and atmospheric pressure in different chambers, a borosilicate glass chamber and a FEP Teflon chamber, was determined to be (2.60 ± 0.42) × 10−16 and (1.54 ± 0.30) × 10−16 cm3 per molecule per s, respectively.12,14 The rate coefficient of the ozonolysis of n-butyl vinyl ether (n-BVE), conducted in a borosilicate glass chamber at similar conditions to the above, was determined to be (2.59 ± 0.52) × 10−16 cm3 per molecule per s.12 Studies on the ozonolysis of methyl vinyl ether and propyl vinyl ether showed that the main intermediate products in these reactions were formaldehyde and esters,15,16 and thus the formation of Criegee Intermediates (CI) in the course of the reactions was speculated, in agreement with the results observed in the ozonolysis of EVE and n-BVE.11,17 However, some of the reaction products were not identified due to limitation of the isolation technique. Moreover, the experimental data were obtained just over a narrow temperature range, 287–298 K, for the EVE ozonolysis and solely at 298 K for n-BVE ozonolysis.
The Criegee mechanism in the ozonolysis of alkenes provides extensive indirect experimental evidence, and is supported by theoretical calculations.18–22 This mechanism involves initial formation of a primary ozonide (POZ) by a 1,3 polar addition across the double bond of the alkene, forming a 1,2,3-trioxolane species.23 The reaction further leads to decomposition into an aldehyde and the proposed CI, a carbonyl oxide. The CI and the aldehyde may then recombine to form a secondary ozonide (SOZ) or react further to form a range of products.24 According to the Criegee mechanism, the general equations for the initial reaction steps of ozone with EVE and n-BVE are as follows:
|
O3 + C2H5OCHCH2 → α(C2H5OCHOO + HCHO) + (1 − α)(C2H5OC(O)H + HCHOO)
| (R1) |
|
O3 + CH3(CH2)3OCHCH2 → α(CH3CH2CH2CH2OCHO + CH2OO) + (1 − α)(CH3CH2CH2CH2OCHOO + HCHO)
| (R2) |
The matrix isolation technique is an effective method to detect unstable reactive intermediates. It has been successfully used to isolate many novel short-lived species and has the potential to be an effective tool for the stabilization of initial intermediates in the ozonolysis of ethers. Ozone and nine different olefins were deposited separately onto a cold (88 K) window, and after the window was warmed (98 to 423 K), two products were formed, presumably a π-complex and 1,2,3-trioxolane (1,2,3-trioxolane only, for C2H4).25 Further warming the window led to the π-complex decomposition, probably to the initial reactants, while POZ gave SOZ and other products. The ozone and ethylene mixtures condensed on a CsI window at 50 K reacted upon diffusion at approximately 80 K in solid xenon to form POZ and SOZ products, and strong infrared absorptions were observed.26 The ethylene POZ fundamentals were also characterized using 13C, 18O, and deuterium isotopic data. POZ and SOZ were reported from the codeposition of ozone and three different olefins (propylene, 2-trans-butene and methylpropene) in a xenon matrix at 80–110 K and a CF3Cl solution at 130–150 K.27 The codeposition onto a 14 K window of ozone with 1,2-dibromoethene and 1,2-dichloroethene in argon matrices has been examined using FTIR and it was believed to follow the Criegee pathway.28 Using matrix isolation, infrared spectroscopy, and theoretical calculations, the reaction between 1,4-cyclohexadiene and ozone was investigated and besides the widely-known Criegee mechanism, a new pathway to this reaction was found to lead to the formation of the benzene–H2O3 complex through dehydrogenation.29
In this study, matrix isolation technique combined with FTIR spectroscopy was used to identify and characterize early intermediate products in the reactions of ozone with EVE and n-BVE in argon matrices. Density functional theory (DFT) calculations were carried out to determine the structures of the reaction intermediate products, the relative energies of different reaction channels and to complement the experiments in assigning the products bands. Most likely reaction mechanisms for these ozonolysis reactions are proposed, and their atmospheric implications are discussed.
