
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon supported Rh nanoparticles for the production of hydrogen and chemicals by the electroreforming of biomass-derived alcohols†
Maria Vincenza Pagliaroab,
Marco Bellinia,
Manuela Bevilacquaa,
Jonathan Filippia,
Maria Gelsomina Follieroab,
Andrea Marchionni a,
Hamish Andrew Miller*a,
Werner Oberhausera,
Stefano Caporalicd,
Massimo Innocentiae and
Francesco Vizza*a
a,
Hamish Andrew Miller*a,
Werner Oberhausera,
Stefano Caporalicd,
Massimo Innocentiae and
Francesco Vizza*a
aCNR-ICCOM, via Madonna del Piano 10, 50019, Sesto Fiorentino (FI), Italy. E-mail: francesco.vizza@iccom.cnr.it
bUniversità di Siena, Dipartimento di Biotecnologie, Chimica e Farmacia, via Aldo Moro 2, Siena 53100, Italy
cConsorzio INSTM, Via Giusti 9, 50121 Firenze, Italy
dCNR-ISC, via Madonna del Piano 10, 50019, Sesto Fiorentino (FI), Italy
eDipartimento di Chimica, Università di Firenze, via della Lastruccia 3, 50019 Sesto Fiorentino (FI), Italy
First published on 2nd March 2017
Abstract
Electroreforming is a low energy cost technology that combines the production of valuable chemicals from biomass-derived alcohols with the evolution of clean hydrogen at low temperature and atmospheric pressure. The selectivity for the desired chemicals is governed by the nature of the anode catalyst. Here we report the synthesis and characterization of a carbon supported nanostructured Rh electrocatalyst. The Rh nanoparticles are shown to be highly dispersed (2.2 nm) and a complete electrochemical study is reported. This Rh/C catalyst exhibits high activity for alcohol electrooxidation (e.g. 5700 A gRh for EG at 80 °C) and when employed with an anion exchange membrane and Pt/C cathode in an electroreformer produces high volumes of hydrogen at low electrical energy input (e.g. 500 mA cm−2 at 0.7 Vcell and Ecost = 9.6 kW h kgH2−1). A complete analysis of the alcohol oxidation products from several renewable alcohols (ethanol, ethylene glycol, glycerol and 1,2-propandiol) shows a selectivity in the formation of valuable chemicals such as lactate and glycolate.
Introduction
Water electrolysis is the most popular alternative to the production of hydrogen from fossil fuels as it is the only route that permits the use of renewable (e.g., photovoltaic, wind, biomass, geothermal) energy sources combined with the production of 99.999% pure hydrogen.1–3 Currently, only a small proportion of the world's hydrogen production (circa 4%) comes from electrolytic water splitting.1 In fact, although water electrolysis is a well-known and consolidated process it does not have a significant commercial impact owing to its high-energy consumption, which, ultimately, makes it economically unattractive. The U.S. Department of Energy (DOE) has highlighted this drawback. Indeed, in 2011, the DOE has set a target that the electrical energy input to an electrolyzer stack should drop from 45 to 43 kW h kg−1 H2 by 2020.4The three most common low temperature water electrolysis technologies may be categorized as follows;5
(i) Alkaline electrolysis with a liquid alkaline electrolyte (typically aqueous KOH).
(ii) Zero gap or advanced alkaline configuration.
(iii) Acidic PEM electrolysis with a proton-conducting polymer electrolyte membrane.
In alkaline electrolyte, water electrolysis proceeds as follows:
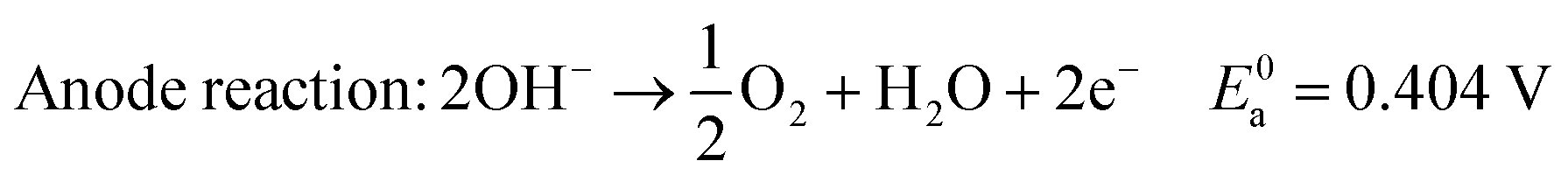 | (1) |
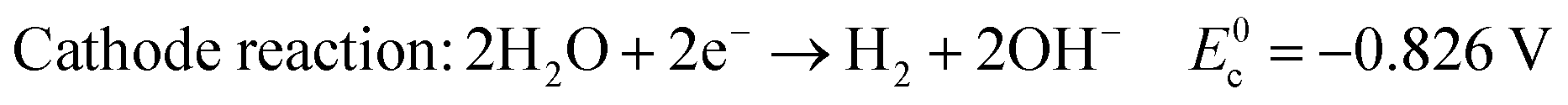 | (2) |
In acid electrolyte:
 | (3) |
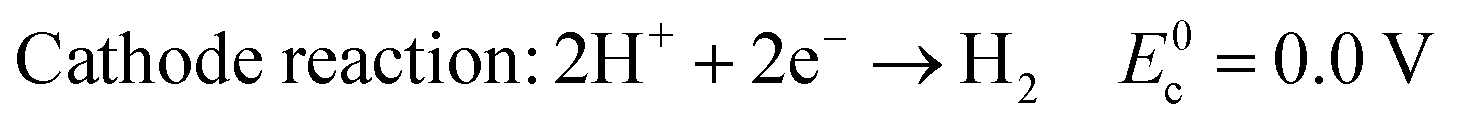 | (4) |
The standard reaction potential for water splitting is 1.23 V, meaning that the process is thermodynamically a non-spontaneous reaction. In practice, to get electrolysis current densities in the range of 1–2 A cm−2 the electrolysis cell potential usually ranges between 1.6 and 2 V.6 Using 1.8 V as a reasonable value, we can calculate that 68.3% of the energy input is consumed by overcoming thermodynamics, while kinetic factors account for only 31.7%. Replacing anodic oxygen evolution with the oxidation of much more readily oxidizable species leads to a significant reduction of the potential required to produce hydrogen. Researchers have demonstrated that this strategy works using so-called sacrificial compounds such as ammonia,7,8 methanol,9 ethanol,10–13 glycerol11,12,14,15 and urea.16 This electrolysis technology combining the oxidation of such substrates with the generation of hydrogen at the cathode are often indicated as “Electrochemical Reforming” or “electroreforming”. Electrolysis occurs at cell potentials lower than 1 V, leading to electrical power savings as compared to conventional electrolytic water splitting. Renewable biomass derived alcohols are attractive substrates for application in electrochemical reforming due to their low toxicity and their potential as part of carbon neutral energy transformation processes.17,18 Included in this group are ethylene glycol19,20 (EG) and glycerol21,22 (G). Ethylene glycol has a volumetric energy density of 5.9 kW h L−1 and can be produced by the heterogeneous hydrogenation of cellulose derivatives.23,24 Glycerol has a volumetric energy density of 6.3 kW h L−1, and is a by-product of biodiesel production and, as such, is inexpensive (0.3 US$ kg−1) and readily available (2.4 million tonnes produced per year).25
Eqn (5)–(7) describe the anode, cathode and overall electrochemical reforming reactions and standard electrode potentials (RHE) respectively for ethanol under alkaline conditions. With ethanol as fuel, the standard reaction potential for hydrogen production is 0.106 V. Therefore, hydrogen evolution is much more favorable when compared to water electrolysis where the standard potential is 1.23 V (RHE).
