
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physicochemical characterization to assess Ni and Zn incorporation into zeotype SAPO-34 nanoparticles synthesized with different mixing methods through ultrasound-promoted crystallization†
F. Marzpour Shalmania,
R. Halladj *a and
S. Askarib
*a and
S. Askarib
aFaculty of Chemical Engineering, Amirkabir University of Technology (Tehran Polytechnic), P. O. Box 15875-4413, Hafez Ave., Tehran, Iran. E-mail: halladj@aut.ac.ir
bDepartment of Chemical Engineering, Science and Research Branch, Islamic Azad University, Tehran, Iran
First published on 19th May 2017
Abstract
The synthesis of transition metal (Me = Ni and Zn) containing SAPO-34 nanoparticles was conducted by applying insonation as the precrystallization treatment. The effects of synthesis parameters on the properties of SAPO-34 products and their derivatives in terms of the crystallinity, size and morphology of particles, structure and hydroxyl functional groups, and stereochemistry of metal ions were investigated to elucidate how the isomorphous substitution or the mere presence of transition metal ions was influenced by the crystallization treatment, mixing method, or the amount and type of metal species. The physicochemical properties of the prepared samples were characterized by various techniques including XRD, SEM, TEM, EDX, UV-Vis DRS, FTIR, and TGA. The results indicated that sonochemically prepared SAPO-34 samples possessed higher crystallinity and uniform crystals with narrow particle size distribution. The nature of the metal species could affect the physicochemical properties of the synthesized samples and Zn-doped SAPO-34 samples showed a lower crystallinity, larger unit cell parameters, and more uniform crystals. Also, it was found that the significant variations in the morphology, size, and uniformity of the metal-doped SAPO-34 crystals were acquired based on the different mixing methods. Meanwhile, the mixing methods played a key role in the incorporation of metal ions into the SAPO-34 framework, and mixing method (II), in which the metal sources were added to the precursor gel prior to the addition of the phosphoric acid, had prominent capability to locate the metal ions, in particular nickel ions, in the SAPO-34 skeleton.
1 Introduction
Porous materials including AlPOs (aluminophosphate molecular sieves) and zeolites (aluminosilicate molecular sieves) have been widely used in industry for many important applications such as separations, gas purification, ion-exchange, and in particular catalysis.1,2 The flexibility of the AlPO system in its ability to substitute other elements into its structure appears to be greater than that of the zeolite system. This feature offers considerable possibilities in designing aluminophosphates of desired catalytic properties.3–5Microporous aluminophosphate (AlPO-n) based materials, where n denotes a particular structure type, consist of alternating tetrahedrally coordinated Al3+ and P5+, which are corner-sharing an oxygen atom.1,6 Some AlPOs have the framework topologies of known zeolites, but many others have unique structures with no zeolite analogue.2,3 Since the structures of AlPOs are electrically neutral, they lack any active sites and usually have no inherent catalytic capabilities.2,7 In order to create the acid sites and to improve the acidic properties, Al and/or P can be partially replaced by silicon (SAPO-n) and/or metals.3,8 The final properties of these kinds of materials are both metal- and structure-dependent.4,9
Among the SAPO structures, SAPO-34 has received a majority of attention because of its various applications in the chemical and petrochemical areas, such as heat thermochemical storage, CO2/CH4 separation, hydrogen purification, and selective catalytic reduction of NOx.1,10–13 Moreover, the mild acidity, shape-selectivity properties by small pore openings, and good thermal/hydrothermal stability of SAPO-34 renders it a best candidate catalyst for the conversion of methanol to olefins (MTO).10,13–17 SAPO-34 with the framework of zeolite CHA topology contains 8-ring channels with a pore diameter of 0.38 nm and ellipsoidal cages of 0.67 nm × 1 nm,1,17–20 which allow the use of SAPO-34 molecular sieve and its derivatives for applications involving the small size molecules.
The isomorphous substitution of transition metal ions into the framework of SAPO-34 can provide additional modifications on the acidic properties and results in the improvement of catalytic properties of SAPO-34, in terms of the activity and selectivity.8,10
Many researches have been conducted to characterize the synthesis of metal-incorporated SAPO-34 structures and to describe the influence of synthesis conditions such as templating agent source,21–23 reaction time24,25 and temperature,13,25,26 and chemical composition of precursor solution7,9,24–31 on the properties of SAPO-34, in terms of the crystalline structure, morphology, textural properties, surface hydroxyl formation, acidic properties, and stereochemistry of metal ions. It has been claimed that a variety of metals can be incorporated into the SAPO-34 structure. Most studies inquire the transition metals, namely Ti,32 Cr,5,32 Mn,5,7,8,21,29,32–34 Fe,7,32,34–36 Co,7,23,25,29,32,34–37 Ni,7,9,21,22,24,26,29,32,35–40 Cu,30–32,41 Zn,27,32,33,42 and Ag,43 while numerable investigations deal with other metal ions such as Na, K, Cs, Mg, Ca, Sr, Ba, Ga, La, Ce, etc.13,17,19,28,32,35,40,43–46
In the above-mentioned references, the conventional hydrothermal method was applied as a predominant strategy for crystallization, but this method possesses a number of disadvantages including uncontrollable nucleation, large crystal size, and long crystallization time.47 It has been understood that the synthesis of AlPOs, zeolites, and other porous materials via sonochemical method, as one of the best crystallization techniques, has overcome the drawbacks detected in using conventional hydrothermal heating. The main advantages of applying ultrasonic waves are: (1) the simplicity of the method, (2) ability to attain fast crystallization, (3) absence of unwanted temperature gradient owing to the direct energy transfer to the synthesis gels, (4) high phase purity, and (5) narrow particle size distribution.11,14,48–51 Ultrasound radiation affects physical or chemical changes due to acoustic cavitation phenomena involving the formation, growth, and implosive collapse of bubbles in the solution. The collapse of the bubbles induces localized extreme conditions: temperature of about 5000–25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K, pressure of roughly 181.8 MPa, and cooling rate in excess of 1011 K s−1.11,14,49,50
000 K, pressure of roughly 181.8 MPa, and cooling rate in excess of 1011 K s−1.11,14,49,50
Inui and Kang39 as well as Kang et al.24 synthesized the Ni containing SAPO-34 crystals and examined the effect of the ultrasonic waves as one of the proper treatments to homogenize the precursor gel. It was found that the application of ultrasonic waves played an important role in controlling the physicochemical properties and performance of the synthesized catalyst. Another notable paper in the field of ultrasound usage was published by Charghand et al. in 2014,46 in which the authors made an attempt to synthesize the nanostructured Ce-doped SAPO-34 via ultrasound irradiation. They concluded that ultrasonic treatment is an appropriate technique for producing smaller crystals with narrow particle size distribution and high specific surface area, resulting in the desired properties of the catalyst.
Isomorphous substitution of nickel into the SAPO-34 skeleton has drawn considerable attention due to its high selectivity toward ethylene in the methanol conversion,3,24,26,29,38 but few studies on the zinc incorporation into the framework of SAPO-34 have been published. One advantage of using zinc ion is that it turned out to be a good candidate for isomorphous substitution.3 We devote attention to commonly and uncommonly used transition metals incorporated in the SAPO-34 structure to evaluate how the behaviors of these two transition metal ions are influenced by the synthesis conditions considered in our research.
Hitherto, there is no report of the synthesis of nanosized metal-incorporated SAPO-34 crystallized within a short period of time in the presence of ultrasound irradiation as the precrystallization treatment. The present study focuses on the investigation of the possibility of incorporation of internal transition metal ions (Me = Ni and Zn) into the chabazite-type silicoaluminophosphate lattice (SAPO-34) by means of sonication process in order to achieve highly crystalline and uniform nanoparticles within a short period of time. Another objective of this article is to seek for the appropriate preparation method and synthesis conditions directed to products with intended properties in terms of the high crystallinity, smaller particles with uniform morphology, and isomorphous substitution of metal ions. For this purpose, a series of SAPO-34 crystals and their derivatives, with Ni and Zn substitution was synthesized via sonochemical-assisted hydrothermal treatment in the presence of TEAOH as the structure-directing agent. The techniques including XRD, SEM, TEM, EDX, UV-Vis DRS, FTIR, and TGA were used to characterize the physicochemical properties of the synthesized samples.
