
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe tracer diffusion coefficient of soft nanoparticles in a linear polymer matrix
Adam E. Imela,
Sahar Rostoma,
Wade Holleyb,
Durairaj Baskarana,
J. W. Maysab and
Mark D. Dadmun *ab
*ab
aDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA. E-mail: dad@utk.edu
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 9th March 2017
Abstract
The diffusion properties of nanoparticles in polymer nanocomposites are largely unknown and are often difficult to determine experimentally. To address this shortcoming, we have developed a novel method to determine the tracer diffusion coefficient of soft polystyrene nanoparticles in a linear polystyrene matrix. Monitoring the interdiffusion of soft nanoparticles into a linear polystyrene matrix provides the mutual diffusion coefficient of this system, from which the tracer diffusion coefficient of the soft nanoparticle can be determined using the slow mode theory. Utilizing this protocol, the role of nanoparticle molecular weight and rigidity on its tracer diffusion coefficient is provided. These results demonstrate that the diffusive behavior of these soft nanoparticles differ from that of star polymers, which is surprising since our recent studies suggest that the nanoparticle interacts with a linear polymer similarly to that of a star polymer. It appears that these deformable nanoparticles mostly closely mimic the diffusive behavior of fractal macromolecular architectures or microgels, where the transport of the nanoparticle relies on the cooperative motion of neighboring linear chains. The less cross-linked, and thus more deformable, nanoparticles diffuse faster than the more highly crosslinked nanoparticles, presumably because the increased deformability allows the nanoparticle to distort and fit into available space.
Introduction
Polymer nanocomposites are produced by dispersing nano-sized particles homogeneously throughout a polymer matrix and have the potential to provide interesting pathways to next generation materials with enhanced mechanical, electrical, and transport properties.1–6 In recent publications, the diffusion of polymer chains in polymer nanocomposites has been the center of attention.7–13 Understanding the diffusion of polymers and nanoparticles in a polymer nanocomposite provides fundamental insight that is needed to understand and control nanocomposite properties, long-term stability, and processability.14–17Current research in the dynamics of polymer nanocomposites has focused on the effect of nanoparticles on the diffusion of the linear polymer chains, with the assumption that the nanoparticles are stationary.9,11,13,18 Most studies that assume the nanoparticles are stationary employ nanoparticles that have hard impenetrable cores, such as inorganic SiO2 nanoparticles. For instance, the diffusion of a polymer chain in a nanocomposite consisting of silica nanoparticles in a matrix of poly(methyl methacrylate) or polystyrene, as well as silica nanoparticles grafted with polystyrene in a polystyrene matrix, have been studied. Each of these studies demonstrate that the diffusion of the polymer chain is inhibited by the presence of the nanoparticle, and the extent of slowing down scales with a constraint parameter that is the ratio of the interparticle distance and two times the radius of gyration of the polymer.7,11–13 The interparticle distances in a nanocomposite is controlled by the size and loading of the nanoparticles. The interparticle distances created by the nanoparticles can be thought of as bottlenecks, where the polymer chains diffusion through the bottlenecks requires the polymer chains to unravel, ultimately decreasing the entropy of the polymer chain. This type of diffusive mechanism is described well by the entropic barrier model.19
The diffusive behavior of linear polymer chains in the presence of soft nanoparticles has also been studied. Contrary to studies that utilize hard nanoparticles, these experiments show that the inclusion of ∼10 nm soft nanoparticles in a polymer matrix increases the diffusion coefficient of the linear polymer chain. This increase is ascribed to an increase in constraint release mechanisms in the reptation of the polymer chain, as with the diffusion of linear polymers in the presence of star polymers.20
The diffusion or motion of nanoparticles in polymer nanocomposites is much less studied, primarily because determining the dynamics of nanoparticles in a polymer matrix is a challenge due to their slow motion relative to the polymer chain and difficulty in tuning the contrast between nanoparticle and matrix. We therefore sought to develop a method to determine the diffusion coefficient of nanoparticles to quantify the mobility of the purported obstacles to polymer chain diffusion in polymer nanocomposites. In particular, we focus on soft organic nanoparticles, which have not been studied as extensively as hard, impenetrable nanoparticles. In these studies, the soft nanoparticles of interest are essentially crosslinked polystyrene molecules that are synthesized through a nano-emulsion technique. Moreover, previously reported results from our group20 indicate that these penetrable soft nanoparticles may be mobile, and that their mobility may impact its interaction with neighboring polymer chains. Here we report the development of a protocol to determine the tracer diffusion coefficient of soft polystyrene nanoparticles in a polymer matrix using neutron reflectivity and provide insight into the role of nanoparticle rigidity on its mobility. The reported results show that the soft nanoparticles are not stationary, as is assumed with inorganic-based nanoparticles, but do move much more slowly than the polymer chains. This result is consistent with our previously reported findings, which indicate that there is an increased complexity in the physics governing the dynamics of all components in soft nanoparticle nanocomposites. Furthermore, we show that the mutual diffusion of the nanoparticle and polymer in a polymer nanocomposite with mobile nanoparticles is described by the slow mode theory of diffusion. This protocol provides a method to analyze the impact of nanoparticle molecular weight and rigidity on its diffusive properties, where these results provide additional insight into the underlying physics that govern the dynamic behavior of these unique additives.
