
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyperbranched conjugated polymers containing 1,3-butadiene units: metal-free catalyzed synthesis and selective chemosensors for Fe3+ ions†
Chuxin Luo,
Yating Liu,
Qi Zhang and
Xuediao Cai *
*
Key Laboratory of Macromolecular Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, P. R. China. E-mail: xdcai@snnu.edu.cn
First published on 21st February 2017
Abstract
Hyperbranched conjugated polymers (P1 and P2) containing 1,3-butadiene repeating units appended with carboxylic ester groups were synthesized in good yield by metal-free catalyzed polymerization of diethyl 4,4′-(5-formyl-1,3-phenylene)(2E,2′E)-bis(but-2-enoate) (M1) or dimethyl 4,4′-(5-formyl-1,3-phenylene)(2E,2′E)-bis(but-2-enoate) (M2). The corresponding P1 and P2 polymers were structurally characterized by NMR and IR spectroscopy. The polymers are highly soluble in common organic solvents. The UV-vis absorption bands of P2 along with its emission spectrum was red-shifted compared to that of P1, indicating the extension of conjugated length. The hyperbranched conjugated polymers were used as ratiometric fluorescence chemosensors for the detection of the biologically important metal ion Fe3+. The corresponding Stern–Volmer constants (Ksv) of P1 and P2 are 2.90 × 104 M−1 and 3.83 × 104 M−1, respectively. Detection limits of 6.99 × 10−7 M for P1 and 6.69 × 10−7 M for P2 were achieved. The neutral polymer P1 or P2 was hydrolyzed to obtain water-soluble hyperbranched conjugated polymer P3. The ability of P3 to detect different Fe3+ ions in aqueous solution was investigated. The results demonstrate that these polymers can serve as sensitive and selective sensors for Fe3+ ions in organic and aqueous solutions.
Introduction
Hyperbranched conjugated polymers have attracted much attention because of their unique properties, such as good solubility, low viscosity, tunable electrical properties and processability, and also because of their potential applications in organic electronics and optoelectronics, including organic electroluminescence,1–5 ion detection,6–8 explosive detection,9–14 disease diagnosis,15 and biological identification.16,17 Various transition-metal-catalyzed polymerization methods have been used for the synthesis of hyperbranched conjugated polymers, such as Suzuki coupling polymerization,18–20 click polymerization,21,22 and Sonogashira coupling polymerization.23,24 These well-established polymerization routes have played an indispensable role in the synthesis of hyperbranched conjugated polymers. However, these methods have drawbacks, such as harsh reaction conditions, tedious synthetic steps, the use of environmentally harmful transition-metal catalysts and solvents. In addition, the trace residues of transition-metal catalysts in polymeric products can seriously affect the efficiency and the life of the resulting devices.25Recently, various linear conjugated polymers were synthesized with transition-metal-free catalysed methods.26–35 However, these methods, including Wittig reaction,4,36 Knoevenagel condensation reaction,11 arylimino-deoxy-trisubstituted reaction,37 and acid-promoted cyclotrimerization reactions,38 were rarely explored to prepare hyperbranched conjugated polymers. In addition, these transition-metal-free catalyzed methods also require harsh reaction conditions and complex synthetic steps. Therefore, in order to expand the application of hyperbranched conjugated polymers, it is desirable to develop efficient polymerization method and new structure under mild reaction conditions by using transition-metal-free catalyst.
In our previous work,39 we have established an efficient transition-metal-free catalyzed polymerization method. Reactions of α,β-unsaturated compounds containing γ-H with aldehydes in ethanol in the presence of 1,8-diazabicyclo[5.4.0]undecene-7-ene (DBU) as the basic catalyst gave a series of linear π-conjugated polymers (Scheme 1), which have alternating arylene and 1,3-butadiene units in the main chain. This method has many advantages, including fewer reaction steps, mild reaction conditions, environmentally friendly solvents (i.e., in ethanol even without purity and protection from oxygen), and no toxic metal byproducts. On the basis of these results, herein we report the synthesis of new hyperbranched conjugated polymers containing carboxylic ester groups appended to repeating 1,3-butadiene units and their uses as sensitive and selective sensors for Fe3+ ions in organic and aqueous solutions. Iron is the second most abundant metal element in nature, and plays very important roles in the whole ecosystem.40 Recently, fluorescence sensors based on conjugated polymers for the detection of Fe3+ have received much attention owing to their signal amplification properties.41–48
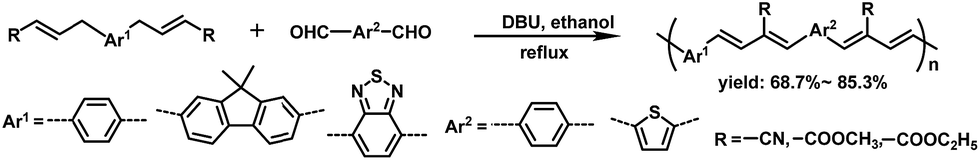 | ||
| Scheme 1 Synthesis of π-conjugated polymers containing 1,3-butadiene units in the main chain. | ||
This special macromolecular structure shows several advantages in Fe3+ detection. First, the carboxylic ester group has been reported to act as chelating group for metal ions, especially for Fe3+ ions. Therefore, neutral polymers P1 and P2 are expected to coordinate with Fe3+ in organic solutions, and the strong association interaction between carboxylic ester groups and Fe3+ ions will cause the fluorescence quenching.49 Second, the carboxylic ester group could be hydrolyzed to obtain water-soluble hyperbranched conjugated polymers. We expected that our synthesized hyperbranched conjugated polymers could be used in the field of selective detection of Fe3+ from environmental and biological samples.
