
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Physico-chemical characteristics, biocompatibility, and MRI applicability of novel monodisperse PEG-modified magnetic Fe3O4&SiO2 core–shell nanoparticles†
Uliana Kostiva,
Vitalii Patsulaa,
Miroslav Šlouf a,
Igor M. Pongracb,
Siniša Škokićb,
Marina Dobrivojević Radmilovićb,
Ivan Pavičićc,
Ivana Vinković Vrčekc,
Srećko Gajovićb and
Daniel Horák*a
a,
Igor M. Pongracb,
Siniša Škokićb,
Marina Dobrivojević Radmilovićb,
Ivan Pavičićc,
Ivana Vinković Vrčekc,
Srećko Gajovićb and
Daniel Horák*a
aInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovského nám. 2, 162 06 Prague 6, Czech Republic. E-mail: horak@imc.cas.cz
bUniversity of Zagreb, School of Medicine, Croatian Institute for Brain Research, Šalata 12, 10000 Zagreb, Croatia
cInstitute for Medical Research and Occupational Health, Analytical Toxicology and Mineral Metabolism Unit, Ksaverska cesta 2, 10000 Zagreb, Croatia
First published on 27th January 2017
Abstract
Monodisperse, superparamagnetic oleic acid-stabilized Fe3O4 nanoparticles of different sizes were prepared by thermal decomposition of Fe(III) oleate. The particles were subsequently coated by silica shells of different thicknesses (yielding Fe3O4&SiO2) using a water-in-oil (w/o) reverse microemulsion technique and/or were decorated with amino groups by reaction with (3-aminopropyl)triethoxysilane. The resulting Fe3O4&SiO2-NH2 nanoparticles were then modified with poly(ethylene glycol) (PEG) via reaction with its succinimidyl ester yielding Fe3O4&SiO2-PEG particles. The in vitro biocompatibility and biosafety of the Fe3O4&SiO2 and Fe3O4&SiO2-PEG particles were investigated in a murine neural stem cell model in terms of oxidative stress response and cell viability, proliferation, and uptake. Finally, the potential of both nanoparticle types for application in magnetic resonance imaging (MRI) visualization was evaluated.
1. Introduction
The outstanding properties of nanometer-scale materials, in particular their small sizes, are very attractive for biomedical applications including diagnostics and therapy of various diseases.1,2 Special attention is paid to particles with magnetic properties, which represent multifunctional materials used for magnetic resonance imaging (MRI) contrast agents,3 drug and gene delivery,4–6 hyperthermia to treat cancer,7 and separation of cells and specific molecules from body liquids.8Magnetic nanomaterials are typically based on ferromagnetic elements such as iron, cobalt, nickel, or their oxides and alloys. The physical and chemical properties of magnetic nanoparticles are strongly associated with their composition, morphology, size, and distribution. Requisite characteristics of the particles can be tailored by selection of a proper preparation method, e.g., co-precipitation,9 spray pyrolysis,10 hydrothermal,11 sol–gel,12 or thermal decomposition. However, magnetic nanoparticles for theranostic applications must meet additional criteria. These involve low toxicity, biocompatibility, proper in vitro and in vivo interactions with living cells, and convenient dosage.13 Among the magnetic nanoparticles, a dominant role is played by iron oxides, mainly maghemite (γ-Fe2O3) and magnetite (Fe3O4), which are known to be nontoxic (biocompatible) and have been approved by the Food and Drug Administration as a contrast agent for MRI.14,15
Monodisperse magnetic nanoparticles with tunable properties are generally prepared by thermal decomposition of metal organic precursors; however, such particles are hydrophobic. Since bioapplications using nanomaterials require particle dispersibility in aqueous media, silica is often used as a coating material.16 Silica is biocompatible, optically transparent, chemically inert, thermally stable, and has a large surface area available for the loading of required drugs.17 In addition, silica can be easily modified with various reactive groups by the addition of appropriate silane derivatives during synthesis. Due to a large specific surface area and a negative charge, silica-coated magnetic nanoparticles have a tendency to adsorb proteins from blood plasma, which leads to undesirable particle aggregation.18 As a result, the particles are rapidly shuttled out of blood circulation and internalized by macrophages and endothelial cells of the reticuloendothelial system, in particular to the liver and spleen.19 To obtain not only a stable colloid without particle aggregation, but also prevent undesirable protein adsorption and prolong half-life time in the blood stream, the nanoparticle surface has to be further modified. The surface coating determines the interaction of the particles with the membranes of living cells. This controls the particle uptake by different cell types and influences biocompatibility, as well as the distribution of the nanoparticles in the tissues of living organisms. Recently, many materials, such as dopamine,20 ascorbic acid,21 peptides,22 mono-23 and polysaccharides,24 and poly(ethylene glycol) (PEG),25 have been proposed for particle coating by physical adsorption or chemical binding. PEG in particular is considered to be a very promising material, that protects the nanoparticles from the immune system, promotes a longer circulation time, and inhibits removal by the reticuloendothelial system. Moreover, PEG is inexpensive and FDA-approved for a wide range of bioapplications.25
The aim of this work is to design, prepare, and characterize monodisperse silica-coated magnetite nanoparticles (Fe3O4&SiO2) with controlled shell thickness. Functionalization of the Fe3O4&SiO2 surface with amino groups (Fe3O4&SiO2-NH2) and PEG (Fe3O4&SiO2-PEG) was another aim. The biocompatibility and biosafety of both Fe3O4&SiO2 and Fe3O4&SiO2-PEG particles were determined using a murine neural stem cell model, while their potential for biomedical use was evaluated by means of stem cell labeling and ex vivo MRI visualization of particles in the mouse brain.
2. Experimental
2.1. Chemicals and materials
Octadec-1-ene (OD), icosane (IS), tetramethyl orthosilicate (TMOS), (3-aminopropyl)triethoxysilane (APTES), Igepal CO-520 [polyoxyethylene(5) nonylphenylether], succinic anhydride, N,N-diisopropylethylamine, and Dulbecco's modified Eagle's medium (DMEM) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Oleic acid (OA), ammonium fluoride, acetone, dichloromethane (distilled), and ethanol were obtained from Lachema (Brno, Czech Republic). Methoxy poly(ethylene glycol) succinimidyl active ester (PEG-NHS; Mw = 2000) was purchased from Rapp Polymere (Tuebingen, Germany). Cellulose dialysis membranes (14 kDa) were obtained from Spectrum Europe (Breda, Netherlands). Fe(III) oleate was prepared according to an earlier report26 Ultrapure Q-water ultrafiltered on a Milli-Q Gradient A10 system (Millipore; Molsheim, France) was used in biological experiments.Molecular Probes LIVE/DEAD™ viability/cytotoxicity and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) kits were from Invitrogen (Fisher Scientific; Hampton, NH, USA). DMEM/F-12, GlutaMAX, B27 factor, penicillin, streptomycin, epidermal growth factor (EGF), and fibroblast growth factor (FGF) were obtained from Life Technologies (Carlsbad, CA, USA). All other chemicals were purchased from Sigma-Aldrich.
