
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A stable and highly efficient visible-light photocatalyst of TiO2 and heterogeneous carbon core–shell nanofibers†
Jing Cheng ,
Yuting Wang,
Yan Xing,
Muhammad Shahid and
Wei Pan*
,
Yuting Wang,
Yan Xing,
Muhammad Shahid and
Wei Pan*
State Key Laboratory of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing, 100084, PR China. E-mail: panw@mail.tsinghua.edu.cn; Fax: +86-10-62771160; Tel: +86-10-62772858
First published on 7th March 2017
Abstract
A novel core–shell heterostructure of TiO2 nanofibers with carbon quantum dots embedded in an amorphous carbon shell has been successfully prepared via a simple electrospinning and impregnation process. Here, carbon quantum dots (CQDs) are designed as sensitizers for the visible-light response and amorphous carbon ensures intimate contact with TiO2. The photocatalytic performance is evaluated by the degradation of rhodamine-B under visible light irradiation. It is found that the composite nanofibers with an appropriate thickness of carbon shell exhibit a stable and highly efficient photocatalytic activity, and the apparent quantum efficiency can reach as high as 52%, which is about 10 times that of pure TiO2 nanofibers. Structural analyses show that the enhanced photocatalytic activity is attributed to the synergistic effect of TiO2, the amorphous carbon thin shell and the CQDs embedded inside. Due to the intimate contact between TiO2 and the carbon shell, the photogenerated electrons can be easily transferred from the CQDs to TiO2 resulting in a longer lifetime of the photogenerated electron–hole pairs and a higher photocatalytic activity. In addition, the unique upconversion properties of the CQDs enables the nanofibers to utilize more solar energy and increase the photocatalytic activity. Also, the carbon shell can induce more oxygen vacancies on the surface of the nanofibers, which can further enhance the photocatalytic activity. The results in this work may be beneficial to the future study of exploring new carbon-based heterostructured materials for visible-light-driven photocatalysts.
1. Introduction
Nowadays, increasing concerns of energy consumption and environmental problems have driven a demand for cost-effective clean energy. As a result, photocatalysts, which convert solar energy into chemical energy, have been extensively investigated.1 Among them, titanium dioxide is the most researched photocatalytic material due to its high stability, low cost and non-toxicity.2–4 Though a large amount of research has been reported on nano-TiO2 since it came to light, there are still some problems that remain to be solved to get improved photocatalytic performance. One is that its wide bandgap (3.2 eV) confines the application of TiO2 to the ultraviolet region, which is only 5% of the total solar spectrum. Furthermore, the photocatalytic efficiency of TiO2 is relatively low due to the high recombination rate of the photogenerated electrons and holes.5–7 To efficiently utilize solar light and increase the lifetime of the photogenerated electron hole pairs, much research has been carried out on surface modification, doping or coupling with other materials to form heterostructures.8–10 Among these approaches, modification of TiO2 with nonmetallic materials is a way to get better photocatalytic performance.11–13As a common nonmetallic material, carbon, for example carbon nanotube, graphene, carbon quantum dot (CQD) and amorphous carbon, shows many excellent characteristics for photocatalysis. It has large electron storage capacity, which can capture the photo-excited electrons and enhance the charge transfer across the interface, resulting in efficient separation of the electron–hole pairs.14 Besides, carbon can assist the adsorption of the pollutant on the surface of photocatalysts, which is beneficial for the photocatalytic process.15 Among the various types of carbon, graphene is one of the most commonly used materials to suppress the recombination of the photogenerated electron–hole pairs.16 However, the contact of graphene with the semiconductor is poor, which is a crucial factor that influences the electron transport.17 So, better contact of carbon with the semiconductor material is desired. Here, a core–shell heterostructure of TiO2 and amorphous carbon embedded with CQDs composite nanofibers is prepared. The amorphous carbon is introduced to increase the contact area of the carbon and TiO2. In addition, CQDs have many advantages over other carbon-based nanostructures and other QDs like CdS QDs18 such as higher photo-induced electron transfer and the up conversion properties. Thus, enhanced photocatalytic activity can be expected on the composite nanofibers.
In recent years, one-dimensional semiconductor nanomaterials, such as nanofibers and nanorods have been extensively investigated because of their potential applications in sensors, solar cells, lithium batteries and so on.19–21 Moreover, the one-dimensional nanostructures show unique properties as promising photocatalysts compared to particles and films. The main benefits include the large surface-to-volume ratio and the short electron–hole diffusion length.22,23 In addition, the light scattering and absorption can be enhanced due to the high length-to-diameter ratio of the one-dimensional nanostructure.24 Lately, electrospinning is found to be a good method to prepare one-dimensional materials. Despite the advantages discussed above, the open structure, easy scale-up, and recoverable characters make the electrospun ultrafine nanofibers suitable for catalysts.25
In present work, heterogeneous carbon coated TiO2 nanofibers are successfully prepared. The core TiO2 nanofibers is fabricated via electrospinning and CQDs embedded in amorphous carbon shell is coated by impregnation process under a relatively low temperature using glucose as carbon precursor. The morphology and structure of the composite nanofibers with different coating thickness are determined. The surface chemical state and photoluminescence properties are characterized. The photocatalytic performance is evaluated by the degradation of rhodamine B (RhB) under visible light irradiation. The mechanism of the enhanced photocatalytic activity of the composite nanofibers is discussed.