Experimental
Materials and instruments
Chemical reagents and different metal salts were purchased from Energy chemical, Meryer or Aladdin and used as received. THF and 1,4-dioxane were distilled from sodium benzophenone ketyl under dry nitrogen immediately prior to the use. 1H and 13C NMR spectra were recorded on a Brüker 600 MHz spectrophotometer in CDCl3 using tetramethylsilane (TMS, δ = 0) as an internal reference. IR spectra were taken on a FT-IR Brüker Tensor 27 spectrometer at a nominal resolution of 2 cm−1 using KBr pellet. UV-vis spectra were collected on Hitachi U-3900 spectrophotometer (Tokyo, Japan). Photoluminescence (PL) spectra were recorded on a Hitachi F-7000 spectrofluorometer (Tokyo, Japan). Thermogravimetric analysis (TGA) measurements were carried out under dry nitrogen on Q1000DSC + LNCS + FACSQ600SDT instruments at 20 °C min−1. The average molecular weight and polydispersity index (PDI) of the polymers were determined using Viscotek gel permeation chromatography (GPC) analysis with THF as the eluent and polystyrene as the standard. Cyclic voltammetry (CV) was conducted on an electrochemistry workstation (CHI 660A, Chenhua Shanghai) with the polymer film on Pt plate as the working electrode, Pt slice as the counter electrode, and saturated calomel electrode (SCE) as a reference electrode in a 0.1 M tetra-n-butylammonium hexafluorophosphate acetonitrile solution at a scan rate of 50 mV s−1. The pH value was measured by using of a MRFS: TOSHNIWAL pH meter.Monomer synthesis
General polymerization
To a flask charged with monomer in ethanol solution, DBU was added. The reaction mixture was stirred under refluxed for 72 h. The reaction mixture was then heated at high temperature (110 °C) for 8 h. The solvent was removed under reduced pressure, and the residue was extracted with CH2Cl2 and washed with HCl solution (2 M, 100 mL × 3) and water. The solvent was removed under reduced pressure.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.63–6.62 (d, J = 5.6 Hz, 2H, CHCH), 6.52–6.50 (d, J = 7.2 Hz, 2H, CHCH), 4.12–4.08 (m, 4H, CH2), 1.23–1.19 (t, 6H, CH3). IR (KBr pellet, cm−1): 3030, 2914, 1740, 1600, 1450, 1350.CH), 6.61–6.59 (d, J = 9.2 Hz, 2H, CHCH), 6.50–6.47 (d, J = 12.8 Hz, 2H, CHCH), 4.09 (s, 6H, CH3). IR (KBr pellet, cm−1): 3004, 2908, 1745, 1650, 1480, 1380.
CH), 6.63–6.62 (d, J = 5.6 Hz, 2H, CHCH), 6.52–6.50 (d, J = 7.2 Hz, 2H, CHCH), 4.12–4.08 (m, 4H, CH2), 1.23–1.19 (t, 6H, CH3). IR (KBr pellet, cm−1): 3030, 2914, 1740, 1600, 1450, 1350.CH), 6.61–6.59 (d, J = 9.2 Hz, 2H, CHCH), 6.50–6.47 (d, J = 12.8 Hz, 2H, CHCH), 4.09 (s, 6H, CH3). IR (KBr pellet, cm−1): 3004, 2908, 1745, 1650, 1480, 1380.Results and discussion
Synthesis and structure characterization
The polymer structures and synthetic routes are shown in Scheme 2. Compound 1 was synthesized according to the published procedures.50 3,5-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzaldehyde (1), diethyl 4,4′-(5-formyl-1,3-phenylene)(2E,2′E)-bis(but-2-enoate) (M1) and dimethyl 4,4′-(5-formyl-1,3-phenylene)(2E,2′E)-bis(but-2-enoate) (M2) were prepared by Pd-catalyzed Suzuki cross-coupling reaction under argon atmosphere. In our previous work, we have established a transition-metal-free polymerization method and successfully synthesized a series of linear conjugated polymers.39 In this work, the hyperbranched conjugated polymers P1 and P2 were synthesized by the simple AB2 protocol based on our reported polymerization method. The polycondensation was carried out in the same way by refluxing in ethanol for 72 h with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the catalyst. The yield of P1 and P2 were 79% and 80%, respectively. This result indicates that the molecular weight is very less affected by the side group. It is worth noting that the carboxylic ester-functional group can be readily obtained without special reaction for the monomer during polymerization. All the polymers displayed excellent solubility in common organic solvents such as chloroform, tetrahydrofuran, dichloromethane and chlorobenzene. The water-soluble polymer P3 was obtained by the hydrolysis of P1 or P2 in a solution of sodium hydroxide in methanol with a yield of 80% (Scheme 3). Table 1 summarized the polymerization results and thermal properties of the polymers. The number-average molecular weight (Mn) of P1 and P2 are 6141 and 6026 with polydispersity index (PDI) of 2.575 and 2.465, respectively. It can be seen that in general, the polymerization afforded conjugated polymers with relatively high molecular weight.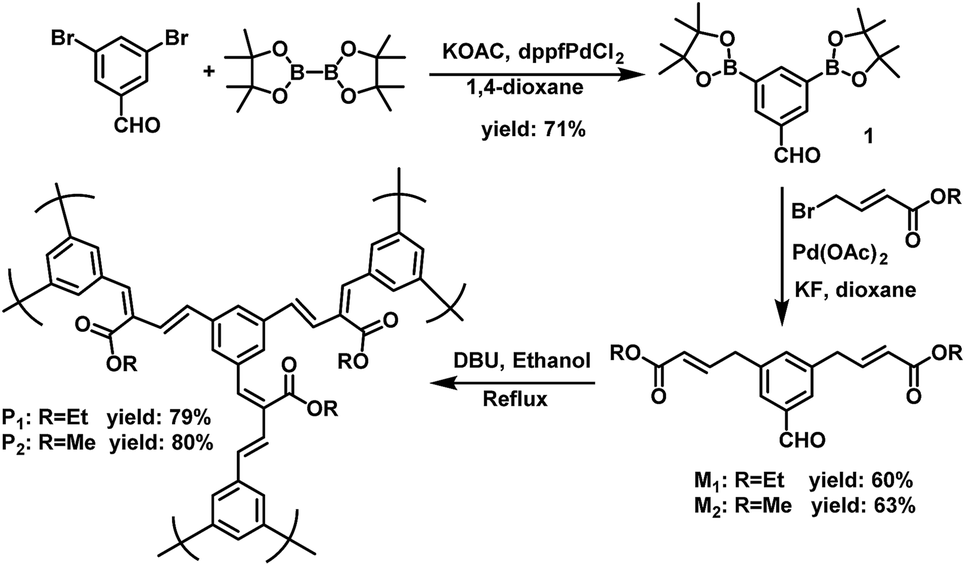 | ||
| Scheme 2 Synthetic routes of monomers and polymers. | ||
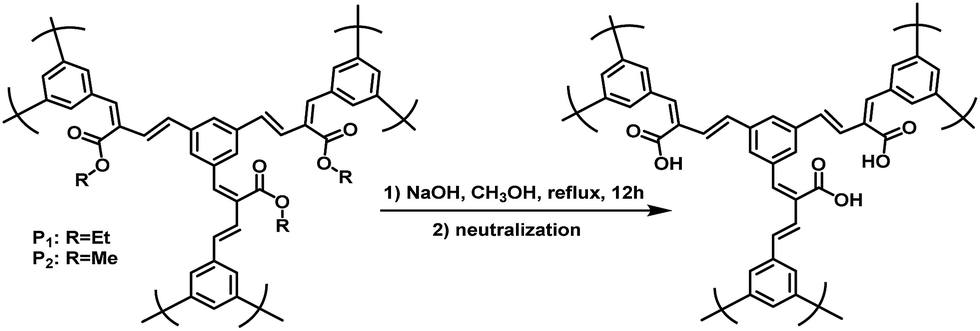 | ||