The telencephalic walls of embryos at gestation day 14.5 were microdissected and dissociated using StemPro Accutase (Life Technologies). Cells were maintained at 37 °C in a humidified 5% CO2 and 95% O2 atmosphere. The expansion medium consisted of DMEM/F-12 with GlutaMAX, 1% N-2 and 2% B-27 supplements, 1% penicillin/streptomycin, 20 ng mL−1 EGF, and 10 ng mL−1 FGF (all Life Technologies). The neurospheres were dissociated and mNSCs were plated in the single-cell state at densities of 2 × 106, 8 × 105, 4 × 104, and 1 × 104 cells per well on 6-, 12-, 24-, and 96-well plates, respectively. All plates were previously coated with aqueous poly(D-lysine) solution (50 μg mL−1; Sigma-Aldrich).
2.2. Synthesis of Fe3O4 nanoparticles
OA-stabilized Fe3O4 nanoparticles were obtained by thermal decomposition of Fe(III) oleate.26 To prepare 6 and 10 nm Fe3O4 nanoparticles, Fe(III) oleate (7.2 g; 8 mmol) was dissolved in a mixture of solvent (38 mL of OD) and stabilizer (4.55 and 3.6 mL of OA, respectively). 20 nm Fe3O4 nanoparticles were obtained analogously in IS (38 mL) with OA (3.6 mL) as a stabilizer. The reaction mixture was degassed at 95 °C for 20 min in vacuum (∼1.3 kPa) and heated at 320 °C for 30 min (in OD) or at 343 °C for 60 min (in IS). After cooling to room temperature (RT), ethanol (100 mL) was added to the reaction mixture, and the resulting particles were separated by a magnet. The nanoparticles were washed ten times with hot ethanol (60–70 °C; 50 mL each) to remove residual solvents and OA. Finally, the Fe3O4 nanoparticles were dispersed in hexane.2.3. Synthesis of Fe3O4&SiO2 and Fe3O4&SiO2-NH2 nanoparticles
Fe3O4&SiO2 and Fe3O4&SiO2-NH2 particles were obtained by a water-in-oil (w/o) reverse microemulsion technique.28,29 To a dispersion of Fe3O4 nanoparticles (30 mg) in hexane (10 mL), Igepal CO-520 (0.5 mL) and 25% NH4OH (0.08 mL) were added, and the mixture was sonicated (Sonopuls sonicator; Bandelin; Berlin, Germany) at RT for 30 min. After addition of TMOS (0.04 mL), the reaction mixture was stirred (600 rpm) at RT for 48 h. To produce Fe3O4&SiO2-NH2 nanoparticles, APTES (0.02 mL) was added, and the reaction was continued at RT for an additional 24 h. Fe3O4&SiO2 and Fe3O4&SiO2-NH2 nanoparticles were precipitated in acetone (10 mL), separated by centrifugation (4000 rpm), and washed with ethanol and water five times each to remove residual surfactant. Finally, the nanoparticles were dialyzed against water at RT for 48 h using Spectra/Por® cellulose membrane (14 kDa; Spectrum; Rancho Dominguez, CA, USA).2.4. PEGylation of Fe3O4&SiO2-NH2 nanoparticles
To PEGylate the Fe3O4&SiO2-NH2 particles (30 mg), they were dispersed in dichloromethane (10 mL), PEG-NHS (40 mg) was added, and the mixture was stirred (200 rpm) at RT for 12 h. Residual amino groups on the particles were reacted with succinic anhydride (10 mg) and N,N-diisopropylethylamine (25 μL) at RT for 1 h with stirring (200 rpm). The resulting Fe3O4&SiO2-PEG particles were precipitated by acetone (5 mL), dispersed in ethanol, purged with Ar for 10 min to remove dichloromethane, separated by centrifugation (4000 rpm), dispersed in water, and finally dialyzed against water at RT for 24 h to remove unreacted PEG.2.5. Physico-chemical characterization of the nanoparticles
| Dn = ∑niDi/∑ni | (1) |
| Dw = ∑niDi4/∑niDi3, | (2) |
2.6. Labeling efficiency of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol mol−1) for 20 min. Cells were counterstained with 0.1% nuclear fast red for 1 min, mounted with HistoMount (Invitrogen), and covered by coverslips. After drying, the cells were viewed using an ECLIPSE E200 light microscope (Nikon Instruments; Tokyo, Japan) under bright field.
:1 mol mol−1) for 20 min. Cells were counterstained with 0.1% nuclear fast red for 1 min, mounted with HistoMount (Invitrogen), and covered by coverslips. After drying, the cells were viewed using an ECLIPSE E200 light microscope (Nikon Instruments; Tokyo, Japan) under bright field.2.7. Biocompatibility and biosafety of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles
2.8. Ex vivo MRI visibility validation of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles in mouse brain
Magnetic resonance imaging (MRI) was performed on a 7 T BioSpec 70/20 USR system (Bruker Biospin; Ettlingen, Germany) with Paravision 6.0.1. software in a configuration using an 86 mm MT0381 transmit volume coil and a 2-element MT0042 mouse brain surface receiver coil. The samples were placed in a suitable plastic tube, which was taped to the receiver coil to lay the region of interest in the center of the field of view (FOV). relaxation times were measured for each particle type at three different concentrations (10, 40, and 100 mg L−1) and host medium. T1, T2, and
relaxation times were measured for each particle type at three different concentrations (10, 40, and 100 mg L−1) and host medium. T1, T2, and 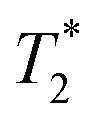 were compared to the results measured on native host media in the absence of the particles. The geometric parameters for all mapping sequences were identical. Two 1 mm thick slices in the FOV center, perpendicular to the phantom tube, were acquired with in-plane isotropic resolution of 133 μm and slice spacing of 0.3 mm. Prior to the main scans, a localizer scan was run, followed by additional local shimming using the MAPSHIM algorithm. This was done to ensure field homogeneity comparable to typical in vivo levels, which increases the relevance of measured
were compared to the results measured on native host media in the absence of the particles. The geometric parameters for all mapping sequences were identical. Two 1 mm thick slices in the FOV center, perpendicular to the phantom tube, were acquired with in-plane isotropic resolution of 133 μm and slice spacing of 0.3 mm. Prior to the main scans, a localizer scan was run, followed by additional local shimming using the MAPSHIM algorithm. This was done to ensure field homogeneity comparable to typical in vivo levels, which increases the relevance of measured  values. The sequence-specific parameters used are listed in Table S1 of ESI.† After acquisition, relaxation maps were calculated using Paravision's built-in post-processing tools (FitInISA macro). Circular regions of interest covering roughly one half of the tube cross-section were placed centrally over each map to avoid artifacts due to abrupt susceptibility changes between the phantom and the plastic tube. One or more shimming iterations were done, until the calculated half-power bandwidth (HPBW) of the proton frequency line was comparable to typical values obtained when applying the MAPSHIM algorithm in vivo on a mouse brain.
values. The sequence-specific parameters used are listed in Table S1 of ESI.† After acquisition, relaxation maps were calculated using Paravision's built-in post-processing tools (FitInISA macro). Circular regions of interest covering roughly one half of the tube cross-section were placed centrally over each map to avoid artifacts due to abrupt susceptibility changes between the phantom and the plastic tube. One or more shimming iterations were done, until the calculated half-power bandwidth (HPBW) of the proton frequency line was comparable to typical values obtained when applying the MAPSHIM algorithm in vivo on a mouse brain. -weighted sequences using sequence-specific parameters (Table S2 in ESI†). Twelve axial slices covering the whole length of the cerebrum were set. The slice thickness was reduced to 0.5 mm, and an in-plane resolution of 80 × 80 μm was chosen for the scans. The reduced voxel size compared to the phantom measurement (>5× smaller volume) and the lower inherent contrast of fixed tissue compared to the live organ was compensated by an increased number of averaging repetitions. No additional post-processing was done on the acquired images.