2. Material and methods
2.1. Chemicals
Ethyl vinyl ether (98%, Aladdin) and n-butyl vinyl ether (analytical standard, Aladdin) were used directly as received. O3 was produced by Tesla coil discharge of O2 (99.999%, Deyang special gas company) and trapped at 77 K to remove residual O2 and trace gases. Argon (99.999%, Deyang special gas company) was used as the matrix gas in all experiments. The mixtures of argon/sample were prepared into a mixing reservoir, which was a 5 L stainless steel vessel, and diluted with argon/sample ratio from 100/1 to 200/1.2.2. Blank experiments
Prior to every codeposition experiments, blank experiments were run on each of the parent compounds, including deposition and annealing. These blanks acted as authentic spectra of the pure starting compounds, allowing for clear identification of product bands as well as an internal standard for the extent of the reaction.2.3. Ozonolysis experiments
All the experiments were performed on a matrix isolation apparatus, using a closed-cycle helium compressor cooled cryostat (PT-SHI-4-5, Janis Research Company, USA) to achieve low temperatures. The cryostat was housed in a vacuum chamber. A pressure gauge (WRG-NW25, Edward, UK) was used to monitor the pressure inside the cryostat chamber. A base pressure of 10−5 mbar was generally recorded at the beginning of an experiment. The vacuum vessel was equipped with diamond windows throughout, and sat in the sample beam of a FTIR spectrometer (Vertex 80v, Bruker) for the duration of the experiment.Twin jet mode was used in the depositing process, in which the Ar/O3 and Ar/samples gas mixtures were fed to the cold window through separate ports in the cold head. A temperature programmed method was used to control reaction time. The two gas samples were deposited from separate jets onto the 14 K cold window at the rate of 2 mmol h−1, allowing for only a very brief mixing time prior to matrix deposition. The matrices were subsequently warmed to 25, 30 and then to 35 K to permit limited diffusion and/or reaction. These matrices were then re-cooled to 14 K and additional spectra were recorded. The spectra were recorded from 500 to 5000 cm−1 averaging 128 scans at a resolution of 0.5 cm−1. The data were analyzed with OPUS 7.2 software. The experiment of argon/sample with a ratio of 200/1 was repeated three times to ensure the results were reproducible. An additional experiment with argon/sample ratio of 100/1 was also conducted.
2.4. Computational methods
Theoretical calculations were carried out on possible intermediates using Gaussian 09 (Revision E.01).30 DFT calculations based on the B3LYP-D3 functional, which includes the dispersion correction,31 were used to locate energy minima, determine structures, and calculate vibrational spectra of all relevant intermediates. The geometry optimizations and vibrational frequency calculations were performed using the aug-cc-pVTZ basis set.32 The relative reaction energies were calculated by subtracting the sum of electronic and zero-point energies of the reactants from those of the products.3. Results and discussion
3.1. Optimized structures and reaction energies
