
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binuclear alkynylplatinum(II) terpyridine complexes with flexible bridges behave as organogelators for several organic solvents†
Ling-Bao Xing *ab,
Fei Qiaob,
Zhao Yuanb,
Jing-Li Zhangb,
Rui-Ying Zhangb,
Shuping Zhuob and
Zi-Yan Zhoub
*ab,
Fei Qiaob,
Zhao Yuanb,
Jing-Li Zhangb,
Rui-Ying Zhangb,
Shuping Zhuob and
Zi-Yan Zhoub
aKey Laboratory of Colloid and Interface Chemistry, Key Laboratory of Special Aggregated Materials, Ministry of Education, Shandong University, Jinan 250100, China. E-mail: lbxing@sdut.edu.cn
bSchool of Chemical Engineering, Shandong University of Technology, Zibo 255049, P. R. China
First published on 3rd March 2017
Abstract
New binuclear alkynylplatinum(II) terpyridyl complexes with flexible bridges have been synthesized and characterized by 1H NMR, mass spectra and elemental analysis. These compounds can gelate a variety of organic solvents via multiple intermolecular interactions such as metal–metal (Pt⋯Pt) and π–π stacking interactions, hydrogen bonding interactions, and van der Waals forces, which were confirmed by ultraviolet-visible (UV-vis) absorption spectroscopy, rheological studies, temperature-dependent 1H NMR spectroscopy, X-ray diffraction (XRD) and Fourier transform infrared (FT-IR) spectroscopy. Moreover, compound 1b with a longer flexible bridge (–O(CH2)6O–) exhibited higher gelation ability than that of compound 1a with a shorter flexible bridge (–O (CH2)4O–). A scanning electron microscopy (SEM) investigation of the xerogels from different organic solvents gave a visual image showing that fibrillar aggregates are entangled in three-dimensional network structures.
1 Introduction
Low molecular weight gelators (LMWGs), which are organized by non-covalent interactions such as hydrogen bonding, π–π interactions, van der Waals forces and dipole–dipole interactions, have shown promise for use in many fields,1–8 including molecular electronics, drug delivery, light harvesting and energy transfer and tissue engineering. Parallel to the growing interest in the development of organogels, there has also been an increasing interest in the investigation of transition–metal complexes as metallogelators,9–18 especially platinum(II) complexes. As a famous transition–metal complex, square-planar platinum(II) polypyridine complexes possess diverse excited states, high-emission quantum yields, and long triplet excited-state lifetimes, which have demonstrated promise for various potential applications,19–30 including photocatalysis, optical sensors, electron transfer, photochromism and so on. Especially, benefiting from intermolecular metal–metal (Pt⋯Pt) and π–π stacking interactions, the complexes have a strong tendency to self-assemble into highly ordered linear chains or oligomeric structures.31–36Yam and Ziessel's group firstly demonstrated that alkynylplatinum(II) terpyridyl complexes are capable of forming stable metallogels and showed drastic color changes during the gel-to-sol phase transition.37,38 Che and coworkers reported a dicationic platinum(II) terpyridyl complex which can form phosphorescent organogels in acetonitrile or in a mixture of acetonitrile and alcohol.39 Yam and coworkers developed a series of alkynylplatinum(II) complexes which were found to show strong luminescence enhancement upon a gel-to-sol phase transition upon increasing the temperature.40 De Cola and coworkers developed a straightforward strategy to obtain a new class of platinum(II) complex by using 2,6-bis(tetrazol-5-yl)pyridine with particular photophysical properties owing to the formation of self-assembled gels.41,42 Tu and coworkers established a method for the visual chiral recognition of (R)- and (S)-binap through an enantiocontrolled breakdown of gels prepared from novel aromatic-linker-steroidal (ALS) type platinum(II) pincer metallogelators.43 Very recently, Yi and coworkers designed and synthesized three alkynylplatinum(II) bipyridyl complexes with two cholesterol groups via alkyl chains and amido bonds, which can self-assemble to gel networks with a well-defined right-handed helical structure.44
In order to further investigate the effect of metal–metal (Pt⋯Pt) and π–π stacking interactions in the self-assembly process of metallogels in organic solvents, herein, we designed and synthesized new type of binuclear alkynylplatinum(II) terpyridyl complexes with flexible bridges (–O(CH2)nO, n = 4 or 6), which were characterized by 1H NMR, mass spectra and elemental analysis. The aromatic platinum(II) unit serves as chromophore, the long alkyl chains were selected as versatile gel-forming segments. Moreover, the amide groups, and binuclear platinum(II) complexes were readily introduced to examine the roles and extent of the metal–metal (Pt⋯Pt) and π–π stacking interactions among the platinum(II) cores, the hydrogen bonding and the van der Waals forces among the amide groups and long alkyl chains as well. It is anticipated that with the cooperative multiple intermolecular interactions, such compounds can self-assemble into long fibers in organic solvents and entangle further to form gels. This expectation was found indeed to be the case. According to the different length of the flexible bridges, 1a can gel aromatic solvents such as benzene, toluene and aniline, while 1b can not only gel aromatic solvents such as benzene, toluene and aniline, but also gel other solvents such as butanol, pentanol, heptane, and 1,2-epoxypropane.
2 Experimental
2.1 Materials
Unless specifically mentioned, all chemicals are commercially available and were used as received. Triethylamine (TEA) were distilled over CaH2 under a nitrogen atmosphere prior to use. 4′-Chloro-2,2′:6′,2′′-terpyridine,45 dodecyloxy-terminal alkyne,46 compound 1,23–27,35 2 (ref. 47) and 3 (ref. 23–27,35) were prepared according to the literature procedures.2.2 General method and instruments
NMR spectra were obtained on a Bruker Avance 400 spectrometer (400 MHz 1H NMR) at room temperature. Chemical shifts (δ) are reported in parts per million (ppm) using the solvent residual signal as an internal reference. Coupling constants (J) are given in hertz. Each resonance multiplicity is described as s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), m (multiplet), or br (broad signal). Variable-temperature 1H NMR experiments were carried out on Bruker DMX 300 spectrometer. Mass spectra (EI, ESI) were obtained in the positive ion mode on a Waters GCT Premier, Waters-QTOF and Bruker Daltonics Microflex spectrometer, respectively. Elemental analyses (CHN) were performed at FLASH EA1112 Elemental Analyzer. UV-vis spectra were obtained on a Shimadzu UV-1601PC spectrophotometer. The solvents for spectroscopic studies were of spectroscopic grade and used as received. Rheological properties were measured by a Thermo Haake RS300 rheometer with cone and plate geometry (35 mm diameter, 0.105 mm cone gap). The frequency spectra were conducted in the linear viscoelastic regime of the samples determined from dynamic strain sweep measurements at 25 °C. Fourier transform infrared (FT-IR) spectra of xerogels were measured at room temperature using a Varian Excalibur 3100 spectrometer. Xerogels were prepared by casting gels of toluene onto a KBr plate, and dried in the air. Field emission scanning electron microscopy (FESEM) images were obtained on a HITACHI S-4300 instrument operating at 10 kV with Au coated. Samples for FESEM measurement were prepared by wiping a small amount of gel onto a silicon wafer followed by naturally evaporating the solvent. Gels in toluene were coated on a glass plate and the solvent was slowly evaporated at room temperature. Then the glass plates with dried gels were fixed on a sample holder and subjected to X-ray diffraction (XRD) analysis at room temperature on Bruker D8 Focus. XRD patterns were recorded at scanning rate of 2° min−1 in the 2θ range of 5° to 60° with Cu-Kα radiation (λ = 1.54178 Å).2.3 Synthesis and characterization
2a. (0.48 g, 0.87 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 8.68 (d, J = 4.6 Hz, 4H), 8.60 (d, J = 7.9 Hz, 4H), 8.03 (s, 4H), 7.84 (m, 4H), 7.32 (dd, J = 7.3, 4.9 Hz, 4H), 4.34 (s, 4H), 2.10 (s, 4H). EI-MS: m/z calcd for C34H28N6O2: 552.2, found: 553.2 [M + 1]+.
2b. (0.53 g, 0.91 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 8.70 (d, J = 3.7 Hz, 4H), 8.63 (d, J = 7.9 Hz, 4H), 8.05 (s, 4H), 7.87 (t, J = 7.1 Hz, 4H), 7.40–7.30 (m, 4H), 4.27 (q, J = 6.5 Hz, 4H), 2.03–1.78 (m, 4H), 1.63 (d, J = 6.9 Hz, 4H). EI-MS: m/z calcd for C36H32N6O2: 580.26, found: 580.26 [M]+.
3a. (0.31 g, 0.28 mmol, 78%). 1H NMR (400 MHz, DMSO) δ 8.76 (d, J = 5.4 Hz, 4H), 8.41 (d, J = 7.8 Hz, 4H), 8.29 (t, J = 7.6 Hz, 4H), 8.16 (s, 4H), 7.95–7.87 (m, 4H), 4.61 (s, 4H), 2.17 (s, 4H). ESI-MS: m/z calcd for [C34H28Cl2N6O2Pt2]2+: 506.045, found: 506.132 [M]2+. Elemental analysis: calcd (%) for C34H28Cl4N6O2Pt2: C, 37.65; H, 2.60; N, 7.75, found: C, 37.58; H, 2.68; N, 7.69.
3b. (0.29 g, 0.26 mmol, 75%). 1H NMR (300 MHz, DMSO) δ 8.27 (t, J = 6.5 Hz, 8H), 8.13 (dd, J = 15.7, 7.8 Hz, 8H), 7.59 (t, J = 6.3 Hz, 4H), 4.49 (s, 4H), 1.89 (s, 4H), 1.58 (s, 4H). ESI-MS: m/z calcd for [C36H32Cl2N6O2Pt2]2+: 520.875, found: 520.561 [M]2+. Elemental analysis: calcd (%) for C36H32Cl4N6O2Pt2: C, 38.86; H, 2.90; N, 7.55, found: C, 38.97; H, 2.95; N, 7.62.
1a. (0.31 g, 0.11 mmol, 62%). 1H NMR (400 MHz, Pyr) δ 9.31 (d, J = 15.2 Hz, 4H), 8.70 (s, 8H), 8.21 (d, J = 8.2 Hz, 4H), 7.72 (s, 4H), 7.63 (d, J = 8.1 Hz, 4H), 7.15 (s, 8H), 4.99 (s, 16H), 4.24 (t, J = 6.3 Hz, 4H), 3.94 (t, J = 6.2 Hz, 8H), 3.73 (d, J = 5.4 Hz, 8H), 2.07–1.81 (m, 8H), 1.70 (dd, J = 31.9, 24.3 Hz, 12H), 1.50–0.94 (m, 96H), 0.81 (t, J = 6.6 Hz, 18H). ESI-MS: m/z calcd for [C144H206N10O12Pt2]2+: 2657.51, found: 2657.75 [M]2+. Elemental analysis: calcd (%) for C34H28Cl2N6O2Pt2: C, 63.65; H, 7.60; N, 5.13, found: C, 63.51; H, 7.65; N, 5.18.
1b. (0.33 g, 0.12 mmol, 66%). 1H NMR (400 MHz, Pyr) δ 9.31 (s, 4H), 8.68 (s, 8H), 8.22 (s, 4H), 7.69 (d, J = 31.6 Hz, 8H), 7.15 (s, 8H), 4.94 (s, 16H), 4.25 (s, 4H), 3.96 (s, 8H), 3.75 (s, 8H), 1.94 (d, J = 27.6 Hz, 8H), 1.69 (d, J = 48.5 Hz, 12H), 1.32 (d, J = 75.1 Hz, 100H), 0.94–0.59 (m, 18H). ESI-MS: m/z calcd for [C146H210N10O12Pt2]2+: 2687.55, found: 2687.63 [M]2+. Elemental analysis: calcd (%) for C146H210Cl2N10O12Pt2: C, 65.25; H, 7.88; N, 5.21, found: C, 65.37; H, 7.80; N, 5.26.
2.4 Gelation test
The formation of metallogels was evaluated using a tube inversion method. A weighed amount of the compound in a selected solvent (0.5 mL) was placed in a glass vial, which was sealed and heated until the compound was dissolved. After the solution was allowed to stand at room temperature for about 12 h, the state of the solution was examined by the “stable to inversion of a test tube” method. The reversibility of the gelation was confirmed by repeated heating and cooling. The critical gelation concentration (CGC) was determined from the minimum amount of gelator required for the formation of gel at room temperature. All experiments for CGC-determinations were run three times for reliable data.If no flow was observed when inverting the vial, a stable gel was formed and noted as gelation (G). If part of the mixture formed a gel but flow was still observed, the phenomenon was recorded as partial gel (PG). If precipitation occurred, P was noted, and if the clear solution was retained, it was marked as soluble (S). If the compound was unable to dissolve, it was noted as insoluble (I).
3 Results and discussion
The synthesis of new gelators based on platinum(II) complexes 1a and 1b were accomplished in high yields by using the Sonogashira reaction (Scheme 1). The starting compound 4′-chloro-2,2′:6′,2′′-terpyridine was directly reacted with 1,6-hexanediol (0.11 g, 0.93 mmol) or 1,4-butanediol with the aid of KOH in DMSO, to give 2a and 2b. After refluxing with K2PtCl4 in the mixture solvents of acetonitrile and water, 3a and 3b were obtained through recrystallization by diffusion of diethyl ether vapor into a solution of the product in acetonitrile. Then 3a and 3b were reacted with dodecyloxy-terminal alkyne in the presence of TEA in DMF under typical Sonogashira reaction conditions to produce target compounds 1a and 1b, which were identified by 1H NMR, electrospray ionization mass spectrometry (ESI MS), and satisfactory elemental analysis.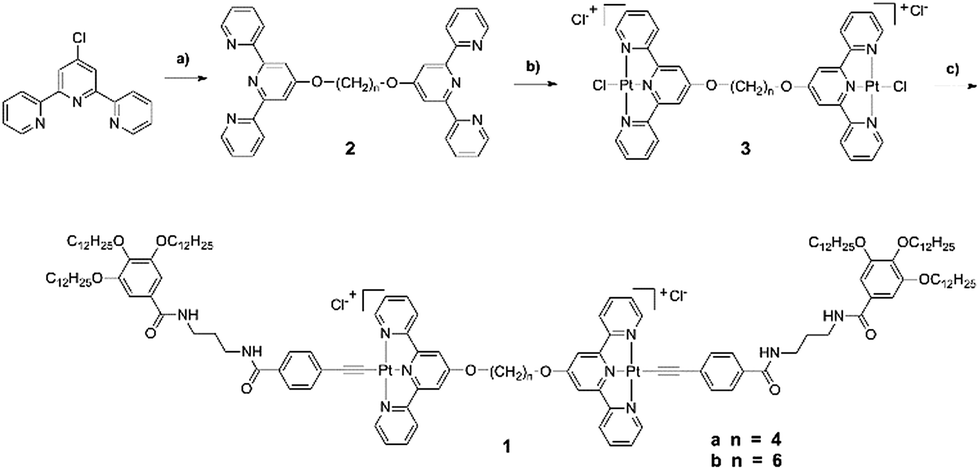 | ||
| Scheme 1 Chemical structures and synthesis routes of 1.a a(a) 1,6-Hexanediol or 1,4-butanediol, DMSO, KOH; (b) K2PtCl4, CH3CN, H2O; (c) dodecyloxy-terminal alkyne, CuI, DMF, TEA. | ||
As shown in Fig. 1, the UV-vis absorption spectra of 1a and 1b in DMSO showed a typical metal-to-ligand charge transfer (MLCT) [dπ(Pt)–π*(terpy)] band with a maximum at 475 nm and 450 nm mixed with an ligand-to-ligand charge transfer (LLCT) [π(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)–π*(tpy)] transition, respectively. The weak electronic absorption from 500–550 nm probably originates from metal-metal-to-ligand charge transfer (MMLCT) [(dσ*–π*)] transition as a result of the possible existence of intermolecular metal–metal and π–π interactions in DMSO solution, which indicates the aggregation of the molecules. The concentration-dependent UV-vis absorption spectral changes as shown in Fig. S1† further demonstrated the aggregation of the molecules with increase of the concentration of 1a and 1b in DMSO solution through the enhancement of the absorption band from 500 to 550 nm. Both 1a and 1b have no emission in DMSO solution at room temperature. This could possibly be ascribed to intramolecular reductive electron-transfer quenching of the 3MLCT state as well as energy transfer to the low-lying 3LLCT state arising from the presence of the electron-rich alkoxy groups on the phenyl ligand.16
C)–π*(tpy)] transition, respectively. The weak electronic absorption from 500–550 nm probably originates from metal-metal-to-ligand charge transfer (MMLCT) [(dσ*–π*)] transition as a result of the possible existence of intermolecular metal–metal and π–π interactions in DMSO solution, which indicates the aggregation of the molecules. The concentration-dependent UV-vis absorption spectral changes as shown in Fig. S1† further demonstrated the aggregation of the molecules with increase of the concentration of 1a and 1b in DMSO solution through the enhancement of the absorption band from 500 to 550 nm. Both 1a and 1b have no emission in DMSO solution at room temperature. This could possibly be ascribed to intramolecular reductive electron-transfer quenching of the 3MLCT state as well as energy transfer to the low-lying 3LLCT state arising from the presence of the electron-rich alkoxy groups on the phenyl ligand.16
 | ||
| Fig. 1 UV-vis absorption spectral changes of compound 1a (2.20 × 10−5 M) and 1b (2.18 × 10−5 M) in DMSO at room temperature. | ||
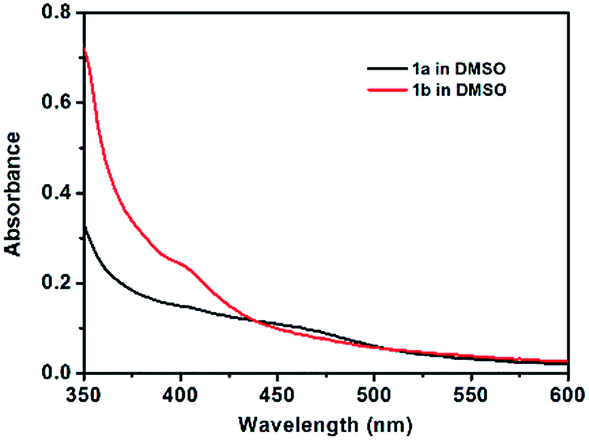
In order to further confirm the formation of gels, the rheological measurements of 1a and 1b in toluene were carried out as shown in Fig. 2. Both the samples of gel 1a and gel 1b in toluene were deformed sinusoidally with frequency f to give the elastic modulus (G′) and viscous modulus (G′′). Meanwhile, the G′ values of the samples were basically invariant with the frequencies, and the G′ values exceeded G′′ values over the entire range of frequencies in samples of gel 1a and gel 1b.
 | ||
| Fig. 2 Rheological properties of 1a (23 mg mL−1) and 1b (20 mg mL−1) in toluene. | ||
The gelation ability of compound 1a and 1b was examined in various solvents by means of the “stable to inversion of a test tube” method. The corresponding critical gelation concentrations (CGC) at room temperature were also measured. As shown in Table 1 and Fig. 3, compound 1a can gel aromatic solvents such as benzene, toluene and aniline; while compound 1b can not only gel aromatic solvents but also butanol, petanol, 1,2-epoxypropane and heptane through the extension of the flexible bridges. These gels were found to be stable in closed tubes for at least 3 months at 25 °C. The better gelation for aromatic solvents relative to the hydrocarbon or halogenated hydrocarbons is probably due to the enhancement of π–π interactions in aromatic solvents.3
| Solvents | 1a | 1b | Solvents | 1a | 1b |
|---|---|---|---|---|---|
| a G: gel; PG: partial gel; P: precipitation; S: soluble; I: insoluble when heated.b The critical gelation concentrations [CGC, mg mL−1] of gelators are shown in the parentheses. | |||||
| Methanol | I | I | Acetonitrile | I | I |
| Ethanol | I | I | Tetrahydrofuran | P | P |
| Butanol | PG | G (22.3) | Heptane | PG | G (21.0) |
| Pentanol | PG | G (19.8) | Cyclohexane | I | I |
| Petroleumether | PG | PG | 1,2-Epoxypropane | PG | G (23.6) |
| Ethyl acetate | I | I | Benzene | G (26.9) | G (23.6) |
| Acetone | I | I | Toluene | G (22.1) | G (20.0) |
| Dimethyl formamide | P | P | Aniline | G (22.7) | G (20.4) |
| Dimethyl sulfoxide | PG | PG | Chloroform | I | I |
 | ||
| Fig. 3 Selected metallogels of 1a in toluene and aniline; and 1b in heptane, benzene, aniline and toluene (from left to right). | ||
The morphology of xerogels obtained from different solvents was characterized by field emission scanning electron microscopy (FESEM) by evaporating the solvent at ambient under identical conditions. For 1a in benzene, due to the serious fusion of the fibers during the evaporation of the solvent, there is no obvious fibrous structures even under high magnification (Fig. 4b and c). In the case of aniline, the xerogels exhibited a closely packed three-dimensional (3D) network structure, which was composed of uniform fibers with diameter about 60 nm extending over micrometers (Fig. 4d and e). As for toluene for 1a, a similar fibrous structures consisted of highly entangled fibers was also observed (Fig. 4f and g). In 1,2-epoxypropane for 1b, a loose packed network composed of long and thick fibers about 100 nm in diameter and even over several micrometers in length were observed (Fig. 5b and c). The xerogels of 1b from butanol were consisted of highly entangled fibers, which were much thinner and longer with some fusion (Fig. 5d and e). In contrast, the xerogels derived from aromatic solvents such as benzene, aniline and toluene exhibited serious fusion of the fibers only under high magnification (Fig. 5f–i). The xerogels of 1b in heptane showed tightly packed and much thicker fibers (Fig. 5l and m).
 | ||
| Fig. 4 Phase transition behavior of the sol (left) and gel (right) phases of 1a (a) in aniline; and FESEM image of xerogels of 1a in different solvents: (b, c) benzene, (d, e) aniline, (f, g) toluene. Samples were prepared by drop-casting the gels on a silicon substrate, drying under air, and coating with Au. | ||
 | ||
| Fig. 5 Phase transition behavior of the sol (left) and gel (right) phases of 1b (a) in toluene; and FESEM image of xerogels of 1b in different solvents: (b, c) 1,2-epoxypropane, (d, e) butanol, (f, g) benzene, (h, i) aniline, (j, k) toluene, (l, m) heptane. Samples were prepared by drop-casting the gels on a silicon substrate, drying under air, and coating with Au. | ||
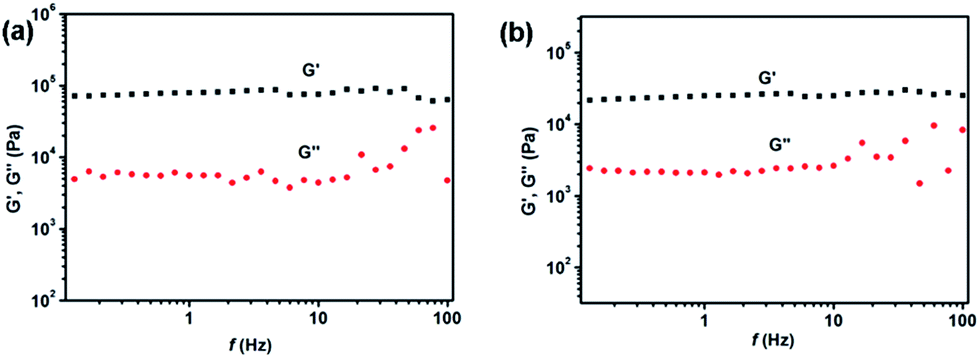
The intermolecular hydrogen bonding between the amide groups was investigated by Fourier transform infrared (FT-IR) spectroscopy, which is an important tool for studying the different non-covalent interactions involved in gelation. As shown in Fig. 6a and b, compared to the non-hydrogen-bonding transmittance band of 1a and 1b in chloroform solution at 3420–3390 cm−1 for vN–H, a remarkable shift of the urea N–H stretching vibration to a lower wavenumber around 3250 cm−1 was observed in xerogels 1a and 1b.48,49 Meanwhile, the absorption bands of the asymmetric (vasym) and symmetric (vsym) CH2 stretching vibrations of xerogels of 1a and 1b were observed at 2922 and 2854 cm−1, which appear at a lower frequency, indicating a strong interaction between the alkyl chains (van der Waals interaction).38 Additionally, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (vCO) and N–H bending (δN–H) bands for the xerogels respectively shifted to lower wavenumber at 1690 cm−1 and higher wavenumber at 1560 cm−1 which further confirmed the existence of hydrogen-bonding in the gel state.36,37
O stretching (vCO) and N–H bending (δN–H) bands for the xerogels respectively shifted to lower wavenumber at 1690 cm−1 and higher wavenumber at 1560 cm−1 which further confirmed the existence of hydrogen-bonding in the gel state.36,37
 | ||
| Fig. 6 The FT-IR spectra 1a (a) and 1b (b) in solid, xerogel (obtained from toluene) and chloroform solution; and XRD pattern of 1a (c) and 1b (d) in solid and xerogel (obtained from toluene). | ||
To probe the roles of the terpyridyl platinum(II) units as well as van der Waals interaction between alkyl chains in the gelation process, the X-rays diffractions (XRD) of the xerogels 1a and 1b obtained from toluene was estimated (Fig. 6c and d). The broad reflection peaks at ∼4.0 Å for 1a and ∼4.2 Å for 1b are related to the π–π stacking distance between the part of terpyridyl platinum(II) complexes. Moreover, the acute peaks at ∼3.08 Å for 1a and ∼3.04 Å for 1b are assigned to the metal–metal (Pt⋯Pt) and π–π stacking interactions of the terpyridyl platinum(II) complexes.9,50,51 On the basis of FT-IR and XRD studies, it is clear that the cooperative interactions of the metal–metal (Pt⋯Pt) and π–π stacking interactions of the terpyridyl platinum(II) complexes, the hydrogen bonding and the van de Waals forces among the amide groups and long alkyl chains can self-assemble molecules of 1a and 1b into long fibers in organic solvents and entangle further to form gels. Meanwhile, the temperature-dependent 1H NMR spectra of 1a and 1b are also carried out as shown in Fig. S2.† With temperature increasing to 80 °C by 10 °C steps, all of the signals in the 1H NMR spectra became sharp and strong, along with the notable splitting, which further indicate the existence of non-covalent interactions between molecules.
4 Conclusions
In summary, we have designed and synthesized new type of binuclear alkynylplatinum(II) terpyridyl complexes bridged by flexible chains (–O(CH2)nO–, n = 4 or 6). Benefiting from the metal–metal (Pt⋯Pt) and π–π stacking interactions of platinum(II) complexes, hydrogen bonding interactions of amide groups, and van der Waals forces of the long alkyl chains, 1a and 1b can form stable metallogels through the entanglement of fibrous aggregates in several organic solvents. Moreover, the extension of the flexible bridges has obvious effect to the formation of the metallogels. The longer of the chain length, the more solvents to form metallogels. The experimental study presented herein could have important potentials for the fabrication of metallogels based on platinum(II) complexes with the aid of the cooperative multiple intermolecular interactions.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21402108), Shandong Natural Science Foundation (ZR2014BQ036), and the China Postdoctoral Science Foundation (2016M590630).Notes and references
- A. Ajayaghosh and S. George, J. Am. Chem. Soc., 2001, 123, 5148–5149 CrossRef CAS PubMed.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS PubMed.
- N. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- S. Banerjee, R. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- X. Chen, Z. Huang, S.-Y. Chen, K. Li, X.-Q. Yu and L. Pu, J. Am. Chem. Soc., 2010, 132, 7297–7299 CrossRef CAS PubMed.
- W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS PubMed.
- S. Bhowmik, B. N. Ghosh, V. Marjomäki and K. Rissanen, J. Am. Chem. Soc., 2014, 136, 5543–5546 CrossRef CAS PubMed.
- L. Sambri, F. Cucinotta, G. D. Paoli, S. Stagni and L. D. Cola, New J. Chem., 2010, 34, 2093–2096 RSC.
- J. Zhang and C.-Y. Su, Coord. Chem. Rev., 2013, 257, 1373–1408 CrossRef CAS.
- J. Wang, Y. Chen, Y. C. Law, M. Li, M. X. Zhu, W. Lu, S. S. Y. Chui, N. Zhu and C. M. Che, Chem.–Asian J., 2011, 6, 3011–3019 CrossRef CAS PubMed.
- H. X. Zhang, M. Kato, Y. Sasaki, T. Ohba, H. Ito, A. Kobayashi, H. C. Chang and K. Uosaki, Dalton Trans., 2012, 41, 11497–11506 RSC.
- A. Y. Y. Tam, K. M. C. Wong, N. Zhu, G. Wang and V. W. W. Yam, Langmuir, 2009, 25, 8685–8695 CrossRef CAS PubMed.
- C. Po, Z. Ke, A. Y.-Y. Tam, H.-F. Chow and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 15735–15744 CrossRef CAS PubMed.
- A. Y. Y. Tam, K. M. C. Wong and V. W. W. Yam, Chem.–Eur. J., 2009, 15, 4775–4778 CrossRef CAS PubMed.
- V. H. Houlding and V. M. Miskowski, Coord. Chem. Rev., 1991, 111, 145–152 CrossRef CAS.
- D. R. McMillin and J. J. Moore, Coord. Chem. Rev., 2002, 229, 113–121 CrossRef CAS.
- S.-W. Lai and C.-M. Che, Top. Curr. Chem., 2004, 241, 27–63 CrossRef CAS.
- I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834–1895 CrossRef CAS PubMed.
- D. Zhang, L. Z. Wu, L. Zhou, X. Han, Q. Z. Yang, L. P. Zhang and C. H. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS PubMed.