2 Experimental
2.1. Sample preparation
The SAPO-34 and metal containing SAPO-34 samples were synthesized by means of sonochemical-assisted hydrothermal treatment based on the procedure in the literatures11,48 with slight modifications. The molar composition of the synthesis gel was 1.0Al2O3:1.0P2O5:0.6SiO2:xMeO:2.0TEAOH:70H2O, for which (x) values and synthesis conditions of different samples are given in Table 1. The source of the framework elements including Al, P, Si, Ni, and Zn were aluminum isopropoxide [Al(OPri)3, Merck], phosphoric acid [H3PO4 85 wt% aqueous solution, Merck], tetraethylorthosilicate [TEOS, Daejung], nickel nitrate [Ni(NO3)2·6H2O, Merck], and zinc sulfate [ZnSO4·7H2O, Merck], respectively. Tetraethylammonium hydroxide [TEAOH 20 wt%, Fluka] was used as a structure-directing agent.
| Sample name | Molar composition of MeO (x value) | Mixing method | Ultrasonic treatment | Heating time conducted in an oven (h) | Phase | Relative crystallinity (%) | Crystallite size (nm) |
|---|---|---|---|---|---|---|---|
| S-1 | — | — | Yes | 5 | SAPO-34 | 100.0 | 53.0 |
| S-2 | — | — | No | 5 | SAPO-34 | 97.6 | 36.0 |
| Ni-1 | 0.05 | Method I | Yes | 1 | Amorphous + SAPO-34 + Al2O3 | 15.9 | 38.9 |
| Ni-2 | 0.05 | Method I | Yes | 2 | SAPO-34 | 49.8 | 47.5 |
| Ni-3 | 0.05 | Method I | Yes | 3 | SAPO-34 | 80.3 | 46.1 |
| Ni-4 | 0.05 | Method I | Yes | 5 | SAPO-34 | 85.0 | 48.2 |
| Ni-5 | 0.10 | Method I | Yes | 5 | SAPO-34 | 90.0 | 41.2 |
| Ni-6 | 0.05 | Method II | Yes | 5 | SAPO-34 | 76.1 | 45.3 |
| Ni-7 | 0.05 | Method III | Yes | 5 | SAPO-34 | 81.2 | 45.5 |
| Ni-8 | 0.05 | Method I | No | 5 | SAPO-34 | 83.6 | 45.9 |
| Zn-1 | 0.05 | Method I | Yes | 1 | Amorphous + SAPO-34 + Al2O3 | 19.3 | 31.7 |
| Zn-2 | 0.05 | Method I | Yes | 2 | SAPO-34 | 52.4 | 44.0 |
| Zn-3 | 0.05 | Method I | Yes | 3 | SAPO-34 | 82.8 | 56.6 |
| Zn-4 | 0.05 | Method I | Yes | 5 | SAPO-34 | 90.4 | 58.6 |
| Zn-5 | 0.10 | Method I | Yes | 5 | SAPO-34 + AlPO4 | 46.5 | 43.2 |
| Zn-6 | 0.10 | Method II | Yes | 5 | SAPO-34 + AlPO4 | 61.4 | 45.5 |
| Zn-7 | 0.05 | Method III | Yes | 5 | SAPO-34 | 65.1 | 45.3 |
| Zn-8 | 0.05 | Method I | No | 5 | SAPO-34 | 89.6 | 41.0 |
In a typical preparation of SAPO-34, the 5.86 g of aluminum isopropoxide was mixed with 21.00 ml of TEAOH and 0.70 ml of distilled water. After stirring the mixture until dissolved completely, 1.80 g of TEOS was added and stirred. Then, 3.30 g of phosphoric acid was added dropwise to the solution. The resulting synthesis gel was further stirred to a uniform reaction mixture.
The metal containing SAPO-34 samples, independently of the transition metal sources and their molar ratio, were prepared following the procedure similar to that adopted for SAPO-34 synthesis but with an extra addition (0.21 or 0.42 g) of metal sources to the mixture. According to the sequence of addition of the metal salts to the synthesis solution, three different mixing methods were investigated:
• Method I: adding metal sources after 1 h of phosphoric acid addition.
• Method II: adding metal sources prior to the addition of the phosphoric acid.
• Method III: mixing metal sources with aluminum isopropoxide and template in the prime step.
For crystallization, regardless of the different mixing methods, the synthesis solution was irradiated by means of Ultrasonic Processor UP200H (Hielscher) with unchangeable frequency of 24 kHz using the titanium sonotrode having a tip diameter of 7 mm, with 300 W cm−2 power intensity (related to 100% amplitude setting). The sonication temperature was controlled at a temperature of 40 °C by using a water bath. After insonation the sample for 15 min as the precrystallization treatment, the mixture was transferred into a Teflon-lined stainless steel autoclave and then heated statically in a conventional oven at 200 °C for various times under autogenous pressure. For comparison, SAPO-34 samples were also prepared by conventional hydrothermal crystallization without using ultrasonic treatment. The synthesis conditions of different SAPO-34 and metal containing SAPO-34 samples as well as their properties acquired from X-ray diffraction patterns are given in Table 1. In both the ultrasound-assisted and the hydrothermal crystallization, the solid product was separated by centrifugation, washed with distilled water, and then dried at 110 °C. The as-synthesized products were calcined at 550 °C for 5.5 h to remove the template molecules and the water trapped within the pores.
2.2. Characterization
The synthesized samples were characterized by several techniques including XRD, SEM, TEM, EDX, UV-Vis DRS, FTIR, and TGA, as presented in Table S1.† For measuring the relative crystallinity from XRD data, the sum of the significant peak heights of the synthesized sample is ratioed against the sum of those peak heights for a standard sample4 after subtraction of the background:
The standard sample is a highly crystalline product which contains no impurity or amorphous phases. The four significant peaks of the synthesized sample are emerged at 2θ = 9.4–9.6, 13.0–13.1, 20.8–21.0, and 31.0–31.2° corresponding to [101], [110], [211], and [401] reflections, respectively. It is worth noting that the slight shifts in the position of the diffraction peaks arise from the metal incorporation that will be discussed in detail in Section 3.1.
Another important parameter extracted from XRD data is the average size of the crystallites in a sample which is estimated by the full width at half-height maximum (FWHM) of the most intense peaks of diffraction pattern through Scherrer method.