Experimental
A series of bilayer samples were prepared on polished silicon wafers, where each sample is composed of deuterated polystyrene (dPS) as the top layer and a layer of the soft nanoparticle as the bottom layer. The intramolecular cross-linked soft polystyrene nanoparticles used in our study were synthesized by a nano-emulsion polymerization, the details of which can be found in Holley et al.21 The dPS, with a number average molecular weight of 535![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, was purchased from Polymer Source and used without further purification. The silicon wafers (2 inches in diameter; 5 or 6 mm thick) were first placed in a ‘piranha’ solution, which consists of 3:1 sulfuric acid:(30%) hydrogen peroxide to remove any organic residue. The silicon wafers were then rinsed with deionized water and dried under a stream of nitrogen gas. The silicon wafers were then placed in an ultraviolet light/ozone cleaner for 15 minutes further removing organic contaminants and reproducibly forming an oxide surface.
000 g mol−1, was purchased from Polymer Source and used without further purification. The silicon wafers (2 inches in diameter; 5 or 6 mm thick) were first placed in a ‘piranha’ solution, which consists of 3:1 sulfuric acid:(30%) hydrogen peroxide to remove any organic residue. The silicon wafers were then rinsed with deionized water and dried under a stream of nitrogen gas. The silicon wafers were then placed in an ultraviolet light/ozone cleaner for 15 minutes further removing organic contaminants and reproducibly forming an oxide surface.
Layers of the nanoparticles were spin cast from toluene solutions with weight percentages that range from 1 wt% to 1.5 wt% relative to the solvent onto 2-inch wide silicon wafers. The dPS top layers were then spin cast onto separate silicon wafers (4 inches in diameter; 1 mm thick), which were also treated with the piranha solution and UV/ozone cleaner as previously described, and then floated off of the silicon wafers onto nanopure water. The floated dPS layers were then picked up by the protonated nanoparticle layers, creating a bilayer. The bilayers were dried in a desiccator under vacuum for a minimum of three days. Before each neutron reflectivity measurement, other than the as cast samples, the samples were annealed under vacuum at 150 °C for varying times. Immediately after annealing, the samples were quenched to room temperature on a frozen aluminum block, halting the interdiffusion process.
The specular reflectivity measurements were conducted at the Spallation Neutron Source in the Oak Ridge National Laboratory utilizing the liquids reflectometer, beam line 4B and the National Institute of Standards and Technology Center for Neutron Research utilizing the Polarized Beam Reflectometer. All reflectivity experiments were conducted in air and at room temperature. The reflectivity is plotted as a function of the momentum transfer vector perpendicular to the surface, qz, where θ is the angle of incidence and λ is the wavelength of the incident neutrons.
|
qz = (4π/λ)sinθ
| (1) |
Modeling of the reflectivity curves was completed using the analysis package MOTOFIT in the IGOR Pro environment. The MOTOFIT procedure uses a slab-model approach with the Abeles matrix method and a non-linear regression extension for the roughness. More detailed discussions concerning the neutron reflectivity technique and the MOTOFIT modeling package are available elsewhere.22–24
