
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrospun nanofiber membranes as ultrathin flexible supercapacitors
W. K. Cheea,
H. N. Lim *ab,
Z. Zainala,
I. Harrisonc,
N. M. Huangd,
Y. Andoue,
K. F. Chongf and
A. Pandikumarg
*ab,
Z. Zainala,
I. Harrisonc,
N. M. Huangd,
Y. Andoue,
K. F. Chongf and
A. Pandikumarg
aDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. E-mail: janetlimhn@gmail.com
bFunctional Device Laboratory, Institute of Advanced Technology, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
cDepartment of Electrical and Electronic Engineering, Faculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK
dCentre of Printable Electronics, Deputy Vice Chancellor Office (Research & Innovation), University of Malaya, 50603 Kuala Lumpur, Malaysia
eGraduate School of Life Science and Systems Engineering, Eco-Town Collaborative R&D Center for the Environment and Recycling, Kyushu Institute of Technology, 2-4 Hibikino, Wakamatsu-ku, Kitakyushu-city, Fukuoka 808-0196, Japan
fFaculty of Industrial Sciences & Technology, Universiti Malaysia Pahang, 26300 Gambang, Kuantan, Pahang, Malaysia
gResearch Institute & Department of Chemistry, SRM University, SRM Nagar, Kattankulathur-603 203, Chennai, India
First published on 20th February 2017
Abstract
A highly flexible electrochemical supercapacitor electrode was developed with a novel metal oxide-reinforced nanofiber electrode by utilizing a solution-based electrospinning technique. The facile fabrication steps involved the introduction of metal precursors into a polymeric solution, which was subjected to an in situ electrospinning process. The electrospun polymeric web with metallic ingredients was then subjected to an oxidative stabilization process that induced the formation of metal oxide nanoparticles within the polymer structure. Finally, the metal oxide nanoparticles incorporated with nanofibers were obtained using a carbonization process, thus converting the polymer backbones into a carbon-rich conductive nanofiber structure. The fabricated nanofibers were decorated and implanted with metal oxide nanoparticles that had a surface-decorated structure morphology due to the solubility of the precursors in the reaction solution. The electrochemical performance of the fabricated metal oxide reinforced with nanofiber electrodes was investigated as an electrochemical system, and the novel morphology significantly improved the specific capacitance compared to a pristine carbon nanofiber membrane. As a result of the uniform dispersion of metal oxide nanoparticles throughout the surface of the nanofibers, the overall capacitive behavior of the membrane was enhanced. Furthermore, a fabricated free-standing flexible device that utilized the optimized nanofiber electrode demonstrated high stability even after it was subjected to various bending operations and curvatures. These promising results showed the potential applications of these lightweight, conductive nanofiber electrodes in flexible and versatile electronic devices.
1. Introduction
The research on flexible electronic devices has expanded to become one of the most popular research fields over the last few decades. The trend of electronics has moved toward the fabrication of devices that are flexible and stretchable. Rapid developments in electronics, such as flexible displays, wearable devices, flexible cellular phones, and energy devices (battery anodes and supercapacitor electrodes), have created a wide demand for flexible devices that theoretically possess excellent flexibility, light weight, softness, high transparency, and good mechanical strength, using integrated circuits that are commonly available today and now possible to obtain.However, despite displaying outstanding performance when tested within their flexibility range, the current flexible devices still show limited bendability when they are subjected to a narrow curvature or high mechanical deformation.1,2 Materials such as nickel foam, aluminum foil, nickel foil, and graphite sheets offer simple flexibility that only allows roll-to-roll production without any assurance of the conformal bonding of the fabricated devices toward the edges. Therefore, there is a great need to fabricate a highly flexible conductive substrate that is able to handle large deformations without breaking apart or experiencing performance failure.3
Electrospinning is one of the most effective techniques for producing non-woven continuous polymer nanofibers with diameters that range from several micrometers down to a few nanometers,4 with characteristics similar to the fibers produced using both electro-spraying and conventional solution dry spinning.5 Non-woven nanofibers commonly have the characteristics of a large surface area and high porosity.6–9
The fascinating characteristics of polymer nanofibers have been utilized in various applications, including biomedical applications involving drug delivery, wound dressing, enzyme immobilization, and tissue engineering.10 These nanofibers have subsequently been introduced in energy storage applications such as lithium ion batteries and as electrode materials for supercapacitors because of their high specific surface area.11–14 Essentially, various polymeric materials have been used as backbone polymers, with polyacrylonitrile (PAN) the most commonly used precursor. Carbon nanofibers (CNF) have been produced via a series of stabilization and carbonization processes using heat treatment in an inert-gas-filled furnace.15
The electrospinning trend has moved toward the introduction of organic/inorganic materials within the nanofibers in order to maximize their characteristic features.16–19 Organic-based materials, including carbon nanotubes, which have been used as electrode materials for supercapacitors as a result of their high specific surface area,15,20 were incorporated into a carbon nanofiber network to boost the electrical conductivity and capacitance behavior of energy storage devices. However, various metal oxides, including nickel oxide,21,22 copper oxide,23 and titanium dioxide have been utilized as nanofillers to enhance the electrochemical performances,24 while also utilizing the sole advantage of the high specific surface area of carbon nanofibers.
With this approach, electrospinning was employed to fabricate continuous non-woven polymer nanofibers with the minimal amount of metal precursors loaded within the polymer solution. The electrospun nanofibers were subjected to oxidative stabilization and carbonization in an inert atmosphere to form carbon nanofibers (CNF). The metal oxides grew along the carbon structure, which then diffused and aggregated into nanoparticles on the nanofibers surface.25 The combination of CNF and metal oxide nanoparticles therefore induced a continuous and conductive network of nanofibers in a membrane, which exhibited superior electrochemical properties, while also retaining a lightweight property and high degree of flexibility. The fabricated membrane had electrochemical properties that outperformed those of carbon cloth, nickel foam, nickel foil, and ITO.