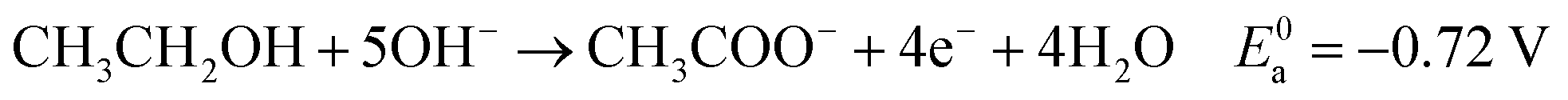 | (5) |
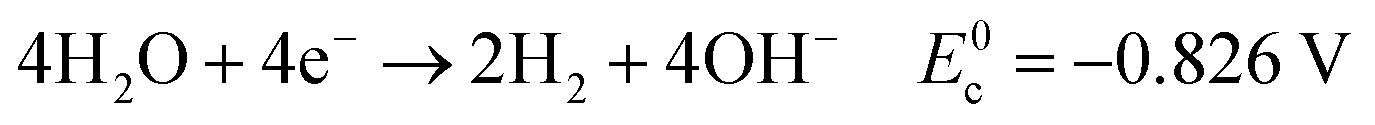 | (6) |
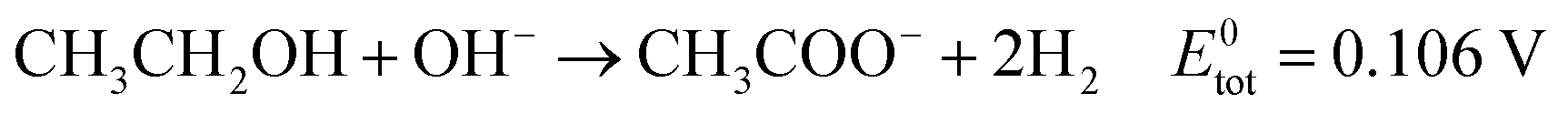 | (7) |
Other poly-alcohols can be oxidized provide not only hydrogen but also valuable chemicals from the formation of partially oxidized intermediates.26–29 For example, 1,2-propandiol can be employed in electroreforming to produce lactate, an industrially relevant chemical.30–33
The efficiency of an electrolyzer is directly related to the electrode materials, in particular to the catalysts deposited onto the surface of the electrodes. The function of these catalysts is to reduce the activation energy for both the anode and cathode reactions. Therefore, a crucial role in improving the effective energetic efficiency of an electrolyzer is played by the electrode materials, because they determine both the energy consumption (at a given reaction rate) and the maximum reaction rate in the cell.
In this paper, we report an electroreformer that couples the partial oxidation of renewable alcohols with hydrogen evolution for the simultaneous and sustainable production of carboxylic compounds and high-purity hydrogen. The electroreformer is assembled with an anode containing nanostructured Rh/C electrocatalyst. This catalyst is composed of highly dispersed Rh nanoparticles supported on carbon, which shows high activity for alcohol electrooxidation. The employment of this catalyst enables high hydrogen production levels at very low energy cost. The paper also reports a net energy analysis of hydrogen production using bio-ethanol (as example) in order to clearly demonstrate the potential of electroreforming technology.
Experimental
Materials
Carbon black (Vulcan XC-72) was purchased from Cabot Corp., USA. The alkaline anion exchange membrane used was the A201 obtained from Tokuyama Corp. (Japan). All metal salts and reagents were purchased from Aldrich and used without further purification. All the solutions were freshly prepared with doubly distilled deionized water.Synthesis of Rh/C anode catalyst
3.8 g of carbon powder (Vulcan XC-72, Cabot corp., USA) was suspended in 640 mL of ethylene glycol and sonicated for 80 min in a 2 L round-bottomed flask. Then an aqueous solution of H2O (160 mL) containing 740 mg of dissolved RhCl3·6H2O (Aldrich), was added dropwise to the suspension under stirring. After this an alkaline solution of NaOH (15.8 g) in H2O (80 mL) and ethylene glycol (200 mL) was introduced to the reactor which was then heated at 120 °C for 3 h under a N2 atmosphere, under vigorous stirring. The mixture was then cooled to room temperature and the solid product was filtered off and washed thoroughly with H2O to neutral pH. The solid product thus obtained was dried under vacuum at 40 °C. Yield 3.67 g. ICP-AES analysis: Rh content = 6 wt% (ICP-AES).Physical characterization
Transmission electron microscopy (TEM) was performed using a Philips CM12 microscope at an accelerating voltage of 100 kV. The microscope was equipped with an EDAX energy dispersive microanalysis system.The BET surface area was determined using an ASAP 2020C Instrument (Micromeritics Corp.).
X-ray photoelectron Spectroscopy (XPS) measurements were performed in a system equipped with a VSW HAC 5000 hemispherical electron energy analyzer and a non-monochromated Mg Kα X-ray source (1253.6 eV). Photoelectron spectra were acquired in the constant-pass-energy mode at Epas = 44 eV, and the overall energy resolution was 1.2 eV measured as a full-width at half maximum (FWHM) of the Ag 3d5/2 line of a pure silver reference. The pressure during the experiment was kept below 2 × 10−9 Torr. No neutralizer was utilized and the spectra energy scale was corrected using the Au 4f7/2 peak. The recorded spectra were fitted using XPS Peak 4.1 software. Gauss–Lorentz curves were used to fit the data after subtraction of a Shirley-type background.