For each nanoparticle, the crosslink density, and thus rigidity of the nanoparticle, is controlled by the amount of divinyl benzene (DVB) crosslinker that is added to the reaction. The nanoparticles studied include NP1, which has 0.80 mol% DVB, NP2 with 1.91 mol% DVB, and NP3 with 4.60 mol% DVB. Interestingly, the increasing amounts of DVB only modify the radius of gyration of the nanoparticles modestly, from 12.9 nm for NP1, to 11.3 nm for NP2 and 9.85 nm for NP3. The morphology of the particles is best described as a nano-gel with homogeneous cross-links producing a distinct core and a fuzzy corona that consist of dangling free chains ends and loops.21
Results and discussion
The first set of experiments serve as proof-of-principle to verify the diffusion of the nanoparticle into the polystyrene matrix. Fig. 1 shows the neutron reflectivity of the dPS/NP1 bilayer as-cast and after it has been annealed for ∼10 hours at 150 °C. The as-cast data has distinct fringes indicating a sharp interface between the bottom (NP1) and top (linear dPS) layers. After nearly 10 hours of annealing the samples at 150 °C in vacuum, the fringes decrease substantially, a distinct trait indicating that the two layers are diffusing into each other. The scattering length density (SLD) profiles of the best fits to the data in Fig. 1 are shown in Fig. 2, where the sharp interface between the layers in the as-cast sample is depicted by a vertical transition around 1000 Å. | ||
| Fig. 1 Representative reflectivity data of the NP1–dPS bilayer, showing the reflectivity and fits of the as-cast sample and after it has been annealed for 10 hours. The data have been scaled for clarity. | ||
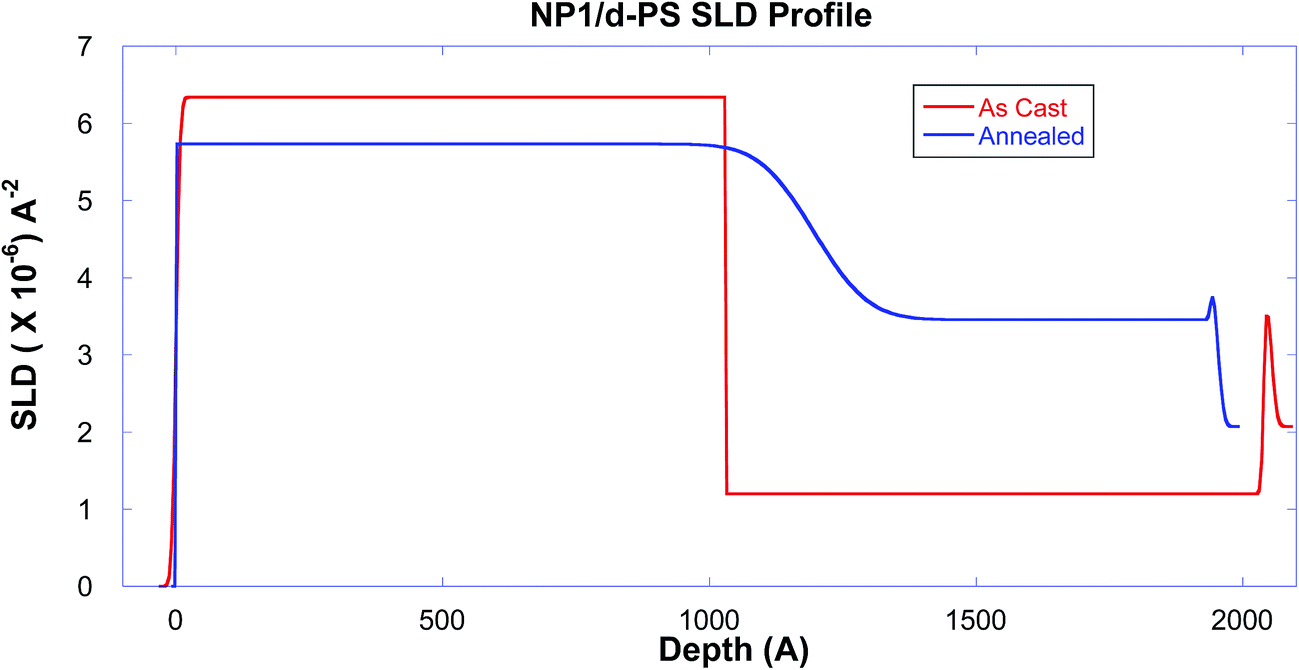 | ||
| Fig. 2 The scattering length density profiles of the NP1 sample, derived from the fits of the reflectivity data as shown in Fig. 1. The observed change clearly demonstrates that the linear polymer chain and nanoparticle inter-diffuse in the bilayer during this annealing. | ||
These experiments were extended to monitor the change in the density profile of the dPS/nanoparticle bilayers for all nanoparticles as a function of annealing time at 150 °C for increased annealing times. In these experiments, all of the nanoparticles diffused into the dPS layer, where the data can be analyzed to determine the diffusion coefficients of NP1, NP2 and NP3.
However, because the nanoparticle and polymer chains diffuse at different rates, the analysis of the data to determine the tracer diffusion coefficient of the nanoparticle is non-trivial. The interdiffusion of the two layers is the result of the mutual diffusion of the nanoparticles diffusing into the linear polymer and vice versa. The mutual diffusion coefficient, DM, of this process is determined from the time evolution of the bilayer interface by fitting the density profile of the bilayer after annealing at time, t, to the one-dimensional solution to Fick's second law of diffusion, shown in eqn (2).25
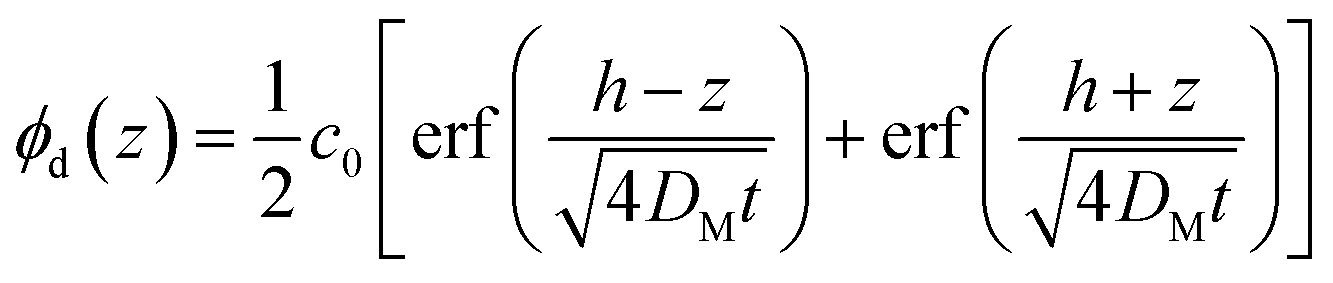 | (2) |
In this equation, t and h are the annealing time in seconds and the initial dPS thickness. Fitting this equation to the dPS density profile provides the mutual diffusion coefficient in the units cm2 s−1. The density profile of the deuterated material, ϕd(z) for a given sample at a given thermal annealing time is determined from the scattering length density profile that results from fitting the reflectivity data, SLDm(z), using eqn (3).