-weighted sequences using sequence-specific parameters (Table S2 in ESI†). Twelve axial slices covering the whole length of the cerebrum were set. The slice thickness was reduced to 0.5 mm, and an in-plane resolution of 80 × 80 μm was chosen for the scans. The reduced voxel size compared to the phantom measurement (>5× smaller volume) and the lower inherent contrast of fixed tissue compared to the live organ was compensated by an increased number of averaging repetitions. No additional post-processing was done on the acquired images.3. Results and discussion
3.1. Magnetite nanoparticles
Superparamagnetic Fe3O4 particles were obtained by OA-stabilized thermal decomposition of Fe(III) oleate. A large advantage of this technique is that it provides monodisperse particles, the size of which can be controlled in the 6–20 nm range by changing the reaction temperature and the concentration of the OA stabilizer. The tunable particle size and uniformity, as well as the reproducibility of the synthesis, are of critical importance for prospective bioapplications. The reaction temperature was controlled by selecting high-boiling solvents, such as OD and IS. As OD has a lower boiling temperature than IS, the reaction in OD produced the smaller particles No. I and II (6 and 10 nm; Fig. 1a and d, respectively) compared to the particles no. III prepared in IS (20 nm; Fig. 1g), which was demonstrated by TEM. While the 10 nm Fe3O4 particles were formed at a lower OA concentration (0.3 mmol mL−1), the 6 nm ones were synthesized in the presence of higher OA concentration (0.38 mmol mL−1). This is because an increased amount of stabilizer (OA) always produces smaller entities because it stabilizes more particles.26,40 | ||
| Fig. 1 Crystal structure of the Fe3O4 nanoparticles. TEM/BF micrographs of (a) 6, (d) 10, and (g) 20 nm Fe3O4 nanoparticles, (b, e and h) the corresponding experimental TEM/SAED diffraction patterns, and (c, f and i) comparison of the radially averaged SAED diffraction patterns with theoretically calculated XRD diffraction patterns of Fe3O4. Each row corresponds to one TEM analysis. The diffraction indices (hkl) given in the right column in bold correspond to diffractions enhanced due to the preferred orientation of the Fe3O4 nanocrystals (zone axis [001]). | ||
The morphology, coating, elemental composition, and crystal structure of the particles were verified by TEM analysis including bright field imaging (TEM/BF), energy-dispersive X-rays analysis (TEM/EDX), and selected area electron diffraction (TEM/SAED). The TEM analysis was supplemented by FTIR measurements. The TEM/BF micrographs (Fig. 1a, d and g) show the nanoparticle sizes as a function of experimental conditions; the SiO2-coating on the nanoparticle surface is seen in Fig. 2.
 | ||
| Fig. 2 TEM micrographs of Fe3O4&SiO2 nanoparticles prepared from starting (d–f) 6, (a–c, g–i) 10, and (j–l) 20 nm Fe3O4 particles. Fe3O4&SiO2 nanoparticles prepared at (a) 40/10, (b) 40/30, (c) 40/80, (d) 40/30, (e) 80/30, (f) 120/30, (g) 20/30, (h) 40/30, (i) 60/30, (j) 50/30, (k) 120/30, and (l) 150/30 μL mg−1 TMOS/Fe3O4 ratios. | ||
TEM/SAED diffraction patterns confirmed that the nanoparticles were the magnetic form of iron oxide, i.e., magnetite (Fe3O4). Fig. 1a, d and g shows the TEM/BF micrographs of nanoparticles with various sizes; the medium column displays the corresponding two-dimensional experimental TEM/SAED diffraction patterns (Fig. 1b, e and h), and the right column compares the one-dimensional, radially averaged SAED diffractograms with theoretically calculated XRD diffractograms of magnetite (Fig. 1c, f and i). The SAED diffraction positions were in excellent agreement with the calculated XRD positions, while the SAED diffraction intensities of several reflections (440 and 400) were slightly different from the calculated XRD intensities (Fig. 1c, f and i). This difference could be attributed to the preferred orientation of the faceted Fe3O4 nanocrystals lying on the electron-transparent carbon film. Diffractions that were stronger in SAED than in XRD were of the (hk0) type, i.e., their last diffraction index was zero. This indicated that the orientation of a significant quantity of the nanocrystals could be characterized by zone axis [001], and the strong diffractions of the SAED pattern came from the zonal planes obeying the Weiss Zone Law (WZL). The general formula of WLS reads: hu + kv + lw = 0, where (h,k,l) are the diffraction indices and (u,v,w) are the indices of the zone axis. In our specific case with zone axis [uvw] = [001], the WZL was reduced to the simple form (l = 0), and as a result the strongest SAED diffractions were (hk0) as discussed above and shown in Fig. 1. For the 6 and 10 nm nanocrystals (Fig. 1a and d), the diffraction (220) was not very strong despite obeying the WZL. This was due to the small nanocrystals (Fig. 1b and e) exhibiting less intensive diffractions than the large ones (Fig. 1h). As a result, the (220) diffraction was partially lost in the intensity of the neighboring primary beam. More details about zone axis, WZL and SAED interpretation can be found elsewhere.41 Here, we simply conclude that the agreement between the experimental SAED and calculated XRD patterns demonstrated that all prepared iron oxide nanoparticles had the Fe3O4 crystal structure.
The diameter of the magnetic nanoparticles in hexane was additionally measured by DLS. The hydrodynamic diameter (Dh) and polydispersity (PD) of No. I, II, and III nanoparticles reached 10, 31, and 41 nm and 0.12, 0.15, and 0.19, respectively, confirming a narrow particle size distribution without any aggregation. The difference between Dh values and diameters calculated from TEM micrographs (Table 1) comes from the fact that direct measurement on TEM yields number-averaged diameters of dried particles (Dn), while DLS yields intensity-averaged diameters of hydrated particles (Dh), which overestimates bigger particles and thus is sensitive to even a small amount of aggregates. Moreover, magnetic nanoparticles prepared by thermal decomposition in organic media adsorbed OA stabilizer on their surfaces, which also contributes to increased Dh; however, since the OA molecules have low atomic numbers in comparison with the iron oxide, they are not visible in the TEM micrographs. It should also be noted that Fe3O4 particles with Dn < 20 nm exhibited superparamagnetic behavior.42 That means that in the absence of an external magnetic field, the particles are dispersed in a solvent forming a colloid; however, they can be attracted by a magnet and magnetically manipulated. Moreover, such a small particle size is convenient for future applications as it avoids particle clearance by the liver and spleen. The magnetic properties of Fe3O4 nanoparticles prepared by thermal decomposition of Fe(III) oleate were described in our earlier publication.26
| No. | Nanoparticles | TMOS/Fe3O4 (μL mg−1) | Dn (nm) | PDI | SiO2 thickness (nm) |
|---|---|---|---|---|---|
| a TMOS – tetramethyl orthosilicate; Dn – number-averaged diameter; PDI – polydispersity index. | |||||
| I | Fe3O4 | — | 6 | 1.01 | — |
| I/1 | Fe3O4&SiO2 | 40/30 | 12 | 1.05 | 3 |
| I/2 | Fe3O4&SiO2 | 80/30 | 14 | 1.03 | 4 |
| I/3 | Fe3O4&SiO2 | 120/30 | 16 | 1.02 | 5 |
| II | Fe3O4 | — | 10 | 1.01 | — |
| II/1 | Fe3O4&SiO2 | 40/10 | 28 | 1.01 | 9 |
| II/2 | Fe3O4&SiO2 | 40/30 | 28 | 1.01 | 9 |
| II/3 | Fe3O4&SiO2 | 40/80 | 18 | 1.01 | 4 |
| II/4 | Fe3O4&SiO2 | 20/30 | 20 | 1.01 | 5 |
| II/5 | Fe3O4&SiO2 | 60/30 | 36 | 1.02 | 13 |
| III | Fe3O4 | — | 20 | 1.01 | — |
| III/1 | Fe3O4&SiO2 | 50/30 | 25 | 1.02 | 2.5 |
| III/2 | Fe3O4&SiO2 | 120/30 | 28 | 1.01 | 4 |
| III/3 | Fe3O4&SiO2 | 150/30 | 30 | 1.01 | 5 |
The structure of the Fe3O4 nanoparticles was analyzed by ATR FTIR spectroscopy (Fig. 3b). Characteristic bands of asymmetric and symmetric CH2 stretching vibrations of OA were observed at 2920 and 2852 cm−1, respectively, and the peak between 595 and 630 cm−1 was ascribed to Fe–O vibrations.
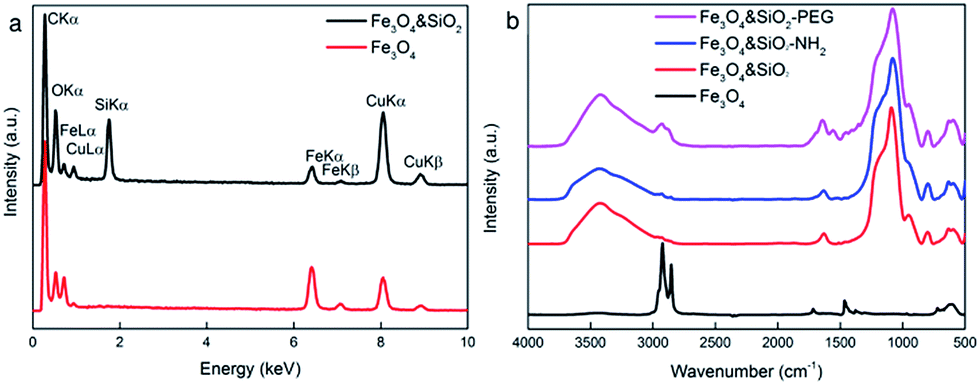 | ||
| Fig. 3 (a) TEM/EDX and (b) ATR FTIR spectra of starting and surface-modified Fe3O4 nanoparticles. | ||
3.2. Surface modification of Fe3O4 with silica
OA-stabilized Fe3O4 particles have the disadvantage that they are hydrophobic. To transfer such particles in water, in which all biological experiments are performed, it is necessary to modify their surface with a hydrophilic compound, such as SiO2. Hence, the starting Fe3O4 nanoparticles were coated with a silica shell by a water-in-oil (w/o) reverse microemulsion method. The reaction is based on the ammonia-catalyzed hydrolysis of TMOS and the condensation of hydrolyzed silica species. The thickness of the silica layer on the particles, which is determinative for internalization in the cells, depends on the concentrations of the starting nanoparticles, silane precursor (TMOS), catalyst (ammonia), and surfactant (Igepal CO-520).43 The silica thickness was thus controlled by changing the TMOS/Fe3O4 ratio in the synthesis, which regulates the degree of hydrolysis and condensation, as well as influencing the particle growth.In the first set of experiments, the optimal iron oxide concentration was determined, that led to formation of core–shell particles with properties satisfying all the requirements imposed on the resulting product. These include formation of homogeneous and robust silica shells around the particles without production of any additional neat silica. The Fe3O4 content in the reaction feed was changed at a constant amount of hexane, NH4OH, Igepal CO-520, and TMOS.
When investigating the effect of the TMOS/Fe3O4 ratio on the particle size and size distribution, the optimal ratio for formation of uniform silica shells on Fe3O4 particles No. II was found to be 40 μL/30 mg (No. II/2 in Table 1; Fig. 2b). At the ratio = 40 μL/10 mg, neat silica particles were also produced (No. II/1 in Table 1; Fig. 2a), while at 40 μL of TMOS/80 mg Fe3O4, inhomogeneous silica shells were formed on the particles (No. II/3 in Table 1; Fig. 2c).
In the second set of experiments, the volume of TMOS in the reaction was varied to control the silica shell thickness on the particles No. I–III, while the other reactants (Fe3O4, hexane, NH4OH, and Igepal CO-520) were kept constant. TEM/EDX measurements proved the presence of silica (Fig. 3a). The silica shell thickness increased from 3 to 5, from 5 to 13, and from 2.5 to 5 nm with TMOS/Fe3O4 ratios increasing from 40/30 to 120/30, from 20/30 to 60/30, and from 50/30 to 150/30 (μL mg−1), respectively (Fig. 2, Table 1). The TEM/EDX spectrum of Fe3O4&SiO2 particles exhibited strong Si peak, while the remaining peaks in the spectra of both Fe3O4 and Fe3O4&SiO2 particles belonged to magnetite (Fe, O) and the supporting carbon-coated copper grid (C, Cu; Fig. 3a). The Fe3O4&SiO2 nanoparticles demonstrated colloidal stability (Dh = 114 nm, PD = 0.14) and negative surface charge (−30 mV) due to the presence of silanol groups on the particle surfaces.
The ATR FTIR spectrum of Fe3O4&SiO2 confirmed the presence of SiO2 shells around the particles, as documented by intensive bands at 950 and 1080 cm−1 (Fig. 3b) ascribed to the symmetric and asymmetric stretching vibrations of Si–OH and Si–O–Si, respectively, while the OA peaks completely vanished. To further functionalize Fe3O4&SiO2 particles, their surfaces were modified by amino groups by reaction with APTES. In the IR spectrum of the Fe3O4&SiO2-NH2 nanoparticles, no significant changes were detected compared to the Fe3O4&SiO2 spectrum probably due to the detection limits of the ATR technique. Intense broad peaks at ∼3430 and 1630 cm−1 were attributed to OH stretching and bending vibrations of adsorbed water.