2. Experimental methods
2.1 Materials
Polyacrylonitrile (PAN, MW: 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Sigma Aldrich, Malaysia. Zinc sulphate heptahydrate (ZnSO4·7H2O) and nickel sulphate hexahydrate (NiSO4·6H2O) were purchased from Merck, Malaysia. N,N-Dimethylformamide (DMF) was obtained from Systerm, Malaysia. Whatman qualitative filter paper was purchased from Sigma Aldrich, Malaysia. Graphite sheet (>99.5%) was obtained from Latech, Singapore.
000) was purchased from Sigma Aldrich, Malaysia. Zinc sulphate heptahydrate (ZnSO4·7H2O) and nickel sulphate hexahydrate (NiSO4·6H2O) were purchased from Merck, Malaysia. N,N-Dimethylformamide (DMF) was obtained from Systerm, Malaysia. Whatman qualitative filter paper was purchased from Sigma Aldrich, Malaysia. Graphite sheet (>99.5%) was obtained from Latech, Singapore.
2.2 Preparation of PAN nanofibers and PAN loaded with metal precursors
A polymer solution of PAN (10 wt%) was prepared by dissolving 1.0 g of PAN in 10 mL of DMF solution and stirring for 24 h. After stirring, the solution was electrospun using an electrospinner setup (Electroris, Nanolab Malaysia) with an applied voltage of 15.0 kV, whereby the solution was used to fill a syringe connected to a flat-tip needle. As a result, the polymer solution was spun out as a polymer nanofiber and collected on a rotating drum. The nanofiber membrane was then collected from the collector after 2 h of spinning. Similarly, PAN reinforced with metal precursors was prepared by dissolving both the PAN powder and a metal precursor (Zn2+ and Ni2+) into a DMF solution. The concentration of metal precursors was fixed at 0.1 M from the total volume of the polymeric solution.2.3 Carbonisation of polymer nanofibers
The carbonization process was carried out using a quartz tube furnace (Thermolyne, Thermo Scientific). The nanofiber was first subjected to oxidative activation by heating it at 280 °C for 1 h under atmospheric air. Subsequently, the carbonization process took place, where the oxidized nanofiber was heated to 800 °C and maintained at that temperature for 1 h under an inert atmosphere. Upon carbonization, a conductive carbon nanofiber (CNF) was obtained. The zinc oxide-reinforced CNF was labeled as CNF-ZnO, and nickel oxide-reinforced CNF was labeled as CNF-NiO.2.4 Characterisation techniques
The surface morphologies and elemental mapping of the nanofibers were analyzed using a field emission scanning electron microscope (FEI Quanta SEM Model 400F) equipped with an energy dispersive X-ray (EDX) accessory. The Fourier transform-infrared (FT-IR) spectra of the samples were recorded in the range of 280–4000 cm−1 using the attenuated total reflectance (ATR) on a Fourier transform infrared spectrophotometer (Perkin Elmer 1650). Raman spectra were recorded using Renishaw's inVia Raman microscope with 532 nm laser excitation. X-ray photoelectron spectra (XPS) were obtained via a Kratos at 450 W and 1.0 eV (Shimadzu).2.5 Preparation of electrochemical cells and measurements
The electrochemical properties of the samples were evaluated using a two-electrode configuration with a potentiostat–galvanostat (Princeton VersaSTAT 3, Princeton Applied Research). The as-prepared carbon nanofibers were then cut into a symmetrical shape with the approximate dimension of 1.0 cm2. A nylon membrane filter was sandwiched between the prepared samples and tightly fitted into an electrochemical cell. The nylon membrane was soaked in liquid electrolyte overnight prior to its use.The specific capacitance was obtained from the galvanostatic charging/discharging mechanism via the slope of the discharge curve according to the following equation:26,27
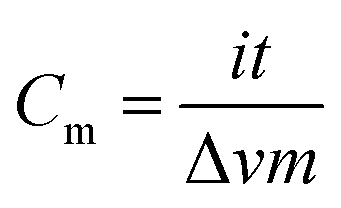 | (1) |
3. Results and discussion
3.1 Effects of carbonisation on morphology of PAN nanofibers
Upon stabilization, the PAN nanofibers changed from white to a yellow-brownish color. Complex reactions occurred throughout the oxidation stabilization process, including cyclization, dehydrogenation, aromatization, oxidation, and crosslinking, which eventually strengthened the physical properties of the nanofibers by forming a ladder structure.28,29 On the other hand, the oxidation process provided the nanofibers with higher stability, allowing them to undergo a high-temperature carbonization treatment by forming bridging links between the fibers by means of oxidative functional groups such as ether and carbonyl groups.30 The PAN nanofibers turned black during the carbonization process (Fig. 1A–C). The electro-spun out nanofibers formed a continuous network by overlapping with each other, resulting in a continuous structure of non-woven nanofibers forming a membrane.31 In the two-step thermal treatment, the PAN nanofibers were successfully carbonized into carbon nanofibers, as proven by the detection of carbon with a very high intensity (Fig. 1D) compared to that of the non-carbonized PAN nanofibers (Fig. 1E). | ||
| Fig. 1 Photograph illustrating effects of carbonization on appearance of (A) pure PAN, (B) CNF-ZnO, and (C) CNF-NiO; EDX measurements of (D) PAN and (E) CNF based on inset images. | ||
3.2 Effects of metal precursor on morphologies of PAN nanofibers and CNF
Fig. 2 illustrates the morphology of the PAN nanofibers loaded with the Zn and Ni precursors upon electro-spinning. Upon the introduction of both precursors, the morphology retained almost a similar structure, with random overlapping nanofibers at various diameters (Fig. 2A and B). After the carbonization process, the CNF that had been reinforced with metal oxide loadings showed a unique morphology, as depicted in Fig. 2C and D, with large amounts of nanoparticles clearly embedded onto the surface of the carbon nanofibers. This unique feature was attributed to the solubility of the metallic precursors in the PAN polymeric solution. During the electrospinning process, the poor miscibility of Zn2+ and Ni2+ ions in the polymeric solution resulted in a two-phase system, leading to a large amount of metal ions depositing on the surface of the polymeric nanofibers after the solvent evaporated. The presence of both elements was strongly evidenced by the EDX spectra, as illustrated in Fig. 2E and F. | ||
| Fig. 2 FESEM images of PAN nanofibers loaded with (A) Zn and (B) Ni metal precursors, CNF nanofibers loaded with (C) ZnO and (D) NiO, and elemental maps of (E) CNF-ZnO and (F) CNF-NiO. | ||
3.3 Chemical composition of nanofibers (NFs)
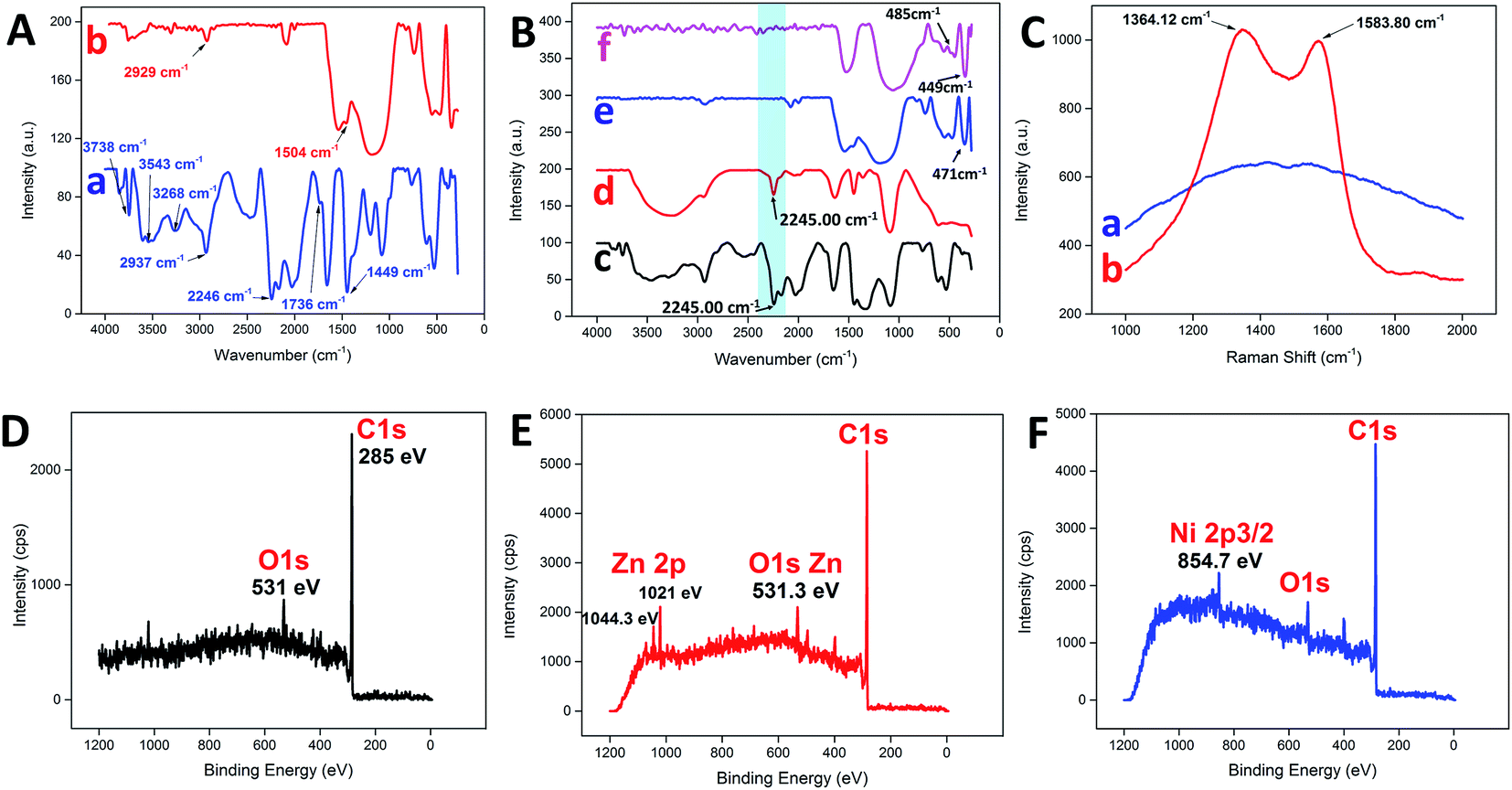
Fig. 3A illustrates the changes in the FTIR spectrum of the PAN NFs before and after the carbonization process. For pure PAN NFs, the main stretching vibration of the nitrile group was assigned to 2246 cm−1,32 whereas the bands at 2937 cm−1 and 1736 cm−1 were assigned to C–H stretching and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, respectively.33 The bending vibration of the methylene group (–CH2–) was detected at 1449 cm−1.4,34 On the other hand, broad bands were detected at 3268 cm−1 and 3543 cm−1, with a sharp band at 3738 cm−1, attributed to the presence of water adsorbed on the surface.35 In contrast, the nitrile group was missing as a consequence of carbonization, leaving behind the C–H and methylene stretching at 2929 cm−1 and 1504 cm−1, respectively. Similarly, the nitrile group stretching at 2246 cm−1 for PAN was still noticeable after the addition of various metal precursors, as shown in Fig. 3B. The addition of metal precursors did not shift the characteristic band of PAN, which confirmed the weak interaction between the main matrix and the precursors.36 However, the typical characteristic vibration of the nitrile group was non-detectable for all the samples after the carbonization process, as highlighted by the blue band in Fig. 3B. The intense band within the region of 3400 cm−1 to 3500 cm−1 may have been due to the presence of adsorbed water.37 Furthermore, some of the characteristic bands corresponding to each of the metal oxides were detected at the lower wavenumber region from the spectra. For CNF-ZnO, the vibration band for Zn–O was detected at 471 cm−1 (Fig. 3B e).38,39 In addition, for CNF-NiO, the bands corresponding to the Ni–O stretching vibration were detected at 485 cm−1 and 449 cm−1 (Fig. 3B f).40,41
O stretching, respectively.33 The bending vibration of the methylene group (–CH2–) was detected at 1449 cm−1.4,34 On the other hand, broad bands were detected at 3268 cm−1 and 3543 cm−1, with a sharp band at 3738 cm−1, attributed to the presence of water adsorbed on the surface.35 In contrast, the nitrile group was missing as a consequence of carbonization, leaving behind the C–H and methylene stretching at 2929 cm−1 and 1504 cm−1, respectively. Similarly, the nitrile group stretching at 2246 cm−1 for PAN was still noticeable after the addition of various metal precursors, as shown in Fig. 3B. The addition of metal precursors did not shift the characteristic band of PAN, which confirmed the weak interaction between the main matrix and the precursors.36 However, the typical characteristic vibration of the nitrile group was non-detectable for all the samples after the carbonization process, as highlighted by the blue band in Fig. 3B. The intense band within the region of 3400 cm−1 to 3500 cm−1 may have been due to the presence of adsorbed water.37 Furthermore, some of the characteristic bands corresponding to each of the metal oxides were detected at the lower wavenumber region from the spectra. For CNF-ZnO, the vibration band for Zn–O was detected at 471 cm−1 (Fig. 3B e).38,39 In addition, for CNF-NiO, the bands corresponding to the Ni–O stretching vibration were detected at 485 cm−1 and 449 cm−1 (Fig. 3B f).40,41
 | ||
| Fig. 3 FTIR spectra of (A) PAN and its carbonized form and (B) comparison of metal precursor-loaded PAN and CNF; (C) Raman spectra of PAN and its carbonized form; wide scans of XPS spectra of (D) CNF, and CNF reinforced with (E) ZnO and (F) NiO. The insets refer to (a) PAN, (b) CNF, (c) PAN-Zn, (d) PAN-Ni, (e) CNF-ZnO, and (f) CNF-NiO. | ||
The Raman spectrum of the PAN NFs was compared to that of CNF (Fig. 3C). The spectrum of CNF had distinguishable D and G bands at 1364.12 cm−1 and 1583.80 cm−1, respectively. The D-band characteristic was attributed to the disordered turbostratic structures or defects,15 whereas the G band correlated to the phonons propagating along the graphitic structure.42 The D and G bands represent the characteristics of disordered carbon and, respectively. The high intensity of the D band compared to the G band indicated that the disordered carbon content was higher than that of the ordered graphite because of the amorphous structure of the CNF as a result of the subsequent stabilization process in air,43 which disrupted the crystallinity of the nanofibers. Moreover, the high intensity of the D and G bands of the CNF compared to the PAN clearly indicated that the PAN NFs were successfully carbonized into a highly carbonaceous compound. An XPS analysis was conducted to distinguish the chemical composition of the prepared carbon nanofibers. A wide scan of the XPS spectra was taken at a range of 0–1200 eV for each sample, with the spectra illustrated. Typically, for pure CNF (Fig. 3D), a distinctive peak was observed at 285 eV, which corresponded to the C 1s of the carbon-based material. Another distinctive peak for O 1s was also detected at 531 eV. For C 1s metal oxide-reinforced nanofibers, the characteristic peaks of ZnO (Fig. 3E) at 2p3/2 and 2p1/2 were detected at 1044.3 eV and 1021 eV, respectively, for CNF-ZnO, which were attributed to Zn(II) being bonded to an oxygen atom to form ZnO.44,45 CNF-NiO (Fig. 3F) also recorded a characteristic peak of Ni 2p5/2 at 754.7 eV, corresponding to NiO,46 along with the corresponding detected peaks for C 1s and O 1s.
3.4 Electrochemical performances of PAN nanofibers and CNF
In the study of the electrochemical performance, both the CNF-NiO and CNF-ZnO membranes were evaluated electrochemically as supercapacitor electrodes, with the use of 6.0 M KOH as the electrolyte in a two-electrode configuration. Identical pieces of CNF-metal oxide membranes were separated by a filter paper (soaked in electrolyte prior to use) and subsequently fitted into an electrochemical cell. Generally, the ideal capacitor behavior shows a perfect square profile in CV measurements, irrespective of the scan rate. A near square rectangular profile was absent from the CV profiles of the pure CNF, as shown in Fig. 4A, reflecting the presence of resistivity within the system, which was demonstrated by a steep discharge loop plausibly contributed by the oxygenated groups present within the nanofibers. CNF-ZnO recorded a pseudo-rectangular-shaped curve and a much broader area under the curve, with no obvious redox peaks observed, as shown in Fig. 4C. The CNF-NiO possessed an almost rectangular-like CV curve regardless of the scan rate (Fig. 4E), which indicated a good capacitance behavior as an energy storage system.47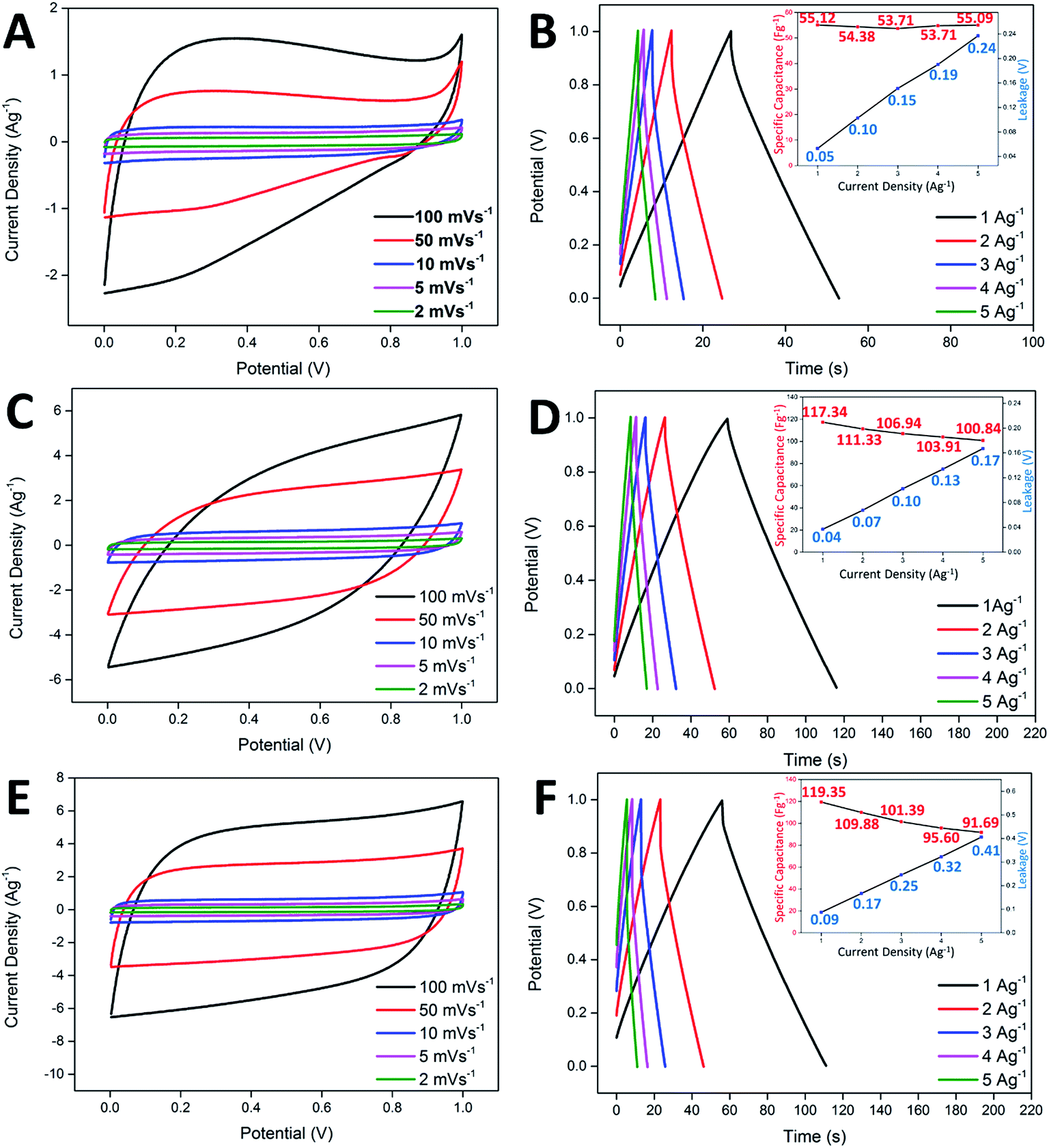 | ||
| Fig. 4 Cyclic voltammograms obtained at various scan rates for (A) blank CNF, (C) CNF-ZnO, and (E) CNF-NiO; galvanostatic charge/discharge analysis of (B) blank CNF, (D) CNF-ZnO, and (F) CNF-NiO. The insets illustrate the specific capacitance and leakage drop as a function of the current density. | ||
Galvanostatic charge/discharge analysis is often applied to mimic the real-field testing of a supercapacitor device, while evaluating the actual performance of the electrochemical system.48 The pure CNF-based supercapacitor showed a specific capacitance of 55.12 Fg−1, with minimal leakage occurring at 0.05 V (Fig. 4B). However, the increase in leakage potential became drastic when the current density also increased. This was attributed to the insulating oxygenated functional groups present within the nanofibers, which created an internal resistance across the electrode/electrolyte interface. Although the CNF-ZnO showed a much higher specific capacitance of 119.35 Fg−1 in 1 Ag−1 compared to the pristine sample, the leakage was prominent (0.09 V) at 1 Ag−1, up to the point that it continued to increase at the highest current applied, where the leakage was recorded at 0.41 V, as shown in the inset of Fig. 4D. This might have been caused by the resistivity characteristic of ZnO.49 The CNF-NiO showed a better performance by recording a specific capacitance of 117.34 at 1 Ag−1 and managed to retain a value of 100.84 Fg−1 even at a high current density of 5 Ag−1. A very minor leakage was detected at 1 Ag−1 (0.04 V) and it remained minimal (0.17 V) even at 5 Ag−1 (inset of Fig. 4F), indirectly supporting the good capacitive behavior portrayed in the CV profiles for CNF-NiO.
The electrochemical impedance spectra recorded for pure CNF, CNF-ZnO, and CNF-NiO are shown in Nyquist plots in Fig. 5A. Two distinctive features are indicated in these plots. First is the high frequency arc caused by a charge transfer resistance (Rct) attributed to the charge-limiting process between the interface of the electrode material and electrolyte,50 which could be obtained from the diameter of the arc.51 Second is the equivalent series resistance (ESR) caused by the resistance of the solution, along with the internal resistance of the electrode. This could be determined from the high-frequency limit, which corresponded to the first intercept on the X-axis of the Nyquist plot.52 As seen in Fig. 5A, the high-frequency arc associated with CNF-ZnO possessed the largest radius, indicating a higher charge transfer resistance than the other samples. In contrast, the CNF-NiO sample exhibited the smallest diameter, indicating a significantly lower resistance and rapid ionic diffusion within the electrode material. The ESR and Rct values of CNF-ZnO (ESR = 0.24 Ω, Rct = 26.39 Ω) were higher than those of blank CNF (ESR = 0.22 Ω, Rct = 10.39 Ω). The presence of Zn ions within the electrospun nanofibers resulted in a conversion that formed large particles of ZnO as an effect of the stabilization heating process, which was responsible for the overall resistive structure.49 On the other hand, the CNF-NiO possessed a significantly smaller Rct, with a slightly higher ESR value (ESR = 0.79 Ω, Rct = 2.48 Ω), which were attributed to the unique performance of the NiO nanoparticles, and thereby facilitated the charge transfer performance of the electrode/electrolyte interface. Nevertheless, all the samples recorded almost ideal vertical behaviors behind the Warburg diffusion region, showing that the total internal structures of the electrodes were completely wetted by the electrolyte.53
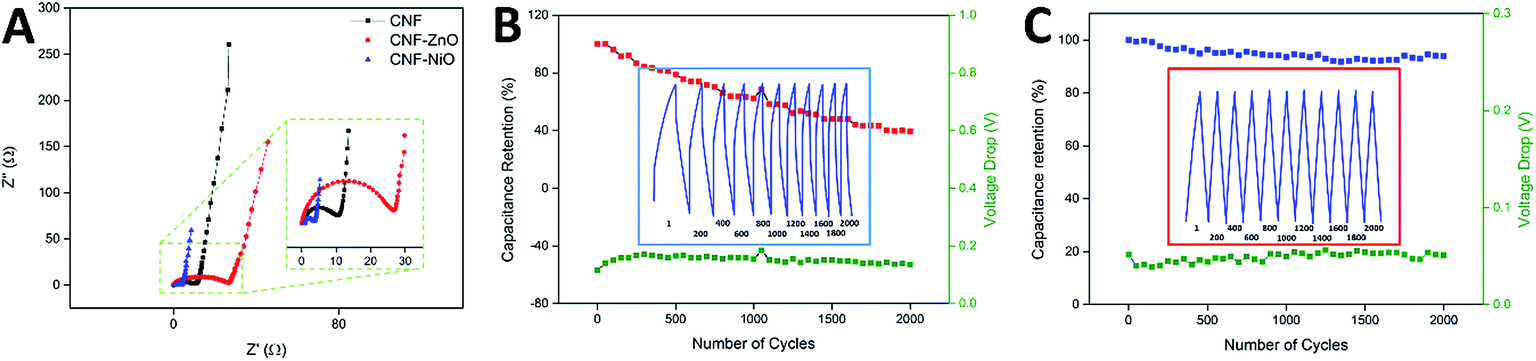 | ||
| Fig. 5 (A) Nyquist plots of pure CNF, CNF-ZnO, and CNF-NiO. Inset shows a magnified view of the high-frequency region of the spectra and cycle stability performances of (B) CNF-ZnO and (C) CNF-NiO supercapacitors. Green plot indicates voltage leakage drop across the charging cycles. | ||
The cyclic performance and capacitance retention of supercapacitors are crucial characteristics for practical energy storage applications. For CNF-ZnO, a capacitance retention of 62.41% was recorded after 1000 continuous charging/discharging cycles, with an average voltage drop of 0.15 V at every discharge cycle (indicated by the green line in Fig. 5B). The major loss in capacitance could have resulted from the leakage that occurred throughout the charge/discharge process, as reflected by the non-symmetrical and distorted discharge slope (Fig. 5B inset), due to the resistivity of the electrode material itself, as depicted by the impedance spectra. The retention had a much lower value of 40% at 2000 cycles, indicating a major degradation and loss of electrode materials, which might have been caused by the conversion of ZnO to aqueous-soluble Zn(OH)2 under a strong alkaline condition.54 On the other hand, CNF-NiO had a very high capacitance retention of approximately 93.60% after 1000 vigorous charging/discharging cycles at 1 Ag−1. The capacitance was retained at 93.90% even after 2000 cycles (Fig. 5C), which was attributed to the very minimal voltage drop of approximately 0.05 V throughout the continuous cycles (green line of Fig. 5C). This showed a very constant capacitive behavior as an effect of the growth of the nickel oxide nanoparticles, which were effectively anchored onto the surface of the CNF and prevented the loss of active materials throughout the influx/outflow of electrolyte ions. Almost symmetrical charge/discharge cycles were obtained, showing the highly stable performance of the electrochemical device.
In order to further evaluate the feasibility of using the fabricated metal oxide-CNF membrane in a realistic application, a free-standing, thin, flexible symmetric supercapacitor was fabricated by integrating CNF-NiO as the electrodes, thin layer graphite sheet/cellophane tape (GSC) as current collectors, and a cellulosic paper as the dielectric in 6 M KOH. The flexible current collector (GSC) was initially prepared by applying a layer of cellophane tape to a graphite sheet and then peeling it off to obtain a thin, conductive graphite layer attached firmly to the tape, as shown in Fig. 6A and B, respectively. A device was then fabricated by sandwiching a KOH-soaked dielectric filter paper with two pieces of identical CNF-NiO attached to GSC layers, as shown in the schematic diagram in Fig. 6C. The synergistic combination of the flexible GSC and CNF-NiO membrane electrodes resulted in a versatile, bendable energy storage device. The fabricated, thin supercapacitor had a stable electrochemical performance with no deviation in the CV profiles (Fig. 6D) even after it was subjected to various flexibility tests, including mechanical bending and roll-up curvature (Fig. 6D b & c), compared to its original state (Fig. 6D, a).
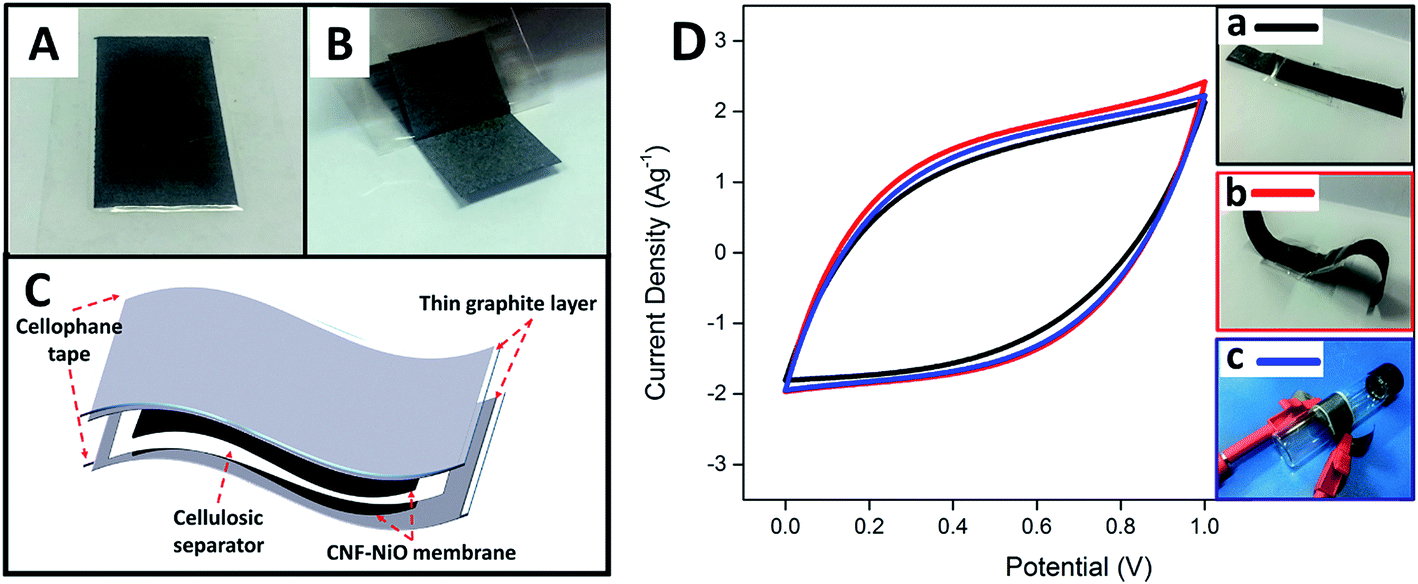 | ||
| Fig. 6 (A) Graphite sheet uniformly covered by cellophane tape, (B) thin layer of graphite/cellophane tape formed immediately upon peeling off tape, (C) schematic diagram of preparation of free-standing supercapacitor device, and (D) CV profiles of free-standing device when subjected to flexibility tests of (a) bending and (b) roll-up curvature, compared to (c) original state. | ||
4. Conclusion
We successfully fabricated metal oxide-reinforced CNF membranes by employing a solution-based mixture of polymer and minute metal precursors via an electrospinning technique, followed by a carbonization method. The CNF showed a large specific surface area, with an average fiber diameter of 326.4 nm and no drastic influence from the introduction of the nanoparticles. Moreover, the electrochemical properties were significantly enhanced, especially in the presence of metal oxides, as distinguished by an XPS analysis. These were uniformly dispersed throughout the CNF matrix, as indicated by the EDX mapping. A unique morphology was shown by having metal oxide nanoparticles embedded within the fiber surface, which contributed to the electrochemical performance of the carbon-based web structure. The fabricated CNF possessed outstanding performance with a specific capacitance of >100 Fg−1 and an exceptionally high retention capability of 93.90% even after 2000 continuous charge/discharge cycles. These lightweight and flexible CNF membranes are potentially applicable in the fabrication of versatile and lightweight flexible electronic devices, as seen by the as-prepared free-standing supercapacitor, which had a high stability performance even after being subjected to bending or being rolled up to a specific curvature. The electrospun novel metal oxide-reinforced CNF membranes are versatile current collectors, not just for supercapacitors, but for all-purpose electronic applications that require exceedingly flexible configurations and geometries.Acknowledgements
This research work was supported by IRU-MRUN (9399901) and Putra Grant IPS (GP-IPS/2014/9449400).References
- H. Yu, L. Ai, M. Rouhanizadeh, D. Patel, E. S. Kim and T. K. Hsiai, J. Microelectromech. Syst., 2008, 17, 1178–1186 CrossRef.
- R. Buchner, K. Froehner, C. Sosna, W. Benecke and W. Lang, J. Microelectromech. Syst., 2008, 17, 1114–1119 CrossRef CAS.
- B. Sun, Y. Z. Long, Z. J. Chen, S. L. Liu, H. D. Zhang, J. C. Zhang and W. P. Han, J. Mater. Chem. C, 2014, 2, 1209 RSC.
- G. Ma, D. Yang and J. Nie, Polym. Adv. Technol., 2009, 20, 147–150 CrossRef CAS.
- D. W. Wang, F. Li, J. P. Zhao, W. C. Ren, Z. G. Chen, J. Tan, Z. S. Wu, I. Gentle, G. Q. Lu and H. M. Cheng, ACS Nano, 2009, 3, 1745–1752 CrossRef CAS PubMed.
- C. Kim, Y. O. Choi, W. J. Lee and K. S. Yang, Electrochim. Acta, 2004, 50, 883–887 CrossRef CAS.
- Z. P. Zhou and X. F. Wu, J. Power Sources, 2014, 262, 44–49 CrossRef CAS.
- M. Inagaki, Y. Yang and F. Kang, Adv. Mater., 2012, 24, 2547–2566 CrossRef CAS PubMed.
- K. Wei and I. Kim, in Electrospun Nanofibers for Energy and Environmental Applications, ed. B. Ding and J. Yu, Springer, Berlin Heidelberg, 2014, ch. 7, pp. 163–181, DOI:10.1007/978-3-642-54160-5_7.
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- B. Liu, X. Hu, H. Xu, W. Luo, Y. Sun and Y. Huang, Sci. Rep., 2014, 4, 4229–4334 Search PubMed.
- Z. Zhou, X.-F. Wu and H. Hou, RSC Adv., 2014, 4, 23622–23629 RSC.
- E. H. Lee, T. M. Lee and B. S. Kim, J. Power Sources, 2014, 255, 335–340 CrossRef CAS.
- H. I. Joh, H. K. Song, C. H. Lee, J. M. Yun, S. M. Jo, S. H. Lee, S. I. Na, A. T. Chien and S. Kumar, Carbon, 2014, 70, 308–312 CrossRef CAS.
- Z. P. Zhou and X. F. Wu, J. Power Sources, 2013, 222, 410–416 CrossRef CAS.
- J. Yuh, L. Perez, W. M. Sigmund and J. C. Nino, Phys. E, 2007, 37, 254–259 CrossRef CAS.
- Y. Yang, H. Y. Wang, X. F. Lu, Y. Y. Zhao, X. Li and C. Wang, J. Mater. Sci. Eng. B, 2007, 140, 48–52 CrossRef CAS.
- J. Li, E. h. Liu, W. Li, X. Y. Meng and S. T. Tan, J. Alloys Compd., 2009, 478, 371–374 CrossRef CAS.