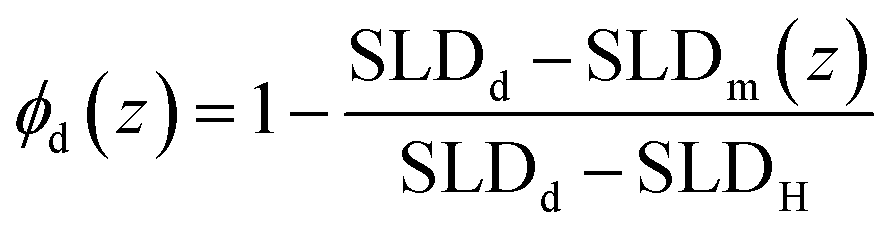 | (3) |
In eqn (3), the subscript d stands for the deuterated PS while the subscript H denotes the protonated nanoparticle and the subscript m represents the SLD of the sample as measured in the experiment. Fig. 3, 4, and 5 illustrate the change in the volume fraction profiles of the NP1, NP2 and NP3 bilayers, respectively, as a function of (short) annealing time. Inspection of these plots shows that all three of the as-cast samples exhibit a nearly vertical transition between layers indicative of a smooth interface. After each annealing time, the interface between the two layers becomes more diffuse for each bilayer, which translates to a broader transition between the layers because of the interdiffusion of the dPS and the nanoparticle.
 | ||
| Fig. 3 The volume fraction profiles of the as-cast and annealed bilayers for the NP1 sample. | ||
 | ||
| Fig. 4 The volume fraction profiles of the as-cast and annealed bilayers for the NP2 sample. | ||
 | ||
| Fig. 5 The volume fraction profiles of the as-cast and annealed bilayers for the NP3 sample. | ||
The behavior of these bilayers is analyzed at low annealing times (<60 minutes) to evaluate the best method to determine the tracer diffusion coefficient of the soft nanoparticle from the mutual diffusion coefficient that is the result of fitting the interdiffusion of the two layers to Fick's second law. The extracted mutual diffusion coefficients after the longest annealing time for this initial study are presented in Table 1.
| DM (×10−17) cm2 s−1 | Dt,slow (×10−17) cm2 s−1 | Dt,fast (×10−16) cm2 s−1 | |
|---|---|---|---|
| NP1 | 1.35 | 5.56 | −4.58 |
| NP2 | 1.81 | 7.31 | −4.33 |
| NP3 | 4.05 | 12.9 | −2.66 |
To extract the tracer diffusion coefficient of the nanoparticle, the mutual diffusion coefficient must be quantitatively related to the tracer diffusion coefficient of each component. In Fick's law of diffusion, eqn (4), the mutual diffusion coefficient is defined as the proportionality between the flux, Ji, and concentration gradient, Δci, or volume fraction.
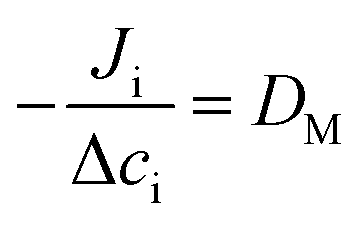 | (4) |
The mutual diffusion coefficients can be related to the tracer diffusion coefficients by noting that the mutual diffusion coefficient is related to the Onsager transfer coefficient DT, in terms of the Flory–Huggins interaction parameter and the volume fractions ϕi of the components as shown in eqn (5).26–28
| DM = 2(χs − χ)ϕ1ϕ2DT | (5) |
The segment–segment interaction parameter χ is zero in our athermal system due to the matrix and nanoparticles being nearly chemically identical and the interaction parameter at the spinodal χs is estimated by eqn (6).26
 | (6) |
In order to obtain the contribution of the motion of each component to the experimentally determined mutual diffusion coefficients, the relationship between the tracer diffusion coefficients of the two components to the Onsager transfer coefficient DT, is required. Two theories have been presented to define this relationship, the fast mode theory and the slow mode theory.29–32
The fast mode theory, also referred to as the ‘vacancy model’, is most often used to describe lower molecular weight systems, where the faster diffusing component controls the overall diffusion of the system.32 The fast mode theory defines the relationship between DT and the tracer diffusion coefficients of the individual components as shown in eqn (7).29,30
| DT = ϕNPDPSNPS + ϕPSDNPNNP. | (7) |
In eqn (7), the subscripts PS and NP denote the property of the linear polystyrene chain and nanoparticles, respectively, and Di denotes the tracer diffusion coefficient of component i. The volume fraction ϕi for each component is set to 0.5 because this is the composition at the inflection point of the slope in the volume fraction profiles. N represents the degree of polymerization of the components, where the molecular weights of the nanoparticles are reported by Holley et al.21 After substituting eqn (5) and (6) in for DT and using the known value of DPS from previous experiments, all variables are known in this equation except the tracer diffusion coefficient of the nanoparticle, DNP.