| Scheme 3 Synthetic routes of P3. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 812
812All the monomers were carefully purified and characterized by 1H and 13C NMR (see Experimental section and ESI† for details). The structures of polymer P1 and P2 were verified by 1H NMR and FT-IR. In the 1H NMR spectrum of P1, the peaks attributable to the aromatic protons were observed at 7.82–7.27 ppm. Another set of olefinic protons were observed at 6.63–6.62 and 6.52–6.50 ppm. The proton signals of ester methylene and methyl for P1 appear at 4.12–4.08 and 1.23–1.19 ppm, respectively (Fig. S6†). In the 1H NMR spectrum of P2, the peak at 4.09 ppm is assigned to the signal from –COOCH3 of P2 (Fig. S7†). Examples of the IR spectra of P1 and P2 are given in Fig. S9.† P1 and P2 shows absorption bands at 3030 and 3004 cm−1 due to their CHCH stretching vibrations. The strong peaks at 1740 cm−1 for P1 and 1745 cm−1 for P2 are associated with their CO groups. As for P3, we see a broad band of strong absorption peaks at 3450 cm−1 due to its OH stretching vibrations of COOH groups. IR spectrum indicated successful synthesis of the target polymers P1, P2 and P3. Because of the poor resolution of related resonances in the 1H NMR spectra of P1 and P2, the important parameter of degree of branching is hardly assessed.
Thermal properties
The thermal properties of the target polymers were obtained by thermogravimetric analysis (TGA) under a nitrogen atmosphere at a heating rate of 20 °C min−1. The TGA diagram of P1 (shown in Fig. S10†) showed a mass loss of 5% starting at 240 °C as the threshold of the thermal decomposition, and a loss of about 50% at 500 °C. On the other hand, the TGA trace of P2 showed that polymer decomposition started at 200 °C and the weight loss was about 55% at 600 °C.Photophysical properties
Fig. 1a and b depict the UV-vis spectra of polymer P1 and P2, respectively, measured in different organic solvents, including CH2Cl2, DMSO, CH3CH2OH, THF, and in solid-state films. P1 and P2 exhibited no significant absorption peaks in most tested organic solvents, while they showed significant absorption peaks at 325 and 330 nm in THF, respectively (Fig. 1). The different absorption features of P1 and P2 in these solvents are due to the different polarity of solvents. It is reported that molecular aggregation in solution exhibited distinct change in the absorption band and can possibly be influenced by the polarity of solvents.16 The polymer P1 in THF solution showed an absorption maximum at 325 nm attributable to phenyl moiety. The solid film of P1 also showed a similar absorption behaviour with an absorption maximum at 327 nm. This result indicates the low aggregation degree of P1 hyperbranched chains in the thin film state. On the other hand, polymer P2 in THF solution showed an absorption peak at 330 nm corresponding to phenyl moiety. The absorption peaks were red-shifted compared to those of P1, suggesting an extension of the conjugated length. It is likely that the substituent has a steric influence on the electronic structure, possibly due to the fact that bulky groups can interrupt the linear π-system, leading to the reduction in the π-conjugated length. The solid film of P2 showed a similar absorption behavior with an absorption maximum at 350 nm. The absorption peaks are red-shifted compared to their solution absorption spectra, which was mainly attributed to the enhanced intermolecular interactions between neighboring molecules in the film state.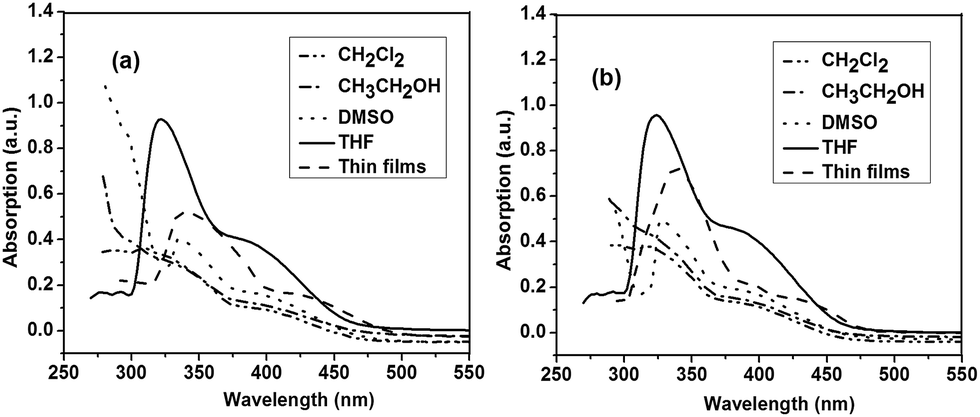 | ||
| Fig. 1 UV-vis spectra of (a) P1 and (b) P2. | ||
The photoluminescent (PLs) spectra of the hyperbranched conjugated polymers in solution excited at the absorption maxima are shown in Fig. 2a and b. As shown in Fig. 2a and b, P1 and P2 exhibited low fluorescence intensity in most organic solvents, while they showed relatively high fluorescence intensity in THF: P1 displayed an emission peak at 410 nm (450 nm in the solid state); P2 showed a peak maximum at 420 nm (460 nm in the solid state). The emission peaks of both P1 and P2 in the solid state are broaden and red-shifted compared with those in THF solution. This result suggests the formation of aggregates in the solid state. The fluorescence quantum yields (ΦF) of the polymers in THF were measured in comparison with quinine sulfate as a standard (in 0.1 M H2SO4 solution, ΦF = 0.55).51 The results are 32% for P1 and 34% for P2. The relatively low quantum yields of P1 and P2 might be caused by imperfect structures presented in the hyperbranched polymers with higher contents of branch units and carboxylic ester groups attached to the main chain, which reduced the conjugated length.