Electrochemical characterization
The electrochemical measurements were performed using a Parstat 2273 potentiostat-galvanostat (Princeton Applied Research) equipped with a Model 616 rotating disk electrode RDE (PAR-Ametek). A 5 mm (A = 0.1963 cm2) Teflon-potted glassy-carbon disk electrode tip (PINE™) was used as substrate for the deposition of the catalyst ink. Before the deposition, the glassy-carbon surface was polished with different CT diamond suspension with a progressively smaller particle size (1 μm, 0.25 μm, 0.1 μm) and finally washed with distilled water. The catalyst ink was prepared by mixing the Rh/C catalyst and distilled water in order to obtain 1 wt% suspension. The ink was sonicated for 1 h in a FALC sonicating bath to obtain a uniform suspension. The catalyst film was prepared by dispersing 8 μL of the catalyst ink on the glassy-carbon electrode. The exact amount of dry catalyst deposited was determined using an analytical balance. The final Rh loading on the electrode was between 5 and 6 μg (25–30 μg cm−2). Each electrode was dried for 30 min before the addition of 2.5 μL of a 0.4 wt% Tokuyama™ OH-type anion exchange resin alcohol solution (AS4). After drying, the catalyst-ionomer coated disk was then mounted on the rotating disk electrode shaft and immersed into the electrolyte solution. The counter electrode was a platinum gauze enclosed in a glass tube with porous bottom.The reference electrode used was commercial Ag/AgCl/KClsat (Princeton Applied Research). All cyclic voltammetry potentials are reported against reversible hydrogen electrode (RHE) calculated taking into account the potential-temperature dependence, as described in the ESI.† The electrochemical experiments were conducted at three temperatures (25, 60 and 80 °C). The CV experiments were undertaken in 2 M aqueous KOH (Sigma-Aldrich, 99.8%) while the alcohol electrooxidation studies were performed in aqueous solutions of 2 M KOH and 2 M alcohol. All the solutions were prepared with Millipore water (18 MΩ cm−1) provided by a Milli-Q Labo apparatus (Nihon Millipore Ltd.). Solutions were deaerated by bubbling high-purity N2 for 30 min before each measurement.
CO stripping voltammograms were performed in aqueous 2 M KOH. As first step, the KOH solution was saturated with bubbling CO for 20 min, after which a fixed voltage of 70 mV (RHE) was applied for 15 min to the working electrode to allow the complete adsorption of a monolayer of CO onto the surface of the catalyst. Then the excess CO in the electrolyte was purged out with bubbling N2 for 20 min. The amount of COads was calculated integrating the respective stripping peak and the corresponding charge was used to extrapolate and calculate the ECSA of the Rh/C catalyst.
Electroreformer cell testing
The MEAs (membrane electrode assemblies) prepared for the electroreformer cell consist of the Rh/C catalyst as anode, a commercial Tokuyama A-201 anion-exchange membrane, and a commercial 40 wt% Pt/C catalyst (Aldrich) as cathode.A dense anode ink was prepared by mixing the desired quantity of powdered catalyst Rh/C with an aqueous dispersion of PTFE (60 wt% PTFE dispersion in H2O obtained from Aldrich) and distilled water. The amount of PTFE added was 5 wt% relative to the dry catalyst weight. The catalyst paste was applied uniformly to a porous nickel foam support (110 PPI HZTYKJ from Heze Tianyu Technology Development Co., Ltd. dimensions 5 cm2). The cathodic ink was prepared in a 5 mL high density polyethylene vial, mixing 200 mg of the commercial Pt (40 wt%)/C in 450 mg of distilled water, 790 mg of 1-propanol and 1.56 g of the ionomer Nafion® (5 wt% in 2-propanol). The mixture was suspended with three pulses of ultrasound, 20 W power at the frequency of 20 kHz (Bandelin Sonor pulse UW 2200 SERIES). Finally this paste was spread onto a carbon cloth W1S1005 (CeTech Co. Ltd.) gas diffusion layer, with a Meyer rod (no. 150) obtaining a 0.4 mg cm−2 Pt loading.