The slow mode theory, on the other hand, is most appropriate for higher molecular weight systems, where mutual diffusion is controlled by the slowest diffusing component in the polymeric system and is applicable to incompressible binary mixtures.31 The slow mode theory correlates the tracer diffusion coefficients of the individual components to the Onsager transfer coefficient as described in eqn (8), where all parameters are identical to those listed in eqn (7).32 Similar to the fast mode theory, substituting eqn (5) and (6) in for DT and using the known value of DPS, all variables are known except the tracer diffusion coefficient of the nanoparticle, DNP
 | (8) |
When the experimentally determined mutual diffusion coefficients were analyzed using the fast mode theory, the analysis results in negative tracer diffusion coefficients of the nanoparticles, as listed in Table 1. Therefore, the fast mode theory does not describe the diffusive behavior of this system because a negative diffusion coefficient is not physically realistic. When the data are analyzed with the slow mode theory, however, positive and reasonable tracer diffusion coefficients emerge for the nanoparticles, as listed in Table 1. The analysis of the data with the slow mode theory, thus provides a physically realistic tracer diffusion coefficient of the nanoparticle, where the fast mode theory does not. Thus, the mutual diffusion of the soft nanoparticle and linear polymer is dominated by the slow nanoparticle diffusion.
The tracer diffusion coefficients of the nanoparticle as a function of (early) annealing time as determined using the slow mode theory are presented in Fig. 6. The tracer diffusion coefficients for the nanoparticles change quite rapidly with annealing time, which indicates that the samples have not entered the diffusive regime, and the analysis above is monitoring the sub-diffusive motions of the nanoparticle. Therefore, further experiments were completed to extend these results into the diffusive region.
 | ||
| Fig. 6 The tracer diffusion coefficient for NP1, NP2 and NP3 plotted as a function of annealing time for short times. | ||
Thus, the interdiffusion of the linear polystyrene and soft polystyrene nanoparticle was monitored for much longer annealing times, past the sub-diffusive state and into the diffusive regime, which is denoted by a constant diffusion coefficient with annealing time. Fig. 7 plots the tracer diffusion of the nanoparticles as determined using the slow mode theory out to annealing times of 63 hours, where the diffusion coefficient becomes constant. The tracer diffusion coefficients of these nanoparticles are thus listed in Table 2.
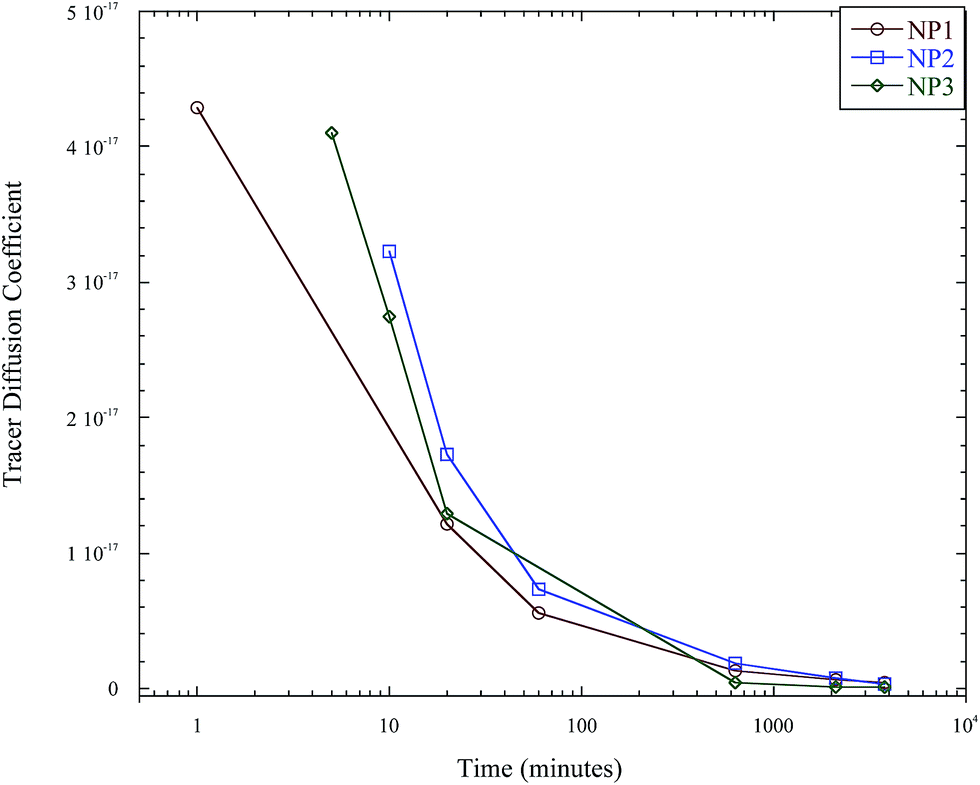 | ||
| Fig. 7 Instantaneous tracer diffusion coefficient of the soft nanoparticles as a function of annealing time. | ||
| % crosslinker | Dt,slow (×10−20) cm2 s−1 | |
|---|---|---|
| NP1 | 0.81 | 39.5 |
| NP2 | 1.91 | 30.8 |
| NP3 | 4.60 | 7.03 |
The collection of this data enables the further analysis of the dependence of the tracer diffusion coefficients of these nanoparticles on molecular weight and crosslink density. Fig. 8 and 9 show the dependence of the soft nanoparticle tracer diffusion coefficient on its molecular weight and crosslink density, respectively. Both an increase in molecular weight and crosslink density decrease the tracer diffusion coefficient of the nanoparticle, with the molecular weight dependence following a DNP ∼ Mw−3.8 dependence and an increase in crosslink density from 1% to 4% decreases NP diffusion by a factor of ∼5.