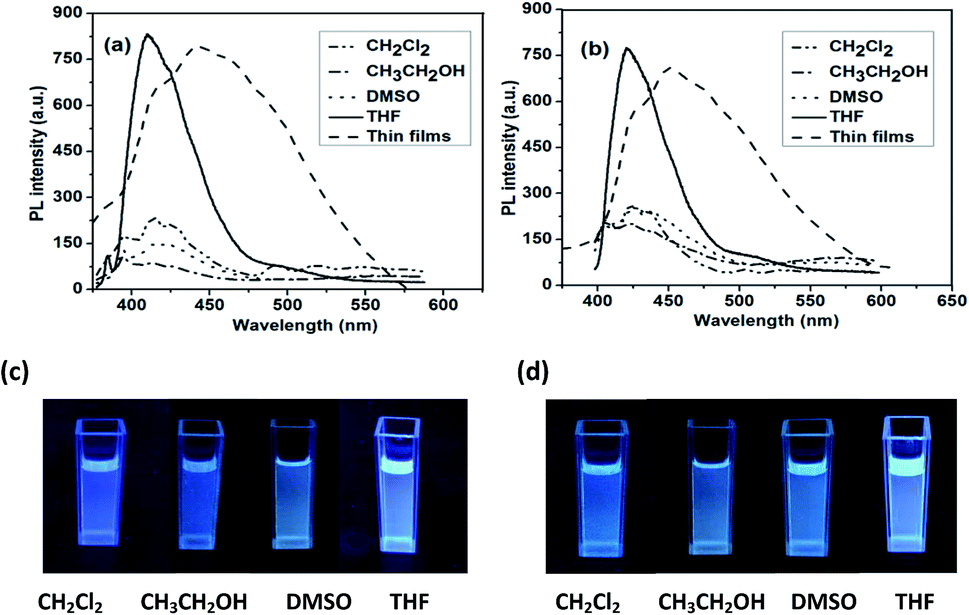 | ||
| Fig. 2 Fluorescence spectra of (a) P1 and (b) P2. Emission images observed for (c) P1 and (d) P2 measured in different solutions. | ||
Electrochemical properties
The electrochemical behaviors of polymers P1 and P2 were investigated by cyclic voltammetry (CV) as shown in Fig. 3 (the results are summarized in Table 2). CV was performed in acetonitrile (0.1 M Bu4NF6) at a scan rate of 50 mV s−1 at room temperature using an SCE as the reference electrode (all potentials reported are versus SCE). A platinum electrode coated with a thin polymer film was used as the working electrode. The onset potentials for oxidation (Eox) are observed to be 0.55 V and 0.54 V for P1 and P2, respectively, and the reduction (Ered) of P1 and P2 were found to be −0.75 V and −0.74 V, respectively. With the formula EHOMO = −e(Eox + 4.29) eV and ELUMO = −e(Ered + 4.29) eV, we estimated the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of the polymer P1 and P2 to be −4.80 eV, −4.79 eV and −3.54 eV, −3.55 eV, respectively. There are no obvious differences in the HOMO and LUMO levels between polymers P1 and P2 due to the same main chain structures and the same end groups.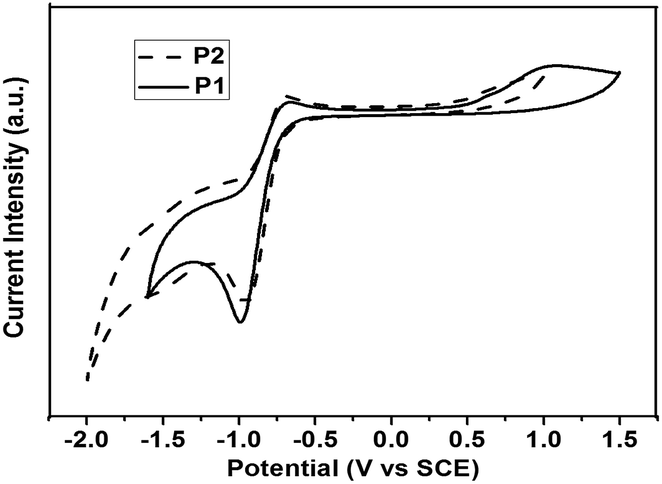 | ||
| Fig. 3 Cyclic voltammograms of the polymer in 0.1 M Bu4NPF6 acetonitrile solution. | ||
| Polymer | Abs. (nm)a | PL (nm)a | HOMOb (eV) | LUMOb (eV) | Egc (eV) |
|---|---|---|---|---|---|
| a THF was used as an the synthesis solvent.b E(HOMO) = −e(Eox + 4.29) eV, E(LUMO) = −e(Ered + 4.29) eV, observed in the cyclic voltammetric analyses.c Eg = E(LOMO) − E(HOMO) (eV). | |||||
| P1 | 325, 380 | 410 | −4.80 | −3.54 | 1.26 |
| P2 | 330, 385 | 420 | −4.79 | −3.55 | 1.21 |
Ion sensing properties
Due to the same main chain of P1 and P2, we chose polymer P1 as a model to investigate its ion sensing property. The results of P2 about ion sensing property were shown in ESI.† The ion sensing properties of polymer P1 was investigated for various metal ions. Variations in the UV-vis absorption and fluorescence spectra of P1 were caused by various competitive metal ions, including Mg2+, Al3+, K+, Fe3+, Cu2+, Ba2+, Ni2+, Zn2+, Mn2+, Pb2+, Ag+, Hg2+, Ca2+, Li+ and Cd2+. The measured concentration of P1 in THF was 1.0 × 10−5 M. The influence of different metal ions ([metal ion] = 1.0 × 10−4 M) on the UV-vis absorption spectra and fluorescence intensity of P1 are shown in Fig. 4a and b. From the UV-vis absorption spectra (Fig. 4a), we can see that P1 showed a significant absorption band centered at about 325 nm upon addition of Fe3+ ions, whereas weaker absorption bands were observed with other ions. Emission quench was observed for P1 upon addition of Fe3+ and Cu2+, and the quench with Fe3+ ions is much more significant compared to