The immediate products of the reactions of ozone with EVE and n-BVE are believed to be the initial intermediates of POZ, CIs and SOZ. Since the POZ formed in these systems are unsymmetrical, cleavage in the second step of the reaction could lead to two different CIs for the ozonolysis of EVE and n-BVE. The structures of the POZ, CIs and SOZ in the ozonolysis of EVE and n-BVE are shown in Fig. 1 and S1,† respectively. The calculated energies relative to the reactants are given in the parentheses. The relative energy values for POZs were calculated to be −50.3 and −60.8 kcal mol−1 for the EVE and n-BVE ozonolysis reactions, respectively, which indicates that the formation of POZs is energetically favorable.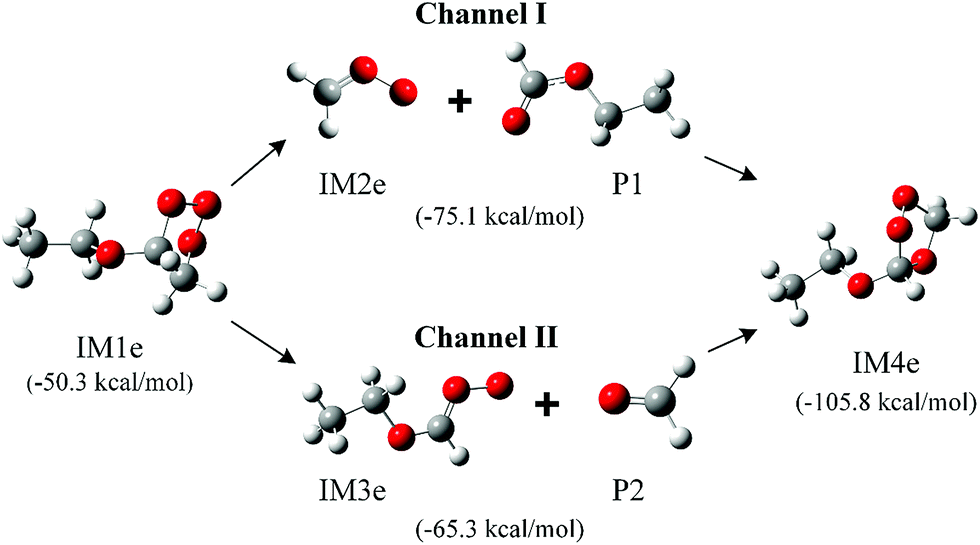 | ||
| Fig. 1 Calculated structures at the B3LYP-D3/aug-cc-pVTZ level for early intermediates in the ozonolysis of EVE. The calculated energy relative to the reactants was corrected by zero-point vibrational energy (ZPVE) and given in parentheses. | ||
For the EVE ozonolysis reaction, the relative energies of CIs from channels I and II were calculated to be −75.1 and −65.3 kcal mol−1, respectively. The relative energy of SOZ was determined to be −105.8 kcal mol−1, which is much lower than the energies of the two CIs. This low energy shows that SOZ is more stable than the CIs and, therefore, could be detected more easily. It was reported using the CCSD(T)/6-31G(d) method that the energy barrier for the formation of CI in channel I is 14.7 kcal mol−1 and the exothermicity value is 0.1 kcal mol−1.33 The dissociation process of channel II, endothermic by 4.8 kcal mol−1, had an energy barrier of 19.0 kcal mol−1. With a lower energy barrier, channel I is likely the dominant reaction pathway. A similar trend was also found in a previous study,11 where ethyl formate and CH2OO were the main intermediates. The formation energies of the CIs from n-BVE ozonolysis were −82.6 and −72.9 kcal mol−1 for channels I and II, respectively. The formation of butyl formate and CH2OO (channel I) was more favorable than the formation of formaldehyde and CH3(CH2)3OCH2OO (channel II). Han et al.17 also calculated the energy barriers towards the formation of CIs in n-BVE ozonolysis and obtained 14.5 and 19.0 kcal mol−1 for channels I and II, respectively. Their results indicate that channel I is more favorable than channel II.
3.2. Reaction mechanism and spectra
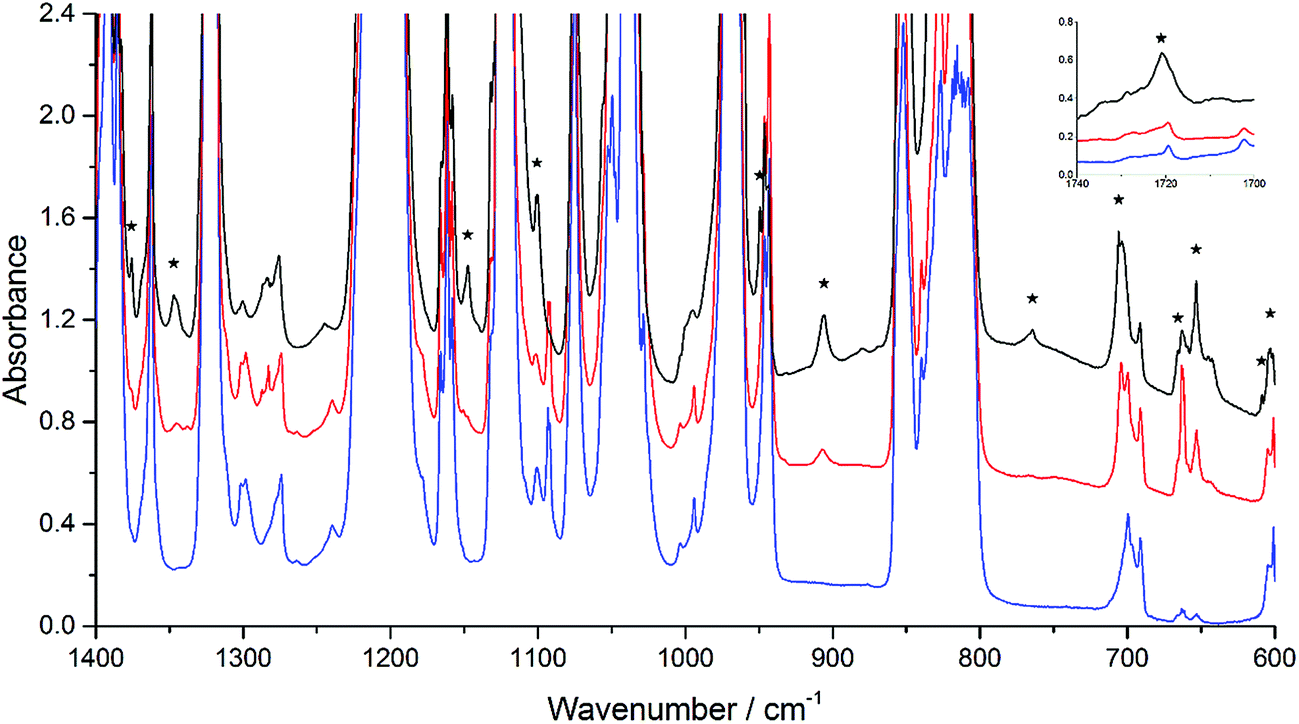 | ||
| Fig. 2 Infrared spectra of the co-deposition of the samples of Ar/EVE = 200 and Ar/O3 = 200. The red line (middle) indicates initial deposition at 14 K and the black line (top) represents the spectrum after annealing to 30 K, compared to a blank spectrum of Ar/EVE = 200 (blue, bottom). Bands marked with an asterisk are the product bands. | ||
| Experimental bands | Calculated bandsa | Assignments |
|---|---|---|
| a Calculated at the B3LYP-D3/aug-cc-pVTZ level of theory. | ||
| 653 | 754 | POZ |
| 666 | 788 | POZ |
| 706 | 812 | Ethyl formate |
| 764 | 754 | POZ |
| 906 | 906 | POZ |
| 949 | 912 | CI |
| 959 | 950 | POZ |
| 1056 | 1023 | Ethyl formate |
| 1040 | SOZ | |
| 1064 | POZ | |
| 1077 | SOZ | |
| 1100 | 1094 | CI |
| 1147 | 1151 | SOZ |
| 1142 | SOZ | |
| 1170 | SOZ | |
| 1171 | CI | |
| 1180 | SOZ | |
| 1201 | CI | |
| 1204 | CI | |
| 1249 | POZ | |
| 1347 | 1288 | SOZ |
| 1375 | 1399 | SOZ |
| 1720 | 1782 | Ethyl formate |