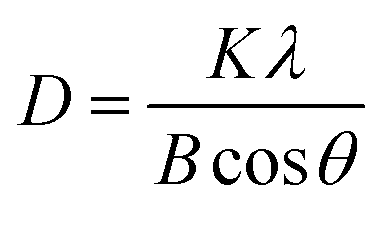
3 Results and discussion
3.1. Powder X-ray diffraction (XRD)
The XRD patterns of the synthesized samples, regardless of some impurity phases, correspond well with the structural pattern of the SAPO-34 chabazite (CHA) structure reported in the ref. 21, 22, 29, 40, 46 and 54, indicating successful synthesis of SAPO-34 structure. Since the metal containing samples retain the integrity of the CHA topology structure, it can be concluded that the metal ions (Ni2+ and Zn2+) and their counter-ions (SO42− and NO3−) have no influence on the structure. On the other hand, no obvious characteristic peaks of the metal oxide phase are detected in the XRD patterns of the Ni- and Zn-containing samples. The possible reason for this observation is the low loading levels of metal ions, but Bing et al.12 reported that the absence of diffraction peaks ascribed to metal species is the evidence for metal incorporation into the framework sites.Fig. 1 illustrates the XRD patterns of nickel and zinc containing SAPO-34 (samples Ni-4 & Zn-4 in Table 1) in comparison with parent SAPO-34 (sample S-1), which were prepared sonochemically through method (I) under the same heating time. It can be seen that by introducing the metal ions into the synthesis gel, the intensity of characteristic peaks, and thus the relative crystallinity of samples is reduced. The results given in Table 1 demonstrate that samples with zinc contents have the lowest relative crystallinity compared with nickel containing samples, probably owing to the fact that the type of metal affects the manner of crystal growth. Furthermore, distortions in the crystalline network can be explained by taking into account the isomorphous substitution of the metals into the SAPO-34 framework, because the difference between the bond lengths of the M–O (M = Zn or Ni) and the typical T–O (T = Si, Al or P) impresses the bond angles between the tetrahedral construction units (MO4 and TO4), creating distortions along the crystalline network and thus, lowering the crystallinity of materials.21,28,55
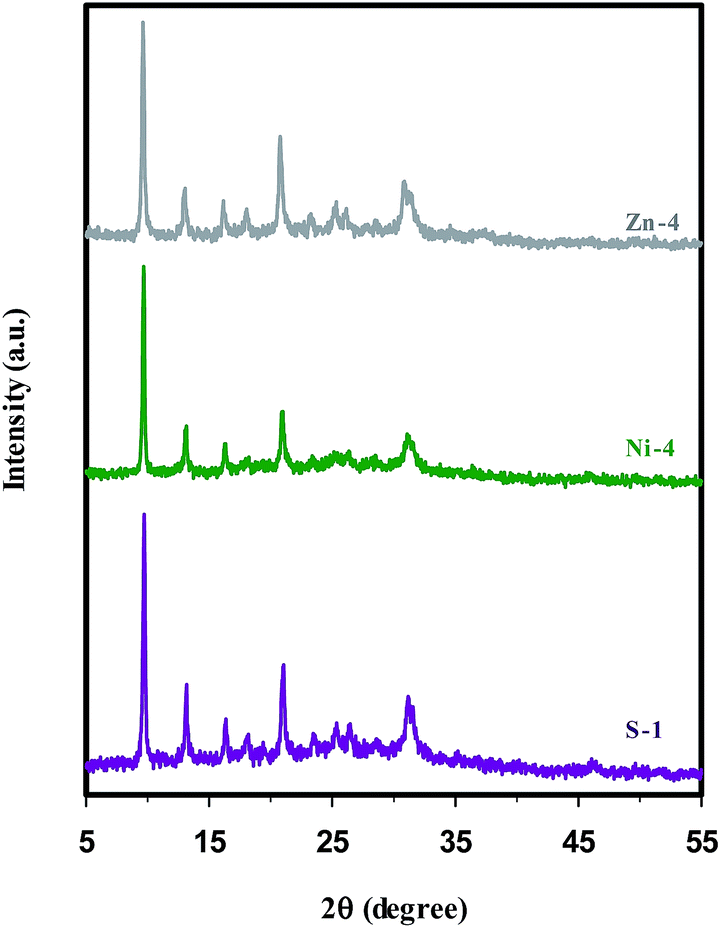 | ||
| Fig. 1 XRD patterns of parent (S-1), nickel (Ni-4), and zinc (Zn-4) containing SAPO-34 samples synthesized sonochemically for 5 h. | ||
The effect of the applied crystallization methods was evaluated by comparing the samples S-1, Ni-4, and Zn-4 synthesized sonochemically and samples S-2, Ni-8, and Zn-8 obtained by conventional hydrothermal crystallization. Although the XRD patterns of samples S-2, Ni-8, and Zn-8 (depicted in Fig. S1† for the sake of brevity) follow the same trends as shown in Fig. 1, the intensity of characteristic peaks and so the crystallinity of samples decreases through conventional hydrothermal method. The higher crystallinity in the presence of ultrasound irradiation implies that the growth of crystalline products is improved by sonication process, presumably due to this fact that abundant nuclei are formed progressively during the ultrasound radiation and grow large by means of the conventional heating.48 Another effect of sonication on nucleation is shortening the induction time between the establishment of supersaturation and the onset of nucleation and crystallization. In addition other postulates suggest that, subsequent rapid local cooling rates occurred after collapsing of bubbles, play a significant role in increasing supersaturation, in which case it should be possible to correlate the number of cavitation and nucleation events in a quantitative way.48 The definitive role of the sonochemical processing in the crystallization rate enhancement was recently proved by investigating the crystallization kinetics of SAPO-34 during the nucleation and growth periods.11
The XRD patterns of metal containing SAPO-34 samples synthesized sonochemically under the same conditions, but with different hydrothermal heating times are shown in Fig. 2. The patterns in Fig. 2(a and b) and the results presented in Table 1 demonstrate that the samples Ni-1 and Zn-1 crystallized for 1 h mainly consist of the amorphous phase. Also, a small amount of Al2O3 impurity cocrystallizes with the SAPO-34 phase. When the samples were crystallized for 2 h (Ni-2 & Zn-2), the SAPO-34 is the sole phase detected. There is an increase in the intensity of characteristic peaks of pure SAPO-34 with increasing crystallization time up to 3 h (samples Ni-3 & Zn-3). Prolonging the heating time to more than 3 h leads to an enhancement in the intensity of the peak emerged at 2θ = 20.8–21°, whereas the intensity of diffraction peak appearing at 2θ = 9.4–9.6° is not greatly affected by crystallization time. Consequently, prolonging the hydrothermal heating time leads to further crystal growth and, thus, an increase in the crystallinity is observed, in accordance with the results reported for parent SAPO-34 samples.11,18,48
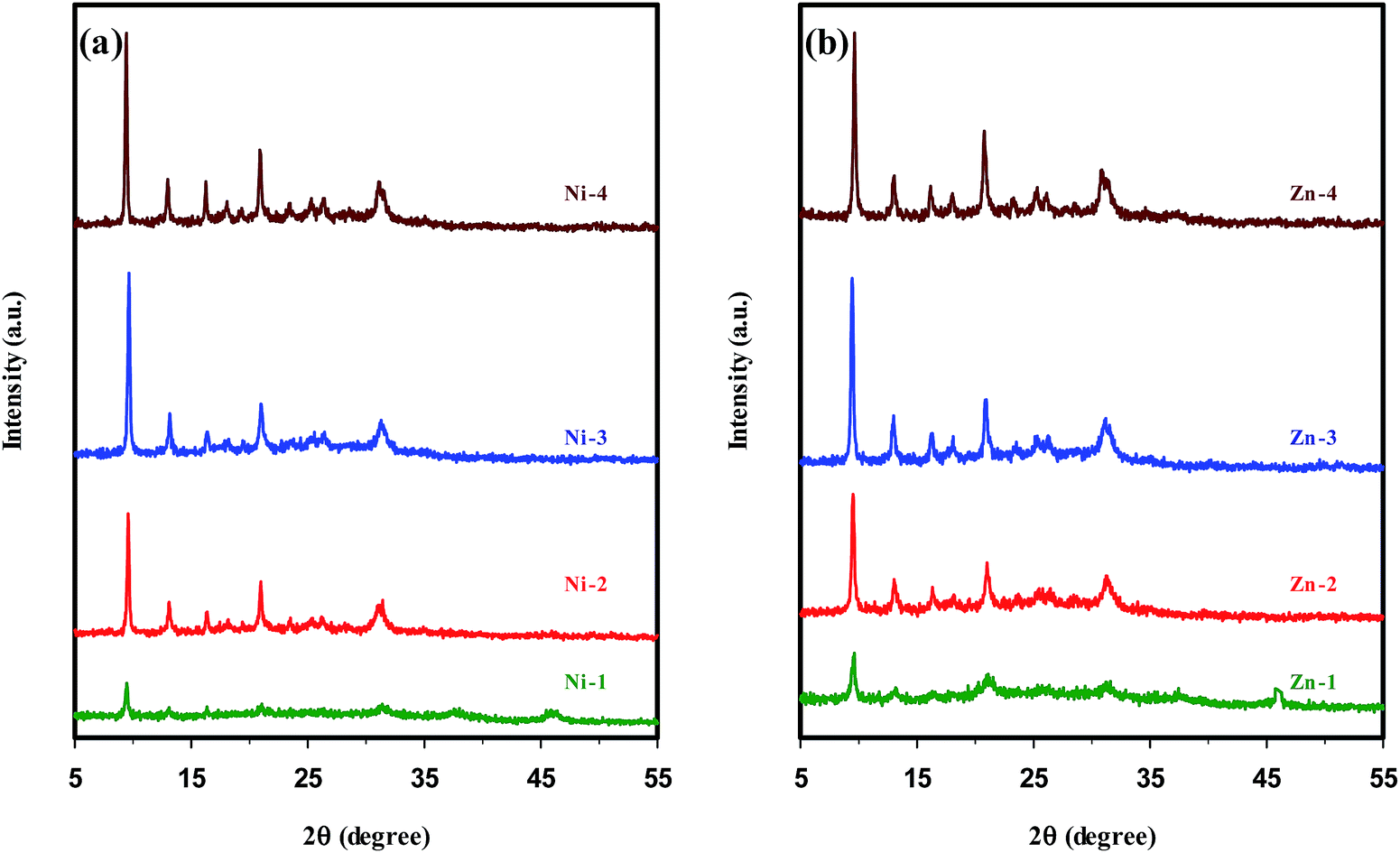 | ||
| Fig. 2 XRD patterns of metal containing SAPO-34 samples synthesized sonochemically with different hydrothermal heating times. | ||
To verify the role of the metal content on the formation of the SAPO-34 crystals, four samples were synthesized sonochemically from the reaction mixture composition of 1.0Al2O3:1.0P2O5:0.6SiO2:xMeO:2.0TEAOH:70H2O, where (x) values were (0.05) and (0.1) for samples (Ni-4 & Zn-4) and (Ni-5 & Zn-5), respectively. The XRD patterns of the samples with different Ni concentrations in Fig. 3(a) manifest the patterns of pure SAPO-34 structure without any impurity. The only difference between them is that the sample Ni-4 with lower nickel content has less crystalline products. In the case of samples with different Zn concentrations depicted in Fig. 3(b), the XRD pattern of the sample Zn-5 with higher zinc content presents lower crystallinity. Also, a considerable amount of impurity identified as the dense phase of AlPO4 with cristobalite structure accompanied by strong reflection at 2θ = 21.54° is observed. The possible reason for the formation of the competing phase AlPO4, which is tangible in the samples with higher Zn concentrations, is the isomorphous substitution of Al and/or P atoms by Zn atoms. We speculate that zinc occupying tetrahedral framework sites increases the presence probability of Al and P atoms in extra-framework positions of SAPO-34 structure; consequently these extra-framework components are prone to produce the AlPO4 phase.