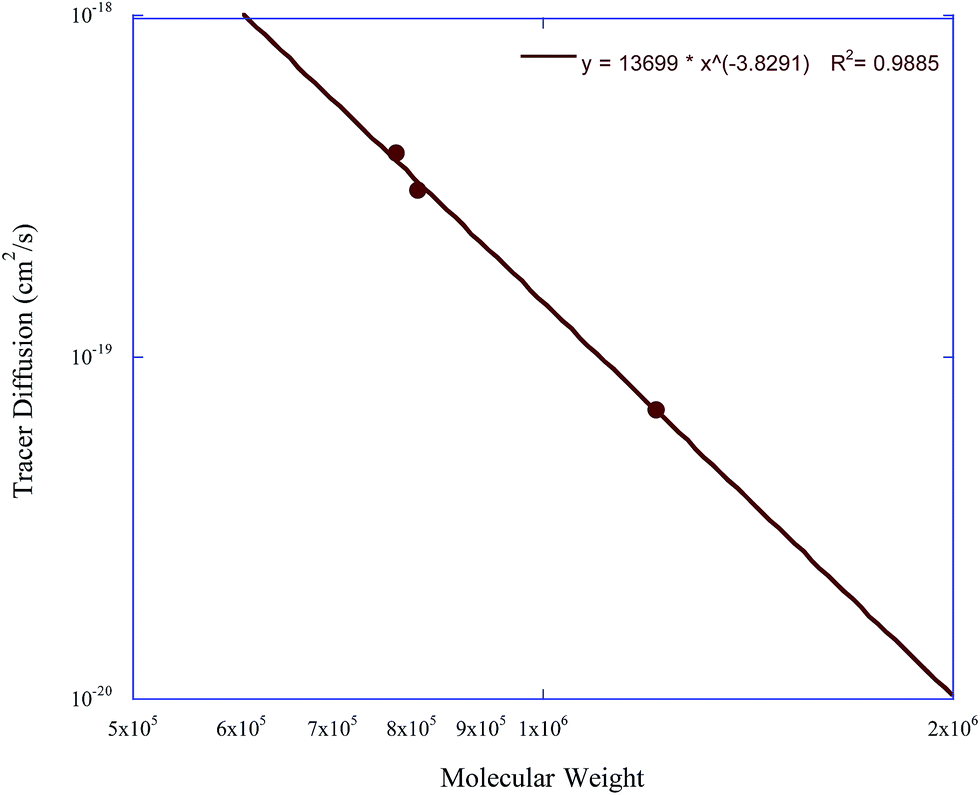 | ||
| Fig. 8 The tracer diffusion coefficient of the soft nanoparticles as a function of molecular weight. | ||
 | ||
| Fig. 9 The tracer diffusion coefficient of the soft nanoparticles as a function of rigidity as quantified by the nominal crosslink density. | ||
It is interesting to observe that the diffusion of the nanoparticle does not follow an exponential dependence on molecular weight that is expected for the diffusion of star polymers,33–35 as our recent studies suggest that the presence of the nanoparticle in a linear polymer matrix interacts with the linear polymer similarly to that of a star polymer.20 It appears that the diffusion of these crosslinked nanoparticles is slower than reptation, but not as slow as the diffusion of star polymers, where their diffusion mimics the expected behavior of fractal macromolecular architectures36 or microgels.33
The understanding of the diffusive behavior of such assemblies invokes the idea of cooperative motion, where the transport of the nanoparticle requires the motion of the linear polymer chains to open a pathway for their transport. The cooperative motion of the linear polymer chain is required, because the nanoparticles are larger than the tube diameter (∼8 nm) of the polystyrene chain.
This interpretation is also consistent with the change in the nanoparticle's diffusion coefficient with crosslink density (i.e. rigidity) as shown in Fig. 9, where it appears that the increased deformability of the lower crosslink density allows the nanoparticle to distort and fit into available space for motion. This mutually cooperative motion of the nanoparticle and linear polymer increases the rate of nanoparticle diffusion as it provides additional pathways for transport.
Comparing the diffusion coefficients of these soft nanoparticles to that of a 10 nm sphere that obeys Stokes Einstein (∼4 × 10−18 cm2 s−1) shows that the rate of diffusion of these crosslinked molecules (0.7–4 × 10−19 cm2 s−1) is less than that of the Stokes Einstein sphere. This difference suggests that there are more complicated interactions between the nanoparticle and surrounding polymer matrix than simple friction/drag. One possibility is that the corona and deformability of the nanoparticle provides a pathway for the two molecules to entangle.