2. Experimental section
TiO2 nanofibers were prepared by electrospinning.2 Tetrabutyl titanate (6 wt%) was dissolved in the mixture of ethanol and acetic acid (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, volume rate). Polyvinylpyrrolidone (PVP, MW = 1300000, 6 wt%) was then added to control the viscosity. After dissolution, a clear transparent precursor solution was obtained and transferred into a hypodermic syringe at a constant flow rate of 1 mL h−1. At an applied high voltage of 12.5 kV, a nonwoven mat was collected on a grounded aluminum foil at an electrode distance of 15 cm. The as-spun nanofibers were then calcined at 550 °C in air for 2 h.
:2, volume rate). Polyvinylpyrrolidone (PVP, MW = 1300000, 6 wt%) was then added to control the viscosity. After dissolution, a clear transparent precursor solution was obtained and transferred into a hypodermic syringe at a constant flow rate of 1 mL h−1. At an applied high voltage of 12.5 kV, a nonwoven mat was collected on a grounded aluminum foil at an electrode distance of 15 cm. The as-spun nanofibers were then calcined at 550 °C in air for 2 h.
The carbon precursor solutions were prepared by the following procedure. In a typical experiment, CQDs source were prepared by a one-step alkali-assisted ultrasonic treatment. Glucose was dissolved in distilled water. NaOH solution was then added to the glucose solution, and the concentration of glucose and NaOH are 0.5 mol L−1. After mixing, the solution was then ultrasonic treated at room temperature. To get different thickness of the carbon shell, we change the ultrasonic time as 1 h, 2 h, and 3 h, respectively. Then, the precursor solution was adjusted to pH = 7 with HCl. Thus, the carbon precursor solutions containing CQDs and other small carbon species with the color of yellow, brown, and dark brown were obtained.
To synthesize the carbon/TiO2 composite nanofibers, 50 mg TiO2 nanofibers were dispersed into 50 mL carbon precursor solution. After that, a few drops of PVP solution (0.4 g mL−1) were added to the solution. Then, the mixture was stirred at 95 °C for 3 h to get the composite nanofibers. The obtained composite nanofibers were then washed with water and ethanol for several times, and then vacuum dried in an oven at 60 °C for 10 h. The samples of the nanofibers of different ultrasonic time are denoted as C1, C2 and C3, respectively.
The photocatalytic activity of the prepared nanofibers was evaluated by the degradation of rhodamine B (RhB) under visible light irradiation of a 300 W Xe lamp with a 400 nm cutoff filter. The total optical power impinging on the solution was obtained as 100 ± 10 mW mL−1, measured by a Digital Power & Energy meter (PM121D, Thorlabs Inc., Newton, NJ). The nanofibers were dispersed in RhB solution (2.5 × 10−5 M, 10 mL) with a catalyst loading of 1 g L−1. The mixture was magnetically stirred in the dark for 2 h to obtain the adsorption and desorption equilibrium of RhB and then loaded in an open beaker and exposed to irradiation with continuous stirring at room temperature. At regular intervals of 5 min, UV-vis absorption spectra of the separated solution were measured. The absorption at 554 nm referring to the concentration of RhB was recorded as a function of irradiation time.
The morphology of the as-prepared nanofibers was characterized by scanning electron microscopy (SEM, JEOL JSM-6460LV, Tokyo, Japan) and transmission electron microscopy (TEM, JEOL-2011, Tokyo, Japan). The crystalline structure of the composite nanofibers was identified by X-ray diffraction (XRD, D/max-2550, Rigaku Co., Tokyo, Japan). X-ray photoelectron spectroscopy (XPS, PHI-5300 ESCA, PerkinElmer, Boston, MA), Raman (LabRAM HR800, Horiba, Kyoto, Japan), and Fourier transform infrared spectroscopy spectrum (FTIR, VERTEX 70v, Bruker, Karlsruhe, Germany) was measured for the analysis of elemental and chemical states of carbon in the nanofibers. UV-visible diffuse reflectance spectra (UV-vis, Shimadzu, UV3600, Tokyo, Japan) were recorded in the diffuse reflectance mode in the scanning range of 300–800 nm. Photoluminescence spectra (PL, FLS-920, Edinburgh Instruments, Livingston, UK) were also measured.