 | ||
| Fig. 3 XRD patterns of metal containing SAPO-34 samples synthesized sonochemically with different metal concentrations in the synthesis gel: (a) nickel and (b) zinc ions. | ||
Ni- and Zn-substituted SAPO-34 samples synthesized by various mixing methods have similar XRD patterns (not depicted for the sake of brevity), but show different relative crystallinity (according to Table 1) due to the distortions in the crystalline network influenced by the type of metal and the isomorphous substitution of metals.
One of the most considerable aspects of this study is isomorphous substitution assessment. In this context, it is worthwhile to point out how the incorporation of metal ions into framework sites is confirmed by XRD results. It is expected that lattice expansions should be observed when metal ions substitute for Al3+ and/or P5+, which is plausibly related to the reason that the radii of T (T = Si, Al, or P) and T–O bond lengths of silicoaluminophosphates (1.5–1.7 Å) are typically smaller than the radii of M (M = metal ions such as Ni or Zn) and M–O bond lengths (1.9–2.0 Å).27,55
Compared with SAPO-34 samples, the XRD patterns of Ni- and Zn-containing SAPO-34 crystals illustrate a slight shift towards the lower angle in the peak positions. As mentioned by Zhang et al.,27 such shift supports the occurrence of true isomorphous substitution of metal ions, due to the increase in the interplanar spacing and thus lattice parameters.
The scrutiny of peak positions as an evidence for the incorporation of metals into the SAPO-34 framework is the qualitative examination. In an attempt to quantify the lattice expansions, the lattice parameters are determined based on the type of crystal system which is hexagonal for SAPO-34 molecular sieve (see “unit cell calculation” p. S4 in the ESI† for more details). Analysis of the diffractograms (by the program UnitCell)5,25 yields the lattice parameters of the unit cell listed in Table 2. It can be seen that there is no lattice expansion for the nickel substituted SAPO-34 sample synthesized with methods (III) (sample Ni-7), whiles other metal containing samples have longer a- and c-axis values, and thus larger unit cell volume than the parent SAPO-34, strongly indicating the expansion of the SAPO-34 structure as well as suggesting that zinc ions, regardless of the different mixing methods, become a constituent of the SAPO-34 framework sites.
| Sample name | Mixing method | Unit cell parameters | ||
|---|---|---|---|---|
| a, b (Å) | c (Å) | volume (Å3) | ||
| S-1 | — | 13.582 | 14.823 | 2367.979 |
| Ni-4 | Method I | 13.615 | 14.883 | 2389.039 |
| Ni-6 | Method II | 13.599 | 14.775 | 2366.304 |
| Ni-7 | Method III | 13.560 | 14.721 | 2344.031 |
| Zn-4 | Method I | 13.688 | 14.809 | 2403.058 |
| Zn-6 | Method II | 13.626 | 14.814 | 2381.897 |
| Zn-7 | Method III | 13.605 | 14.846 | 2379.739 |
3.2. Scanning electron microscopy (SEM) & transmission electron microscopy (TEM)
The scanning electron microscopy (SEM) images of parent SAPO-34, Ni-, and Zn-containing SAPO-34 samples are shown in Fig. 4–6, respectively, representing that the morphology of the crystals depends on the different synthesis conditions. In order to evaluate the effect of the mixing method, crystallization treatment, and metal types and contents on the size and shape of the synthesized crystals, all samples employed in the morphology analysis were taken from a reaction point at the top of the crystallization process, i.e. the maximum heating time. | ||
| Fig. 4 SEM images of parent SAPO-34 samples synthesized sonochemically (S-1) and hydrothermally (S-2). | ||
 | ||
| Fig. 5 SEM images of Ni containing SAPO-34 samples crystallized for 5 h: (a) mixing method I (low nickel content), (b) mixing method I (high nickel content), (c) mixing method II, (d) mixing method III, and (e) mixing method I (low nickel content) in the absence of insonation. | ||
 | ||
| Fig. 6 SEM images of Zn containing SAPO-34 samples crystallized for 5 h: (a) mixing method I (low zinc content), (b) mixing method I (high zinc content), (c) mixing method II, (d) mixing method III, and (e) mixing method I (low zinc content) in the absence of insonation. | ||
In Fig. 4, the SEM micrograph of parent SAPO-34 sample (S-1) indicates intergrowth of cubic particles with dimensions of ∼200–500 nm to form ball-shaped aggregates with a broad size distribution (around 0.8–2 μm). Introducing 0.05 moles of nickel ion into the synthesis gel of sample Ni-4 exerts no change on the intergrowth morphology (Fig. 5(a)), but leads to the formation of smaller aggregates with sizes ranging between 0.9 and 1.6 μm, presumably due to an increase in the number of nuclei.28,55 As depicted in Fig. 5(b), when the nickel content in the synthesis gel increases from 0.05 to 0.1, the smaller agglomerated particles with less uniform morphology compared with sample Ni-4 are obtained. On the other hand, the SEM result in Fig. 6(a) signifies that the Zn doping alters the structural topology and the sample Zn-4 is composed of spherical as well as cubic aggregates. Fig. 6(b) displays that by further increasing the zinc content to 0.1 (sample Zn-5), nonuniform crystals with undefined morphology are formed due to the presence of AlPO4 impurity phase, in accordance with the result of XRD analysis discussed earlier.
The SEM images of nickel containing samples synthesized sonochemically under the same crystallization time, but with different mixing methods (samples Ni-4, Ni-6, and Ni-7) are shown in Fig. 5(a, c, and d). Despite their similar intergrowth morphology, the ball-shaped aggregates obtained by using methods (II) and (III) are smaller than that prepared by method (I). Furthermore, the constituent particles of agglomerates in each of these samples have different sizes and sample Ni-4 contains larger and less uniform constituent particles, owing to the fact that larger crystallites, which are estimated by Scherrer method and exhibited in the last column of Table 1, are created through method (I).