An additional twin jet experiment was conducted by changing the argon/EVE ratio to 100, while the argon/ozone ratio was held constant. The experiment was conducted to provide some variations in the concentration, while maintaining relatively concentrated conditions to increase the likelihood of secondary reactions. The same product bands described above were observed, and the observed band intensities varied directly with the concentrations used in the experiment. The infrared spectra at different concentrations are shown in Fig. S2.†
Some studies on the reaction of EVE with O3 have documented the formation of POZ and SOZ arising from the ozonolysis of the CC double bonds.11,33 The products of this reaction suggest that the Criegee mechanism may be taking place in this system, although the two CIs have not been observed. The possible pathway generating POZ and CI was consistent with the known, low activation barrier (∼2.5 kcal mol−1) pathway for the reaction of ozone with EVE.33 This reaction was believed to lead initially to the POZ, with an exothermicity on the order of 51 kcal mol−1. If the energy was not removed sufficiently, the reaction proceeded through a second barrier leading to decomposition of POZ and formation of CIs. For small alkenes, the barrier separating the POZ from the CI was ∼18 kcal mol−1, substantially less than the exothermicity of the initial reaction.24 The CI could either be stabilized in the matrix or undergo a recombination process to form the SOZ. The most likely step in a subsequent reaction would be decomposition by CO2 elimination from the carbonyl oxide moiety of the CI.24 However, this possibility could be eliminated by comparing experimental data to theoretical calculations33 and literature spectra.34 Further reactions leading to stable final products were not expected to occur within the matrix due to the nature of the physical environment (low temperature) and the known stability of SOZ.
While POZ, CI, and SOZ are potential, reasonable intermediate products in the EVE ozonolysis, definitive identification and band assignments are challenging. The assignments could be achieved by comparing the vibrations of functional groups obtained from theoretical calculations to their infrared absorption characteristic peaks (Table 1). A good fit between experiment and theory was best achieved by invoking the presence of all three species (POZ, CI, and SOZ) in the argon matrix. This was in agreement with the observation that the peaks located at 653, 666, 906 and 959 cm−1 grew considerably more than the others did, indicating the presence of multiple species. Due to some bands such as 1040, 1046, 1077, 1142, 1170, 1171, 1180, 1201, 1204 and 1249 cm−1 that were overlapping with parent bands or other product bands, good quantitative intensities could not be obtained for all the product bands. Nonetheless, the bands measured precisely could be reasonably assigned to POZ, CI and SOZ in the light of position and growth rate with annealing. Such a set of assignments accounted for nearly all the calculated vibrational bands of these three species with calculated intensities greater than 10 km mol−1. The only calculated band with I > 10 km mol−1 that was not observed was predicted to fall very close to the experimentally observed strong parent bands.