3. Result and discussion
Fig. 1a shows the SEM image of the composite nanofibers. It can be seen that the nanofibers form a three-dimensional open structure, while the diameters of the nanofibers range from 50 to 250 nm. The surface of the nanofibers is quite smooth because of the carbon coating. High resolution TEM (HRTEM) is carried out to study the microstructure of the composite nanofibers. Fig. 1b–d show the HRTEM images of the composite nanofibers, the TEM images of pure TiO2 nanofibers are shown in Fig. S1.† The HRTEM images of C1, C2 and C3 show the well-developed interfaces between carbon and the TiO2 nanofibers. It can be seen that a shell of a thin carbon layer is formed on the TiO2 nanofiber core. And as the ultrasonic time increases, the thickness of the carbon shell increases, which is about 1 nm, 2 nm and 5 nm for C1, C2 and C3, respectively. The electron dispersive spectrometer (EDS) line scanning and mapping results further confirms the formation of the carbon shell (Fig. S2†). The inset of Fig. 1d shows the precise structure of the carbon shell of C3. The lattice fringes with d spacing of around 0.34 nm agree well with the (002) spacing of the graphitic carbon, which is the typical structural of the CQDs.26,27 The diameter of the CQDs ranges from 2 nm to 5 nm. Thus, the as prepared carbon shell is a structure of an amorphous carbon embedded with CQDs. | ||
| Fig. 1 (a) SEM images of the composite nanofibers; (b–d) HRTEM images of C1, C2, and C3. The inset shows the HRTEM of CQDs of C3. | ||
The proposed formation schematic for the synthesis of the composite nanofibers is shown in Fig. 2. CQDs are derived from the dehydration and polycondensation of glucose molecules under ultrasonic wave. With the increasing time of the ultrasonication, glucose first nucleates and further grows at the nucleus surface. The longer the ultrasonic time, the larger the size of CQDs, which is corresponded to the photoluminescence spectra of the CQDs precursor solutions as shown in Fig. S3.† Partially carbonized organic species are also existed in the solution, which are the carbon source of the amorphous carbon of the composite nanofibers. Thus, with the existence of PVP, composite nanofibers with different thickness of the carbon shell are obtained.
 | ||
| Fig. 2 Schematic illustration for the synthesis of the composite nanofibers. | ||
To further investigate the structure of the carbon shell, Raman and FTIR spectra were measured (Fig. 3). Fig. 3a shows the Raman results. The peak at around 1340 cm−1 corresponds to the D-band relating to the disorder induced in the carbon structure, while the peak at 1610 cm−1 corresponds to the G-band attributing to the vibration of sp2 carbon vibration.28 The relative intensity of D to G peaks (ID/IG) can refer to the disorder or graphitization degree of carbon based hybrids.29 ID/IG are about 0.94 for C1, C2 and C3, respectively, suggesting the existence of both disordered graphitic form and graphitic domains,30 which is in accord with the CQDs embedded in amorphous carbon structure. Except for the typical D-band and G-band of carbon, the Raman spectra of the as-prepared composite nanofibers also show some incompletely carbonized organic matter, which may result from the partially carbonized species from glucose. Fig. 3b shows the FTIR spectra of pure TiO2 nanofibers and the as-prepared composite nanofibers. The broad peaks at 3426 and 1633 cm−1 are correspond to the water and hydroxyl groups absorbed on the surface.31 The differences between pure TiO2 nanofibers and the composite nanofibers are due to the coated carbon. The absorption peaks at around 1050 cm−1 are attributed to the C–C stretching vibration, while the peaks at around 1456 cm−1 are ascribed to the C–H deformation vibration.32 Absorptions due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C
O, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and C–H stretching vibration modes are located at around 1716 cm−1, 2343 cm−1, and 2900 cm−1. The results imply that carbon exists as functional groups in the nanocomposites.
C and C–H stretching vibration modes are located at around 1716 cm−1, 2343 cm−1, and 2900 cm−1. The results imply that carbon exists as functional groups in the nanocomposites.
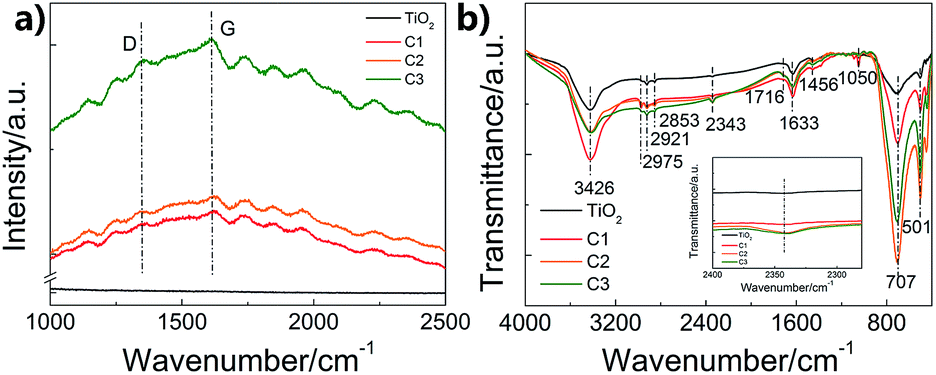 | ||
| Fig. 3 Raman and FTIR spectra of the prepared composite nanofibers. (a) Raman; (b) FTIR. | ||
The phase structures of the nanofibers were studied by XRD, shown in Fig. 4. It is found that both pure TiO2 nanofibers and the composite nanofibers exhibit the diffraction peaks of anatase and rutile TiO2. And as the ultrasonic time increases, the ratio of the rutile phase to anatase phase increases. Anatase phase tend to transform to rutile phase during the coating process. Moreover, as shown in the inset of Fig. 4, the (200) peak of TiO2 shifts to the smaller 2θ angle direction, indicating the increase in lattice constant, which may due to the coating of carbon on the surface of the TiO2 nanofibers. The graphitic carbon peak at 25.6° is too weak to see. Notably, the decrease of the intensity from pure TiO2 nanofibers to C1, C2, and C3 may be due to the presence of glucose, which deteriorated the crystallinity.33
 | ||
| Fig. 4 XRD patterns of pure TiO2 nanofibers and C1, C2, C3. The inset shows the 2θ shift of (200) peak of TiO2. | ||
In order to investigate the chemical states of C, O and Ti in the composite nanofibers, XPS spectra were measured (Fig. 5). Fig. 5a shows the concentration of C, O and Ti atoms on the surface of the nanofibers. It is found that carbon content increases as the shell thickness increases. The C 1s signal of pure TiO2 nanofiber is caused by the hydrocarbon from the XPS instrument itself.34 Fig. 5b shows the XPS spectra of Ti 2p. The Ti 2p binding energy of all the nanofibers are located around 458.6 eV (Ti 2p3/2) and 464.3 eV (Ti 2p1/2) without any shift, indicating a normal state of Ti4+ in the nanofibers.35 C 1s spectra are shown in Fig. 5c, the main contribution at 284.8 eV is ascribed to the delocalized alternant hydrocarbon, while the two peaks at 286.4 and 288.5 eV are due to the C–O and CO, respectively.36 The OH and CO groups observed in XPS are from the partially dehydrated carbohydrates during the ultrasonic process. As shown in Fig. 5d, the spectra of O 1s can be divided into three peaks around 529.9, 531.6, and 533.1 eV, corresponding to the binding energy of the lattice oxygen, oxygen vacancies, and absorbed oxygen species, respectively.37 The concentration of surface oxygen species can be estimate according to the photoelectron cross-sections and mean path. According to Fig. 5d, it is found that the concentrations of oxygen vacancies for C1, C2, and C3 are 14.5%, 20.8%, and 27.3%, while the concentration of the absorbed oxygen species are 9.5%, 5.3%, and 16.1%, respectively. It suggests that the surface oxygen vacancies increase as the carbon shell becomes thicker. However, the concentration of the absorbed oxygen species doesn't increase with the thickness of the carbon shell, which may because that the carbon of C1 is ultrathin and more active.
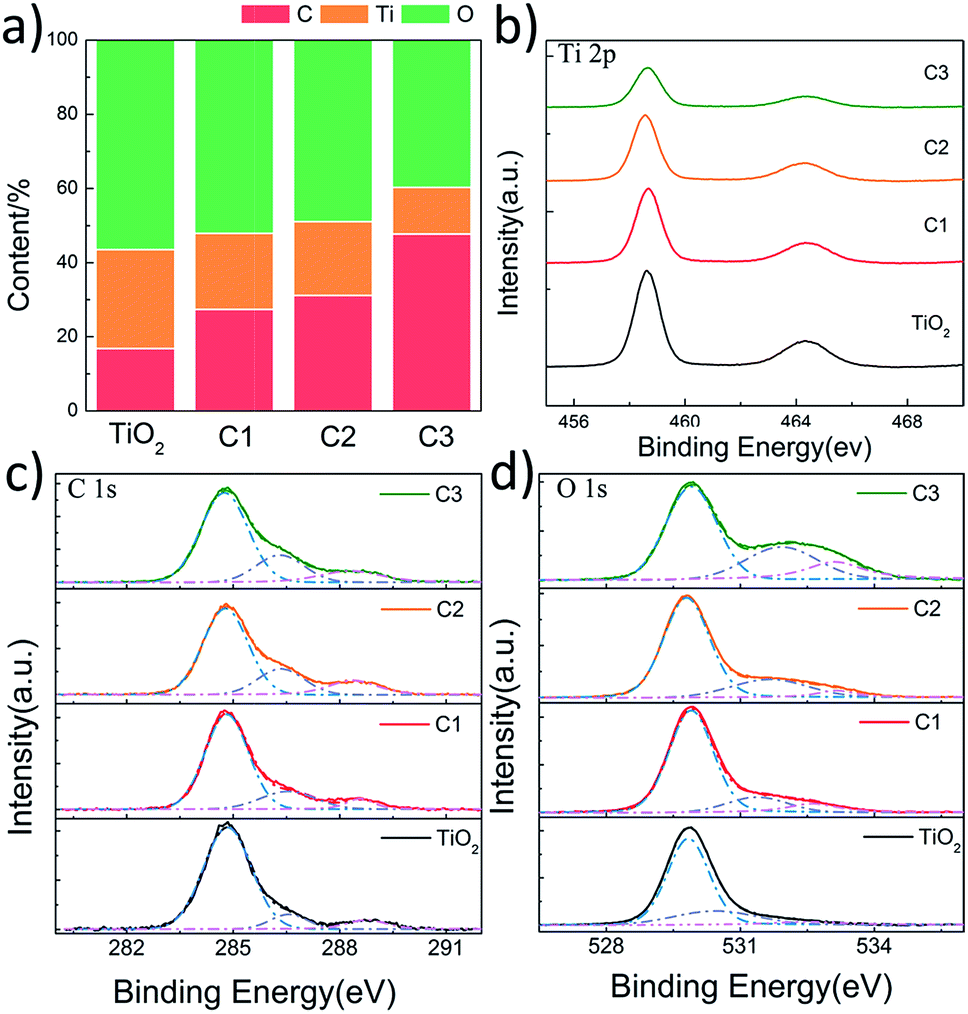 | ||
| Fig. 5 XPS results of pure TiO2 nanofibers, C1, C2 and C3. (a) The content of surface C, Ti, and O; (b–d) Ti 2p, C 1s, and O 1s scan, respectively. | ||