The SEM results of the Zn-doped SAPO-34 samples prepared with different mixing methods (samples Zn-4, Zn-6, and Zn-7 in Fig. 6(a, c, and d)) imply that the morphologies, sizes, and homogeneities of crystals can be controlled by changing the mixing sequence of the metal salts to the synthesis solution. As mentioned earlier, sample Zn-4 prepared with method (I) is composed of spherical as well as cubic aggregates. By applying method (II), the obtained crystals in sample Zn-6 tend to be spherical aggregates instead of cubic agglomerates. It deserves mentioning that the crystals with irregular shape in this sample are assigned to AlPO4 impurity phase. Unfortunately, sample Zn-6 synthesized by method (II) has a broad particle size distribution (0.2–1.4 μm). Using method (III) not only decreases the sizes of spherical aggregates but also improves the size distribution of the agglomerated particles (0.4–0.9 μm). Besides, Zn-containing SAPO-34 samples crystallized with methods (II) and (III) (samples Zn-6 and Zn-7) have aggregates with spherical and nanosized constituent particles of ca. 50–100 nm diameter, unlike the sample Zn-4 comprising constituent particles with nonuniform morphology and dimensions of ∼90–500 nm.
The comparison of SEM photographs of samples S-1, Ni-4, and Zn-4 synthesized sonochemically with samples S-2, Ni-8, and Zn-8 synthesized hydrothermally under the same crystallization time (5 h) demonstrates the presence of an amorphous phase and, thus, the incomplete crystallization in the absence of insonation process. In addition, narrow particle size distributions in samples Ni-4 & Zn-4 compared with heterogeneous distribution of particles in samples Ni-8 & Zn-8 clarify the advantage of ultrasound irradiation. The improvement in the crystallinity as well as the particle size distribution can be explained by this fact that when ultrasound is applied, the formation of a greater number of nuclei results in the presence of large number of structures limiting the inappropriate growth of particles.49
The TEM image is used to study smaller particles of samples due to its higher resolution. Fig. 7 illustrates the TEM micrographs of the parent SAPO-34, Ni-, and Zn-containing SAPO-34 samples synthesized with method (II), which are selected as the representative TEM images of the synthesized products. As depicted in Fig. 7(a), intergrowth of cubic particles is observed for sample S-1. The sample Ni-6 shows aggregates composed of cubic particles of around 60–300 nm (Fig. 7(b)), while sample Zn-6 presents the constituent particles of 30–100 nm. Consequently, the TEM micrograph of samples further confirmed the results acquired from the SEM images.
 | ||
| Fig. 7 TEM images of (a) parent SAPO-34, (b) Ni-, and (c) Zn-containing SAPO-34 samples synthesized with method (II). | ||
3.3. Energy dispersive X-ray spectroscopy (EDX)
Table 3 represents the results of the elemental composition of the synthesis gel and the final products examined by EDX analysis. The metal composition in the final products is found to be affected by the nature of metal used and the content of metal ion in the synthesis gel. It is not possible to substitute all of the nickel loaded into the framework positions, in contrast to Zn-doped SAPO-34 samples. Besides, independently of the metal sources, the samples obtained by using methods (II) and (III) have higher metal contents than those prepared by method (I).| Sample name | Gel composition (mol%) | Product composition (mol%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Al | Si | P | Me | P/Al | Al | Si | P | Me | (Si + P)/(Al + Me) | |
| S-1 | 0.4348 | 0.1304 | 0.4348 | — | 1.0 | 0.5585 | 0.1257 | 0.3158 | — | 0.79 |
| Ni-4 | 0.430 | 0.129 | 0.430 | 0.011 | 1.0 | 0.5594 | 0.1286 | 0.3110 | 0.0010 | 0.78 |
| Ni-5 | 0.426 | 0.128 | 0.426 | 0.020 | 1.0 | 0.4944 | 0.1380 | 0.3558 | 0.0119 | 0.98 |
| Ni-6 | 0.430 | 0.129 | 0.430 | 0.011 | 1.0 | 0.4998 | 0.1459 | 0.3501 | 0.0042 | 0.98 |
| Ni-7 | 0.430 | 0.129 | 0.430 | 0.011 | 1.0 | 0.4814 | 0.1709 | 0.3459 | 0.0018 | 1.07 |
| Zn-4 | 0.430 | 0.129 | 0.430 | 0.011 | 1.0 | 0.5274 | 0.1223 | 0.3227 | 0.0276 | 0.80 |
| Zn-5 | 0.426 | 0.128 | 0.426 | 0.020 | 1.0 | 0.4888 | 0.1247 | 0.3316 | 0.0549 | 0.84 |
| Zn-6 | 0.426 | 0.128 | 0.426 | 0.020 | 1.0 | 0.4786 | 0.1163 | 0.3552 | 0.0499 | 0.89 |
| Zn-7 | 0.430 | 0.129 | 0.430 | 0.011 | 1.0 | 0.4820 | 0.1152 | 0.3540 | 0.0488 | 0.88 |
The silicon contents and Si/Al molar ratios of the synthesized samples, except for sample Ni-7, show no considerable deviation from those of the starting gels, implying that almost all of the Si loaded in the synthesis gel is included in the obtained product. Presumably, the higher Si/Al ratio than 0.3 means incorporating more Si content into the SAPO-34 framework and/or less Al content which can be considered as an evidence for metal ions substituted into the framework sites through replacing some Al atoms. Consequently, it is expected that method (III) is apt to result in isomorphous substitution of heteroatoms such as Si and Ni. Also, it is observed that the amount of phosphorous content in the products is lower than the actual loading and, indeed, the P/Al molar ratio of the obtained samples is smaller than 1.0, which indicates an incomplete incorporation of phosphorous element.7
On the other hand, it is known that the molar ratio of (Si + P)/Al less than or equal to 1.0 can prove more decisively the Si incorporation via SM2 substitution mechanism, i.e. the silicon contents are introduced in the framework by phosphorous replacement. It is worth noting that the theoretically possible silicon substitution mechanisms (SM) in SAPOs are Si for Al (SM1), Si for P (SM2), two Si atoms for one Al and one P, simultaneously, (SM3).56 Regardless of the metal species, the final compositions of the synthesized samples manifest that the molar ratio of (Si + P)/Al is less than 1 except in case of sample Ni-7. It is likely that the Si undergoes dual substitution mechanisms (SM2 + SM3) in sample Ni-7.
In metal containing SAPO systems, Al and/or P can be partially replaced by Si and/or Me ions. It is noteworthy that finding incontrovertible conclusions about the substitution of metal ions from chemical analysis data alone is difficult, due to the low loading levels of metal ions, the presence of extra-framework elements, or the occurrence of different substitution mechanisms simultaneously.3 However, Xu et al.32 proposed that if the molar ratio of (Si + P)/(Me + Al) is more than or equal to 1.0, the incorporation should be considered as a partial replacement of Al by metal ions. If this ratio is less than 1, metal ion substitution into the framework can be fulfilled by replacing Si. The molar ratio of (Si + P)/(Me + Al) in the last column of Table 3 reveals that a partial of tetrahedrally coordinated Al atoms is replaced by nickel ions during the synthesis of sample Ni-7 (as expected and mentioned earlier), but metal ions in other samples are probably incorporated into the framework by Si as well as Al replacements.