The electroreformer test cell fixture was purchased from Scribner-Associates (USA). The MEAs were assembled by mechanically pressing together the anode, cathode and membrane described above within the cell hardware. The cell temperature was regulated at 60 °C using a Scribner 805e fuel cell station. The aqueous fuel solution (30 cc of 2 M KOH and 2 M alcohol) was delivered to the anode at 1 mL min−1. Voltage scans and galvanostatic experiments were carried out using an ARBIN BT-2000 5A 4-channel instrument. Polarization experiments were recorded by applying a linear voltage ramp with a 10 mV s−1 scan rate between 0.2 to 0.8 V.
Alcohol electrooxidation selectivity study
Chronopotentiometry experiments were performed by applying a constant electrolysis current of 125 mA to the cell at 60 °C until the voltage reached the cut off value of 0.65 V. The hydrogen produced at the cathode was monitored by a Bronkhorst mass-specific hydrogen-calibrated flowmeter. The fuel solutions after the constant current experiments were quantitatively and qualitatively analyzed by 13C{1H} NMR spectroscopy and HPLC. A UFLC Shimadzu Chromatograph equipped with refraction index detector (RID) was used; the column is a GRACE-Alltech OA-1000 Organic Acids (300 mm × 6.5 mm), thermostated at 35 °C. The eluent is 0.01 N H2SO4; and the eluent flow is 0.8 mL min−1. NMR spectra were acquired with a with a Bruker Advance DRX 400 spectrometer. Chemical shifts (δ) are reported in ppm relative to TMS (1H and 13C NMR spectra). Deuterated solvents (Sigma-Aldrich) used for NMR measurements were dried with activated molecular sieves; 1,4-dioxane was used as internal standard for product quantification.Results and discussion
Catalyst synthesis and characterization
The Rh/C catalyst was prepared by electro-less reduction in aqueous media of Vulcan XC-72-impregnated with RhCl3 using ethylene glycol at 120 °C for 3 h under inert atmosphere. The morphology and distribution of the supported metal particles was evaluated by transmission electron microscopy (TEM in Fig. 1). The particle size distribution of the Rh/C catalyst is also shown and has a mean diameter value of 2.2 nm.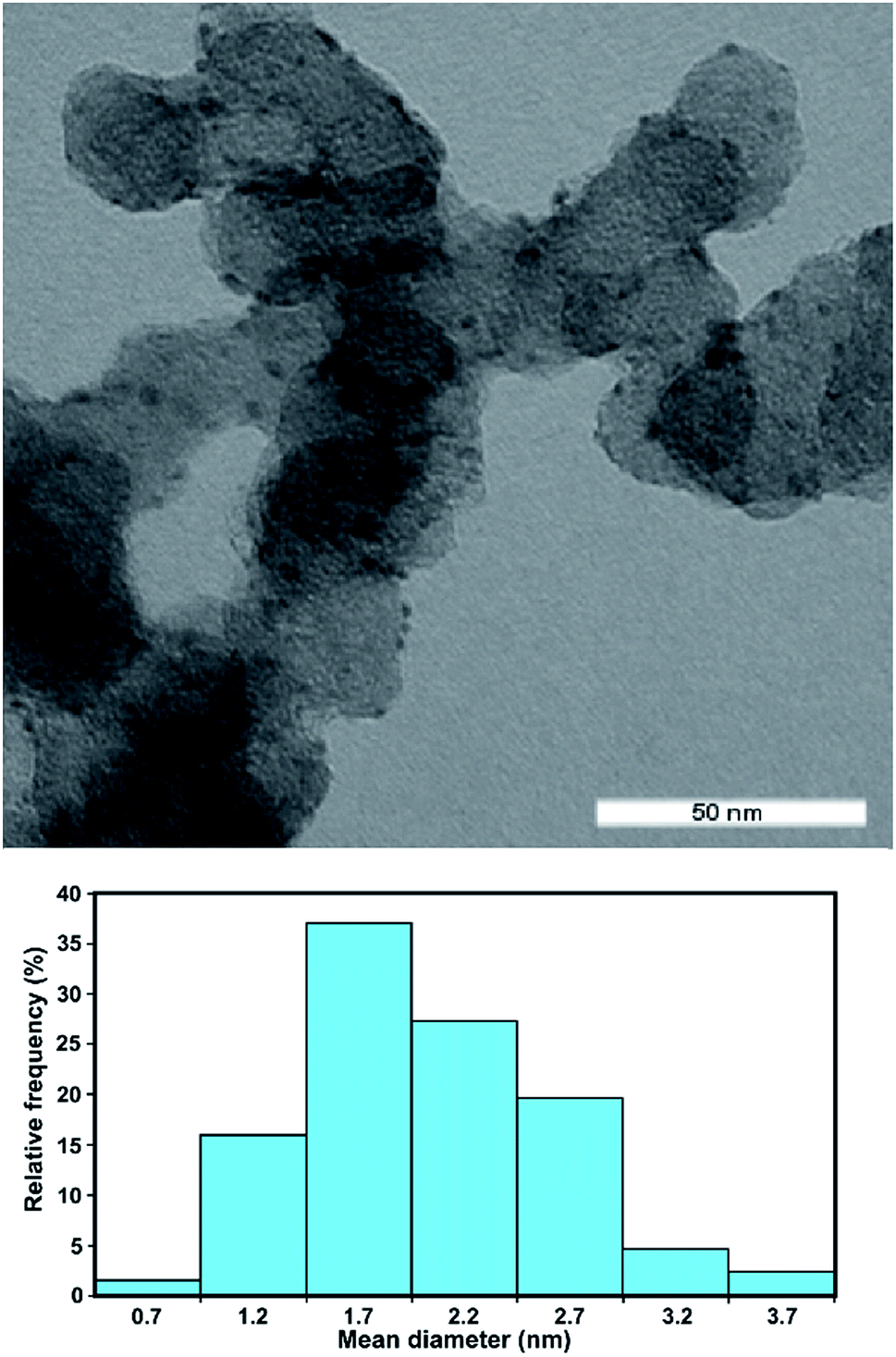 | ||
| Fig. 1 TEM image and histogram of particle distribution versus diameter for Rh/C. | ||
The crystalline structure of the catalyst was also investigated by X-ray powder diffraction (XRD). The XRD pattern (Fig. SI†) does not reveal the presence of a metallic Rh phase, consistent with the formation of small nanoparticles of Rh in agreement with the TEM analysis. The Brunauer–Emmett–Teller (BET) surface area was calculated as 195 m2 g−1.
The surface composition of the Rh/C sample was also studied using X-ray photoelectron spectroscopy (XPS). The fitting of the data obtained shows the presence of three peaks (from the binding energy values) attributable to Rh(0) 307.3 eV, Rh2O3 308.8 eV (ref. 34–36) and a satellite peak of Rh2O3 311.5 eV (Fig. 2). The latter species was recently assigned by Boronin and coworkers.37 This analysis shows that rhodium oxide is the major component of the surface of the nanoparticles with Rh metal accounting for only around 35%. Such a structure would lead to the XRD pattern that showed no visible trace for metallic Rh crystallites.
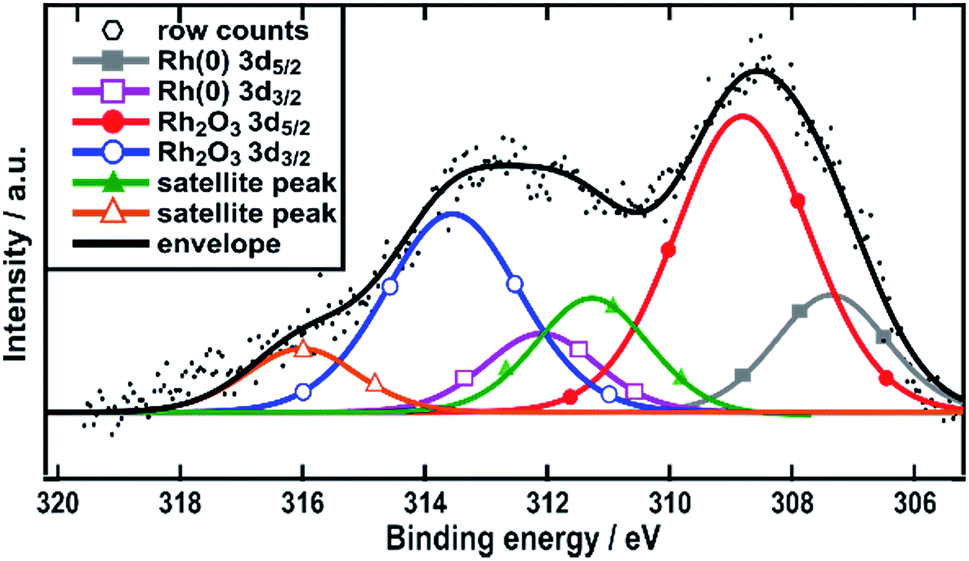 | ||
| Fig. 2 XPS spectra in the Rh 3d region. Spectra are referenced with respect to adventitious carbon at 284.8 eV. | ||
Electrochemical properties
The electrochemical activity of Rh/C for alcohol electrooxidation was investigated by linear sweep voltammetry in electrochemical cells maintained at three different temperatures (25, 60 and 80 °C). For all experiments, the Rh loading on the glassy carbon substrate was between 25 and 35 μgRh cm−2. We first studied the electrochemical behavior by cyclic voltammetry (CV) in 2 M KOH purged with N2 (Fig. 3).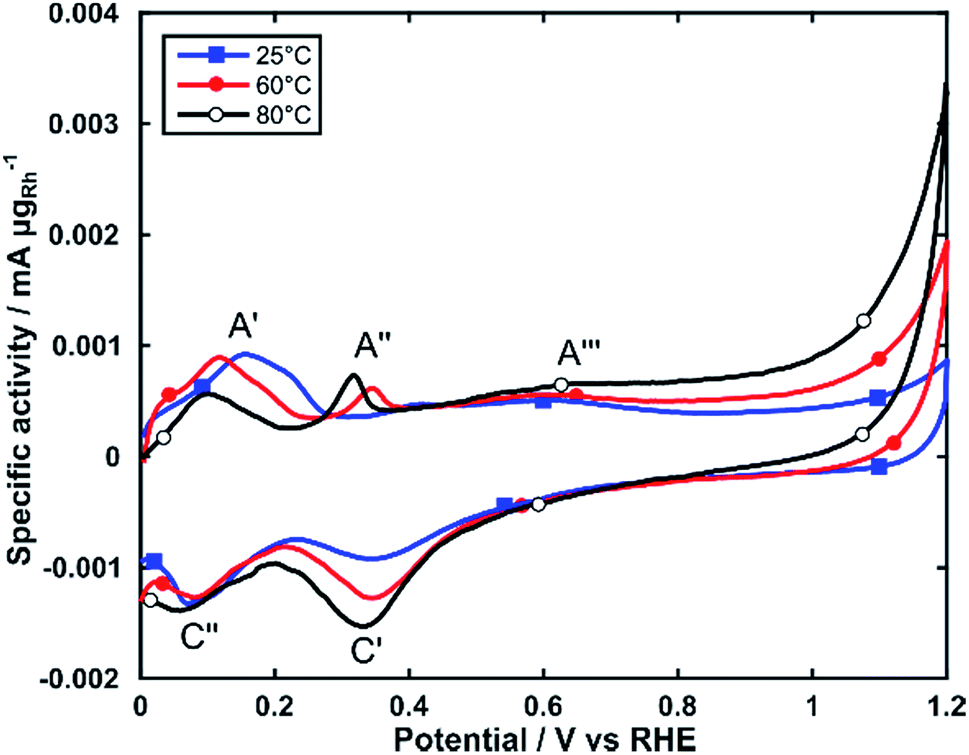 | ||
| Fig. 3 CVs of Rh/C in N2 saturated 2 M KOH. Scan rate: 10 mV s−1. | ||
As reported in the literature the CV exhibits a number of anodic and cathodic transitions that can be assigned to Rh centered transitions.38,39 On the positive potential scan (0 to 1.2 V), peak A′ represents the oxidative desorption of hydrogen; peak A′′ relates to the formation of adsorbed OH species on the Rh nanoparticle surface and peak A′′′ is the onset of the formation of Rh oxide species. On the cathodic return scan peak C′ corresponds to the electroreduction of the oxide layer, and peak C′′ to the electrosorption of H-adatoms.