The photocatalytic performances of the composite nanofibers with different thickness of carbon coating are evaluated by the degradation of RhB under visible light irradiation. Catalyst loading is 1 g L−1. The total optical power (TOP) impinging on the solution is 100 ± 10 mW mL−1. The normalized optical density change of RhB at the maximal absorption peak 554 nm is plotted in Fig. 6a as a function of time, where C0 is the concentration of RhB after the adsorption and desorption equilibrium before irradiation. For comparison, P25 and CQDs are used as references. As shown in Fig. 6a, the commercial TiO2 powders (Degussa P-25) show no photodegradation of RhB because for P25, TiO2 can be activated only under UV radiation. It is found that when carbon is coated on the TiO2 nanofibers, a remarkable enhancement in photocatalytic activity is observed. C2 has the best photocatalytic activity, and RhB can be almost completely degraded when irradiated under visible light for 20 min. The visible-light-driven photocatalytic activity of pure TiO2 nanofibers may due to the surface oxygen vacancies, which is in accord with the XPS results. It is indicated from Fig. 6b that the photocatalytic activity of C2 composite nanofibers shows good repeatability and stability in 4-time cycle tests, for which RhB can still be almost completely degraded when irradiated under visible light for 20 min. Moreover, the RhB adsorbed on the surface of the nanofibers also prove to be removed totally, indicating that the decrease of RhB concentration is due to the photocatalytic degradation rather than adsorption (see FTIR results, ESI Fig. S4†).
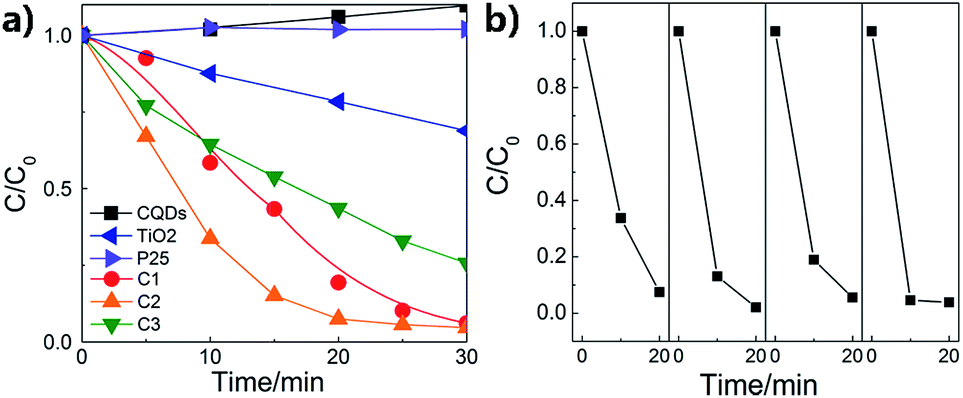 | ||
| Fig. 6 (a) Photodegradation of RhB by the composite nanofibers; (b) the repeatability tests of C2. | ||
The photodegradation of RhB can be considered as a pseudo-first-order reaction.38,39 The rate constant k can be calculated by the formula as follows:
| C = C0e−kt | (1) |
 | (2) |
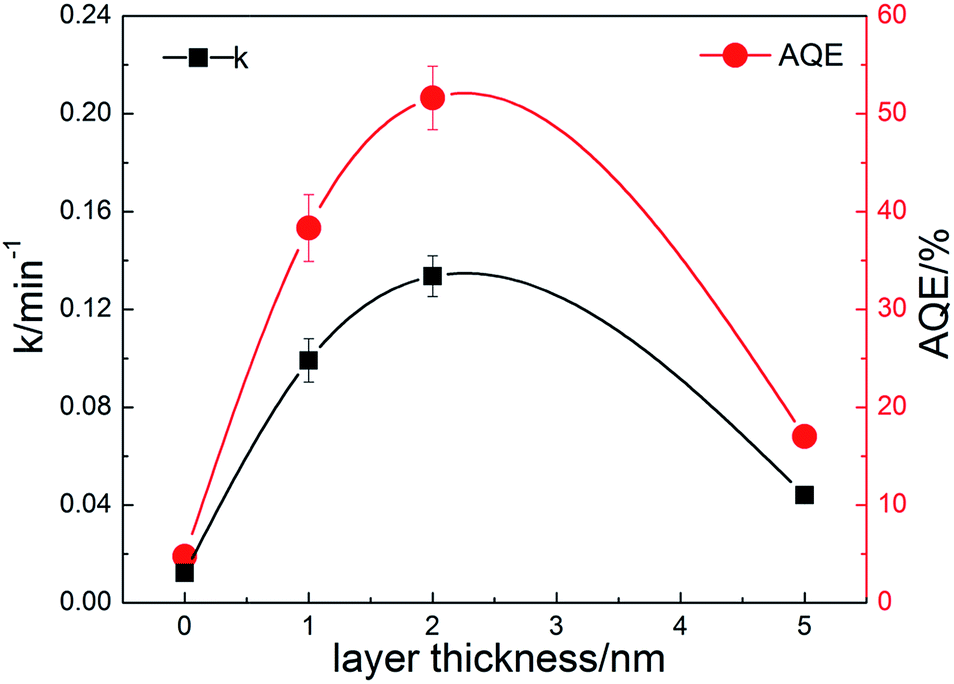 | ||
| Fig. 7 Degradation rate constants and apparent quantum efficiencies of the prepared composite nanofibers. | ||
To investigate the mechanism of the enhanced photocatalytic activity of the heterostructure, we have further studied the origin of the visible light absorption of the nanofibers and the effect of the composite carbon structure on the optical properties. Fig. 8a shows the UV-vis diffuse reflection spectra of pure TiO2 and the composite nanofibers. It can be seen that enhanced absorption for C1, C2 and C3 in the visible light range is observed. The band gaps can be estimated from the reflection spectra using the Tauc plot (inset of Fig. 8a).42 Obviously, the band gap of pure TiO2 nanofiber (3.13 eV) is higher than the carbon coated nanofibers, and the bandgaps for C1 (3.12 eV), C2 (3.08 eV) and C3 (3.04 eV) are further narrowed by the increase of carbon loading. Moreover, the middle inset of Fig. 8a indicates that the color of the nanofibers turns to grey after carbon coating. There are probably two reasons accounting for this: one is due to the carbon coating induced surface defects, which are found to have a significant effect on the electrical and optical properties of semiconductor materials;43 another is related to the increased content of rutile phase (bandgap 3.0 eV) in the nanofibers, corresponding to the former XPS and XRD results. Fig. 8b shows the up conversion PL spectra of CQDs with excitations from 700 to 900 nm. It is indicated that the emissions are located at visible light range of 350–600 nm (some are cut by the filter). Thus, the embedded CQDs have obvious up conversion PL behavior, and it can absorb near infrared light and then emit light in the region of 350–450 nm, which can promote electrons from the valance band to the conduction band of TiO2 with rutile or anatase phase. As a result, CQDs in the carbon shell can efficiently utilize the broad spectrum of the sunlight and enhance the photocatalytic activity. Fig. 8c shows the PL spectra of the nanofibers with excitation wavelength of 300 nm. Although the band–band PL phenomenon cannot be found, all the samples exhibit a wide PL signal at the range of 350–600 nm. There are four PL peaks at about 420, 440, 460, and 520 nm, respectively. These PL signals are attributed to excitonic PL, which mainly result from the surface oxygen vacancies and some interface traps of radiative defects at the boundaries between carbon and TiO2.44 The signal of pure TiO2 nanofibers is too weak to see, due to the relatively low density of the surface defects. In addition, the weakest PL emission intensity of C2 is also observed in the composite nanofibers, illustrating that C2 has the best separation rate of photo-induced charges, which enhances the photocatalytic activity. In compared with C2, C3 with the thicker carbon shell shows a relatively stronger emission density and lower photocatalytic activity, which may due to the fast recombination of the photogenerated electron–hole pairs on the carbon sites that are not at the interface. To further understand the recombination process in the nanofibers, we have measured the time-resolved photoluminescence spectra, shown in Fig. 8d. The laser wavelength is 405 nm. It can be seen that a very fast relaxation is observed, which can be fitted into two exponential decay components of the fast recombination of excitons and a relatively long-lasting emission.45 The fitting results are shown in the inset of Fig. 8d. Obviously, C2 shows much slower PL decay and the longest average lifetime (0.69 ns), which is about 2 times of pure TiO2 (0.39 ns) and 3 times of C3 (0.23 ns), indicating an increase of long-lived excited states. These may be originated from the electron trapping by the surface defects and the heterostructure of carbon coating. As a result, an appropriate loading of carbon is beneficial for increasing the amount of long-lived charge trapping states. It's not that the thicker the carbon shell, the better the photocatalytic activity. There are probably two reasons accounting for this: one is that the contaminant molecules have to diffuse through the carbon shell before reaching the surface of the TiO2; another is that the light arriving at the surface of TiO2 are weakened because of the scattering of the carbon shell.46
 | ||
| Fig. 8 (a) UV-visible diffuse reflection spectra of the composite nanofibers, the inset shows the colour of the sample and the corresponding plots of (αhν)2 vs. photon energy (hν); (b) up conversion properties of the CQDs; (c) photoluminescence spectra of the composite nanofibers; (d) photoluminescence decay of the composite nanofibers, the inset shows the fitting results. | ||
On the basis of the above results, the mechanism of the enhanced photocatalytic activity in the heterostructure of TiO2 and amorphous carbon embedded with CQDs composite nanofibers is schematically shown in Fig. 9. When the catalysts are illuminated by visible light with photon energy higher than the band gap of TiO2 and CQDs, electrons in the valance band can be excited to the conduction band with the generation of the same amount of holes. In the heterostructure of the composite nanofibers, the electrons will transfer from CQDs to the electron-trapping site of amorphous carbon and then injected into the conduction band of TiO2. The good contact with the amorphous carbon (a-C), CQDs, and TiO2 also promotes the separation process. In addition, surface oxygen vacancies can further trap the photogenerated electrons and help releasing holes from the catalyst. As a result, the lifetime of the photogenerated electron–hole pairs will be increased, leading to an enhanced photocatalytic activity. The photoinduced holes are apt to react with surface-bound H2O or OH− to produce the hydroxyl radical species (˙OH), while the dissolved oxygen molecules (O2) react with the electrons to yield superoxide radical anions (˙O2−), both are extremely strong oxidant for the mineralization of organic compounds. On the other hand, we can also discuss the mechanism of the enhanced photocatalytic activity from the aspects of the optical character. The up conversion properties make CQDs absorb near infrared light and emit shorter wavelength light which can excite TiO2 to form the photogenerated electron–hole pairs and increase the photocatalytic activity.
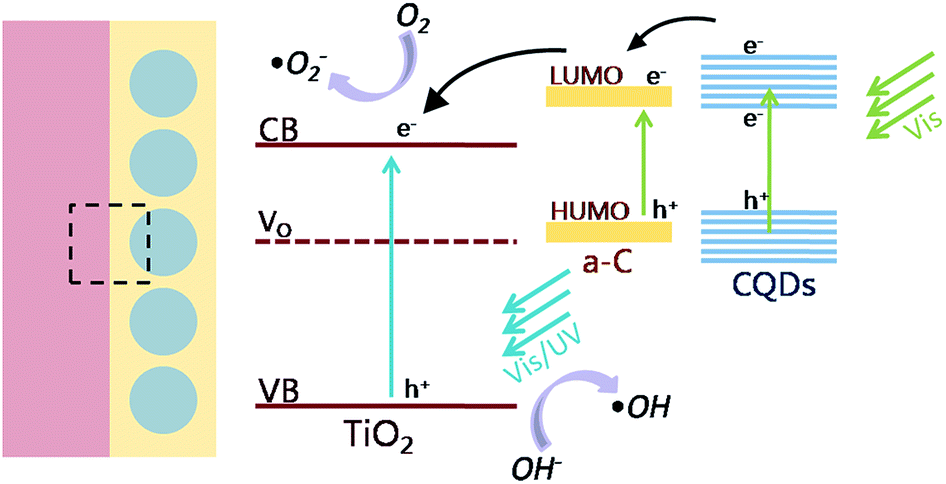 | ||
| Fig. 9 Schematic illustration of the photocatalytic mechanism of the heterostructure. | ||
4. Conclusions
In summary, a novel core–shell heterostructure of TiO2 nanofibers and heterogeneous carbon is successfully prepared via a simple electrospinning and impregnation process. A shell of CQDs embedded amorphous carbon has been coated on the surface of TiO2 nanofibers. The obtained core–shell nanofibers show an enhanced photocatalytic activity in the degradation of rhodamine-B under visible light irradiation. The nanofibers with a thickness of carbon shell of 2 nm exhibit a stable and highly efficient photocatalytic activity. The apparent quantum efficiency can reach as high as 52%, which is about 10 times of pure TiO2 nanofibers. The enhanced photocatalytic activity is attributed to the efficient separation of the photogenerated electrons and holes due to the unique heterostructure of TiO2, CQDs and amorphous carbon. The intimate contact of TiO2 and the carbon shell further enhances the charge transfer process. Moreover, the up conversion property of CQDs enables the composite nanofiber to utilize more solar energy to increase the photocatalytic activity. Thus, the photocatalytic activity of the composite nanofibers is efficiently improved. The results in this work may be beneficial to the future study of exploring the carbon-based heterostructured materials for visible-light-driven photocatalyst.Acknowledgements
This study was supported by the Natural Science Foundation of China (Grant No. 51323001).References
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834 CAS.
- H. P. Li, W. Zhang and W. Pan, J. Am. Ceram. Soc., 2011, 94, 3184 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler Jr, Science, 2002, 297, 2243 CrossRef CAS PubMed.
- A. Ajmal, I. Majeed, R. N. Malik, H. Idriss and M. A. Nadeem, RSC Adv., 2014, 4, 37003 RSC.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798 CrossRef CAS PubMed.
- Y. Zhang, Z. Jiang, J. Huang, L. Y. Lim, W. Li, J. Deng, D. Gong, Y. Tang, Y. Lai and Z. Chen, RSC Adv., 2015, 5, 79479 RSC.
- X. Wang, T. Li, R. Yu, H. Yu and J. Yu, J. Mater. Chem. A, 2016, 4, 8682 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoke and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- X. B. Chen, Y. B. Lou, A. C. S. Samia, C. Burda and J. L. Gole, Adv. Funct. Mater., 2005, 15, 41–49 CrossRef CAS.
- L. Hu, Y. Zhang, S. Zhang and B. Li, RSC Adv., 2016, 6, 43098 RSC.
- D. E. Gu, Y. Lu, B. C. Yang and Y. D. Hu, Chem. Commun., 2008, 21, 2453 RSC.
- S. Sun, M. Sun, Y. Fang, Y. Wang and H. Wang, RSC Adv., 2016, 6, 13063 RSC.
- A. Kongkanand and P. V. Kamat, ACS Nano, 2007, 1, 13 CrossRef CAS PubMed.