3.4. UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS)
The UV-Vis diffuse reflectance spectroscopy, applied to opaque solid state materials including powders and surfaces, is known to be a sensitive probe for the recognition of the valence state and the stereochemistry of transition metal ions, leading eventually to the confirmation of metal ions existence in the framework and/or extra-framework positions of metal containing zeolites and zeotype materials.57There are six complex structures of Ni2+ according to the coordination number 3 to 6. The configuration of complexes with odd coordination number, i.e. trigonal planar (3-), trigonal bipyramidal (5-), and square pyramidal (5-coordinate) geometries, is found to be uncommon states.58 For the 4-coordinate Ni2+ complexes, both square planar and tetrahedral configurations are identified. The last structure of Ni2+ is the octahedral configuration corresponded to the maximum coordination number of Ni2+.58,59
The UV-Vis DR spectra of both nickel- and zinc-containing SAPO-34 samples together with the parent SAPO-34 sample are profiled in Fig. 8. The introduction of nickel ion into the synthesis gel through method (I) and (III) presents three absorption bands centering around 400, 630, and 756 nm. As declared by Cotton et al.,58 the DRS bands at both 400 and 756 nm indicate the presence of octahedral nickel species, whiles the band at 630 nm is characteristic of tetrahedral coordinated Ni2+ ions in product and of the incorporation of nickel into the SAPO-34 framework. The spectrum of sample Ni-6 shows that the absorption bands related to the octahedral coordinated Ni (i.e. bands at 400 and 756 nm) disappear. Therefore, it can be concluded that Ni ions are located in the SAPO-34 skeleton by applying method (II). On the other hand, the comparison of UV-Vis spectra of sample Ni-4 and Ni-8 illustrates a decrease in the intensity of bands assigned to octahedrally coordinated Ni, implying that the incorporation of Ni into the SAPO-34 framework is promoted by sonication process.
 | ||
| Fig. 8 UV-Vis diffuse reflectance spectra of samples synthesized sonochemically for 5 h: (a) parent and Zn-containing SAPO-34 samples, and (b) Ni-containing SAPO-34 samples. | ||
The spectrum of sample Zn-4 in Fig. 8, which is selected as the representative UV-Vis DR spectra of the zinc containing products, demonstrates that Zn-doped SAPO-34 samples do not present any absorption band belonging to Zn species, because the crystal field splitting energy (CFSE, 10Dq parameter, or Δ0) for Zn2+ with d10 configurations is zero.60 Therefore, the UV-Vis DRS analysis is incapable to specify the framework or extra-framework position of Zn2+ ions in SAPO-34 structure.
In the spectra of all samples, regardless of the different synthesis conditions, there is an almost sharp band centered at 250 nm. The absorption band in the short-wavelengths (<350 nm), which is inherent in different types of zeolites and zeotype materials, originates from the Al–O charge transfer transitions.45,61
3.5. Fourier transform infrared spectroscopy (FTIR)
IR spectroscopy is a widely used technique to determine the framework structures, adsorbed species, and the acidity of zeolites and zeotype materials, because the vibrational spectrum of these materials originates from several contributions, namely, the vibrations from the framework of the material, from the charge-balancing cations, and from the relatively isolated groups, such as the surface OH groups.62 The FTIR spectra of parent SAPO-34 and different metal-containing SAPO-34 samples are shown in Fig. 9, exhibiting IR bands similar to those reported for SAPO-34-type materials.13,19,22,35,46 | ||
| Fig. 9 FTIR spectra of parent SAPO-34 sample in comparison with metal containing SAPO-34 samples: (a) nickel and (b) zinc species. | ||
In the framework or fingerprint region (400–1300 cm−1), the bands detected around 490, 640, 875, 1000–1200 cm−1 are related to T–O bending of TO4 tetrahedral, T–O bending in double-6 rings as the external linkage vibrations, symmetric stretching of TO4 tetrahedral, and asymmetric stretching of TO4 tetrahedral, respectively.12,32,40,62
The vibration band at 1640 cm−1 observed in all samples is assigned to H–O–H bending motion of adsorbed water.12,13,22,63 Weaker transmittance at 2360 cm−1 is due to the asymmetric stretching vibration of CO2 which is physically adsorbed from the surrounding.13,62
In the hydroxyl region (3000–4000 cm−1), a broad stretching band between 3200 and 3600 cm−1 is ascribed to bridging hydroxyl groups, Si–OH–Al, which is responsible for the Brønsted acidity of SAPO-34.13,17,22 The broad feature of this band is opined to arise from the two reasons: firstly, there may be an overlap of two bands belonging to OH stretching vibrations of free water as well as bridging hydroxyl groups.63,64 Secondly, the strong hydrogen bonding formed by the presence of OH groups lowers the stretching frequency, sometimes dramatically, and broadens the OH bands. It should be noted that the number and strength of hydrogen bonds differ according to the chemical environments and the intermolecular interactions, such that the half widths may become 300–500 cm−1 for OH stretches.64
It is worthwhile to mention that in addition to bridging Si–OH–Al groups, zeolitic materials contain four other hydroxyl groups: lattice termination silanol (Si–OH) groups at ∼3740 cm−1, OH groups occurring at defect sites (hydroxyl nests) at ∼3720 cm−1, OH groups attached to extra-framework T atom-containing species at ∼3680 cm−1, and OH groups attached to multivalent cations that compensate the negative charge of the framework at ∼3520–3580 cm−1.62 In SAPO-34 structure, Si–OH, P–OH, and Si–OH–Al groups are the most probable vibration bands, but the first two groups have much lower acidity than Si–OH–Al group.40,56
It is expected that the charge-compensating cations within the channels and cages of microporous material have an influence on the position of framework bands.62 The IR spectra depicted in Fig. 9(a) reveal that the introduction of nickel ions into the SAPO-34 synthesis gel by means of methods (I) & (II) (samples Ni-4 and Ni-6) increases the band widths of asymmetrical stretching mode as well as hydroxyl vibrations, in accordance with the results of Kondratov et al.65 who reported that the wavenumber of asymmetrical stretching band shifted upon substitution of mono- and divalent cations, while much smaller shifts were observed for the symmetrical stretching bands and the bending modes. This observation can be explained by the structural inhomogeneities caused by metal incorporation.62 A possible explanation for the further band broadening in the hydroxyl region of samples Ni-4 & Ni-6 is the appearance of another IR band belonging to OH groups attached to cations that compensate the negative charge of the framework.
On the other hand, the intensity of the IR band scales with the dipole moment as well as the concentrations of particular type of molecule or functional group in a sample.62 Since the intensity of the hydroxyl vibration reflects particularly the nature of the Si–OH–Al configuration, the low transmittance of the respective IR band in samples Ni-4 & Ni-6 implies a decrease in the concentration of Si–OH–Al groups, and thus can provide evidence for structural substitution of Ni element for Al atoms. Furthermore, the absence of intense peak of bridging hydroxyl groups in these samples may be the result of the presence of low surface Brønsted acid sites.
Fig. 9(b) illustrates that the aforementioned band broadening is observed inconsiderably in the spectra of Zn-containing SAPO-34 samples. Nevertheless, it cannot be certainly concluded that Zn ions are located in the extra-framework sites of SAPO-34, because IR band broadening is indirect criteria to verify isomorphous substitution of metal ions into silicoaluminophosphate frameworks.3 The appearance of a new band around 720 cm−1 in sample Zn-6 can be attributed to the Me–O–T bond32 formed by the incorporation of zinc ions into the framework of SAPO-34. This band becomes visible for samples with higher concentration of zinc ions in the synthesis gels.
3.6. Thermo-gravimetric analysis (TGA)
The TGA data of the uncalcined SAPO-34 and different metal containing SAPO-34 samples synthesized sonochemically are presented in Fig. 10 and Table 4, indicating that the greatest amount of total weight loss during heating to 750 °C is around 24% as well as the temperatures of weight losses are higher in the uncalcined metal containing SAPO-34 samples than parent SAPO-34.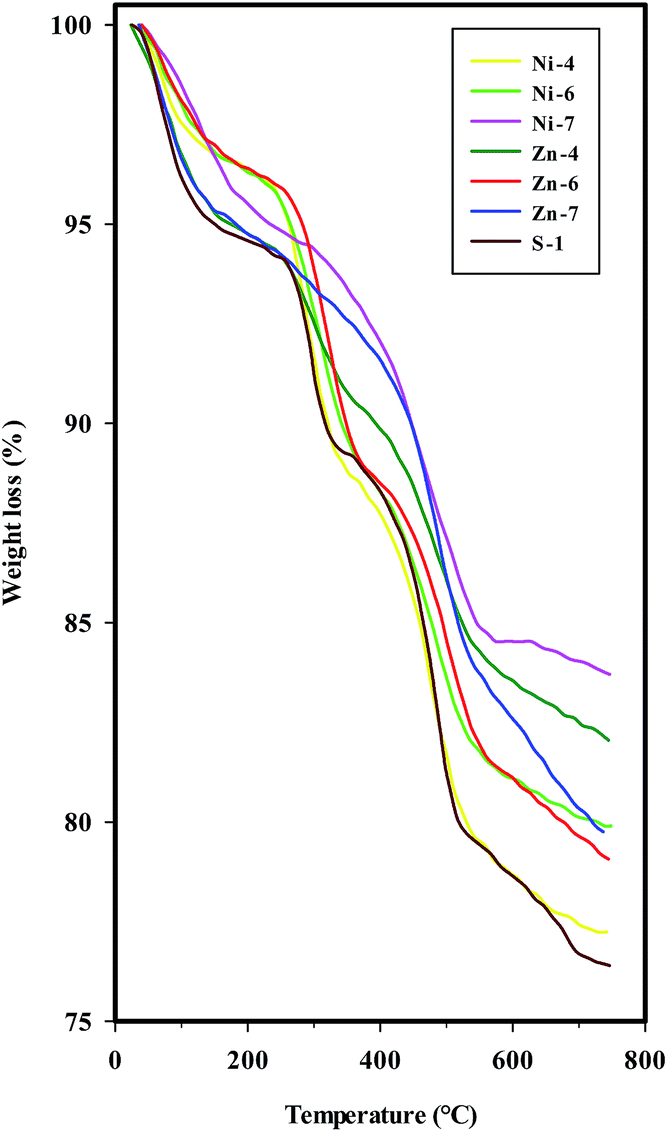 | ||
| Fig. 10 TGA curves of the uncalcined SAPO-34 and different metal containing SAPO-34 samples synthesized sonochemically. | ||
| Sample name | Weight loss stages | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | ||||
| Temperature range (°C) | Weight loss (%) | Temperature range (°C) | Weight loss (%) | Temperature range (°C) | Weight loss (%) | |
| Ni-4 | 25–101 | 2.5 | 240–331 | 6.7 | 430–529 | 6.8 |
| Ni-6 | 25–151 | 3.3 | 260–360 | 5.9 | 432–537 | 5.3 |
| Ni-7 | 25–179 | 4 | 302–552 | 9.5 | Alongside stage 2 | |
| Zn-4 | 25–145 | 5 | 260–353 | 3.4 | 417–539 | 5 |
| Zn-6 | 25–134 | 3 | 273–367 | 6.3 | 459–552 | 4.9 |
| Zn-7 | 25–120 | 4 | 410–532 | 7.4 | Alongside stage 2 | |
| S-1 | 25–139 | 5 | 257–326 | 4.6 | 416–518 | 7 |
TGA curves exhibit three weight loss stages for the parent SAPO-34, Ni-, and Zn-containing SAPO-34 samples synthesized with mixing methods (I) and (II), i.e. samples S-1, Ni-4, Ni-6, Zn-4, and Zn-6. The first weight loss in the low temperature range with an endothermic process is assigned to the water desorption which is observable by the peak at 1640 cm−1 in FTIR spectra.
The second and third weight losses, both accompanied by an exothermic effect, are attributed to the oxidative decomposition of organic template, suggesting that the template of TEAOH in the channel of SAPO-34 and metal containing SAPO-34 samples may have different chemical environment. The template decomposition can be related to the dimensions of the channel system, the nature of the template, and the template–framework interaction.8,44 By considering the strong template–framework interaction between protonated template and negatively charged framework, the second weight loss can be attributed to the removal of neutral template molecular, while the third weight loss may be due to the decomposition of charged template molecular in the channel of uncalcined molecular sieves.8,34 In the case of metal containing SAPO-34 samples prepared with methods (III) (samples Ni-7 & Zn-7), the second and third weight loss stages are merged, probably due to the different negatively charged framework in samples obtained by mixing methods (III).
4 Conclusions
The transition metal (Me = Ni and Zn) containing SAPO-34 nanoparticles were synthesized by applying insonation as the precrystallization treatment within a short period of time. Also, the possibility of incorporation of Ni and Zn ions into the SAPO-34 framework sites and seeking the appropriate preparation method and synthesis conditions directed to products with intended properties were inquired. The results elucidated that the sonication process as well as synthesis conditions, especially the mixing method and the metal types, affected (appreciably) the physicochemical properties of SAPO-34 products and their derivatives in terms of (regarding) the crystalline structure, size and morphology of particles, structure and hydroxyl functional groups, and stereochemistry of metal ions. The sonochemically prepared SAPO-34 samples possessed higher crystallinity and uniform crystals with narrow particle size distribution, which confirms the advantage of sonication process through crystallization. The combination of various characterization techniques and analyses revealed that the nature of the metal species could affect the physicochemical property of synthesized sample because of its effect on the manner of crystal growth. In comparison with Ni-doped SAPO-34 samples, the Zn-doped SAPO-34 samples showed a lower crystallinity, larger unit cell parameters, and more uniform crystals. The significant variations in the morphology, size, and uniformity of the metal-doped SAPO-34 crystals were acquired based on the mixing methods employed to prepare metal containing precursor gels. Meanwhile, the mixing methods played a key role in the incorporation of metal ions into the SAPO-34 framework, and mixing method (II), in which the metal sources were added to the precursor gel prior to the addition of the phosphoric acid, had the prominent capability to locate the metal ions, in particular nickel ions, in the SAPO-34 skeleton.References
- J. Čejka, A. Corma and S. Zones, Zeolites and Catalysis, Synthesis, Reactions and Applications, Wiley-VCH, Weinheim, 2010, vol. 1 Search PubMed.
- B. Chen and Y. Huang, Microporous Mesoporous Mater., 2009, 123, 71–77 CrossRef CAS.
- M. Hartmann and L. Kevan, Res. Chem. Intermed., 2002, 28, 625–695 CrossRef CAS.
- R. Szostak, Molecular Sieves: Principles of Synthesis and Identification, Springer Science + Business Media, New York, 1989 Search PubMed.
- N. Rajić, D. Stojaković, S. Hočevar and V. Kaučič, Zeolites, 1993, 13, 384–387 CrossRef.
- B. M. Weckhuysen, R. R. Rao, J. A. Martens and R. A. Schoonheydt, Eur. J. Inorg. Chem., 1999, 565–577 CrossRef CAS.
- F. C. Sena, B. F. de Souza, N. C. de Almeida, J. S. Cardoso and L. D. Fernandes, Appl. Catal., A, 2011, 406, 59–62 CrossRef CAS.
- Y. Wei, Y. He, D. Zhang, L. Xu, S. Meng, Z. Liu and B. L. Su, Microporous Mesoporous Mater., 2006, 90, 188–197 CrossRef CAS.
- M. A. Djieugoue, A. M. Prakash and L. Kevan, J. Phys. Chem. B, 2000, 104, 6452–6461 CrossRef CAS.
- F. Marzpour Shalmani, S. Askari and R. Halladj, Rev. Chem. Eng., 2013, 29–2, 99–122 Search PubMed.
- F. Marzpour Shalmani, R. Halladj and S. Askari, Ultrason. Sonochem., 2016, 29, 354–362 CrossRef CAS PubMed.
- L. Bing, X. Liu and B. Zhang, J. Mater. Sci., 2016, 51, 1476–1483 CrossRef CAS.
- E. Aghaei and M. Haghighi, J. Porous Mater., 2015, 22, 187–200 CrossRef CAS.
- S. Askari and R. Halladj, J. Solid State Chem., 2013, 201, 85–92 CrossRef CAS.
- S. Askari, R. Halladj and M. Sohrabi, Microporous Mesoporous Mater., 2012, 163, 334–342 CrossRef CAS.
- S. Askari, S. Kashi Kalhori, R. Halladj and N. Najafi, Prog. React. Kinet. Mech., 2016, 41–3, 1–9 Search PubMed.