Vibrational modes and assignments of the bands to CIs were particularly diagnosed by comparing some modes from theoretical calculations with similar modes in other similar structures reported in the literature. One such mode for the CI was the CO stretching mode in ethyl formate, observed at 1720 cm−1, which agreed well with the calculated position of 1782 cm−1. The calculated absorption of CO stretching mode in formaldehyde is 1831 cm−1. The absorption of the POZ in this region was the weak H–C–H scissoring mode, determined to be 1522 cm−1 with low intensity. No vibrational mode for SOZ was expected in this region. Thus, the observation of the product band at 1720 cm−1 would require the presence of a carbonyl-containing species, making the fit to CI reasonable, with the systematic 62 cm−1 overestimation by theoretical calculations. The CO mode in different molecules was observed at 1733, 1742, 1740 and 1715–1722 cm−1 in cyclopentene, cyclopentadiene, 1,3-cyclohexadiene and ethylene, respectively.24,29,35
Some CI bands could be indicated from the great change of the peaks at different temperature. Calculations of bands for two CIs indicated that each had one very intense mode, the O–O stretching mode in the 900–1000 cm−1 region. Only one peak at 949 cm−1 was observed in this region. Though not obvious at 14 K, this peak appeared at 25 K and then, grew significantly as the matrix was annealed to 30 K. The peak could be definitively assigned to formaldehyde-O-oxide. Meanwhile, the characteristic intense O–O stretching modes of the two possible CI structures were computed to be 912 cm−1 for formaldehyde-O-oxide (IM2e) and 880 cm−1 for C2H5OCHOO (IM3e). The band position of formaldehyde-O-oxide was closer to the experimental peak (949 cm−1) than the peak of C2H5OCHOO. The O–O stretching mode in gaseous formaldehyde-O-oxide, measured at 298 K by a step-scan FTIR spectrometer, was located at 908 cm−1.36 The difference between this band position and the 949 cm−1 obtained in the current study is likely assigned to the difference in temperatures between the two experiments, which have different effect on the matrix. Band positions in other systems were also found to change with respect to change in temperature.37 Thus, these results support the identification of the CI and show that channel I is the dominating path in the ozonolysis of EVE.
The interesting part of the POZ molecule was the –O–O–O– group, and the information on the binding in this group could be obtained through comparison to similar molecules containing the trioxygen linkage. The relative intensities and positions between the two fundamentals for O3 and O3− showed that the weak 906 cm−1 band and the strong 764 cm−1 band were assigned to the symmetric and antisymmetric O–O–O stretching mode, respectively.38,39 A similar comparison could be made with the spectrum of CF3–O–O–O–CF3 (ref. 40) and 1,2,3-trioxolan.26 The O–O–O modes in different molecules showed that the O–O stretching mode of POZ is lower than those in ozone and inorganic ozonides. The O–O–O bonds in POZ are weak σ bonds, particularly relative to the “standard” O–O single bond fundamental at 880 cm−1 in hydrogen peroxide.41 The ease of POZ to decompose by breaking an O–O bond was also indicative of weak O–O bonds. From the calculations, the most intense absorption of EVE is the O–O–O antisymmetric stretching vibration, which was measured to be at 754 cm−1.
Recombination of CI and ethyl formate within the matrix environment may lead to the formation of SOZ. Calculations predicted that the very intense band at 1151 cm−1 was the symmetric H–C–H bending vibration of the trioxolane ring. The only candidate in this region was a product band at 1147 cm−1. This band was not observed at 14 K, but it appeared at 25 K and grew significantly at 30 K. However, the most intense computed band at 1077 cm−1, namely, the antisymmetric C–O–C stretching band of SOZ, was overlapped with other vibrational modes of the parent molecules, and it could not be identified in the experimental spectra.
Besides, the peaks at 653, 666, and 959 cm−1 were attributed to the –CH2 asymmetric bending, O–O–O symmetric bending, and O–C–O asymmetric stretching in POZ, respectively. The peaks at 706 and 1056 cm−1 were assigned to the –CH2 asymmetric bending vibration and C–C–O asymmetric stretching vibration in ethyl formate. The –CH2 wagging vibration in SOZ was observed at 1375 cm−1, while the –CH2 twisting vibration was at 1347 cm−1. With the help of the calculations, several other weak bands were also tentatively assigned (Table 1).
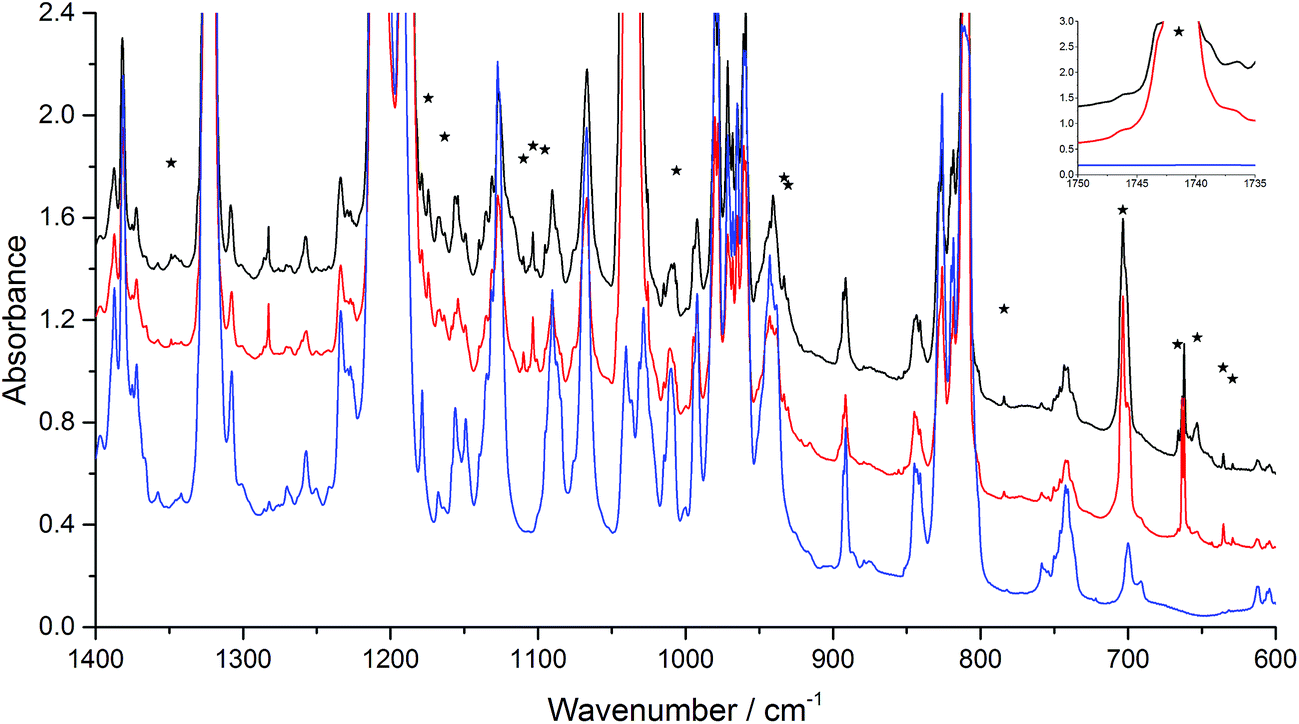 | ||
| Fig. 3 Infrared spectra of the co-deposition of the samples of Ar/n-BVE = 200 and Ar/O3 = 200. The red line (middle) indicates initial deposition at 14 K and the black line (top) represents the spectrum after annealing to 30 K, compared to a blank spectrum of Ar/n-BVE = 200 (blue, bottom). Bands marked with an asterisk are the product bands. | ||
| Experimental bands | Calculated bandsa | Assignments |
|---|---|---|
| a Calculated at the B3LYP-D3/aug-cc-pVTZ level of theory. | ||
| 635 | 640 | CI |
| 653 | 660 | SOZ |
| 666 | 673 | CI |
| 703 | 694 | POZ |
| 784 | 753 | POZ |
| 930 | 903 | POZ |
| 933 | 912 | CI |
| 1006 | 979 | POZ |
| 1021 | SOZ | |
| 1103 | 1068 | CI |
| 1072 | POZ | |
| 1109 | 1080 | SOZ |
| 1154 | 1153 | SOZ |
| 1163 | 1145 | POZ |
| 1174 | 1171 | SOZ |
| 1187 | 1205 | Butyl formate |
| 1177 | POZ | |
| 1189 | SOZ | |
| 1193 | POZ | |
| 1199 | SOZ | |
| 1742 | 1782 | Butyl formate |
On the basis of the Criegee mechanism and the results for the EVE/ozone system, likely candidates for the bands of initial products were POZ, CI, and SOZ from n-BVE ozonolysis. Band assignments were made with the comparison to the EVE ozonolysis and with the help of computational results. The band observed at 1742 cm−1 indicated the formation of CO group, which is in agreement with the calculated CO stretching of butyl formate at 1782 cm−1. The O–O stretching mode of the CI was observed at 933 cm−1, which grew significantly after annealing to 30 K. The agreement between calculated and observed band positions is very good, though some bands were overlapped by parent absorptions, supporting the formation and stabilization of CI in the n-BVE ozonolysis. Alternative explanations involving products from fragmentation could include 1-butanol formed through CO2 elimination from the CI. However, this possibility can be excluded by comparing the spectrum obtained to authentic argon matrix spectrum of 1-butanol available in the literature.42
For the POZ, particularly characteristic and intense bands were observed at 784 and 930 cm−1. The two most intense bands were determined to be the antisymmetric O–O–O stretching mode at 753 cm−1 and the symmetric O–O–O stretching mode at 903 cm−1. Similarly, the O–O stretching mode in the ring of POZ were significantly lower than those in ozone38 and in inorganic ozonide.39 The summary of the vibrations in similar structures are provided in Table 3. The comparison shows that the O–O–O bonds in the POZ from n-BVE ozonolysis are relatively weak σ bonds, which may easily be broken and lead to further reactions. This agreement for the two most distinct modes is a strong support to the identification of POZ. In addition, two other peaks were found in the experimental spectrum, the O–O–O symmetric bending vibration at 703 cm−1 and the C–C–C symmetric stretching vibration at 1163 cm−1.
Two intense bands observed at 1109 and 1154 cm−1 were assigned to the SOZ, while the two most intense bands of the SOZ from calculations were obtained at 1080 and 1153 cm−1. These bands support the identification of SOZ in the n-BVE ozonolysis reaction. The spectroscopic properties presented in this study support the observation and characterization of POZ, CI and SOZ in the reaction of ozone with n-BVE, hence providing direct evidence for the Criegee mechanism.
4. Conclusions
Matrix isolation technique combined with FTIR was used to identify and characterize the direct intermediate products formed during the EVE and n-BVE ozonolysis reactions. DFT calculations based on the B3LYP-D3/aug-cc-pVTZ level of theory were performed to determine the most likely structures, spectra, and relative energies of the direct products in these reactions. By combining experimental observations with theoretical calculations, the product bands were successfully assigned to the primary ozonides, Criegee intermediates, and secondary ozonides. Our findings demonstrate that the ozonolysis of EVE and n-BVE predominantly follow the Criegee mechanism, and the dominant pathways in these reactions lead to the formation of carbonyl oxides and corresponding esters. The current results further provide significant information to better understand the processes of the atmospheric degradation of vinyl ethers.Acknowledgements
This work was supported by National Natural Science Foundation of China (21577080, 91644214, 21407095) and Shandong Provincial Natural Science Foundation, China (ZR2014BQ013).Notes and references
- S. G. Van Ornum, R. M. Champeau and R. Pariza, Chem. Rev., 2006, 106, 2990–3001 CrossRef CAS PubMed.
- H. Singh, Y. Chen, A. Staudt, D. Jacob, D. Blake, B. Heikes and J. Snow, Nature, 2001, 410, 1078–1081 CrossRef CAS PubMed.
- F. Fehsenfeld, J. Calvert, R. Fall, P. Goldan, A. B. Guenther, C. N. Hewitt, B. Lamb, S. Liu, M. Trainer and H. Westberg, Global Biogeochem. Cycles, 1992, 6, 389–430 CrossRef CAS.
- A. Mellouki, T. J. Wallington and J. Chen, Chem. Rev., 2015, 115, 3984–4014 CrossRef CAS PubMed.
- A. Mellouki, G. Le Bras and H. Sidebottom, Chem. Rev., 2003, 103, 5077–5096 CrossRef CAS PubMed.
- M. Kalberer, J. Yu, D. R. Cocker, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2000, 34, 4894–4901 CrossRef CAS.
- U. Pöschl, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef PubMed.
- M. Scarfogliero, B. Picquet-Varrault, J. Salce, R. Durand-Jolibois and J.-F. Doussin, J. Phys. Chem. A, 2006, 110, 11074–11081 CrossRef CAS PubMed.
- G. Thiault and A. Mellouki, Atmos. Environ., 2006, 40, 5566–5573 CrossRef CAS.
- M. He, H. Wang, X. Sun, Q. Zhang and W. Wang, J. Theor. Comput. Chem., 2009, 8, 261–277 CrossRef CAS.
- G. Thiault, R. Thévenet, A. Mellouki and G. Le Bras, Phys. Chem. Chem. Phys., 2002, 4, 613–619 RSC.
- S. Zhou, I. Barnes, T. Zhu, I. Bejan and T. Benter, J. Phys. Chem. A, 2006, 110, 7386–7392 CrossRef CAS PubMed.
- L. Wang, M. Ge and W. Wang, Chem. Phys. Lett., 2009, 473, 30–33 CrossRef CAS.
- E. Grosjean and D. Grosjean, J. Atmos. Chem., 1999, 32, 205–232 CrossRef CAS.
- B. Klotz, I. Barnes and T. Imamura, Phys. Chem. Chem. Phys., 2004, 6, 1725–1734 RSC.
- S. Zhou, I. Barnes, T. Zhu, B. Klotz, M. Albu, I. Bejan and T. Benter, Environ. Sci. Technol., 2006, 40, 5415–5421 CrossRef CAS PubMed.
- D. Han, H. Cao, Y. Sun, R. Sun and M. He, Chemosphere, 2012, 88, 1235–1240 CrossRef CAS PubMed.
- W. T. Chan and I. P. Hamilton, J. Chem. Phys., 2003, 118, 1688–1701 CrossRef CAS.
- M. F. Hendrickx and C. Vinckier, J. Phys. Chem. A, 2003, 107, 7574–7580 CrossRef CAS.
- I. Ljubić and A. Sabljić, J. Phys. Chem. A, 2002, 106, 4745–4757 CrossRef.
- J. M. Anglada, R. Crehuet and J. M. Bofill, Chem.–Eur. J., 1999, 5, 1809–1822 CrossRef CAS.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- M. Clay and B. S. Ault, J. Phys. Chem. A, 2010, 114, 2799–2805 CrossRef CAS PubMed.
- M. D. Hoops and B. S. Ault, J. Am. Chem. Soc., 2009, 131, 2853–2863 CrossRef CAS PubMed.
- L. A. Hull, I. C. Hisatsune and J. Heicklen, J. Am. Chem. Soc., 1972, 94, 4856–4864 CrossRef CAS.
- C. K. Kohlmiller and L. Andrews, J. Am. Chem. Soc., 1981, 103, 2578–2583 CrossRef CAS.
- L. Andrews and C. K. Kohlmiller, J. Phys. Chem., 1982, 86, 4548–4557 CrossRef CAS.
- R. J. H. Clark and L. J. Foley, J. Phys. Chem. A, 2002, 106, 3356–3364 CrossRef CAS.
- L. Pinelo, A. D. Gudmundsdottir and B. S. Ault, J. Phys. Chem. A, 2013, 117, 4174–4182 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2013 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. Han, H. Cao, Y. Sun and M. He, Struct. Chem., 2011, 23, 499–514 CrossRef.
- A. J. Barnes and H. E. Hallam, Trans. Faraday Soc., 1970, 66, 1932–1940 RSC.
- U. Samuni, R. Fraenkel, Y. Haas, R. Fajgar and J. Pola, J. Am. Chem. Soc., 1996, 118, 3687–3693 CrossRef CAS.
- Y.-T. Su, Y.-H. Huang, H. A. Witek and Y.-P. Lee, Science, 2013, 174–177 CrossRef CAS PubMed.
- A. S. Hansen, L. Du and H. G. Kjaergaard, J. Phys. Chem. Lett., 2014, 5, 4225–4231 CrossRef CAS PubMed.
- L. Andrews and R. C. Spiker Jr, J. Phys. Chem., 1972, 76, 3208–3213 CrossRef CAS.
- R. C. Spiker Jr and L. Andrews, J. Chem. Phys., 1973, 59, 1851–1862 CrossRef.
- J. D. Witt, J. R. Durig, D. Des Marteau and R. M. Hammaker, Inorg. Chem., 1973, 12, 807–810 CrossRef CAS.
- R. L. Redington, W. B. Olson and P. C. Cross, J. Chem. Phys., 1962, 36, 1311–1326 CrossRef CAS.
- M. Kansiz, J. R. Gapes, D. McNaughton, B. Lendl and K. C. Schuster, Anal. Chim. Acta, 2001, 438, 175–186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01011g |
| This journal is © The Royal Society of Chemistry 2017 |