- J. Shi, J. Zheng, P. Wu and X. Ji, Catal. Commun., 2008, 9, 1846 CrossRef CAS.
- P. Shao, J. Tian, W. Shi, S. Gao and F. Cui, J. Mater. Chem. A, 2015, 3, 19913 CAS.
- X. Cheng, H. Liu, Q. Chen, J. Li and P. Wang, Carbon, 2014, 66, 450 CrossRef CAS.
- N. Qin, Y. Liu, W. Wu, L. Shen, X. Chen, Z. Li and L. Wu, Langmuir, 2015, 31, 1203–1209 CrossRef CAS PubMed.
- A. I. Hochbaum, R. Chen, R. D. Delgado, W. Liang, E. C. Garnett, M. Najarian, A. Majumdar and P. Yang, Nature, 2008, 451, 163 CrossRef CAS PubMed.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425 CrossRef CAS PubMed.
- B. Tian, X. Zheng, T. J. Kempa, Y. Fang, N. Yu, G. Yu, J. Huang and C. M. Lieber, Nature, 2007, 449, 885 CrossRef CAS PubMed.
- H. Wu, W. Pan, D. Lin and H. Li, J. Adv. Ceram., 2012, 1, 2 CrossRef CAS.
- W. Han, Appl. Phys. Lett., 2010, 96, 183112 CrossRef.
- M. S. Park, G. X. Wang, Y. M. Kang, D. Wexler, S. X. Dou and H. K. Liu, Angew. Chem., Int. Ed., 2007, 119, 764 CrossRef.
- D. D. Lin, H. Wu, R. Zhang and W. Pan, Chem. Mater., 2009, 21, 3479 CrossRef CAS.
- D. S. Bethune, C. H. Klang, M. S. Devries, G. Gorman, R. Savoy and R. Beyers, Nature, 1993, 363, 605 CrossRef CAS.
- Z. H. Kang, E. B. Wang, L. Gao, S. Y. Lian, M. Jiang, C. W. Hu and L. Xu, J. Am. Chem. Soc., 2003, 125, 13652 CrossRef CAS PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud’homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef CAS PubMed.
- M. Liu, L. Gan, W. Xiong, Z. Xu, D. Zhu and L. Chen, J. Mater. Chem. A, 2014, 2, 2555 CAS.
- S. R. Jian, Y. T. Chen, C. F. Wang, H. C. Wen, W. M. Chiu and C. S. Yang, Nanoscale Res. Lett., 2008, 3, 230 CrossRef CAS.
- J. M. Yu, L. Z. Zhang, Z. Zheng and J. C. Zhao, Chem. Mater., 2003, 15, 2280 CrossRef CAS.
- T. Tsumura, N. Kojitani, H. Umemura, M. Toyoda and M. Inagaki, Appl. Surf. Sci., 2002, 196, 429 CrossRef CAS.
- W. J. Ren, Z. H. Ai, F. L. Jia, L. Z. Zhang, X. X. Fan and Z. G. Zou, Appl. Catal., B, 2007, 69, 138 CrossRef CAS.
- J. G. Yu, J. F. Xiong, B. Cheng and S. W. Liu, Appl. Catal., B, 2005, 3, 211 CrossRef.
- Z. Song, J. Hrbek and R. Osgood, Nano Lett., 2005, 5, 1327 CrossRef CAS PubMed.
- E. M. Rockafellow, X. X. Fang, B. G. Trewyn, K. Schmidt-Rohr and W. S. Jenks, Chem. Mater., 2009, 21, 1187 CrossRef CAS.
- A. G. Marrani, F. Caprioli, A. Boccia, R. Zanoni and F. Decker, J. Solid State Electrochem., 2014, 18, 505 CrossRef CAS.
- Z. Y. Liu, D. D. Sun, P. Guo and J. O. Leckie, Nano Lett., 2007, 7, 1081 CrossRef CAS PubMed.
- Q. Wan, T. H. Wang and J. C. Zhao, Appl. Phys. Lett., 2005, 87, 083105 CrossRef.
- A. Salinaro, A. V. Emeline, J. Zhao, H. Hidaka, V. K. Ryabchuk and N. Serpone, Pure Appl. Chem., 1999, 71, 321–335 CrossRef CAS.
- I. E. Wachs, S. P. Phivilay and C. A. Roberts, ACS Catal., 2013, 3, 2606 CrossRef CAS.
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914 CrossRef CAS.
- S. C. Erwin, L. Zu, M. I. Haftel, A. L. Efros, T. A. Kennedy and D. J. Norris, Nature, 2005, 436, 91 CrossRef CAS PubMed.
- L. Jing, Y. Qu, B. Wang, S. Li, B. Jiang, L. Yang, W. Fu, H. Fu and J. Sun, Sol. Energy Mater. Sol. Cells, 2006, 90, 1773 CrossRef CAS.
- M. V. Dozzi, C. D'Andrea, B. Ohtani, G. Valentini and E. Selli, J. Phys. Chem. C, 2013, 117, 25586 CAS.
- M. Inagaki, F. Kojin, B. Tryba and M. Toyoda, Carbon, 2005, 43, 1652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00546f |
| This journal is © The Royal Society of Chemistry 2017 |