- T. Hajiashrafi, A. Nemati Kharat, A. Dauth, A. R. Lewis and J. A. Love, React. Kinet., Mech. Catal., 2014, 113, 585–603 CrossRef CAS.
- F. Marzpour Shalmani, R. Halladj and S. Askari, Powder Technol., 2012, 221, 395–402 CrossRef.
- S. Tian, S. Ji, D. Lü, B. Bai and Q. Sun, J. Energy Chem., 2013, 22, 605–609 CrossRef CAS.
- S. Askari, A. Bashardoust Siahmard, R. Halladj and S. Miar Alipour, Powder Technol., 2016, 301, 268–287 CrossRef CAS.
- M. Amirhosseini, S. Askari and R. Halladj, J. Exp. Nanosci., 2016, 11, 1032–1043 CrossRef CAS.
- P. Sadeghpour and M. Haghighi, Particuology, 2014, 19, 69–81 CrossRef.
- S. Aghamohammadi and M. Haghighi, Chem. Eng. J., 2015, 264, 359–375 CrossRef CAS.
- M. Kang, M. H. Um and J. Y. Park, J. Mol. Catal. A: Chem., 1999, 150, 195–203 CrossRef CAS.
- N. Rajić, V. Kaučič and D. Stojaković, Zeolites, 1990, 10, 169–173 CrossRef.
- M. Inoue, P. Dhupatemiya, S. Phatanasri and T. Inui, Microporous Mesoporous Mater., 1999, 28, 19–24 CrossRef CAS.
- Y. Y. Zhang, L. Chen, Y. F. Li, Y. Q. Deng, C. T. Au and S. F. Yin, React. Kinet., Mech. Catal., 2015, 115, 691–701 CrossRef CAS.
- M. Kang and C. T. Lee, J. Mol. Catal. A: Chem., 1999, 150, 213–222 CrossRef CAS.
- D. R. Dubois, D. L. Obrzut, J. Liu, J. Thundimadathil, P. M. Adekkanattu, J. A. Guin, A. Punnoose and M. S. Seehra, Fuel Process. Technol., 2003, 83, 203–218 CrossRef CAS.
- R. Martínez-Franco, M. Moliner, C. Franch, A. Kustov and A. Corma, Appl. Catal., B, 2012, 127, 273–280 CrossRef.
- J. Wang, D. Fan, T. Yu, J. Wang, T. Hao, X. Hu, M. Shen and W. Li, J. Catal., 2015, 322, 84–90 CrossRef CAS.
- L. Xu, Z. Liu, A. Du, Y. Wei and Z. Sun, Stud. Surf. Sci. Catal., 2004, 147, 445–450 CrossRef CAS.
- Y. Chen, Y. Wu, L. Tao, B. Dai, M. Yang, Z. Chen and X. Zhu, J. Ind. Eng. Chem., 2010, 16, 717–722 CrossRef CAS.
- Y. Wei, D. Zhang, L. Xu, F. Chang, Y. He, S. Meng, B. Su and Z. Liu, Catal. Today, 2008, 131, 262–269 CrossRef CAS.
- M. Sedighi, M. Ghasemi, M. Sadeqzadeh and M. Hadi, Powder Technol., 2016, 291, 131–139 CrossRef CAS.
- M. Kang, J. Mol. Catal. A: Chem., 2000, 160, 437–444 CrossRef CAS.
- M. J. van Niekerk, J. C. Q. Fletcher and C. T. O'Connor, Appl. Catal., A, 1996, 138, 135–145 CrossRef CAS.
- M. Kang and T. Inui, J. Mol. Catal. A: Chem., 1999, 144, 329–335 CrossRef CAS.
- T. Inui and M. Kang, Appl. Catal., A, 1997, 164, 211–223 CrossRef CAS.
- M. Salmasi, Sh. Fatemi and A. Taheri Najafabadi, J. Ind. Eng. Chem., 2011, 17, 755–761 CrossRef CAS.
- F. Gao, E. D. Walter, N. M. Washton, J. Szanyi and C. H. F. Peden, ACS Catal., 2013, 3, 2083–2093 CrossRef CAS.
- K. Wang, G. Cao, G. J. Kennedy, M. Afeworki, R. E. Bare and R. B. Hall, J. Phys. Chem. C, 2011, 115, 18611–18617 CAS.
- D. L. Obrzut, P. M. Adekkanattu, J. Thundimadathil, J. Liu, D. R. Dubois and J. A. Guin, React. Kinet. Catal. Lett., 2003, 80, 113–121 CrossRef CAS.
- L. Zhang, J. N. Primera-Pedrozo and A. J. Hernández-Maldonado, J. Phys. Chem. C, 2010, 114, 14755–14762 CAS.
- J. Lu, X. Wang and H. Li, React. Kinet. Catal. Lett., 2009, 97, 255–261 CrossRef CAS.
- M. Charghand, M. Haghighi and S. Aghamohammadi, Ultrason. Sonochem., 2014, 21, 1827–1838 CrossRef CAS PubMed.
- S. Askari, S. Miar Alipour, R. Halladj and M. H. Davood Abadi Farahani, J. Porous Mater., 2013, 20, 285–302 CrossRef CAS.
- S. Askari and R. Halladj, Ultrason. Sonochem., 2011, 19, 554–559 CrossRef PubMed.
- M. Pankaj Ashokkumar, Theoretical and Experimental Sonochemistry: Involving Inorganic Systems, Springer, Dordrecht, Heidelberg, London, New York, 2011 Search PubMed.
- C. N. R. Rao, A. Müller and A. K. Cheetham, The Chemistry of Nanomaterials, Synthesis, Properties and Applications, Wiley-VCH, Germany, 2004 Search PubMed.
- J. Geng, L. Jiang and J. Zhu, Sci. China: Chem., 2012, 55, 2292–2310 CrossRef CAS.
- Y. Waseda, E. Matsubara and K. Shinoda, X-Ray Diffraction Crystallography, Introduction, Examples and Solved Problems, Springer, Heidelberg, Dordrecht, London, New York, 2011 Search PubMed.
- F. H. Chung and D. K. Smith, Industrial Applications of X-Ray Diffraction, Marcel Dekker, Inc., New York & Basel, 2000 Search PubMed.
- B. M. Lok, C. A. Messina, R. L. Patton, R. T. Gajek, T. R. Cannan and E. M. Flanigen, US Patent 4,440,871, Union Carbide Corporation, 1984 Search PubMed.
- M. Strauss, G. A. V. Martins, G. Berlier, S. Coluccia, L. Marchese and H. O. Pastore, Microporous Mesoporous Mater., 2014, 187, 135–144 CrossRef CAS.
- Z. Zhu, M. Hartmann and L. Kevan, Chem. Mater., 2000, 12, 2781–2787 CrossRef CAS.
- B. Imelik and J. C. Vedrine, Catalyst Characterization: Physical Techniques for Solid Materials, Springer Science + Business Media, New York, 1994 Search PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley India Pvt. Limited, 6th edn, 2007 Search PubMed.
- C. J. Ballhausen, Introduction to Ligand Field Theory, McGraw-Hill, 1962 Search PubMed.
- D. S. McClure, J. Phys. Chem. Solids, 1957, 3, 311–317 CrossRef CAS.
- A. Nezamzadeh-Ejhieh and S. Khorsandi, J. Ind. Eng. Chem., 2014, 20, 937–946 CrossRef CAS.
- S. M. Auerbach, K. A. Carrado and P. K. Dutta, Handbook of Zeolites Science and Technology, Marcel Dekker, Inc., New York & Basel, 2003 Search PubMed.
- R. A. Nyquist and R. O. Kagel, Infrared Spectra of Inorganic Compounds, Academic Press, New York and London, 1971 Search PubMed.
- D. W. Mayo, F. A. Miller and R. W. Hannah, Course Notes on the Interpretation of Infrared and Raman Spectra, John Wiley & Sons, Inc., Hoboken, New Jersey and Canada, 2003 Search PubMed.
- V. K. Kondratov, L. F. Lopatova and V. G. V. Vykhristyuk, Russ. J. Phys. Chem., 1979, 53, 1796 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00272f |
| This journal is © The Royal Society of Chemistry 2017 |