Fig. 10 compares the diffusion coefficients of nanoparticles with the diffusion of their linear analogues. All three nanoparticles are three orders of magnitude slower than their linear analogs, further documenting their more complicated dynamics due to their distinctive crosslinked morphology.
 | ||
| Fig. 10 Comparison of the tracer diffusion coefficient of the soft nanoparticles to the tracer diffusion coefficients of linear polymers with the same molecular weight. | ||
To tease out the effect of crosslinking on the diffusion of the nanoparticle and exclude molecular weight effects, the ratio of the diffusion coefficient of the linear analogue and that of the soft NP (Dlinear/DNP) is plotted as a function of nominal crosslink density, divinylbenzene concentration, [DVB], in Fig. 11. The increase in this ratio with crosslink density further validates the impact of the deformability or softness of the nanoparticle on its mobility. Even once any molecular weight effects are accounted for, the increased rigidity of the nanoparticle translates into a slowing down of the nanoparticle motion. Clearly, the deformability of the nanoparticle improves its ability to diffuse in the polymer matrix.
 | ||
| Fig. 11 The ratio of the tracer diffusion coefficient of the linear polymer to that of the soft nanoparticle as a function of crosslink density. | ||
Conclusion
We have developed and utilized a novel method to determine the tracer diffusion coefficient of soft polystyrene nanoparticles in a polymer matrix. This method correlates the tracer diffusion coefficient of the soft nanoparticle and linear polymer chain to the experimentally determined mutual diffusion coefficient applying the fast and slow mode theories of diffusion.Ultimately, these results show that the slow mode diffusion theory describes the nanoparticle diffusion. This result also illustrates that the soft polystyrene nanoparticles are not stationary but the nanoparticle center of mass motion is significantly slower than that of an equivalent linear polystyrene chain. Further analysis indicates that an increase in molecular weight or crosslink density (i.e. rigidity) decreases the tracer diffusion coefficient of the nanoparticle. The nanoparticle diffusion differs from that of star polymers, in that it does not follow an exponential dependence on molecular weight. Rather, the diffusion of these nanoparticles more closely mimics the expected diffusion behavior of fractal macromolecular architectures or microgels, where the motion of the nanoparticle requires a cooperative motion of the polymer chain to diffuse. This interpretation is also consistent with the observation that the nanoparticle diffusion rate decreases with crosslink density, where the increased deformability of the lightly crosslinked molecules provides increased conformational freedom that translates to additional pathways for transport.
Acknowledgements
This research is supported by the Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering. The support of the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy, who sponsors the Oak Ridge National Laboratory Spallation Neutron Source is gratefully acknowledged. We also acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce for use of the Polarized neutron reflectometer.References
- J. Jordan, K. I. Jacob, R. Tannenbaum, M. A. Sharaf and I. Jasiuk, Experimental trends in polymer nanocomposites—a review, Mater. Sci. Eng., A, 2005, 393, 1–11 CrossRef
.
- D. Ciprari, K. Jacob and R. Tannenbaum, Characterization of Polymer Nanocomposite Interphase and Its Impact on Mechanical Properties, Macromolecules, 2006, 39, 6565–6573 CrossRef CAS
- U. Khan, K. Ryan, W. J. Blau and J. N. Coleman, The effect of solvent choice on the mechanical properties of carbon nanotube–polymer composites, Compos. Sci. Technol., 2007, 67, 3158–3167 CrossRef CAS
- P. Banerjee, et al., Plasmon-induced electrical conduction in molecular devices, ACS Nano, 2010, 4, 1019–1025 CrossRef CAS PubMed
- C. Mattevi, et al., Evolution of Electrical, Chemical, and Structural Properties of Transparent and Conducting Chemically Derived Graphene Thin Films, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS
- F. Puosi and D. Leporini, Scaling between relaxation, transport, and caged dynamics in polymers: from cage restructuring to diffusion, J. Phys. Chem. B, 2011, 115, 14046–14051 CrossRef CAS PubMed
- C. Lin, et al., Do Attractive Polymer–Nanoparticle Interactions Retard Polymer Diffusion in Nanocomposites?, Macromolecules, 2013, 46, 4502–4509 CrossRef CAS
- J. Choi, M. J. A. Hore, N. Clarke, K. I. Winey and R. J. Composto, Nanoparticle Brush Architecture Controls Polymer Diffusion in Nanocomposites, Macromolecules, 2014, 47, 2404–2410 CrossRef CAS