that with Cu2+. No fluorescence reduce was observed upon treatment with other metal ions. This result indicates the good selectivity of P1 toward the sensing of Fe3+. According to Swager's group reported, if the absorption peak of UV-vis spectra was red-shifted, the mechanism of quenching is consistent with the static quenching by a charge transfer complex.49 For P1 the addition of 1.0 × 10−4 M Fe3+ shifts the absorption onset from 325 nm to 334 nm (Fig. 4a). For P2 shows the similar phenomenon from 330 nm to 340 nm (Fig. S12†). Therefore, these results indicate that the fluorescence quenching induced by Fe3+ occurs mainly via static quenching mechanism owing to the strong association interaction between the Fe3+ ions and the receptors, carboxylic ester groups (the proposal mechanism was shown in Fig. S11†).52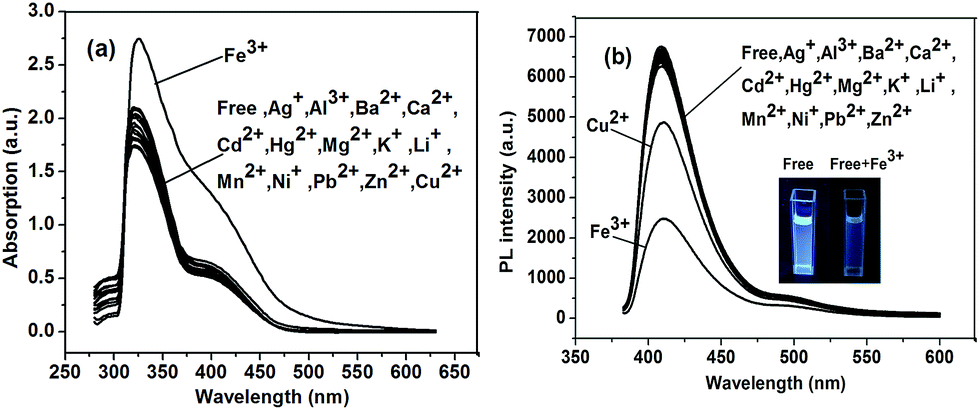 | ||
| Fig. 4 (a) UV-vis absorption spectra of P1; (b) fluorescence spectra and images of P1. | ||
In order to quantitatively measure the effect of metal cations on fluorescence intensity, we calculated the relative fluorescence intensity (I/I0), in which I0 is the fluorescence intensity without addition of quencher and I is the fluorescence intensity after the addition of metal cations. The I/I0 values of P1 and P2 for Mg2+, Al3+, K+, Ba2+, Ni2+, Zn2+, Mn2+, Pb2+, Ag+, Hg2+, Ca2+, Li+ and Cd2+ are almost 1. However, this value of P1 is 0.72 and 0.36 for Cu2+ and Fe3+, respectively; this value of P2 is 0.72 and 0.41 for Cu2+ and Fe3+, respectively. Conspicuously, the highest fluorescence quenching effect of both P1 and P2 was observed in the presence of Fe3+ ions.
Fig. 5 shows the fluorescence change of P1 upon titration with Fe3+ ion ([Fe3+] = 0 to 120 × 10−6 M). With addition of Fe3+ ions, the fluorescence intensity of P1 at 425 nm continuously reduced. The corresponding Stern–Volmer constants (Ksv) were calculated from the Stern–Volmer relationships (eqn (1)),53 where [Q] is the concentration of the quencher (i.e., Fe3+).
| I0/I = 1 + Ksv[Q] | (1) |
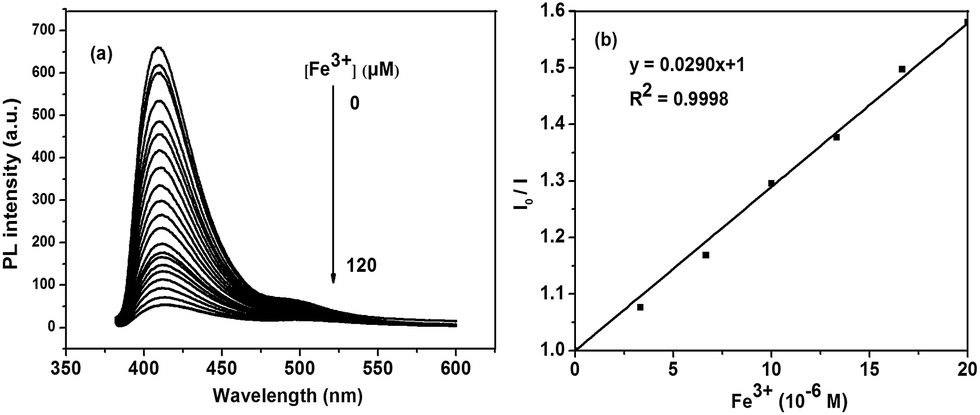 | ||
| Fig. 5 (a) Fluorescence response of P1 to Fe3+ in THF solution; (b) the concentration line chart of P1. | ||
At low concentration (0 to 20 × 10−6 M), I0/I increases linearly with the concentration of quencher. The corresponding Stern–Volmer constant (Ksv) were calculated to be 2.90 × 104 M−1 for P1 and 3.83 × 104 M−1 for P2 (shown in ESI S13†). On the basis of these relationships and 3 × δblank/k (where δblank is the standard deviation of the blank solution and k is the slop of the calibration plot), the detection limit was determined by blank standard method and was calculated to be 6.99 × 10−7 M for P1 and 6.69 × 10−7 M for P2. These numbers indicated that both P1 and P2 were highly sensitive to Fe3+ ions.
It is well known that large side groups attached to the polymer backbone will reduce the inter polymer aggregation. We investigated the fluorescent quenching of P1 and P2 in THF solution upon the addition of Fe3+ ions. The absorption and emission spectra, and the quantum yield of P1 and P2 in THF solution are very similar, which implied the change of side groups did not have much influence on the main chain of the polymer. However, Fe3+-induced quenching is much less efficient for P1 (Ksv = 2.90 × 104 M−1) than P2 (Ksv = 3.83 × 104 M−1), which indicates the bulky side groups indeed stifle interpolymer aggregation and lower the response to metal ions as expected.54
It is reported that the interference from other metal ions will influence the fluorescence quenching assay for a specific metal ion when the sensor is used in complex media. The selectivity of polymers for Fe3+ in the presence of excesses various metal ions was investigated (Fig. 6, and S14† for P2). Upon addition of a mixture of metal ions including Mg2+, Al3+, K+, Cu2+, Ba2+, Ni2+, Zn2+, Mn2+, Pb2+, Ag+, Hg2+, Ca2+, Li+ and Cd2+ (concentration of each metal ion was 1.0 × 10−4 M), the fluorescence intensity of P1 has negligible change. However, when Fe3+ (1.0 × 10−4 M) was added to the mixture solution, the fluorescence intensity could be quenched up to 62% of P1 and 57% for P2.
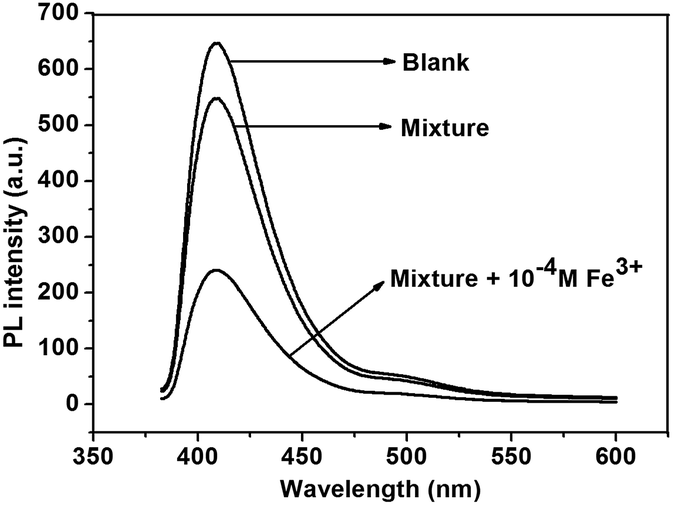 | ||
| Fig. 6 Fluorescence change of P1 upon titration of metal ion mixtures (concentration of each metal ion was 10−4 M) with or without Fe3+ in THF solution. | ||
Ion sensing properties of water-soluble polymer P3
Water-soluble hyperbranched conjugated polymer P3 was obtained by hydrolysis of neutral polymer P1 or P2. The structure of P3 was determined by IR (Fig. S9†), showing a strong broad absorption band at 3450 cm−1 attributed to the OH stretching vibration of –COOH group in P3.Polymer P3 was also examined as a sensor for metal cations. As shown in Fig. 7, dramatic quenching of the emission by Fe3+ was observed, while almost no change could be found in the presence of other cations (a small quenching was observed with Cu2+), indicating the good selectivity of P3 for Fe3+ in aqueous solution.
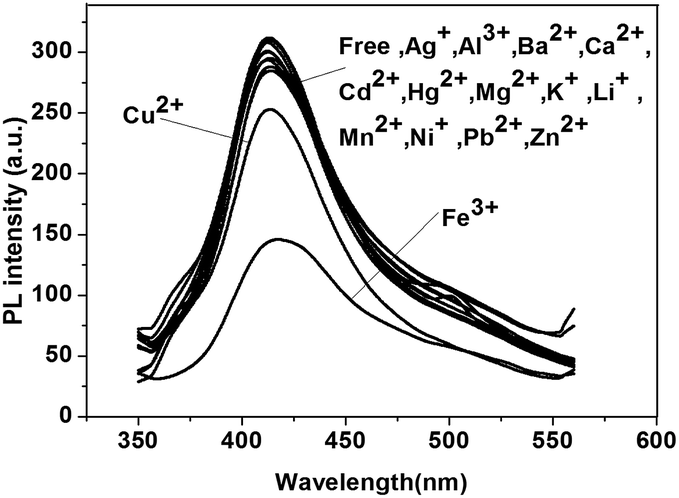 | ||
| Fig. 7 The fluorescence spectra of P3. | ||
Fig. S15† showed the titration of P3 with Fe3+ in aqueous solution. The fluorescence intensity at 415 nm exhibited a good linear relationship with the concentration of Fe3+ (0–40 μM). The corresponding Stern–Volmer constants (Ksv) was calculated to be 1.17 × 104 M−1, and the detection limit was determined to be 5.86 × 10−6 M. According to the results, water-soluble P3 could be considered as a selective detector of Fe3+ ion in aqueous environment.
In order to investigate the pH effect on the fluorescent properties of P3, we measured the fluorescence in 10 mM NaH2PO4–Na2HPO4 buffer solutions with a pH range of 2 to 11. As show in Fig. 8, at basic level (pH ≥ 7), P3 exhibits similar emission behavior with the emission maximum located approximately at 415 nm, which is nearly identical to its neutral polymer P1 and P2 in THF. This result indicates that the polymer isolates well in basic solutions. As the pH decreases to 2, a break point is observed where the absorption maximum shifts from 415 to 400 nm. A further decrease in the pH results in significant reduction of the emission intensity of P3. This pH-dependent fluorescence property indicates that P3 may have two different forms under different pH conditions.6 At high pH, the majority of the carboxylic acid groups are deprotonated (the anionic form). The electrostatic repulsions between anionic carboxylate groups surrounding the polymer backbones reduce the unfavorable aggregation of the polymer chains. While at low pH, the carboxylate groups can be partially neutralized, leading to decreased charge density and thus a decrease in the inter chain electrostatic repulsion. On one hand, P3 may aggregate through hydrogen bonding between carboxylic acid groups and aromatic π–π stacking of hydrophobic backbones. On the other hand, the carboxylic acids make the conjugated main chain twisted and thus decrease the conjugated length. Therefore, at low pH, the emission maxima show blue shift.
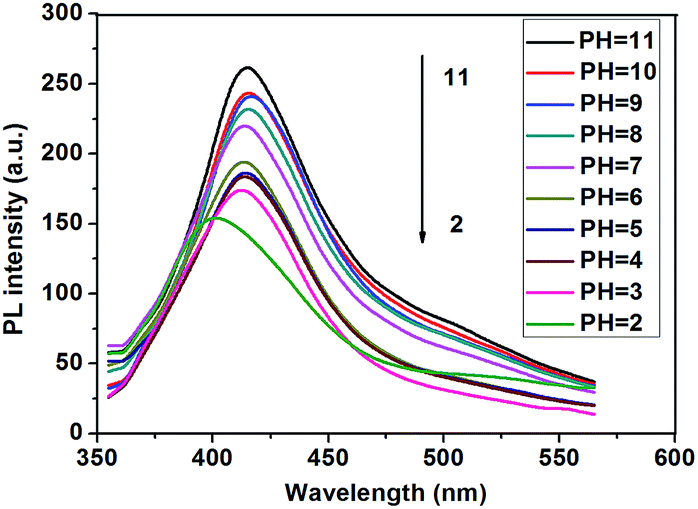 | ||
| Fig. 8 pH-Dependent emission spectra of P3. | ||
Conclusion
In summary, we have synthesized functional hyperbranched polymers containing carboxylic ester groups attached to 1,3-butadiene repeating units. Synthesized polymers P1 and P2 are soluble in common organic solvents and are thermally stable. The neutral hyperbranched conjugated polymers can be hydrolyzed to obtain water-soluble hyperbranched conjugated polymer P3. The fluorescence intensity of these hyperbranched polymers had a good linear response with Fe3+ ions over other ions, indicating their potential uses as ratiometric fluorescence chemosensor for Fe3+ ions in organic and aqueous solutions.Acknowledgements
This work was supported by National Science Basic Research Plan in Shaanxi Province of China (2016JM2019) and the National Key Research Project MOST (2016YFA0202400).Notes and references
- X.-Y. Cao, X.-H. Zhou, H. Zi and J. Pei, Macromolecules, 2004, 37, 8874–8882 CrossRef CAS.
- Y. Xin, G.-A. Wen, W.-J. Zeng, L. Zhao, X.-R. Zhu, Q.-L. Fan, J.-C. Feng, L.-H. Wang, W. Wei, B. Peng, Y. Cao and W. Huang, Macromolecules, 2005, 38, 6755–6758 CrossRef CAS.
- J. Sun, J. Yang, C. Zhang, H. Wang, J. Li, S. Su, H. X. Xu, T. Zhang, Y. Wu, W.-Y. Wong and B. Xu, New J. Chem., 2015, 39, 5180–5188 RSC.
- K. R. Prashanth Kumar, M. G. Murali and D. Udayakumar, Des. Monomers Polym., 2014, 17, 7–18 CrossRef CAS.
- W. Wu, R. Tang, Q. Li and Z. Li, Chem. Soc. Rev., 2015, 44, 3997–4022 RSC.
- B. Bao, X. Zhan and L. Wang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3431–3439 CrossRef CAS.
- W. Ding, H. Zhang, J. Xu, Y. Wen, J. Zhang, H. Liu, Y. Yao and Z. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(23), 3694–3700 CrossRef CAS.
- H. Li, X. Wu, Y. Xu, H. Tong and L. Wang, Polym. Chem., 2014, 5, 5949–5956 RSC.
- L. Chen, Y. Gao, Y. Fu, D. Zhu, Q. He, H. Cao and J. Cheng, RSC Adv., 2015, 5, 29624–29630 RSC.
- Y. Xu, X. Wu, Y. Chen, H. Hang, H. Tong and L. Wang, Polym. Chem., 2016, 7, 4542–4548 RSC.
- X.-S. Ma, Y.-Z. Cui, Y.-Q. Ding, F.-R. Tao, B. Zheng, R.-H. Yu and W. Huang, Sens. Actuators, B, 2017, 238, 48–57 CrossRef CAS.
- B. Zheng, Y. Li, F. Tao, Y. Cui and T. Li, Sens. Actuators, B, 2017, 241, 357–363 CrossRef CAS.
- R. Hu, J. W. Y. Lam, J. Liu, H. H. Y. Sung, I. D. Williams, Z. Yue, K. S. Wong, M. M. F. Yuend and B. Z. Tang, Polym. Chem., 2012, 3, 1481–1489 RSC.
- W. Z. Yuan, R. Hu, J. W. Y. Lam, N. Xie, C. K. W. Jim and B. Z. Tang, Chem.–Eur. J., 2012, 18, 2847–2856 CrossRef CAS PubMed.
- D. Ding, G. Wang, J. Liu, K. Li, K.-Y. Pu, Y. Hu, J. C. Y. Ng, B. Z. Tang and B. Liu, Small, 2012, 8(22), 3523–3530 CrossRef CAS PubMed.
- H. Ejima, H. Matsuno and T. Serizawa, Langmuir, 2010, 26, 17278–17285 CrossRef CAS PubMed.
- D. Wang, T. Zhao, X. Zhu, D. Yan and W. Wang, Chem. Soc. Rev., 2015, 44, 4023–4071 RSC.
- X. Wu, H. Li, Y. Xu, H. Tong and L. Wang, Polym. Chem., 2014, 6, 2305–2311 RSC.