CVs obtained at increasing temperatures (60 and 80 °C) show significant changes to the intensity and potential of these transitions. Peak A′ shifts by approximately 0.1 V towards more negative potentials, while decreasing in intensity. Peak A′′ increases in intensity and shifts about 75 mV towards more negative potentials. This behavior indicates an increased ability for the electrooxidation of alcohols, as it shows that RhOHads species form at lower potentials (A′′). The RhOHads species are those responsible for alcohol electrooxidation, analogously to the PdOHads species formed onto palladium electrocatalysts, albeit formed at lower potential for Rh.
The electrochemically active surface area (ECSA) of the Rh nanoparticles was determined by integrating the charge of the electrooxidation peak of an adsorbed monolayer of COads in CO stripping voltammograms (Fig. 4).40 The ECSA normalized by the mass of Rh was calculated as 75 m2 gRh−1.
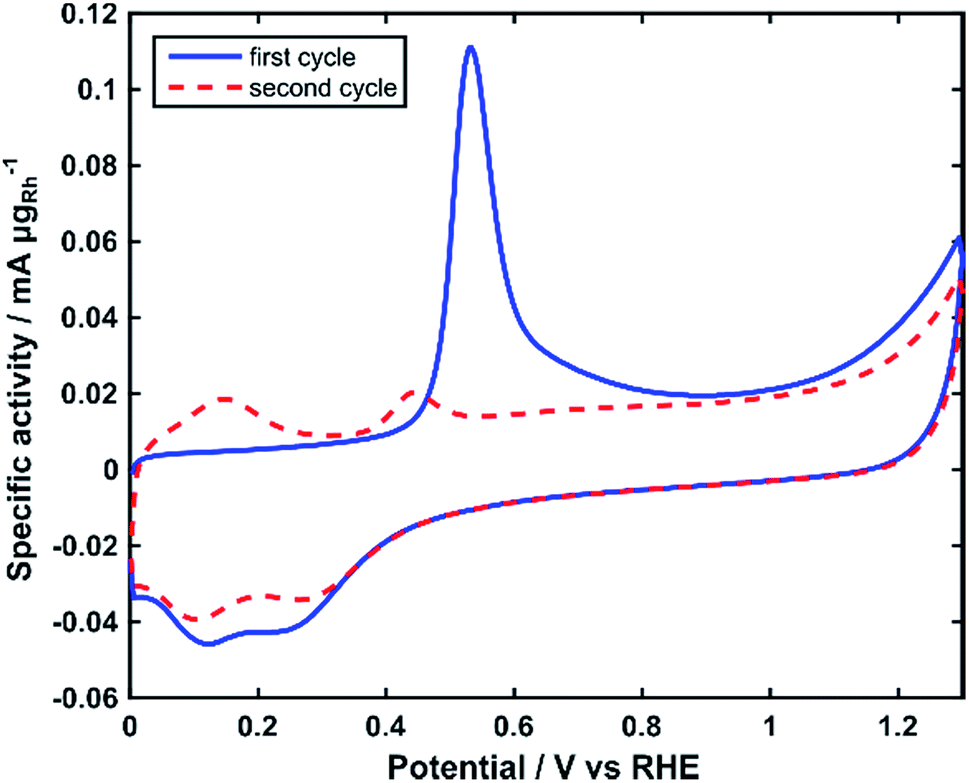 | ||
| Fig. 4 CO-stripping voltammograms of Rh/C in 2 M KOH. Scan rate: 10 mV s−1. | ||
The electrochemical activity of Rh/C towards the oxidation of alcohols in alkaline media was investigated by cyclic voltammetry at 25 °C, 60 °C and 80 °C, in degassed 2 M aqueous KOH and 2 M of either ethanol (EtOH), 1,2-propandiol (1,2-P), glycerol (G) or ethylene glycol (EG) (Fig. 5). Relevant electrochemical data is also listed in Table 1. In Fig. 5, we show the onset region (0.2–0.6 V RHE) of the forward scan for each alcohol at all three temperatures (25, 60 and 80 °C).
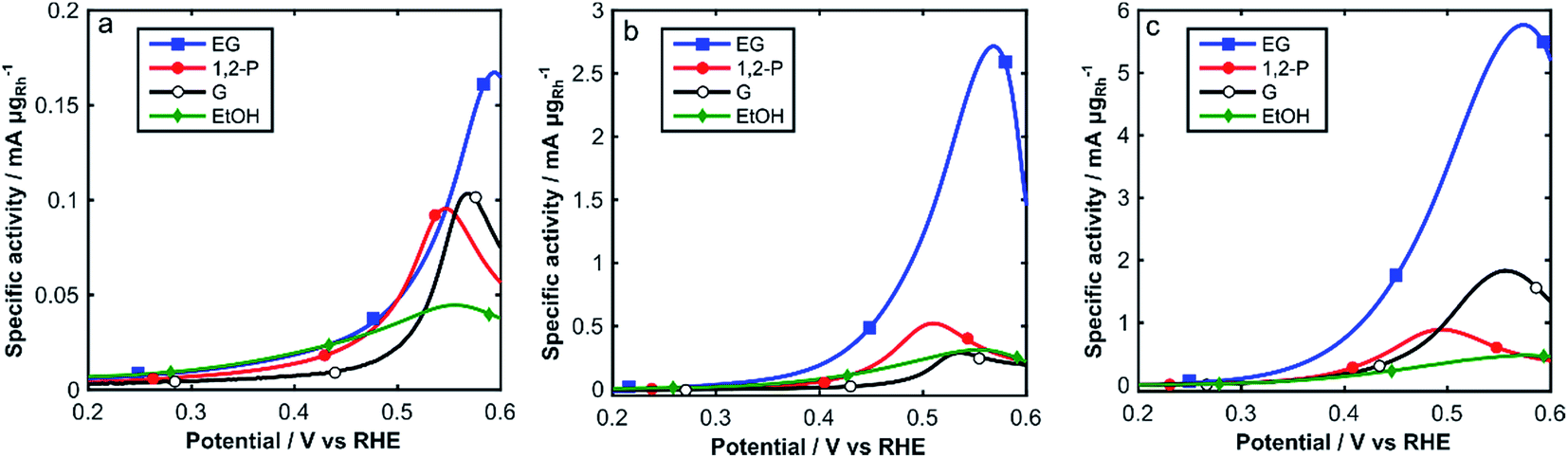 | ||
| Fig. 5 CVs of Rh/C in N2 sat. 2 M KOH and 2 M alcohol solutions: (a) 25 °C, (b) 60 °C and (c) 80 °C (0.2–0.6 V RHE). Scan rate: 10 mV s−1. | ||
| Fuel | T (°C) | Eonset (V vs. RHE) | Eforward peak (V vs. RHE) | Jpeaka (mA mgRh−1) | Jpeak/ECSAb (mA cm−2) |
|---|---|---|---|---|---|
| a Current normalized on total Rh metal loading.b Current normalized on Rh electrochemical surface area (ECSA) i.e. 75 m2 gRh−1. | |||||
| EtOH | 25 | 0.34 | 0.56 | 45 | 0.060 |
| 1,2-P | 25 | 0.35 | 0.55 | 95 | 0.127 |
| EG | 25 | 0.33 | 0.59 | 167 | 0.223 |
| G | 25 | 0.46 | 0.57 | 103 | 0.137 |
| EtOH | 60 | 0.32 | 0.56 | 320 | 0.427 |
| 1,2-P | 60 | 0.33 | 0.53 | 521 | 0.695 |
| EG | 60 | 0.32 | 0.57 | 2728 | 3.637 |
| G | 60 | 0.44 | 0.54 | 287 | 0.383 |
| EtOH | 80 | 0.30 | 0.59 | 500 | 0.667 |
| 1,2-P | 80 | 0.31 | 0.49 | 886 | 1.155 |
| EG | 80 | 0.28 | 0.57 | 5767 | 7.689 |
| G | 80 | 0.31 | 0.55 | 1839 | 2.452 |
From Fig. 5 and Table 1, we can observe low oxidation onset potentials, which generally decrease as the temperature increases from 25 °C to 80 °C, whereas the specific current densities dramatically increase with temperature for all alcohols, up to 35 times for EG. The intensity of the forward peaks in the CVs is determined by the progressive coverage of the Rh surface with oxide or partially oxidized poisoning species. In the complete CVs, (Fig. S2†) for ethanol a change of slope at 0.7 V is observed, caused by different adsorption types of ethanol on Rh.41 The enhanced electrocatalytic activity observed at higher temperatures (60–80 °C) can be ascribed to the accelerated kinetics of alcohols oxidation, which it is also favored by an anticipated formation of RhOHads species at lower potentials as observed in Fig. 3.
Electrochemical-reforming experiments
Nickel foam (5 cm2) coated with Rh/C and using PTFE as binder was used as anode electrode (1 mgRh cm−2) in the electroreformer (ER) cell equipped with an anion exchange membrane (Tokuyama A201) and a Pt/C on carbon cloth cathode (0.4 mgPt cm−2). The anode compartment of the cell was fed by recycling 30 mL of aqueous solutions of alcohol 2 M and 2 M KOH. At a cell temperature of 60 °C the performance was measured using potentiodynamic scans and galvanostatic experiments. For each alcohol, three batches of fuel were run consecutively in the same cell.Polarization curves were obtained in the potential range from 0.2 to 0.7 V at 10 mVs−1. Fig. 6a and Table 2 show that the electroreformer performs best with EtOH and EG reaching current densities of 492 mA cm−2 and 416 mA cm−2, respectively at a cell voltage of 0.7. With the longer chain alcohols, 1,2-P and G, the current density obtained at 0.7 V were 367 mA cm−2 and 113 mA cm−2 respectively.
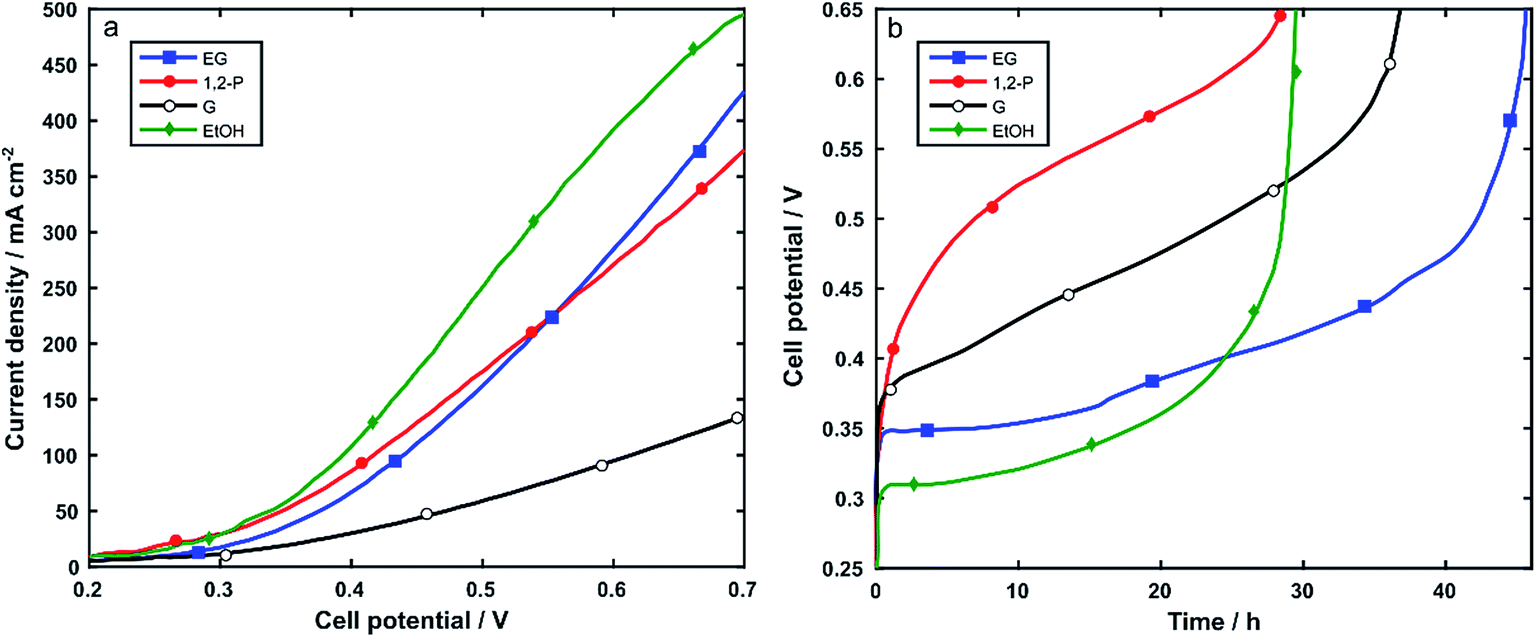 | ||
| Fig. 6 (a) Potentiodynamic cycles of the electroreformer fueled with: 2 M aqueous alcohol solutions in 2 M KOH. Linear potential ramp, 10 mV s−1. Cell temperature 60 °C; (b) galvanostatic curves at 125 mA, fueled with 30 cc of 2 M aqueous alcohol solutions in 2 M KOH. Cell temperature 60 °C. | ||
| Fuel | Jmax @ 700 mV (mA cm−2) | Conversion (%) [mmol] | H2 production (Nm3 m−2) | Energy consumption (kW h kg−1 H2) | Selectivity% |
|---|---|---|---|---|---|
| EtOH | 492 | 57 [34.2] | 3 | 9.6 | Acetate (100) |
| 1,2-P | 367 | 55 [33.0] | 2.8 | 14.4 | Lactate (98) |
| Carbonate (1) | |||||
| Acetate (1) | |||||
| EG | 416 | 89 [53.4] | 5.4 | 11.0 | Glycolate (76) |
| Carbonate (16) | |||||
| Oxalate (5) | |||||
| Formate (3) | |||||
| G | 113 | 72 [43.2] | 4.3 | 12.6 | Glycerate (46) |
| Tartronate (23) | |||||
| Carbonate (18) | |||||
| Formate (8) | |||||
| Glycolate (3) | |||||
| Oxalate (2) |
In all cases, the cell performance was stable over the three batch experiments indicating no loss in performance during electrocatalysis. In order to study both the stability and the electrocatalytic activity over time as well as the electrooxidation product selectivity, the electroreformer was subjected to galvanostatic experiments where 30 mL of solutions of 2 M alcohol and 2 M KOH were recycled at the anode electrode and the cell was set at a constant total current of 125 mA. Each batch was stopped when the cell potential reached the value of 0.65 V. The same set-up was run with three consecutive batches of fresh alcohol solutions. No decrease in activity was observed over the course of the three batches, indicating the stability of the catalyst over time. Fig. 6b and Table 2 summarize the data obtained at 60 °C.
The fuel exhausts were quantitatively and qualitatively analyzed by 13C{1H} NMR spectroscopy and HPLC. The results summarized in Table 2 show the fuel conversion and the distribution of the oxidation products identified and quantified by NMR and HPLC analysis.
With ethanol, the only product obtained is acetate with no evidence of C–C bond breaking and formation of carbonate. The cells fed with EG and 1,2-P yielded circa 76% glycolate and 98% of lactate, respectively. Glycerol oxidation leads to significant C–C bond scission, resulting in a mixture of products including glycerate, tartronate and also oxalate, glycolate, formate and carbonate, all from C–C bond breaking reactions. No trace of the secondary alcohol oxidation products of G such as dihydroxyacetone, hydroxypiruvate, or mesoxalate was observed. As expected, at the cell working potentials no oxygen evolution at the anode was detected.
The amount of hydrogen evolved during each experiment was calculated from the number of moles of electrons exchanged in the electroreformer (see ESI†) and measured experimentally with an hydrogen-calibrated flowmeter; the values were consistent indicating a 100% faradaic efficiency for the cathodic process. The energy consumption was then evaluated from the integration of the instantaneous charging power over the experimental duration time. This value, expressed in kW h, is reported with respect to the amount of hydrogen produced in kilograms (i.e. kW h kgH2−1). Table 2 shows that the amount of electrical energy required for H2 production for all alcohols is in the range of 10–14 kW h kgH2−1.
Our recent reports of alcohol electroreforming with Pd based anode catalysts showed electrical energy consumption for the same levels (i.e. same current density) of H2 production in the region of 18–24 kW h kg−1.30,31 Hence, the use of Rh/C as anode catalyst significantly reduces the energy cost of electroreforming. In particular, we can calculate that the use of ethanol (9.6 kW h kg−1 H2) results in an electrical energy saving of 35.4 kW h kg−1 H2 as compared with that reported by the DOE for PEM water electrolyzer stacks as of 2011 (45 kW h kg−1 H2).4 The reason of this drop in energy consumption is due to the lower operating potentials for alcohol electrooxidation caused by the cathodic shift of the onset of the formation of RhOHads species, that are responsible for the alcohol electrooxidation process itself.
One may now consider if from an overall energetic point of view the exploitation of alcohols such as ethanol for hydrogen production by electroreforming is actually worthy of consideration. According to the DOE the output energy density for bioethanol is 7.4 kW h kgEtOH−1. From our calculations reported in the SI, we conclude that the production of bioethanol with an Energy Return of the Energy Invested (EROEI) larger than 3.1 is required in order to obtain a net energy saving with respect to current water electrolysis technology (47 kW h kg−1 H2). The DOE target for electrolyzer stack electric energy input for 2020,4 43 kW h kg−1 H2, is instead met for a bio-ethanol EROEI larger than 3.6. Hence, hydrogen production by the electroreforming of bio-ethanol is energetically convenient depending on the production source of the alcohol e.g. using sugarcane that is commonly reported to have an EROEI of 8 and higher42 from cellulose with an EROEI potentially up to 35 depending on production methods.43
Conclusions
In this paper, examples of electrooxidation of biomass-derived alcohols such as EtOH, EG, G and 1,2-P have been studied in an electroreformer containing Rh nanoparticles supported on carbon as the anode electrocatalyst, equipped with an anion exchange membrane and a Pt/C on carbon cloth cathode. The oxidation of alcohols was first investigated in electrochemical half-cells at room temperature and at 60–80 °C in alkaline media. The results highlighted the excellent activity of Rh/C in terms of peak current densities (as high as 5700 A gRh−1 for EG at 80 °C) and low onset of potentials.When employed as anode catalyst in electroreformer cells high current densities were obtained at cell potential well below 1 V, resulting in significant energy saving when compared to traditional water electrolyzers. The amount of electrical energy required for H2 production for all alcohols is in the range of 10–14 kW h kgH2−1. These values are the lowest compared to those reported up to now in the literature. In particular, 9.6 kW h kgH2−1 from EtOH results in an electrical energy saving of 35.4 kW h kgH2−1 as compared with that reported by the DOE for PEM electrolyzer.
In terms of oxidation product selectivity, this Rh/C catalyst was selective for EtOH electrooxidation to acetate (100%) and 1,2-P oxidation to lactate (98%). From EG and G a more complex mixture of partial oxidation products was obtained. In summary, the electroreformer reported in this paper provides not only hydrogen production but also valuable chemicals from the large variety of products that can be obtained by the electrooxidation of renewable alcohols. The remarkable electrocatalytic activity of Rh/C employed as anode catalyst can be attributed to both the high dispersion of the metal nanoparticles as well as the intrinsic properties of nanostructured metallic Rh that shows low onset potentials for alcohol electrooxidation in half cells and low cell working potential during electroreforming.