- J. S. Meth, et al., Excluded volume model for the reduction of polymer diffusion into nanocomposites, J. Phys. Chem. B, 2013, 117, 15675–15683 CrossRef CAS PubMed
- J. T. Kalathi, U. Yamamoto, K. S. Schweizer, G. S. Grest and S. K. Kumar, Nanoparticle Diffusion in Polymer Nanocomposites, Phys. Rev. Lett., 2014, 112, 108301 CrossRef PubMed
- J. Choi, et al., Universal Scaling of Polymer Diffusion in Nanocomposites, ACS Macro Lett., 2013, 2, 485–490 CrossRef CAS
- S. Gam, et al., Polymer diffusion in a polymer nanocomposite: effect of nanoparticle size and polydispersity, Soft Matter, 2012, 8, 6512 RSC
- S. Gam, et al., Macromolecular Diffusion in a Crowded Polymer Nanocomposite, Macromolecules, 2011, 44, 3494–3501 CrossRef CAS
- M. P. Stevens, Polymer additives: II. Chemical and aesthetic property modifiers, J. Chem. Educ., 1993, 70, 535 CrossRef CAS
- M. P. Stevens, Polymer additives: Part I. Mechanical property modifiers, J. Chem. Educ., 1993, 70, 444–448 CrossRef CAS
- M. P. Stevens, Polymer additives: III. Surface property and processing modifiers, J. Chem. Educ., 1993, 70, 713 CrossRef CAS
- P. Rouse, A Theory of the Linear Viscoelastic Properties of Dilute Solutions of Coiling Polymers, J. Chem. Phys., 1953, 21, 1272–1280 CrossRef CAS
- C. Lin, K. Ohno, N. Clarke, K. I. Winey and R. J. Composto, Macromolecular Diffusion through a Polymer Matrix with Polymer-Grafted Chained Nanoparticles, Macromolecules, 2014, 47, 5357–5364 CrossRef CAS
- M. Muthukumar, Entropic barrier model for polymer diffusion in concentrated polymer solutions and random media, J. Non-Cryst. Solids, 1991, 131–133, 654–666 CrossRef CAS
- B. Miller, A. Imel, D. W. Holley, D. Baskaran, J. W. Mays and M. D. Dadmun, The Role of Nanoparticle Rigidity on the Diffusion of Linear Polystyrene in a Polymer Nanocomposite, Macromolecules, 2015, 48, 8369–8375 CrossRef CAS
- D. W. Holley, M. Ruppel, J. W. Mays, V. S. Urban and D. Baskaran, Polystyrene nanoparticles with tunable interfaces and softness, Polymer, 2014, 55, 58–65 CrossRef CAS
- A. Nelson, Co-refinement of multiple-contrast neutron/X-ray reflectivity data using MOTOFIT, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS
- T. Russell, A. Karim, A. Mansour and G. Felcher, Specular Reflectivity of Neutrons by Thin Polymer Films, Macromolecules, 1988, 21, 1890–1893 CrossRef CAS
- T. Russell, X-ray and neutron reflectivity for the investigation of polymers, Mater. Sci. Rep., 1990, 5, 171–271 CrossRef CAS
- P. Mills, P. F. Green, C. J. Palmstrom, J. W. Mayer and E. J. Kramer, Polydispersity Effects on Diffusion in Polymers: Concentration Profiles of D-Polystyrene Measured by Forward Recoil Spectrometry, J. Polym. Sci., Part B: Polym. Phys., 1986, 24, 1–9 CrossRef CAS
- M. J. Arlen, M. D. Dadmun and W. A. Hamilton, Using neutron reflectivity to determine the dynamic properties of a copolymer in a homopolymer matrix, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3235–3247 CrossRef CAS
- R. J. Composto, E. J. Kramer and D. M. White, Mutual Diffusion in the Miscible Polymer Blend Polystyrene/Poly(xylenyl ether), Macromolecules, 1988, 21, 2580–2588 CrossRef CAS
- P. F. Green, in Diffusion in Polymers, ed. P. Neogi, 1996, pp. 251–298 Search PubMed
- H. H. Kausch and M. Tirrell, Polymer Interdiffusion, Annu. Rev. Mater. Sci., 1989, 19, 341–377 CrossRef CAS
- E. Kim, E. J. Kramer, J. O. Osby and D. J. Walsh, Mutual Diffusion and Thermodynamics in the Blends of Polystyrene and Tetramethylbisphenol-a Polycarbonate, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 467–478 CrossRef CAS
- A. Z. Akcasu, G. Naegele and R. Klein, Remarks on the “fast” and “slow” mode theories of interdiffusion, Macromolecules, 1995, 28, 6680–6683 CrossRef CAS
- A. Z. Akcasu, The “fast” and “slow” mode theories of interdiffusion in polymer mixtures: resolution of a controversy, Macromol. Theory Simul., 1997, 6, 679–702 CrossRef CAS
- M. Antonietti and H. Sillescu, Diffusion of Intramolecular Cross-Linked and Three-Arm-Star Branched Polystyrene Molecules in Different Matrices, Macromolecules, 1986, 19, 798–803 CrossRef CAS
- F. R. Colley, S. A. Collins and R. W. Richards, Tracer diffusion of four arm polystyrene star molecules into linear and star polymer matrices from nuclear reaction analysis, J. Mater. Chem., 2003, 13, 2765–2770 RSC
- N. Clarke, F. R. Colley, S. A. Collins, L. R. Hutchings and R. L. Thompson, Self-Diffusion and Viscoelastic Measurements of Polystyrene Star Polymers, Macromolecules, 2006, 39, 1290–1296 CrossRef CAS
- M. Fuchs and K. S. Schweitzer, Mode-coupling theory of the slow dynamics of polymeric liquids: fractal macromolecular architectures, J. Chem. Phys., 1997, 106, 347–375 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |