
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dual-functional UiO-66/TiO2 composite for water treatment and CO2 capture†
Yang Wang*,
Huijin Liu,
Manling Zhang,
Wubiao Duan and
Bo Liu *
*
Department of Chemistry, School of Science, Beijing Jiaotong University, Beijing 100044, China. E-mail: boliu@bjtu.edu.cn
First published on 14th March 2017
Abstract
UiO-66/TiO2 composites were fabricated via self-assembly using a solvothermal method. The composites exhibits high efficiency and stability for removing rhodamine B and methyl blue under strongly acidic conditions via a simple regeneration process. The participation of the UiO-66 structure in TiO2 was found to reduce the electron–hole recombination but did not change its adsorption ability on dyes, which leads to its enhanced photocatalytic properties under visible light radiation. In addition, the introduced TiO2 in MOFs was firstly found to increase the number of active sites, which is beneficial for CO2 capture. Therefore, it can be suggested that porous MOFs combined with photoactive semiconductors may introduce a new class of dual-functional materials for use in water treatment and CO2 capture.
Introduction
Degrading organic contaminants in water and removing CO2 in air have long been treated as separate disciplines. On the one hand, it has been recognized that the public perception of water quality is greatly influenced by coloured organics.1 In addition, it is difficult to degrade contaminants because they are very stable to light and oxidation, which has led to the need for more efficient methods or materials.2 To date, the two main ways to deal with pollutants are photocatalysis and adsorption using materials such as TiO2, Bi2WO6, clays, and activated carbon.3–6 On the other hand, the growing problem of excessive emissions of carbon dioxide has been driving the development of CO2 capture technology. Over the past few decades, metal oxides, ionic liquids, and metal–organic frameworks (MOFs) have been studied to capture CO2 and purify natural gas.7,8 However, whether semiconductors combined with MOFs may enhance the CO2 capture properties still remains unknown.Note that while dealing with pollutants in water, the contact between the dye and catalyst is imperative and is related to the degradation efficiency, especially most of the times when the concentration of the pollutants in water is low. Therefore, porous MOFs with excellent adsorption abilities seem to be a good choice to first gather pollutants, which can make the next photocatalytic process more sufficient. Metal–organic frameworks (MOFs) are crystalline porous materials, which are well-known for their various applications, especially in the field of heterogeneous catalysis and separation and storage of gases even in the photocatalytic field.9–12 Actually, MOFs with photocatalytic abilities have already been reported for a long time; MIL-125, for example, was first synthesized by Dan-Hardi and was found to be photoactive.13 Recently, Vinogradov prepared the depleted heterojunction MIL-125/TiO2, which was used in solar cells and showed excellent photocatalytic properties.14 However, the requirement of the high purity of raw materials used in the synthetic procedure, which require extremely anhydrous conditions and have weak stability in a strongly acidic environment, has limited their applications in the water purification field. As for the adsorption of MOFs, Hamon first studied hydrogen sulfide adsorption using the MIL-53(Al, Cr, Fe), MIL-47(V), MIL-100(Cr), and MIL-101(Cr) metal–organic frameworks at room temperature. Then, MIL-101 for the adsorptive removal of methyl orange from an aqueous solution was suggested by Haque.15,16 UiO-66, first synthesized by cava, was found to possess a high surface area and is highly stable under various conditions and has already been proved to have the ability to adsorb organic compounds.17,18 As a consequence, it is reasonable to expect that a more stable and efficient MOF-photocatalyst composite material may be formed using UiO-66 hybridized with a photocatalyst.
Due to its environmental safety, low-cost, and non-toxicity, TiO2 has been studied more than other photocatalysts; however, the relatively wide band gap (3.2 eV) of TiO2 is still a problem while utilizing visible light; it is well-known that an appropriate amount of transition metal ions doped into TiO2 can introduce electron capture centers and may as well change the crystallinity of TiO2, resulting in the formation of some defects that decrease the electron/hole recombination to enhance the abilities of the catalyst.19–21 Moreover, when used to capture CO2, the Ti–O bond in the lattice is more likely to increase the active sites available for the absorption of carbon dioxide. Based on this, a dual-functional heterojunction was designed. Herein, UiO-66 is expected to efficiently adsorb dyes in the dark and then degrade them in the light. TiO2 was introduced to increase the active sites in UiO-66 to capture CO2.
In this study, a tunable functionalized material comprising UiO-66/TiO2 (UT series) was prepared for the first time. On the one hand, we controlled the composite growth process and obtained different morphologies. On the other hand, among the heterojunctions, the initial shape and the final shape were found to possess interesting abilities, which were superior to the pure materials in both the photocatalysis and gas adsorption fields. Furthermore, we observed that the functions could be tuned by changing the ratio of the two components. As is known, this type of dual-functional material with tunable efficiency and using photoactive semiconductor as the added active site to enhance the CO2 capture ability has not been reported to date.
Experimental
Synthesis of the UiO-66/TiO2 series
UT0.05, UT19, UT49, and UT99 were synthesized according to the Zr/Ti ratio of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:19, 1:49, and 1:99, respectively, using the same procedure as reported in literature. Taking UT49 as an example, 3.92 g TiO2 was added to a solution comprising 0.233 g ZrCl4 (1 mmol), 0.161 g 1,4-benzenedicarboxylic acid (1 mmol), and 0.167 mL HCl (2 mmol) in 25 mL N,N-dimethylformamide (DMF). After stirring for 12 h, the mixture was transferred to a Teflon-lined autoclave (50 mL) and heated at 120 °C for 24 h. When the solvothermal reaction was completed, the mixture was cooled down to room temperature and the resulting white solid powder was recovered by filtration, washed three times with DMF to remove the unreacted organic ligand, and then washed three times with methanol. UiO-66 was prepared using the same procedure without adding P25. As a contrast, 3.92 g P25 dissolved in 25 mL of DMF was also transferred to a 50 mL Teflon-lined autoclave for the solvothermal reaction. Finally, the solid products were placed in a vacuum oven at 150 °C for 12 h.22
:1, 1:19, 1:49, and 1:99, respectively, using the same procedure as reported in literature. Taking UT49 as an example, 3.92 g TiO2 was added to a solution comprising 0.233 g ZrCl4 (1 mmol), 0.161 g 1,4-benzenedicarboxylic acid (1 mmol), and 0.167 mL HCl (2 mmol) in 25 mL N,N-dimethylformamide (DMF). After stirring for 12 h, the mixture was transferred to a Teflon-lined autoclave (50 mL) and heated at 120 °C for 24 h. When the solvothermal reaction was completed, the mixture was cooled down to room temperature and the resulting white solid powder was recovered by filtration, washed three times with DMF to remove the unreacted organic ligand, and then washed three times with methanol. UiO-66 was prepared using the same procedure without adding P25. As a contrast, 3.92 g P25 dissolved in 25 mL of DMF was also transferred to a 50 mL Teflon-lined autoclave for the solvothermal reaction. Finally, the solid products were placed in a vacuum oven at 150 °C for 12 h.22
Characterization
TEM and HR-TEM images were obtained using a FEI Tecnai G2F20 transmission electron microscope. SEM and EDAX mapping images were obtained using a Hitachi S4700 apparatus. Powder X-ray diffraction (Rigaku Ultima IV) was used to characterize the crystallinity and chemical composition of the materials studied. The intensity data were obtained over a 2θ range of 20–80° and the step size was 0.025° with the time duration per step of 0.8 s. A spectrum 100 FT-IR spectrometer was used to obtain the FT-IR data. The specific surface area and pore distribution were calculated using a Micromeritics Instrument Corporation Tristar II 3020 at 77 K. The pore diameters were determined by the BJH method. The chemical components of the UT series were examined by X-ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALAB 250Xi). The photoluminescence (PL, FLS 980) emission spectra of the samples were obtained to investigate the recombination of photogenerated electron–hole pairs. UV-vis diffuse reflectance spectroscopy (DRS, Cary 5000) was used to measure the light absorption of the samples. ICP-MS data was obtained using a NexION 300X ICP-MS. About 0.1 g of sample was dissolved in a solution of 2 mL HF, 2 mL HNO3, and 6 mL HCl for 2 h first and then diluted 10, 100, and 1000 times for the detection steps to obtain the specific results. EPR analysis was carried out using a Bruker E500 spectrometer. The solid catalyst was mixed with an ice-cooled DMPO solution in the presence or absence of an aqueous solution of RhB to capture ˙OH and a methanol solution to capture O2˙−. The mixture was quickly transferred into a glass capillary tube and examined using EPR spectroscopy at room temperature with/without visible light irradiation (500 W Xe lamp).Contaminant removal tests and CO2 capture evaluation
The contaminant removing tests were investigated upon treatment with RhB and MB. In a typical experiment, a 45 mL solution of RhB or MB and 100 mg of the powders were placed in a 50 mL beaker. The suspensions were magnetically stirred in the dark for 30 min to establish an adsorption–desorption equilibrium. Then, a 500 W Xe arc lamp with a UV cut-off filter was used as the visible light source. The test results were analyzed using UV-vis spectroscopic measurements. When the tests were completed, the residual samples were washed three times with water and ethanol and dried at 80 °C for regeneration. Moreover, the mechanism experiments were performed by adding 4 μL of IPA, 0.0054 g of BQ, and 0.0146 g of EDTA to the abovementioned system. CO2 and CH4 adsorption–desorption isotherms under static conditions were obtained using a Micromeritics Instrument Corporation Tristar II 3020 at 298 K. The samples were degassed at 120 °C for 8 h without further activation.Results and discussion
In the present study, the synthesis of the UT composite series was carried out using a hydrothermal method. As a contrast, commercial P25 was treated under the same conditions. In the last step, all the samples were washed three times with methanol. The formation of UiO-66 on the TiO2 particles is clearly seen in Fig. 1(a) and (b). The prepared materials only exhibit a long range-ordered structure of TiO2 at (101), which could be an evidence of the existence of two substances. Upon changing the ratio, different morphologies were obtained, which are shown in Fig. 1(c–e) and S1.† The initial shape (UT49), as shown in Fig. 1(c), can be attributed to agglomerated TiO2 particles without a regular shape. Upon increasing the amount of UiO-66, a layered structure of TiO2 can be seen in Fig. 1(d). Fig. 1(e) shows the final shape, which illustrates that UiO-66 crystal domains are present. Actually, due to the different diffractions of the two crystals, it was not hard to prove the formation of UiO-66. Fig. 1(f) also shows the XRD patterns of the UT series with an increasing molar ratio of Ti; the intensities of the characteristic diffraction peaks of UiO-66 from UT0.05 (Zr:Ti = 19:1) to UT99 (Zr:Ti = 1:99) decreased. Moreover, the FT-IR spectrum (Fig. 1(g)) shows the in- and out-of-phase stretching modes of the carboxylate group at 1399 and 1583 cm−1, respectively, in the composite corresponding to UiO-66, which did not show in TiO2 spectrum.17 Together with the EDAX mapping images, shown in Fig. S2,† all the conformations proved the formation of the UiO-66/TiO2 heterojunction. However, due to the large molar ratio of Ti and Zr, it was not possible to cover the TiO2 particles with UiO-66 when the morphology was in its initial shape (shown in Fig. 1 (c) and 2(a)). Interestingly, taking UT49 (Zr:Ti = 1:49) as an example, the specific surface area of the composite still doubled (108 m2 g−1) as compared to that of the TiO2 powder (about 50 m2 g−1), which is shown in Fig. 1(h) and S3(b).† These can be attributed to the UiO-66 between the TiO2 particles. The IV type curves with a H3 hysteresis loop suggests large amount of TiO2 in the UT49 and UT19 samples. Moreover, the transformed morphology was confirmed by Fig. 1(i) and S3.† The UT0.05 sample (726 m2 g−1) exhibited a type I curve. In addition, a different pore diameter distribution was observed, which suggests an ordered mesoporous morphology that is very different from UT49 and can be attributed to the UiO-66 whose specific surface area reached 908 m2 g−1 (Fig. S4†).23–26
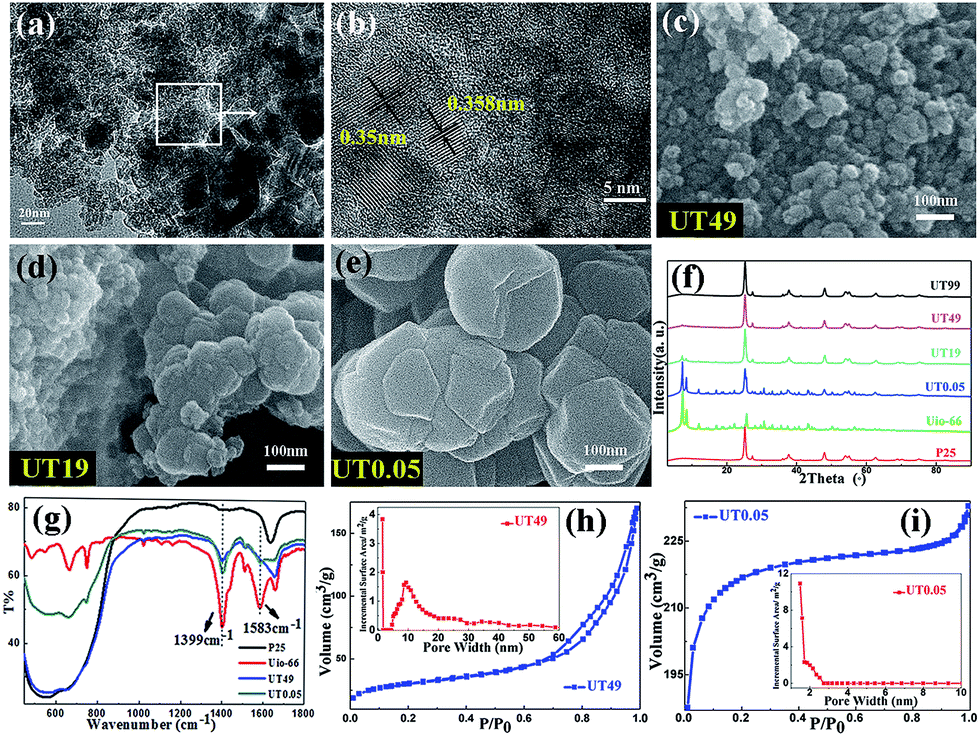 | ||
| Fig. 1 (a) The TEM image of the UT49 sample; (b) the HRTEM image of the UT49 sample; (c–e) SEM images of UT49 (the initial shape), UT19 (the intermediate shape), and UT0.05 (the final shape) samples; (f) the XRD patterns of the P25, UiO-66, UT0.05, UT19, UT49, and UT99 heterojunctions; (g) the FT-IR spectra of the P25, UiO-66, UT49, and UT0.05 samples and (h and i) the BET surface area and pore distribution of the UT49 and UT0.05 samples. | ||
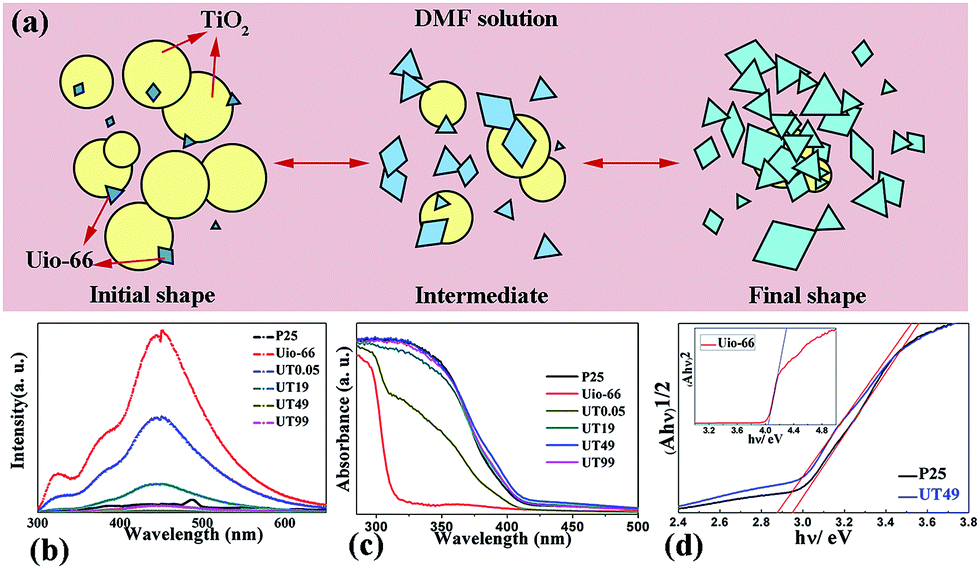 | ||
| Fig. 2 (a) The growth mechanism of the UT series of heterojunctions. The initial shape was TiO2 domains and the UiO-66 crystals were small on or out of the TiO2 particles, which exhibit excellent photocatalytic properties. As the process proceeded, the crystals grew bigger and bigger and finally, the UiO-66 crystals domains and the function-transformation occurred, which become a better CO2 capture sorbent. (b) The PL spectra and decay curves of the P25, UiO-66, UT0.05, UT19, UT49, and UT99 heterojunctions; the lowest curve was the UT49 sample. (c) The diffuse-reflectance UV-vis spectra of the P25, UiO-66, UT0.05, UT19, UT49, and UT99 samples. (d) The figure shows the relationship between the transformed Kubelka–Munk function versus the light energy. (Ahν)0.5 versus hν for P25 and UT49 and the inset shows (Ahν)2 versus hν for UiO-66. | ||
The exact transformation process was proposed as shown in Fig. 2(a). As the amount of UiO-66 increased, the TiO2 particles were covered with UiO-66, which may lead the function to greatly transform. Photoluminescence (PL) spectroscopy is widely used to follow the irradiative recombination of photogenerated charge carriers in TiO2 and to determine the energetic distribution of mid-band gap states.27 Fig. 2(b) shows the PL spectrum of the prepared samples. It can be seen that pure UiO-66 shows the highest peak, which illustrates that the recombination of the electrons and holes was high. As the TiO2 component increased, the recombination process was greatly impaired, especially when the two components reached a specific value, the recombination was lowest (UT49). The diffuse-reflectance UV-vis spectrum of the samples is presented in Fig. 2(c). We observed a slight red-shift, which was attributed to surface defects, most likely oxygen vacancies formed due to the substitution of Ti by Zr.28 Fig. 2(d) and the inset shows the plots of the Kubelka–Munk remission function. It can be seen that the band gaps of UiO-66, P25, and UT49 are 4.10, 2.92, and 2.88 eV, respectively. It is not hard to envisage that the reduced band gap and recombination of electron–hole pairs on UT49 may lead to a better photocatalytic performance.
Fig. 3 shows that that the samples mainly contained four elemental species: C, O, Ti, and Zr. The photoelectron spectra of O 1s electrons show the presence of two components corresponding to the two different chemical states of oxygen: lattice oxygen and oxygen in the surface functional groups.29 The high resolution C 1s spectrum is shown in Fig. 3(b), and the main peak is observed at 284.8 eV, which corresponds to the sp2 carbon atoms. However, the other weak peaks at 286.4 eV and 288.7 eV were assigned to the epoxy/hydroxyl groups (C–O) and carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), respectively, which correspond to the terephthalic acid in UiO-66.30–32 The Ti 2p3/2 and Ti 2p1/2 binding energies are ascribed to the Ti4+ oxidation state, and the values of Zr 3d5/2 and Zr 3d3/2 indicates that Zr is in the Zr4+ state. The reported binding energies are 458.8 and 183 eV for pure Ti 2p3/2 and pure Zr 3d5/2, respectively.33 Lucky et al. found that while introducing Zr into the TiO2 matrix, the binding energies for both Ti 2p3/2 and Zr 3d5/2 shifted towards a lower energy, which was also found in our experiments.34 Due to the similarity of the Ti4+ and Zr4+ ionic radii, Zr4+ may partially substitute Ti4+ in the TiO2 lattice when the sample was prepared using ZrCl4 during the solvothermal process. This phenomenon could also be observed in Fig. 1(b). The interplanar distance has two values, 0.35 nm for TiO2 and 0.358 nm, attributed to the substituted situation.
O), respectively, which correspond to the terephthalic acid in UiO-66.30–32 The Ti 2p3/2 and Ti 2p1/2 binding energies are ascribed to the Ti4+ oxidation state, and the values of Zr 3d5/2 and Zr 3d3/2 indicates that Zr is in the Zr4+ state. The reported binding energies are 458.8 and 183 eV for pure Ti 2p3/2 and pure Zr 3d5/2, respectively.33 Lucky et al. found that while introducing Zr into the TiO2 matrix, the binding energies for both Ti 2p3/2 and Zr 3d5/2 shifted towards a lower energy, which was also found in our experiments.34 Due to the similarity of the Ti4+ and Zr4+ ionic radii, Zr4+ may partially substitute Ti4+ in the TiO2 lattice when the sample was prepared using ZrCl4 during the solvothermal process. This phenomenon could also be observed in Fig. 1(b). The interplanar distance has two values, 0.35 nm for TiO2 and 0.358 nm, attributed to the substituted situation.
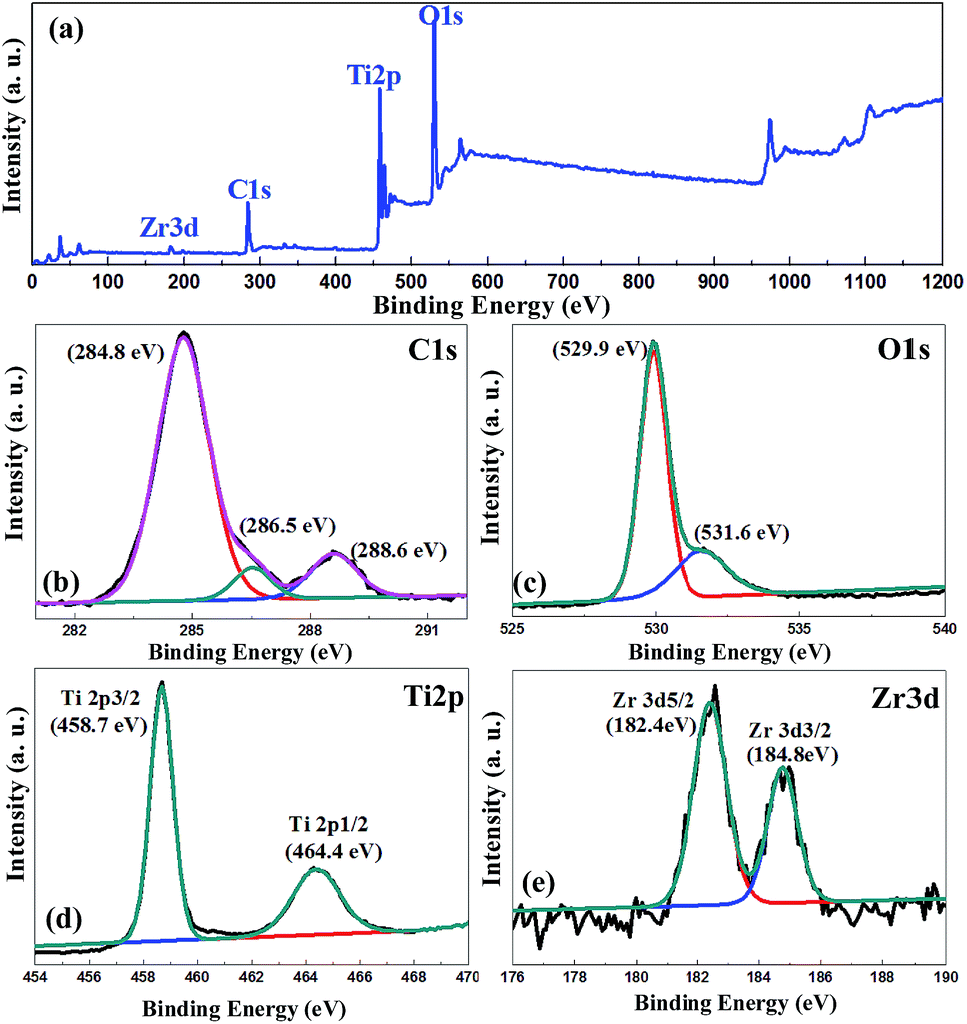 | ||
| Fig. 3 (a) The wide survey scans. The (b) C 1s, (c) O 1s, (d) Ti 2p, and (e) Zr 3d XPS spectra of the MT49 heterojunction. | ||
To prove that the UiO-66/TiO2 heterostructures are good materials to efficiently remove contaminants, two typical organic dyes, MB and RhB, were tested. All the sample tests were carried out under the same conditions for comparison. The degradation efficiency of RhB and MB under visible light irradiation is shown in Fig. 4(a and b). Under dark conditions, the pure UiO-66 absorption ability was the best, which could be attributed to its high surface area. On the contrary, TiO2 showed the lowest ability. As the TiO2 component was increased, the absorption showed a small decrease in the composite; however, the photocatalytic efficiency was enhanced to maximum when the ratio of the two substances reached a certain value. Compared to the lowest adsorptive ability of TiO2, it is believed that the strong attraction with dyes was mainly due to the UiO-66 component and excellent photocatalytic properties were shown by the heterojunction, which are presented in Fig. 5. As for the treatment of MB, it could accomplish the absorption process in a very short time, which is shown in Fig. 4(d), with the efficiency reaching 80% within 10 minutes. Then, under light radiation, the degradation of MB continued to carry on. The stability of the materials was also investigated and is presented in Fig. 4(c and d). Under neutral and strongly acidic conditions (pH = 3), the removal of RhB and MB was retained. Furthermore, after a simple regeneration process, the material retained its high efficiency, which shows potential for industrial application. As for the CO2 capture ability, UT0.05 (Zr:Ti = 19:1) was tested and CO2 and CH4 were selected for the study. As shown in Fig. 4(e), the maximum CO2 uptake was 46 mL g−1, which was a significant enhancement when compared to that of pure TiO2 (4 mL g−1) and UiO-66 (30 mL g−1). Moreover, UT49 was tested and a CO2 uptake of 5 mL g−1 was observed, which illustrates the function transformation from UT49 to UT0.05. This may suggest that photocatalytic semiconductors could be used to improve the CO2 capture ability. Note that the transition state of the composite (UT19) was also observed. It showed both good photocatalytic and CO2 capture abilities but lower than the best results, which are presented in Fig. 4(b) and (e). The compromised abilities in the two functions suggest that the transformation process was accomplished step by step, which is described in Fig. 2(a). To further explore the materials function in gas separations, CH4 was also tested, as shown in Fig. 4(d), with the maximum uptake less than 10 mL g−1; the great adsorption difference of the two gases can illustrate the UT series' prospect in nature gas purification field. Moreover, the specific ratio of Zr and Ti was detected using ICP-MS to examine the actual ratio to eliminate the possible errors in the preparation procedure. As shown in Table S1,† the actual values are listed, except for UT0.05, and the actual ratio of all the samples correspond to the theoretical values.
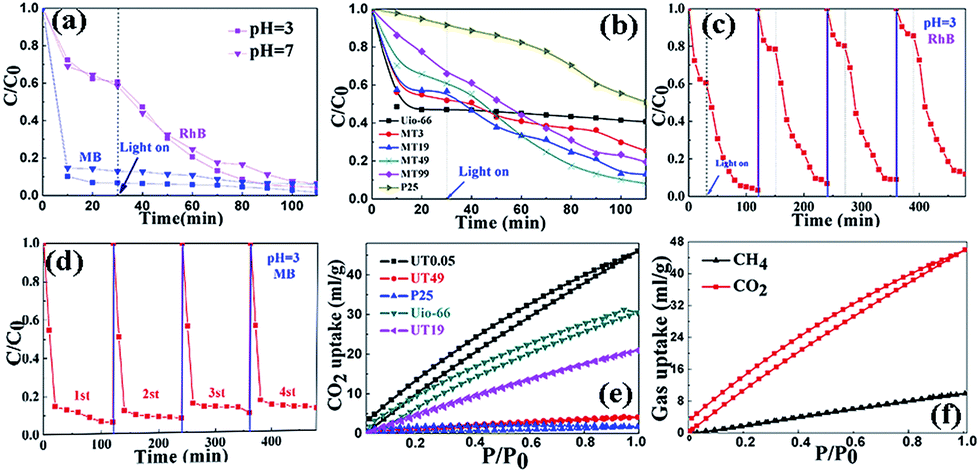 | ||
| Fig. 4 (a) The effects of removing RhB and MB in water using UT49 at pH = 7 and pH = 3. It can be clearly seen that the removal of RhB was mainly via photocatalysis and MB was via adsorption. (b) A comparison of the removal of RhB from a neutral solution with or without visible light irradiation using the P25, UiO-66, UT0.05, UT19, UT49, and UT99 samples. (c and d) The removal effects using UT49 to remove RhB and MB under acidic conditions and cycled four times after a simple regeneration process. (e) The CO2 capacity of the P25, UiO-66, UT0.05, UT19, and UT49 heterojunctions, which illustrate the function transformation process. (f) A comparison of CO2 and CH4 uptake, which may be used to introduce a more efficient material for use in natural gas purification. | ||
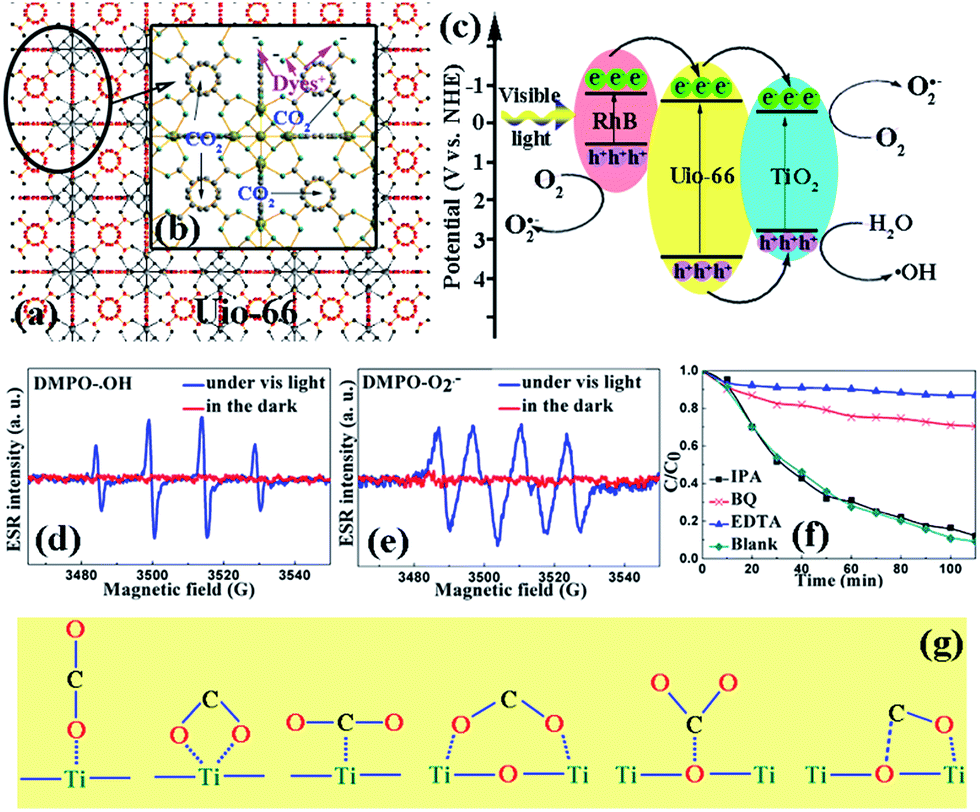 | ||
| Fig. 5 (a and b) The proposed adsorption mechanism for both CO2 capture and dye removal. CO2 was adsorbed in the tetrahedral cages of UiO-66 and the cationic dyes interacted with the –COO− groups. (c) The photocatalytic mechanism on the heterojunctions. (d and e) The DMPO-˙OH adduct in a RhB solution and the DMPO–O2˙− adduct in methanol with/without visible light irradiation using the UT49 heterojunction. (f) A comparison of the RhB solutions with added IPA, BQ, and EDTA and the blank test. (g) The six enhanced CO2 active interaction sites for the UT series of composites compared to pure UiO-66. | ||
Due to the molecular structure of the dyes being big, the dyes were most likely to absorb on the external surface of the composite. Therefore, the dual-functional mechanism was proposed using two steps, as shown in Fig. 5(a); the high efficiency adsorption of the dyes was attributed to the electrostatic attraction between the adsorbents and dyes. The exposed –COO− on the external surface is more attractive for cationic dyes.35 Then, under light irradiation, the higher conduction band edge of UiO-66 will lead to the transfer of photoexcited electrons into the conduction band of TiO2 through the quantum tunneling effect,36 then the h+ can react with H2O to form ˙OH free radicals or directly participate the degradation progress and e− reacts with O2 adsorbed on the surface to form O2˙− free radicals, which are all involved in the degradation of RhB. As shown in Fig. 5(d) and (e), we used DMPO to capture ˙OH and O2˙− and remarkable 1:2:2:1 quadruple EPR peaks for the DMPO-˙OH adduct in the RhB solution and a 1:1:1:1 DMPO–O2˙− adduct in methanol solution were observed, which confirmed the proposed mechanism. Furthermore, it was necessary to find out which factor of the three species, ˙OH, O2˙−, and h+, directly functioned in the process. Herein, isopropyl alcohol (IPA), benzoquinone (BQ), and ethylenediaminetetraacetic acid (EDTA) were introduced into the degradation process to attempt to trap ˙OH, O2˙−, and h+, respectively.37–41 As shown in Fig. 5(f), there was no significant effect on the degradation rate when IPA was added, thus suggesting that ˙OH is not a dominant active species in the degradation process. However, the degradation rate significantly decreased after the introduction of BQ and EDTA, which were able to quench O2˙− and h+. Thus, O2˙− and h+ were identified as the major active species in the decomposition of the dyes. Moreover, the results implied that the dyes can also be oxidized by the photogenerated h+ in the UiO-66/TiO2 system.
The enhanced CO2 capture abilities mechanism is described in Fig. 5(b) and (g). In general, CO2 is preferentially adsorbed in the tetrahedral cages of UiO-66(Zr), which are shown in Fig. 4(b). Moreover, the material saturated with CO2 contains an average of four CO2 molecules in this cage in an arrangement with two CO2 molecules interacting with the two –OH functions, which has been reported by Chevreau.42 In our case, TiO2 was introduced to UiO-66, which led the specific surface area of the composite to decrease. However, the six possible active sites added in the composite, as shown in Fig. 5(g), which can be divided into monodentated carbonates and bidentated carbonates, led to an increase in the CO2 uptake.43,44 Especially, when the ratio reached a special value, UT0.05 in our system, the maximum adsorption ability was obtained.
Conclusions
In summary, the UiO-66/TiO2 composites were synthesized and found to accomplish two functions for the first time. HR-TEM, SEM, XRD, FT-IR, ICP-MS, and BET analysis confirmed the UiO-66 and TiO2 components. During the growth progress, we found that the initial form of the UT series (UT49) could efficiently remove RhB and MB under vis-light irradiation. The durable ability was also tested. It was found that the composites possessed high efficiency after four cycles in a strongly acidic environment with a simple regeneration process. Their abilities in photocatalysis are good and could be explained by the XPS, PL, and UV-vis spectra. Moreover, the final form of the composite (UT0.05) was also tested and showed superior ability in CO2 capture and gas separation as a sorbent. Thus, the MOF/photocatalyst composites could be a new type of material for both photocatalysis and adsorption to achieve multifunctional properties.References
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- E. Haque, J. E. Lee, I. T. Jang, Y. K. Hwang, J. S. Chang, J. Jegal and S. H. Jhung, J. Hazard. Mater., 2010, 181, 535–542 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- J. Tian, Y. Sang, G. Yu, H. Jiang, X. Mu and H. Liu, Adv. Mater., 2013, 25, 5075–5080 CrossRef CAS PubMed.
- M. Alkan, Ö. Demirbaş, S. Çelikçapa and M. Doğan, J. Hazard. Mater., 2004, 116, 135–145 CrossRef CAS PubMed.
- K. Kadirvelu, M. Kavipriya, C. Karthika, M. Radhika, N. Vennilamani and S. Pattabhi, Bioresour. Technol., 2003, 87, 129–132 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hareb and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 CAS.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- R. B. Getman, Y. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed.
- J. Wang, C. Wang and W. Lin, Metal–organic frameworks for light harvesting and photocatalysis, ACS Catal., 2012, 2, 2630–2640 CrossRef CAS.
- L. Yang, G. Fang, J. Ma, E. Ganz and S. S. Han, Cryst. Growth Des., 2014, 14, 2532–2541 CAS.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- A. V. Vinogradov, H. Zaake-Hertling, E. Hey-Hawkins, A. V. Agafonov, G. A. Seisenbaeva, V. G. Kesslerd and V. V. Vinogradov, Chem. Commun., 2014, 50, 10210–10213 RSC.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. D. Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed.
- E. Haque, J. E. Lee, I. T. Jang, Y. K. Hwang, J. Chang, J. Jegal and S. H. Jhung, J. Hazard. Mater., 2010, 181, 535–542 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13851–13852 CrossRef PubMed.
- Q. Chen, Q. He, M. Lv, Y. Xu, H. Yang, X. Liu and F. Wei, Appl. Surf. Sci., 2015, 327, 77–85 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- Y. Liu, S. Zhou, J. Li, Y. Wang, G. Jiang, Z. Zhao, B. Liu, X. Gong, A. Duan, J. Liu, Y. Wei and L. Zhang, Appl. Catal., B, 2015, 168, 125–131 CrossRef.
- S. Kim, Y. Lee, S. Hong, M. Jang and W. Ahn, Catal. Today, 2015, 245, 54–60 CrossRef CAS.
- Z. Hu, M. Khurana, Y. H. Seah, M. Zhang, Z. Guo and D. Zhao, Chem. Eng. Sci., 2015, 124, 61–69 CrossRef CAS.
- F. Liu, W. Li, B. Liu and R. Li, J. Mater. Chem. A, 2013, 1, 8037–8044 CAS.
- C. Wang, X. Liu, J. P. Chen and K. Li, Sci. Rep., 2015, 16613–16621 CrossRef CAS PubMed.
- L. Shi, T. Wang, H. Zhang, K. Chang and J. Ye, Adv. Funct. Mater., 2015, 25, 5360–5367 CrossRef CAS.
- X. L. Wang, Z. C. Feng, J. Y. Shi, G. Q. Jia, S. A. Shen, J. Zhou and C. Li, Phys. Chem. Chem. Phys., 2010, 12, 7083–7090 RSC.
- P. Zhang, Y. L. Yu and E. J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 4622–4629 CAS.
- J. Lukáč, M. Klementová, P. Bezdička, S. Bakardjieva, J. Šubrt, L. Szatmáry, Z. Bastl and J. Jirkovský, Appl. Catal., B, 2007, 74, 83–91 CrossRef.
- X. F. Yang, J. L. Qin, Y. Jiang, K. M. Chen, X. H. Yan, D. Zhang, R. Li and H. Tang, Appl. Catal., B, 2015, 166–167, 231–240 CrossRef CAS.
- Z. Jin, W. Duan, B. Liu, X. Chen, F. Yang and J. Guo, Appl. Surf. Sci., 2015, 356, 707–718 CrossRef CAS.
- Y. H. Yang, E. Z. Liu, H. Z. Dai, L. M. Kang, H. T. Wu, J. Fan, X. Y. Hu and H. C. Liu, Int. J. Hydrogen Energy, 2014, 39, 7664–7671 CrossRef CAS.
- I. R. Galindo, T. Viveros and D. Chadwick, Ind. Eng. Chem. Res., 2007, 46, 1138–1147 CrossRef CAS.
- R. A. Lucky, Y. Medina-Gonzalez and P. A. Charpentier, Langmuir, 2010, 26, 19014–19021 CrossRef CAS PubMed.
- Q. Chen, Q. He, M. Lv, Y. Xu, H. Yang, X. Liu and F. Wei, Appl. Surf. Sci., 2015, 327, 77–85 CrossRef CAS.
- Y. Li, A. Pang, C. Wang and M. Wei, J. Mater. Chem., 2011, 21, 17259–17264 RSC.
- Y. Zhou, Y. Zhang, M. Lin, J. Long, Z. Zhang, H. Lin, J. Wu and X. Wang, Nat. Commun., 2015, 6, 9340–9348 Search PubMed.
- X. Yan, X. Zhu, R. Li and W. Chen, J. Hazard. Mater., 2016, 303, 1–9 CrossRef CAS PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 CAS.
- M. Pera-Titus, V. Garca-Molina, M. A. BaÇos, J. Gimnez and S. Esplugas, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS.
- H. Chevreau, W. Liang, G. J. Kearley, S. G. Duyker, D. M. D'Alessandro and V. K. Peterson, J. Phys. Chem. C, 2015, 119, 6980 CAS.
- S. Chowdhury, G. K. Parshetti and R. Balasubramanian, Chem. Eng. J., 2015, 263, 374–384 CrossRef CAS.
- L. Mino, G. Spoto and A. M. Ferrari, J. Phys. Chem. C, 2014, 118, 25016 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00028f |
| This journal is © The Royal Society of Chemistry 2017 |