Acknowledgements
We gratefully acknowledge the Ente Cassa di Risparmio di Firenze for the project EnergyLab and Hydrolab 2.0.References
- J. Mergel, M. Carmo and D. Fritz, Transition Renewable Energy Syst., 2013, 425–450, DOI:10.1002/9783527673872.
- A. Ursua, L. M. Gandia and P. Sanchis, Proc. IEEE, 2012, 100, 410–426 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- Fuel Cell Technologies Office, Fuel cells, Fuel Cell Technologies Office Multi-year Research, Development and Demonstration Plan of the US Department of Energy, 2011 Search PubMed.
- S. Marini, P. Salvi, P. Nelli, R. Pesenti, M. Villa, M. Berrettoni, G. Zangari and Y. Kiros, Electrochim. Acta, 2012, 82, 384–391 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Merge and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- M. Schalenbach, M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 14921–14933 CrossRef CAS.
- F. Vitse, M. Cooper and G. G. Botte, J. Power Sources, 2005, 142, 18–26 CrossRef CAS.
- T. Take, K. Tsurutani and M. Umeda, J. Power Sources, 2007, 164, 9–16 CrossRef CAS.
- C. Lamy, T. Jaubert, S. Baranton and C. Coutanceau, J. Power Sources, 2014, 245, 927–936 CrossRef CAS.
- V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Lavacchi, A. Marchionni, F. Vizza and P. K. Shen, ChemSusChem, 2010, 3, 851–855 CrossRef CAS PubMed.
- A. Caravaca, F. M. Sapountzi, A. de Lucas-Consuegra, C. Molina-Mora, F. Dorado and J. L. Valverde, Int. J. Hydrogen Energy, 2012, 37, 9504–9513 CrossRef CAS.
- A. Caravaca, A. de Lucas-Consuegra, A. B. Calcerrada, J. Lobato, J. L. Valverde and F. Dorado, Appl. Catal., B, 2013, 134, 302–309 CrossRef.
- A. T. Marshall and R. G. Haverkamp, Int. J. Hydrogen Energy, 2008, 33, 4649–4654 CrossRef CAS.
- S. Kongjao, S. Damronglerd and M. Hunsom, J. Appl. Electrochem., 2011, 41, 215–222 CrossRef CAS.
- W. Yan, D. Wang and G. G. Botte, Appl. Catal., B, 2012, 127, 221–226 CrossRef CAS.
- A. Brouzgou, A. Podias and P. Tsiakaras, J. Appl. Electrochem., 2013, 43, 119–136 CrossRef CAS.
- E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3, 1499–1528 CrossRef CAS.
- V. Livshits, A. Philosoph and E. Peled, J. Power Sources, 2008, 178, 687–691 CrossRef CAS.
- D. Kaplan, L. Burstein, Y. Rosenberg and E. Peled, J. Power Sources, 2011, 196, 8286–8292 CrossRef CAS.
- L. Xin, Z. Y. Zhang, Z. C. Wang and W. Z. Li, ChemCatChem, 2012, 4, 1105–1114 CrossRef CAS.
- A. Marchionni, M. Bevilacqua, C. Bianchini, Y. X. Chen, J. Filippi, P. Fornasiero, A. Lavacchi, H. Miller, L. Q. Wang and F. Vizza, ChemSusChem, 2013, 6, 518–528 CrossRef CAS PubMed.
- N. Ji, T. Zhang, M. Y. Zheng, A. Q. Wang, H. Wang, X. D. Wang and J. G. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS PubMed.
- M. Y. Zheng, A. Q. Wang, N. Ji, J. F. Pang, X. D. Wang and T. Zhang, ChemSusChem, 2010, 3, 63–66 CrossRef CAS PubMed.
- J. Van Gerpen, Fuel Process. Technol., 2005, 86, 1097–1107 CrossRef CAS.
- V. Bambagioni, M. Bevilacqua, C. Bianchini, J. Filippi, A. Lavacchi, A. Marchionni, F. Vizza and P. K. Shen, ChemSusChem, 2010, 3, 851–855 CrossRef CAS PubMed.
- J. de Paula, D. Nascimento and J. Linares, J. Appl. Electrochem., 2015, 45, 689–700 CrossRef CAS.
- S. Gonzalez-Cobos, S. Baranton and C. Coutanceau, ChemElectroChem, 2016, 3, 1694–1709 CrossRef.
- Z. Y. Zhang, L. Xin, J. Qi, D. J. Chadderdon, K. Sun, K. M. Warsko and W. Z. Li, Appl. Catal., B, 2014, 147, 871–878 CrossRef CAS.
- Y. X. Chen, A. Lavacchi, H. A. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 4036–4041 CAS.
- H. A. Miller, M. Bellini, F. Vizza, C. Hasenohrl and R. D. Tilley, Catal. Sci. Technol., 2016, 6, 6870–6878 CAS.
- D. J. Chadderdon, L. Xin, J. Qi, B. Brady, J. A. Miller, K. Sun, M. J. Janik and W. Z. Li, ACS Catal., 2015, 5, 6926–6936 CrossRef CAS.
- M. Bellini, M. Bevilacqua, J. Filippi, A. Lavacchi, A. Marchionni, H. A. Miller, W. Oberhauser, F. Vizza, S. P. Annen and H. Grutzmacher, ChemSusChem, 2014, 7, 2432–2435 CrossRef CAS PubMed.
- M. H. M. T. Assumpção, R. M. Piasentin, P. Hammer, R. F. B. De Souza, G. S. Buzzo, M. C. Santos, E. V. Spinacé, A. O. Neto and J. C. M. Silva, Appl. Catal., B, 2015, 174–175, 136–144 CrossRef.
- Q. He, S. Mukerjee, B. Shyam, D. Ramaker, S. Parres-Esclapez, M. J. Illán-Gómez and A. Bueno-López, J. Power Sources, 2009, 193, 408–415 CrossRef CAS.
- M. A. Montero, J. L. Fernández, M. R. Gennero de Chialvo and A. C. Chialvo, J. Power Sources, 2014, 254, 218–223 CrossRef CAS.
- L. S. Kibis, A. I. Stadnichenko, S. V. Koscheev, V. I. Zaikovskii and A. I. Boronin, J. Phys. Chem. C, 2016, 120, 19142–19150 CAS.
- Z. Cataldi, R. O. Lezna, M. C. Giordano and A. J. Arvia, J. Electroanal. Chem. Interfacial Electrochem., 1989, 261, 61–75 CrossRef CAS.
- M. Łukaszewski, H. Siwek and A. Czerwiński, Electrochim. Acta, 2007, 52, 4560–4565 CrossRef.
- M. A. Montero, M. R. Gennero de Chialvo and A. C. Chialvo, J. Power Sources, 2015, 283, 181–186 CrossRef CAS.
- S. Y. Shen, T. S. Zhao and J. B. Xu, Int. J. Hydrogen Energy, 2010, 35, 12911–12917 CrossRef CAS.
- J. Goldemberg, Science, 2007, 315, 808–810 CrossRef CAS PubMed.
- C. A. S. Hall, B. E. Dale and D. Pimentel, Sustainability, 2011, 3, 2413–2432 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00044h |
| This journal is © The Royal Society of Chemistry 2017 |