- J. Sun, J. Yang, C. Zhang, H. Wang, J. Li, S. Su, H. Xu, T. Zhang, Y. Wu, W.-Y. Wong and B. Xu, New J. Chem., 2015, 39, 5180–5188 RSC.
- H. T. Nguyen, L.-T. T. Nguyen, T. T. Nguyen, A. T. Luu and T. V. Le, J. Polym. Res., 2014, 21(552), 2–11 Search PubMed.
- D. Li, X. Wang, Y. Jia, A. Wang and Y. Wu, Chin. J. Chem., 2012, 30, 861–868 CrossRef CAS.
- J. Wang, J. Mei, E. Zhao, Z. Song, A. Qin, J. Z. Sun and B. Z. Tang, Macromolecules, 2012, 45, 7692–7703 CrossRef CAS.
- C. P. Sen, V. D. Goud, R. G. Shrestha, L. K. Shrestha, K. Ariga and S. Valiyaveettil, Polym. Chem., 2016, 7, 4213–4225 RSC.
- J. Qu, M. Shiotsuki, N. Kobayashi, F. Sanda and T. Masuda, Polymer, 2007, 48, 6481–6490 CrossRef CAS.
- J. Oriou, F. Ng, G. Hadziioannou, G. Garbay, M. Bousquet, L. Vignau, E. Cloutet and C. Brochon, Polym. Chem., 2014, 5, 7100–7108 RSC.
- M. R. Raj and S. Anandan, RSC Adv., 2013, 3, 14595–14608 RSC.
- S. Chhatre, S. Agarkar, V. Dhas, S. Nagane, V. Thakare, S. Ogale and P. Wadgaonkar, J. Mater. Chem., 2012, 22, 23267–23271 RSC.
- M. Yoon, H. W. Yoon, A. J. Kim, R. S. Ree, Y.-S. Gal, J. W. Lee, J. H. Lee and S.-H. Jin, Mol. Cryst. Liq. Cryst., 2011, 550, 304–310 CrossRef CAS.
- B. J. Laughlin and R. C. Smith, Macromolecules, 2010, 43, 3744–3749 CrossRef CAS.
- S. Barik, T. Bletzacker and W. G. Skene, Macromolecules, 2012, 45, 1165–1173 CrossRef CAS.
- S. Barik and W. G. Skene, Polym. Chem., 2011, 2, 1091–1097 RSC.
- S. Dufresne and W. G. Skene, J. Phys. Org. Chem., 2012, 25, 211–221 CrossRef CAS.
- A. Upadhyay and S. Karpagam, Polym. Bull., 2016, 73, 2741–2760 CrossRef CAS.
- M. S. Maji, T. Pfeifer and A. Studer, Chem.–Eur. J., 2010, 16, 5872–5875 CrossRef CAS PubMed.
- M. Bourgeaux and W. G. Skene, Macromolecules, 2007, 40, 1792–1795 CrossRef CAS.
- Q. He, F. Bai, J. Yang, H. Lin, H. Huang, G. Yu and Y. Li, Thin Solid Films, 2002, 417, 183–187 CrossRef CAS.
- Y. Sun, Y. Sun, Q. Pan, G. Li, B. Han, D. Zeng, Y. Zhang and H. Cheng, Chem. Commun., 2016, 52, 3000–3002 RSC.
- X.-Y. Cao, W.-B. Zhang, J.-L. Wang, X.-H. Zhou, H. Lu and J. Pei, J. Am. Chem. Soc., 2003, 125, 12430–12431 CrossRef CAS PubMed.
- X. Cai, Y. Liu, T. Lu, R. Yang, C. Luo, Q. Zhang and Y. Chai, Macromol. Rapid Commun., 2016, 37, 2005–2010 CrossRef CAS PubMed.
- J. Y. Cabon, P. Giamarchi and A. L. Bihan, Anal. Chim. Acta, 2010, 664, 114–120 CrossRef CAS PubMed.
- B. Muthuraj, S. Hussain and P. K. Iyer, Polym. Chem., 2013, 4, 5096–5107 RSC.
- G. Saikia and P. K. Iyer, Macromolecules, 2011, 44, 3753–3758 CrossRef CAS.
- J. Y. Xu, Y. Zhou, G. F. Cheng, S. X. Liu, M. T. Dong and C. B. Huang, Luminescence, 2015, 30, 451–456 CrossRef CAS PubMed.
- C. J. Qin, Y. X. Cheng, L. X. Wang, X. B. Jing and F. S. Wang, Macromolecules, 2008, 41, 7798–7804 CrossRef CAS.
- P. Li, Y. Zhao, L. Yao, H. Nie and M. Zhang, Sens. Actuators, B, 2014, 191, 332–336 CrossRef CAS.
- W. C. Ding, J. K. Xu, Y. P. Wen, H. Zhang and J. J. Zhang, J. Photochem. Photobiol., A, 2016, 314, 22–28 CrossRef CAS.
- X. F. Wu, B. W. Xu, H. Tong and L. X. Wang, Macromolecules, 2010, 43, 8917–8923 CrossRef CAS.
- H. Zhang, G. Zhang, J. K. Xu, Y. P. Wen, B. Y. Lu, J. Zhang and D. W. Ding, Sens. Actuators, B, 2016, 230, 123–129 CrossRef CAS.
- Q. Zhou and T. M. Swager, J. Am. Chem. Soc., 1995, 117, 7017–7018 CrossRef CAS.
- K. Ono, R. Aizawa, T. Yamano, S. Ito, N. Yasuda, K. Johmoto, H. Uekusa and N. Iwasawa, Chem. Commun., 2014, 50, 13683–13686 RSC.
- D. Nagaraja, R. M. Melavanki, N. R. Patil, H. S. Geethanjali and R. A. Kusanur, Luminescence, 2015, 30, 495–502 CrossRef CAS PubMed.
- G. Zhou, Y. Cheng, L. Wang, X. Jing and F. Wang, Macromolecules, 2005, 38, 2148–2153 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publishers: New York, 1999 Search PubMed.
- H. Tong, L. Wang, X. Jing and F. Wang, Macromolecules, 2002, 35, 7169–7171 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00540g |
| This journal is © The Royal Society of Chemistry 2017 |