Fe3O4&SiO2-NH2 particles had a rather low positive charge (12 mV), which was not sufficient for efficient colloidal stability; Dh = 210 nm and PD = 0.25 documented the tendency of these particles to form aggregates.44
3.3. Surface modification of Fe3O4&SiO2 nanoparticles with PEG
To minimize the toxicity of the silica-coated particles and to render them invisible to the reticuloendothelial system, i.e., to prevent their uptake by different cell types (macrophages and NSCs) and to increase the blood circulation time, the Fe3O4&SiO2-NH2 nanoparticles were modified with PEG, which is well-known for hydrophilicity and nonionogenicity,25 by reaction with its succinimidyl ester. The modification was documented by DLS and ATR FTIR spectroscopy. The Fe3O4&SiO2-PEG nanoparticles demonstrated good colloidal stability (Dh = 43 nm, PD = 0.20), although the total negative surface charge was relatively low (−5 mV). ATR FTIR spectroscopy of the Fe3O4&SiO2-PEG nanoparticles revealed new intensive peaks at 2925 and 2850 cm−1, which were attributed to the asymmetric and symmetric stretching CH2 vibrations (Fig. 3b).3.4. Labelling efficiency and uptake of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles by murine neural stem cells
Superparamagnetic iron oxide nanoparticles are one of the most promising tools for labelling and tracking various cells by MRI.45,46 The labelling efficiency and cellular uptake of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles were evaluated by Prussian blue staining, flow cytometry, quantitative ICP-MS determination of total Fe cellular content, and TEM.The simple method of Prussian blue staining demonstrated that Fe3O4&SiO2-PEG nanoparticles were not taken up by the mNSCs and labelling was very efficient with Fe3O4&SiO2 (Fig. 4). Flow cytometry and TEM confirmed these results and clearly showed that mNSCs did not internalize a significant amount of Fe3O4&SiO2-PEG nanoparticles (Fig. 5 and 6). The uptake of Fe3O4&SiO2 was dose-dependent. Moreover, Fe3O4&SiO2 labelled more than 50% of the mNSC population at a 20-fold lower concentration (200 mg L−1) than commercial dextran-coated nanomag®-D-spio particles.27 TEM images clearly demonstrated that Fe3O4&SiO2 particles were located both in the cytoplasm and membrane-bound vesicles after uptake into mNSCs (Fig. 6c and d).
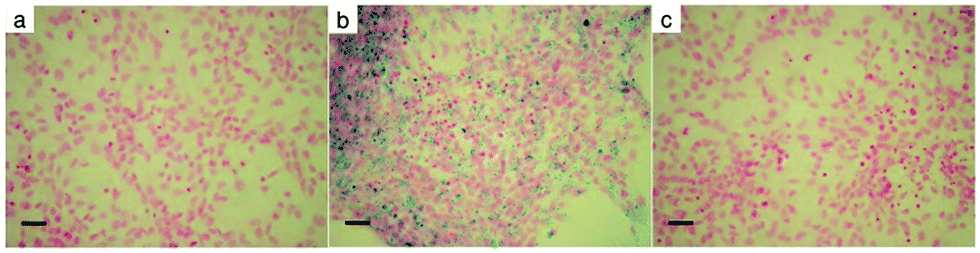 | ||
| Fig. 4 Light micrographs of Prussian blue-stained (a) unlabeled, (b) Fe3O4&SiO2-, and (c) Fe3O4&SiO2-PEG-labeled mNSCs (20 μg of particle per mL) for 24 h. Nuclear fast red staining showed the nuclei. Scale bar 50 μm. | ||
 | ||
| Fig. 5 Labeling efficiency of mNSCs by Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles analyzed by flow cytometry. mNSCs were exposed to different concentrations of particles for 24 h. Labeling efficiency, expressed as the mean of three independent experiments conducted in 5 replicates, was calculated as the percentage of the increase of the side-scattered light of the laser beam (SSC) relative to negative controls. Error bars represent standard deviations. Fe content in mNSCs was measured by ICP-MS. Non-treated cells (control) contained 0.23 ± 0.03 mg Fe per L. | ||
 | ||
| Fig. 6 TEM micrographs of (a) non-treated mNSCs (control), (b) mNSCs treated with Fe3O4&SiO2-PEG (no nanoparticles detected) for 24 h, (c) dispersed and agglomerated Fe3O4&SiO2 in a large endosome, and (d) agglomerated Fe3O4&SiO2 in cytoplasm. | ||
3.5. Biocompatibility and biosafety evaluation of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles
The unique and desirable specific features of superparamagnetic nanoparticles may potentially induce cytotoxic effects and should be evaluated for each novel particle type, addressing biocompatibility and biosafety concerns associated with their usage.38 Fe3O4&SiO2 nanoparticles were found in endosomes of mNSCs (Fig. 6), assuming the transfer of particles to lysosomes and the release of free iron ions into the cell cytoplasm after lysosomal degradation.47,48 In the cytosol, nanoparticle-derived Fe ions are stored in ferritin, catalyze ROS generation and/or are used for Fe-dependent cellular processes.49In this study, the response of mNSCs to treatment with Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles was evaluated by determination of cytotoxicity. This included cell viability and survival/mortality tests and investigation of oxidative stress, i.e., production of ROS, and study of changes in intracellular GSH levels and mitochondrial membrane potential. Fe3O4&SiO2-PEG nanoparticles, which were not actually taken up by the cells, had no cytotoxic effect (Fig. 7). Even the highest concentration (200 mg L−1) of Fe3O4&SiO2-PEG nanoparticles did not significantly affect either the survival or viability of mNSCs.
 | ||
| Fig. 7 Effect of different concentrations of Fe3O4&SiO2 nanoparticles on mortality, survival, and viability of mNSCs relative to control after 24 h exposure. Control cells were cultivated in particle-free exposure media. Cell viability was measured by the MTT assay. Cell survival/mortality was determined by flow cytometry. All data, calculated as percentages of the values in control cells, were expressed as the mean of three independent experiments conducted in five replicates. Error bars represent standard deviations. * – statistically significant difference (P < 0.05) versus control. | ||
For Fe3O4&SiO2 particles, only their lowest concentrations had no influence on the survival and viability of mNSCs. When the lowest possible concentration to achieve efficient cell labelling (20 mg L−1) was used, Fe3O4&SiO2 particles decreased cell viability by more than 50% compared to the control, while cell mortality increased by less than 20% (Fig. 7). It is interesting to note that the survival of mNSCs treated with 200 mg of Fe3O4&SiO2 per L was ca. 50% compared to the control (Fig. 7), while the same concentration of Fe3O4&SiO2 labelled ca. 50% of live cells (Fig. 5). This means that the number of labelled mNSCs was almost the same for 20 and 200 mg of Fe3O4&SiO2 per L, indicating that the optimal particle dose for stem cell labelling was 20 rather than 200 mg L−1. This was also supported by ICP-MS determination of the total Fe content in treated compared to control mNSCs (Fig. 5). A ten-fold increase in Fe3O4&SiO2 concentration applied to mNSCs, i.e., from 20 to 200 mg L−1, did not proportionally increase the Fe content in the cells. As expected, Fe levels in mNSCs treated with Fe3O4&SiO2-PEG nanoparticles were similar to those in the control cells (∼0.23 mg Fe per L). However, the increased value (0.8 mg Fe per L) in cells treated with 200 mg of Fe3O4&SiO2-PEG per L indicated that some nanoparticles were strongly attached to the cell surface.
Oxidative stress in mNSCs treated with Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles was evaluated using DCFH-DA, MBCl, and DiOC6 fluorescent dyes (Fig. 8). The ROS level significantly increased after a 4 h treatment of mNSCs at 20 and 200 mg of Fe3O4&SiO2 per L, while the increase was significant only for the highest concentration of Fe3O4&SiO2-PEG (Fig. 8). These results clearly showed that disturbances in cell functions leading to ROS production may occur either upon internalization of the nanoparticles or even upon contact with them.
 | ||
| Fig. 8 Effect of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles on the levels of reactive oxygen species (measured by DCFH-DA staining), GSH (measured by MBCl staining), and changes in mitochondrial membrane potentials (measured by DiOC6 staining) in mNSCs. Cells were exposed to different concentrations of nanoparticles for 4 h. Negative control cells were cultivated in nanoparticle-free exposure media, and positive controls were treated with 100 μM hydrogen peroxide. The data, expressed as the mean of three independent experiments conducted in five replicates, were calculated as percentages relative to negative controls. Error bars represent standard deviations. * – statistically significant difference (P < 0.05) compared to negative controls. | ||
The response of mNSCs to treatment with Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles was further evaluated by using the major endogenous antioxidant scavenger, GSH, as a biomarker.50 An alteration in the GSH level is an elegant direct measure for the adaptive cellular response to oxidative damage.51 Significant depletion of GSH in mNSCs was observed for the highest dose of Fe3O4&SiO2, whereas all doses of Fe3O4&SiO2-PEG particles caused the opposite effect, i.e., a significant increase in the GSH level after 4 h (Fig. 8). These observations revealed that mNSCs activated antioxidative defense mechanisms when exposed to Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles. Depletion in GSH levels is considered an indirect measure for persistent oxidative stress caused by efficient degradation of the ROS, while increased GSH indicates triggering of a preventive cellular mechanism at an early stage of redox stress.
Measurements of changes in the mitochondrial membrane potential (MMP) provided additional evidence of oxidative stress response in mNSCs by Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles (Fig. 8). Redox stress may perturb mitochondrial function in several ways, including dissipation or an increase in MMP. Changes in MMP were measured using the voltage-dependent dye DiOC6 in non-quenching mode at a nanomolar concentration. In addition, treatment of mNSCs was performed before staining. Using this experimental setup, a decrease in dye fluorescence compared to control cells implies depolarized (less negative) mitochondria, which took a lower amount of dye. In contrast, higher fluorescence in treated compared to control cells refers to hyperpolarized (more negative) mitochondria. Treatment of mNSCs with Fe3O4&SiO2 for 4 h induced a dose-dependent decrease in MMP compared to control cells (Fig. 8), thus leading to the mitochondrial depolarization. Only the lowest dose of Fe3O4&SiO2 did not induce significant mitochondrial depolarization. Similar behavior has already been described for cells exposed to magnetite nanoparticles.52 It is well-known that dissipation of MMP can be associated with ROS production and may be a prerequisite step for cell death by apoptosis or necrosis.53 As expected, Fe3O4&SiO2-PEG nanoparticles, which were not internalized in the cells, did not significantly change MMP. In contrast, treatment of mNSCs with hydrogen peroxide as the positive control (Fig. 6) caused mitochondrial membrane hyperpolarization in mNSCs, as described previously.38 Again, similarly to the uncoated, D-mannose-, and poly(L-lysine)-coated maghemite nanoparticles,38 our results showed that iron oxides may perturb cellular functions. Here, it was clearly demonstrated that surface coating has an important role in the design of nanoparticles enabling different modalities of biocompatibility and applicability in medicine.
3.6. MRI validation of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticle visibility in mouse brain ex vivo
 relaxation time were observed (Table 2). It should be noted, however, that
relaxation time were observed (Table 2). It should be noted, however, that 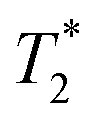 values are not a measure of tissue relaxation only, but of the local field homogeneity as well, which in turn depends on the quality of shimming and can vary in the same sample from one scan session to another.55 The worse the shimming, the shorter the
values are not a measure of tissue relaxation only, but of the local field homogeneity as well, which in turn depends on the quality of shimming and can vary in the same sample from one scan session to another.55 The worse the shimming, the shorter the 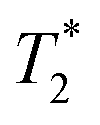 time. Thus, the
time. Thus, the  values (Table 2) are not to be taken as absolute reference values; rather, their purpose is to provide the basis for estimating the minimal particle concentration for sufficient contrast and reliable MRI detection in in vivo/ex vivo conditions. A
values (Table 2) are not to be taken as absolute reference values; rather, their purpose is to provide the basis for estimating the minimal particle concentration for sufficient contrast and reliable MRI detection in in vivo/ex vivo conditions. A  value around one half of the typical
value around one half of the typical  of the surrounding tissue in a given application should be sufficient to produce negative contrast in T2- and
of the surrounding tissue in a given application should be sufficient to produce negative contrast in T2- and  -weighted images, even if the injected particles spread over a larger volume. Depending on the application and the injected contrast agent volume, the concentration may be even lower. For comparison, typical T2 times in a mouse brain in vivo are approximately 35–40 ms when measured in a 7 Tesla instrument,54 while
-weighted images, even if the injected particles spread over a larger volume. Depending on the application and the injected contrast agent volume, the concentration may be even lower. For comparison, typical T2 times in a mouse brain in vivo are approximately 35–40 ms when measured in a 7 Tesla instrument,54 while 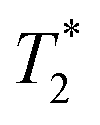 times are shorter, typically 15–20 ms. Thus, a measured T2 and
times are shorter, typically 15–20 ms. Thus, a measured T2 and  and 10 ms, respectively, on a phantom are reliable indicators of the required MRI visibility of magnetic particles in the mouse brain.
and 10 ms, respectively, on a phantom are reliable indicators of the required MRI visibility of magnetic particles in the mouse brain.
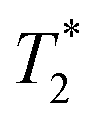 relaxation times of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles suspended in 20 vol% glycerol
relaxation times of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles suspended in 20 vol% glycerol
| Particles | Concentration (mg L−1) | T1 (ms) | T2 (ms) |
|
|---|---|---|---|---|
| a The value was outside the reliable fitting range of the measurement; HPBW – half-power bandwidth. | ||||
| Fe3O4&SiO2-PEG | 0 (native) | 1605 ± 8 | 46.7 ± 0.3 | 16.7 ± 1.7/20 |
| 10 | 1630 ± 13 | 28.8 ± 0.1 | 13.0 ± 1.0/29 | |
| 40 | 1655 ± 8 | 13.1 ± 0.1 | <10/39a | |
| 100 | 1621 ± 19 | <10a | <10/55a | |
| Fe3O4&SiO2 | 0 (native) | 1610 ± 6 | 47.8 ± 0.3 | 22.6 ± 0.5/20 |
| 10 | 1633 ± 4 | 27.6 ± 0.1 | 14.5 ± 1.0/36 | |
| 40 | 1608 ± 5 | 14.3 ± 0.1 | <10/78a | |
| 100 | 1591 ± 9 | <10a | <10/61a | |

The effect of increasing Fe3O4&SiO2-PEG particle concentration in 20 vol% glycerol on the negative contrast is illustrated in Fig. 9. Similar behavior was observed for Fe3O4&SiO2 nanoparticles (Table 2). The effect of shimming on the  relaxation time is visible in the rightmost column representing
relaxation time is visible in the rightmost column representing  maps, where the particle-doped phantom did not display steady values across the whole cross-section of the tube, unlike in the T1 and T2 maps. This effect was augmented for higher particle concentrations, as the decreased relaxation time resulted in reduced signal strength, thus making good shimming much harder to realize.
maps, where the particle-doped phantom did not display steady values across the whole cross-section of the tube, unlike in the T1 and T2 maps. This effect was augmented for higher particle concentrations, as the decreased relaxation time resulted in reduced signal strength, thus making good shimming much harder to realize.
 | ||
Fig. 9 Maps of measured T1, T2, and 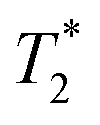 relaxation times for 10 (top row), 40 (middle row), and 100 mg of Fe3O4&SiO2-PEG particles per L of 20 vol% glycerol (bottom row). The uneven color in relaxation times for 10 (top row), 40 (middle row), and 100 mg of Fe3O4&SiO2-PEG particles per L of 20 vol% glycerol (bottom row). The uneven color in  maps reflects local magnetic field inhomogeneity that shimming was unable to correct for. maps reflects local magnetic field inhomogeneity that shimming was unable to correct for. | ||
3.7. MRI visualization of Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles in mouse brain ex vivo
Both Fe3O4&SiO2 and Fe3O4&SiO2-PEG nanoparticles (40 mg L−1) were clearly visible in MRI images, as seen from the comparison between the T2- and -weighted images of adult mouse brain ex vivo with injected particles (Fig. 10). As expected, the contrast was much more pronounced in the
-weighted images of adult mouse brain ex vivo with injected particles (Fig. 10). As expected, the contrast was much more pronounced in the  -weighted images. The contrast created by iron oxide particles in the surrounding tissue results from the fact that the iron oxide core disturbs the local magnetic field creating magnetic inhomogeneity, to which acquisition schemes based on gradient echo sequences (for
-weighted images. The contrast created by iron oxide particles in the surrounding tissue results from the fact that the iron oxide core disturbs the local magnetic field creating magnetic inhomogeneity, to which acquisition schemes based on gradient echo sequences (for 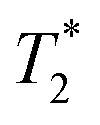 -weighting) are much more susceptible.55
-weighting) are much more susceptible.55
 | ||
Fig. 10 T2- (top row) and 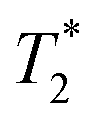 -weighted images (bottom row) of adult mouse brain ex vivo with (a) Fe3O4&SiO2-PEG and (b) Fe3O4&SiO2 nanoparticles injected on the left side and control host medium (20 vol% glycerol) injected on the right side. -weighted images (bottom row) of adult mouse brain ex vivo with (a) Fe3O4&SiO2-PEG and (b) Fe3O4&SiO2 nanoparticles injected on the left side and control host medium (20 vol% glycerol) injected on the right side. | ||
4. Conclusions
Fe3O4 nanoparticles with uniform size and well-defined spherical shape were synthetized by thermal decomposition of Fe(III) oleate. Monodispersity of the nanoparticles ensures their uniform physical and chemical properties. By simple variation of the reaction parameters, such as the temperature and concentration of the stabilizer, the particle size was tuned from 6 to 20 nm. To facilitate particle dispersibility in aqueous media, Fe3O4 was coated by silica via reverse microemulsion technique. Some Fe3O4&SiO2 particles were also functionalized with aminosilica to allow facile conjugation of PEG-NHS through amide bond formation. Fe3O4&SiO2 nanoparticles had a core–shell structure with a homogeneous silica layer on the surface. The thickness of the silica shell was controlled by selection of the reaction parameters, such as the TMOS/Fe3O4 ratio and volume of TMOS added. Fe3O4&SiO2-PEG nanoparticles exhibited good colloidal stability in aqueous media, as confirmed by DLS and ζ-potential measurements.The uptake of Fe3O4&SiO2-PEG nanoparticles by mNSCs was inferior to that of Fe3O4&SiO2, in agreement with reports that PEG can protect the particles from the immune cell system. A biocompatibility evaluation showed that Fe3O4&SiO2-PEG and Fe3O4&SiO2 nanoparticles did not induce cytotoxic effects, although slight disturbances in cell function leading to ROS production, mitochondrial depolarization, and activation of antioxidative defense mechanisms may occur either upon internalization or even contact with the nanoparticles. Both types of nanoparticles at concentrations of 40 mg L−1 provided sufficient contrast for MRI, indicating their possible applications in cell labeling or as circulating contrast particles.
Acknowledgements
This project was supported by the Czech Science Foundation (No. 17-04918S) and EU FP7 grant GlowBrain (REGPOT-2012-CT2012-316120). This article was based upon work from COST Action (CM1403), supported by COST (European Cooperation in Science and Technology). The MR imaging was done at GlowLab Multimodal Imaging Facility, University of Zagreb School of Medicine, Croatia. We are grateful to Lada Brkić Ahmed, Željka Punčec, Ivan Alić, and Hrvoje Mlinarić for their valuable help during cell and MRI experiments. The authors also acknowledge support from the RECOOP HST Association and the participating Cedars-Sinai Medical Center and the Charles University in Prague, Department of Physical and Macromolecular Chemistry, for the opportunity of Uliana Kostiv's and Vitalii Patsula's doctoral studies. Electron microscopy at the Institute of Macromolecular Chemistry was supported through project POLYMAT LO1507 (Ministry of Education, Youth and Sports of the CR, program NPU I).Notes and references
- G. Schmid, Nanoparticles: from theory to application, Wiley, 2004 Search PubMed.
- E. C. Wang and A. Z. Wang, Integr. Biol., 2014, 6, 9–26 RSC.
- T. H. Shin, Y. Choi, S. Kim and J. Cheon, Chem. Soc. Rev., 2015, 44, 4501–4516 RSC.
- G. Unsoy, R. Khodadust, S. Yalcin, P. Mutlu and U. Gunduz, Eur. J. Pharm. Sci., 2014, 62, 243–250 CrossRef CAS PubMed.
- J. Dobson, Gene Ther., 2006, 13, 283–287 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- D. Serantes, K. Simeonidis, M. Angelakeris, O. Chubykalo-Fesenko, M. Marciello, M. P. Morales, D. Baldomir and C. Martinez-Boubeta, J. Phys. Chem. C, 2004, 118, 5927–5934 CrossRef.
- N. Ohnishi, H. Furukawa, H. Hideyuki, J. M. Wang, C. An, E. Fukusaki, K. Kataoka, K. Ueno and A. Kondo, Nanobiotechnology, 2006, 2, 43–49 CrossRef CAS.
- D. Horák, M. Babič, P. Jendelová, V. Herynek, M. Trchová, Z. Pientka, E. Pollert, M. Hájek and E. Syková, Bioconjugate Chem., 2007, 18, 635–644 CrossRef PubMed.
- R. Strobel and S. Pratsinis, Direct synthesis of maghemite, magnetite and wüstite nanoparticles by flame spray pyrolysis, Adv. Powder Technol., 2009, 20, 190–194 CrossRef CAS.
- M. Viswanathiah, K. Tareen and V. Krishnamurthy, J. Cryst. Growth, 1980, 49, 189–192 CrossRef CAS.
- T. Sugimoto and K. Sakata, J. Colloid Interface Sci., 1992, 152, 587–590 CrossRef CAS.
- R. Bardhan, S. Lal, A. Joshi and N. J. Halas, Acc. Chem. Res., 2011, 44, 936–946 CrossRef CAS PubMed.
- R. Weissleder, D. D. Stark, B. L. Engelstad, B. R. Bacon, C. C. Compton, D. L. White, P. Jacobs and J. Lewis, AJR, Am. J. Roentgenol., 1989, 152, 167–173 CrossRef CAS PubMed.
- C. Tassa, S. Y. Shaw and R. Weissleder, Acc. Chem. Res., 2011, 44, 842–852 CrossRef CAS PubMed.
- W. Y. Rho, H. M. Kim, S. Kyeong, Y. L. Kang, D. H. Kim, H. Kang, C. Jeong, D. E. Kim, Y. S. Lee and B. H. Jun, J. Ind. Eng. Chem., 2014, 20, 2646–2649 CrossRef CAS.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef PubMed.
- X. He, H. Nie, K. Wang, W. Tan, X. Wu and P. Zhang, Anal. Chem., 2008, 80, 9597–9603 CrossRef CAS PubMed.
- J. P. Almeida, A. L. Chen, A. Foster and R. Drezek, Nanomedicine, 2011, 6, 815–835 CrossRef CAS PubMed.
- L. Wang, Z. Yang, J. Gao, K. Xu, H. Gu, B. Zhang, X. Zhang and B. A. Xu, J. Am. Chem. Soc., 2006, 128, 13358–13359 CrossRef CAS PubMed.
- L. X. Chen, T. Liu, M. C. Thurnauer, R. Csencsits and T. Rajh, J. Phys. Chem. B, 2002, 106, 8539–8546 CrossRef CAS.
- N. Nitin, L. LaConte, O. Zurkiya, X. Hu and G. Bao, J. Biol. Inorg Chem., 2004, 9, 706–712 CrossRef CAS PubMed.
- R. J. Wydra, P. G. Rychahou, B. M. Evers, K. W. Anderson, T. D. Dziubla and J. Z. Hilt, Acta Biomater., 2015, 25, 284–290 CrossRef CAS PubMed.
- C. I. Covaliu, D. Berger, C. Matei, L. Diamandescu, E. Vasile, C. Cristea, V. Ionita and H. Iovu, J. Nanopart. Res., 2011, 13, 6169–6180 CrossRef CAS.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- V. Patsula, E. Petrovský, J. Kovářová, R. Konefal and D. Horák, Colloid Polym. Sci., 2014, 292, 2097–2110 CAS.
- I. M. Pongrac, M. Dobrivojević, L. Brkić Ahmed, M. Babič, M. Šlouf, D. Horák and S. Gajović, Beilstein J. Nanotechnol., 2016, 7, 926–936 CrossRef CAS PubMed.
- U. Kostiv, O. Janoušková, M. Šlouf, N. Kotov, H. Engstová, K. Smolková, P. Ježek and D. Horák, Nanoscale, 2015, 7, 18096–18104 RSC.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- J. L. Lábár, Ultramicroscopy, 2005, 103, 237–249 CrossRef PubMed.
- W. Kraus and G. Nolze, J. Appl. Crystallogr., 1996, 29, 301–303 CrossRef CAS.
- S. Gražulis, A. Daškevič, A. Merkys, D. Chateigner, L. Lutterotti, M. Quirós, N. R. Serebryanaya, P. Moeck, R. T. Downs and A. Le Bail, Nucleic Acids Res., 2012, 40, D420–D427 CrossRef PubMed.
- R. M. Zucker and K. M. Daniel, Methods Mol. Biol., 2012, 906, 497–509 CAS.
- W. Overton, Cytometry, 1988, 9, 619–626 CrossRef CAS PubMed.
- I. Vinković Vrček, I. Pavičić, T. Crnković, D. Jurašin, M. Babič, D. Horák, M. Lovrić, L. Ferhatović, M. Ćurlin and S. Gajović, RSC Adv., 2015, 5, 70787–70807 RSC.
- M. Birringer, D. Lington, S. Vertuani, S. Manfredini, D. Schartau, M. Glei and M. Ristow, Free Radical Biol. Med., 2010, 49, 1315–1322 CrossRef CAS PubMed.
- D. Stevenson, D. Wokosin, J. Girkin and M. H. Grant, Toxicol. In Vitro, 2002, 16, 609–619 CrossRef CAS PubMed.
- I. M. Pongrac, I. Pavičić, M. Milić, L. Brkić Ahmed, M. Babič, D. Horák, I. Vinković Vrček and S. Gajović, Int. J. Nanomed., 2016, 11, 1701–1715 CAS.
- P. R. Hof, W. G. Young, F. E. Bloom, P. V. Belichenko and M. R. Celio, Comparative Cytoarchitectonic Atlas of the C57BL/6 and 129/Sv Mouse Brains, Elsevier, 2000 Search PubMed.
- V. Patsula, L. Kosinová, M. Lovrić, L. Ferhatovic Hamzić, M. Rabyk, R. Konefal, A. Paruzel, M. Šlouf, V. Herynek, S. Gajović and D. Horák, ACS Appl. Mater. Interfaces, 2016, 8, 7238–7247 CAS.
- K. W. Andrews, D. J. Dyson and S. R. Keown, Interpretation of Electron Diffraction Patterns, Plenum Press, 1967 Search PubMed.
- V. Patsula, M. Moskvin, S. Dutz and D. Horák, J. Phys. Chem. Solids, 2016, 88, 24–30 CrossRef CAS.
- C. L. Chang and H. S. Fogler, Langmuir, 1997, 13, 3295–3307 CrossRef CAS.
- U. Kostiv, I. Kotelnikov, V. Proks, M. Šlouf, J. Kučka, H. Engstová, P. Ježek and D. Horák, ACS Appl. Mater. Interfaces, 2016, 8, 20422–20431 CAS.
- J. W. Bulte, I. D. Duncan and J. A. Frank, J. Cereb. Blood Flow Metab., 2002, 22, 899–907 CrossRef PubMed.
- M. Modo, D. Cash, K. Mellodew, S. C. Williams, S. E. Fraser, T. J. Meade, J. Price and H. Hodges, NeuroImage, 2002, 17, 803–811 CrossRef PubMed.
- A. S. Arbab, L. B. Wilson, P. Ashari, E. K. Jordan, B. K. Lewis and J. A. Frank, NMR Biomed., 2005, 18, 383–389 CrossRef CAS PubMed.
- X. Jing, L. Yang, X. Duan, B. Xie, W. Chen, Z. Li and H. Tan, Jt., Bone, Spine, 2008, 75, 432–438 CrossRef PubMed.
- M. C. Hohnholt, M. Geppert and R. Dringen, Acta Biomater., 2011, 7, 3946–3954 CrossRef CAS PubMed.
- H. Sies, Free Radical Biol. Med., 1999, 27, 916–921 CrossRef CAS PubMed.
- G. G. Xiao, M. Wang, N. Li, J. A. Loo and A. E. Nel, J. Biol. Chem., 2003, 278, 50781–50790 CrossRef CAS PubMed.
- M. Könczöl, S. Ebeling, E. Goldenberg, F. Treude, R. Gminski, R. Gieré, B. Grobéty, B. Rothen-Rutishauser, I. Merfort and V. Mersch-Sundermann, Chem. Res. Toxicol., 2011, 24, 1460–1475 CrossRef PubMed.
- L. Galluzzi, N. Larochette, N. Zamzami and G. Kroemer, Oncogene, 2006, 25, 4812–4830 CrossRef CAS PubMed.
- D. N. Guilfoyle, V. V. Dyakin, J. O'Shea, G. S. Pell and J. A. Helpern, Magn. Reson. Med., 2003, 49, 576–580 CrossRef PubMed.
- D. W. McRobbie, E. A. Moore, M. J. Graves and M. R. Prince, MRI From Picture To Proton, Cambridge University Press, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00224f |
| This journal is © The Royal Society of Chemistry 2017 |