- X. Han, L. Z. Wu, G. Si, J. Pan, Q. Z. Yang, L. P. Zhang and C. H. Tung, Chem.–Eur. J., 2007, 13, 1231–1239 CrossRef CAS PubMed.
- Q. Z. Yang, L. Z. Wu, H. Zhang, B. Chen, Z. X. Wu, L. P. Zhang and C. H. Tung, Inorg. Chem., 2004, 43, 5195–5197 CrossRef CAS PubMed.
- J. Ding, K. Feng, C.-H. Tung and L.-Z. Wu, J. Phys. Chem. C, 2010, 115, 833–839 Search PubMed.
- X. Y. Liu, X. Han, L. P. Zhang, C. H. Tung and L. Z. Wu, Phys. Chem. Chem. Phys., 2010, 12, 13026–13033 RSC.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS PubMed.
- W. Lu, B. X. Mi, M. C. W. Chan, Z. Hui, C. M. Che, N. Zhu and S. T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS PubMed.
- J. Thaddeus, Q. M. Wang, Y. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef PubMed.
- K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed.
- W. Lu, V. Roy and C. M. Che, Chem. Commun., 2006, 3972–3974 RSC.
- Y. Chen, K. Li, W. Lu, S. S. Y. Chui, C. W. Ma and C. M. Che, Angew. Chem., Int. Ed., 2009, 48, 9909–9913 CrossRef CAS PubMed.
- C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136–12143 CrossRef CAS PubMed.
- L.-B. Xing, S. Yu, X.-J. Wang, G.-X. Wang, B. Chen, L.-P. Zhang, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2012, 48, 10886–10888 RSC.
- N. Liu, B. Wang, W. Liu and W. Bu, Chem. Commun., 2011, 47, 9336–9338 RSC.
- A. Y.-Y. Tam, K. M.-C. Wong, G. Wang and V. W.-W. Yam, Chem. Commun., 2007, 2028–2030 RSC.
- F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J.-P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659–2662 CrossRef CAS PubMed.
- W. Lu, Y. C. Law, J. Han, S. S. Y. Chui, D. L. Ma, N. Zhu and C. M. Che, Chem.–Asian J., 2008, 3, 59–69 CrossRef CAS PubMed.
- A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253–6260 CrossRef CAS PubMed.
- C. A. Strassert, C.-H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS PubMed.
- L. De Cola, N. K. Allampally and C. Strassert, Dalton Trans., 2012, 41, 13132–13137 RSC.
- T. Tu, W. Fang, X. Bao, X. Li and K. H. Dötz, Angew. Chem., Int. Ed., 2011, 50, 6601–6605 CrossRef CAS PubMed.
- Y. Mao, K. Liu, L. Meng, L. Chen, L. Chen and T. Yi, Soft Matter, 2014, 10, 7615–7622 RSC.
- E. C. Constable and M. D. Ward, J. Chem. Soc., Dalton Trans., 1990, 1405–1409 RSC.
- X.-J. Wang, L.-B. Xing, W.-N. Cao, X.-B. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Langmuir, 2011, 27, 774–781 CrossRef PubMed.
- U. S. Schubert, C. Eschbaumer, O. Hien and P. R. Andres, Tetrahedron Lett., 2001, 42, 4705–4707 CrossRef CAS.
- J. Puigmartí-Luis, A. Minoia, P. del Pino, G. Ujaque, C. Rovira, A. Ledós, R. Lazzaroni and D. Amabilino, Chem.–Eur. J., 2006, 12, 9161–9175 CrossRef PubMed.
- J. Puigmartí-Luis, Á. Pérez del Pino, E. Laukhina, J. Esquena, V. Laukhin, C. Rovira, J. Vidal-Gancedo, A. G. Kanaras, R. J. Nichols, M. Brust and D. B. Amabilino, Angew. Chem., Int. Ed., 2008, 47, 1861–1865 CrossRef PubMed.
- B. Mohr, G. Wegner and K. Ohta, J. Chem. Soc., Chem. Commun., 1995, 995–996 RSC.
- J. Wang, Y. Chen, Y. C. Law, M. Li, M. X. Zhu, W. Lu, S. S. Y. Chui, N. Zhu and C. M. Che, Chem.–Asian J., 2011, 6, 3011–3019 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00208d |
| This journal is © The Royal Society of Chemistry 2017 |