- D. Lin, H. Wu, R. Zhang and W. Pan, Chem. Mater., 2009, 21, 3479–3484 CrossRef CAS.
- Y. W. Ju, G. R. Choi, H. R. Jung and W. J. Lee, Electrochim. Acta, 2008, 53, 5796–5803 CrossRef CAS.
- B. Vidhyadharan, N. K. M. Zain, I. I. Misnon, R. A. Aziz, J. Ismail, M. M. Yusoff and R. Jose, J. Alloys Compd., 2014, 610, 143–150 CrossRef CAS.
- A. M. Al-Enizi, A. A. Elzatahry, A. M. Abdullah, M. A. AlMaadeed, J. Wang, D. Zhao and S. Al-Deyab, Carbon, 2014, 71, 276–283 CrossRef CAS.
- B. Vidhyadharan, I. I. Misnon, R. A. Aziz, K. P. Padmasree, M. M. Yusoff and R. Jose, J. Mater. Chem. A, 2014, 2, 6578–6588 CAS.
- S. Nagamine, S. Ishimaru, K. Taki and M. Ohshima, Mater. Lett., 2011, 65, 3027–3029 CrossRef CAS.
- Z. P. Zhou, X. F. Wu and H. Fong, Appl. Phys. Lett., 2012, 100, 023115 CrossRef.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 CAS.
- D. Zhang, H. Yan, Y. Lu, K. Qiu, C. Wang, C. Tang, Y. Zhang, C. Cheng and Y. Luo, Nanoscale Res. Lett., 2014, 9, 139 CrossRef PubMed.
- Z. Bashir, Carbon, 1991, 29, 1081–1090 CrossRef CAS.
- S. Dalton, F. Heatley and P. M. Budd, Polymer, 1999, 40, 5531–5543 CrossRef CAS.
- M. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421–1432 CrossRef CAS.
- C. Kim and K. S. Yang, Appl. Phys. Lett., 2003, 83, 1216–1218 CrossRef CAS.
- Y. Aykut, B. Pourdeyhimi and S. A. Khan, J. Appl. Polym. Sci., 2013, 130, 3726–3735 CrossRef CAS.
- Q. Wang, Y. Du, Q. Feng, F. Huang, K. Lu, J. Liu and Q. Wei, J. Appl. Polym. Sci., 2013, 128, 1152–1157 CrossRef CAS.
- D. Zhang, A. B. Karki, D. Rutman, D. P. Young, A. Wang, D. Cocke, T. H. Ho and Z. Guo, Polymer, 2009, 50, 4189–4198 CrossRef CAS.
- R. F. Ribeiro, L. C. Pardini, N. P. Alves, B. Júnior and C. A. Rios, Polimeros, 2015, 25, 523–530 Search PubMed.
- F. Dong, Z. Li, H. Huang, F. Yang, W. Zheng and C. Wang, Mater. Lett., 2007, 61, 2556–2559 CrossRef CAS.
- L. Wang, Y. Yu, P. C. Chen, D. W. Zhang and C. H. Chen, J. Power Sources, 2008, 183, 717–723 CrossRef CAS.
- X. Yang, C. Shao, H. Guan, X. Li and J. Gong, Inorg. Chem. Commun., 2004, 7, 176–178 CrossRef CAS.
- S. Maensiri, P. Laokul and V. Promarak, J. Cryst. Growth, 2006, 289, 102–106 CrossRef CAS.
- N. Dharmaraj, P. Prabu, S. Nagarajan, C. Kim, J. Park and H. Kim, Mater. Sci. Eng., B, 2006, 128, 111–114 CrossRef CAS.
- H. Guan, C. Shao, S. Wen, B. Chen, J. Gong and X. Yang, Inorg. Chem. Commun., 2003, 6, 1302–1303 CrossRef CAS.
- V. Barranco, M. Lillo Rodenas, A. Linares Solano, A. Oya, F. Pico, J. Ibañez, F. Agullo-Rueda, J. Amarilla and J. Rojo, J. Phys. Chem. C, 2010, 114, 10302–10307 CAS.
- Y. H. Hsu, C. C. Lai, C. L. Ho and C. T. Lo, Electrochim. Acta, 2014, 127, 369–376 CrossRef CAS.
- Y.-P. Zhu, T.-Y. Ma, T.-Z. Ren, J. Li, G.-H. Du and Z.-Y. Yuan, Appl. Catal., B, 2014, 156–157, 44–52 CrossRef CAS.
- C. H. Kim and B.-H. Kim, J. Power Sources, 2015, 274, 512–520 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- M. Zhi, A. Manivannan, F. Meng and N. Wu, J. Power Sources, 2012, 208, 345–353 CrossRef CAS.
- W. K. Chee, H. N. Lim, I. Harrison, K. F. Chong, Z. Zainal, C. H. Ng and N. M. Huang, Electrochim. Acta, 2015, 157, 88–94 CrossRef CAS.
- Z. Li, P. Liu, G. Yun, K. Shi, X. Lv, K. Li, J. Xing and B. Yang, Energy, 2014, 69, 266–271 CrossRef CAS.
- Y.-L. Chen, Z.-A. Hu, Y.-Q. Chang, H.-W. Wang, Z.-Y. Zhang, Y.-Y. Yang and H.-Y. Wu, J. Phys. Chem. C, 2011, 115, 2563–2571 CAS.
- D. P. Dubal, S. H. Lee, J. G. Kim, W. B. Kim and C. D. Lokhande, J. Mater. Chem., 2012, 22, 3044–3052 RSC.
- Y. S. Lim, Y. P. Tan, H. N. Lim, N. M. Huang, W. T. Tan, M. A. Yarmo and C.-Y. Yin, Ceram. Int., 2014, 40, 3855–3864 CrossRef CAS.
- E. J. Ra, E. Raymundo-Piñero, Y. H. Lee and F. Béguin, Carbon, 2009, 47, 2984–2992 CrossRef CAS.
- S. Xu and Z. L. Wang, Nano Res., 2011, 4, 1013–1098 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |