
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the surface chemistry of graphene flakes: new strategies for selective oxidation†
Mariana P. Araújoa,
O. S. G. P. Soaresb,
A. J. S. Fernandesc,
M. F. R. Pereira b and
C. Freire*a
b and
C. Freire*a
aREQUIMTE/LAQV, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/n, 4169-007 Porto, Portugal. E-mail: acfreire@fc.up.pt; Fax: +351 22 0402 695; Tel: +351 22 0402 0590
bLaboratório de Catálise e Materiais (LCM), Laboratório Associado LSRE-LCM, Departamento de Engenharia Química, Faculdade de Engenharia, Universidade do Porto, Rua Dr Roberto Frias s/n, 4200-465 Porto, Portugal
cInstituto de Nanoestruturas, Nanomodelação e Nanofabricação (I3N), Departamento de Física, Universidade de Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal
First published on 3rd March 2017
Abstract
To accomplish a rational tuning of the surface chemistry of graphene flakes (GF), four different one-step protocols towards the selective oxidation of GF were performed, using different oxidants: nitric acid, potassium permanganate/sulfuric acid, ozone and 3-chloroperbenzoic acid. The characterization of the resulting materials confirmed the successful preparation of oxidized GF with C/O atomic ratios varying in the range of 21.2–4.9, with distinct types of oxygen functionalities. While the oxidation of GF with nitric acid exclusively promotes the introduction of carboxylic groups and carbonyl/quinones, 3-chloroperbenzoic acid is responsible for the introduction of epoxyl groups and carboxylic anhydrides, potassium permanganate favours the introduction of epoxyl and hydroxyl groups and some content of carboxylic anhydrides, and ozone promotes predominantly the introduction in graphene structure of epoxyl groups, carboxylic anhydrides, phenols, quinones and lactones, and in a lesser extension carbonyl groups in α-substituted ketones and aldehydes if the oxidation is performed in the solid phase, or hydroxyl groups and a moderate content of carbonyl groups and aldehydes if GF are in a water dispersion. Furthermore, this work highlighted the possibility of identifying and distinguishing labile groups, namely hydroxyl and epoxyl groups, which are predominant in the structure of GF oxidized with potassium permanganate/sulfuric acid, ozone and 3-chloroperbenzoic acid. This is the first comprehensive study on the fine tuning of the surface of oxidized GF and a major contribution for the rational design of graphene composites since the application of these specific strategies can be useful in the anchoring of other molecules or nanoparticles.
1. Introduction
Graphene, an extended honeycomb network of sp2-hybridised carbon atoms and the first close-packed two-dimensional crystalline material isolated in nature,1–4 has outstanding electronic, optical, thermal and mechanical properties such as ambipolar electric field effect,1 extremely high mobility of charge carriers,1,5 high values of thermal conductivity,6 specific surface area,7 high chemical stability and high optical transmittance.7,8 Due to these remarkable properties, a great number of potential applications involving this nanomaterial have been observed and forecasted in several areas, such as electronics, energy conversion and storage or biotechnology.9,10Despite the great potential applications of graphene, it possesses zero band gap and it is not very reactive. For most applications, graphene needs to be integrated with different functional materials; hence, the introduction of various chemical functionalities in graphene's backbone is mandatory in order to promote the formation of graphene composites with other nanomaterials and molecules.11 Moreover, an obstacle to graphene processing and physical handling is its low dispersibility in both aqueous and organic solutions. Due to strong tendency to cluster together via π–π stacking and van der Waals interactions, graphene sheets can form irreversible agglomerates or restack to form graphite.4 Thus, functionalization of graphene without damaging its electronic structure is crucial, in order to overcome its chemical inertness and to maximize its application.
In this context, most of the studies involve graphene oxide (GO), prepared through drastic oxidative conditions, or the material obtained upon its reduction (r-GO).2 Graphene oxide, which is commonly prepared via chemical oxidation of graphite and subsequent exfoliation of graphite oxide, has been exhaustively studied due to the possibility of immobilizing different nanomaterials onto GO surface, by taking advantage of its oxygen functionalities richness.12,13
Typically, graphene oxide has a carbon–oxygen ratio of 1.5 to 2.5 and can be produced by different chemical methods, being the most common the one proposed by Hummers and Offeman,14 which derived from the method of Staudenmaier, and the Brodie method.15 According to the production processes and conditions, this material can have very distinct compositions and structures.16
The final structure of GO is highly dependent on differences in the starting materials (such as the graphite source) and the preparation methods, which complicates the exact identification and distribution of oxygen functional groups (usually, hydroxyls, epoxides, carbonyls, carboxylic acids, quinones and lactones) of the obtained structures.4,17 Currently, the knowledge of the exact bonding configuration and location of the oxygen chemical groups of GO is lacking, despite the fact that graphite oxide was firstly prepared 150 years ago.18 In the last decades, several possible graphite (and graphene) oxide models were suggested, based on several techniques such as nuclear magnetic resonance, X-ray diffraction, Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy and theoretical calculations, e.g. those proposed by Hofmann, Ruess, Scholz–Boehm and Nakajima–Matsuo, and more recently by Lerf–Klinowski, Dékány and Ajayan.18–24 Ruoff et al. reported for the first time the synthesis of a 100% 13C-labelled graphite oxide for 13C MAS NMR analysis.25 It highlighted some structural and spatial features: it was found that hydroxyl and epoxide carbons are bonded to each other, and carbonyl groups are separated from the most of the sp2, hydroxyl and epoxide groups. Furthermore, unattributed NMR signals were described, which Ajayan suggested to be due to the presence of lactols.24
Although the high diversity of oxygen functionalities in GO is advantageous to promote the anchoring of nanoparticles or other chemical entities, its high degree of oxidation is responsible for considerable disruption of the electronic π delocalization, even after reduction treatments, since the flow of charge carriers in reduced GO remains partially disrupted, severely affecting its electrochemical behaviour.26–28
Herein, the aim of the work was the selective oxidation of graphene flakes by four different one-step strategies in order to accomplish a rational tuning of the surface chemistry of graphene. In method A, nitric acid – one of the reagents employed in the Brodie method15 – was used as oxidant. In method B, graphene flakes were treated with potassium permanganate/sulfuric acid, which are typically used in Hummers–Offeman method.14,29 In method C, the oxidation was promoted by ozone – to our knowledge, very few works have been reported concerning the modification of graphene-based materials with ozone30–32 and most of the existing ones are focused on the graphene modification with ozone obtained through aggressive oxidation methods, such as the Hummers–Offeman method.30,31 Finally, in method D, graphene flakes were oxidized with 3-chloroperbenzoic acid (m-CPBA), which was only used in the epoxidation of carbon nanotubes33 and graphite.34 To achieve a full understanding on the effect on graphene structure, type of oxygen functional groups introduced during the oxidation protocols, composition, morphology and textural properties, a combination of several techniques were used. Finally, the electrochemical behaviour of the different oxidized graphene flakes was studied by cyclic voltammetry in the presence of [Fe(CN)6]3−/4−, in order to evaluate the influence of the oxygen surface groups on the electron transfer properties of oxidized graphene flakes. To the best of our knowledge, this is the first work on the fine tuning of graphene surface by selectively introducing distinct oxygen functional groups.
2. Experimental
2.1. Reagents and solvents
All the reagents and solvents used during the experimental work were used as received. Graphene flakes (GF) were commercially obtained from Graphene Technologies (Lot #GTX-7/6-10.4.13). Nitric acid (>65%, Merck), hydrogen peroxide solution 30 wt% in H2O (ACS reagent, Aldrich), potassium permanganate (ACS reagent, Merck), sulphuric acid (95–97% Merck), 3-chloroperbenzoic acid (m-CPBA, ≤77% Aldrich), dichloromethane (99.99% Fisher) and hydrochloric acid (37% Fisher) were used in the oxidation of commercial GF. All the filtrations were performed through a polyamide membrane (Whatman, 45 μm). In the electrochemical studies, potassium hexacyanoferrate (III) (ACS reagent, Merck) and potassium chloride (99.999% suprapur, Merck) were used and for the preparation of solutions Milli-Q water with a resistivity of 18.2 MΩ was used.2.2. Preparation of the oxidized graphene flakes
Original graphene flakes (denoted as p-GF) were submitted to several oxidation treatments using different experimental conditions, after being sonicated for 10 minutes:2.3. Physicochemical characterization
X-ray photoelectron spectroscopy (XPS) was performed at Centro de Materiais da Universidade do Porto, Portugal (CEMUP), in a Kratos AXIS Ultra HSA spectrometer using monochromatized Al Kα radiation (1486.6 eV). The powdered materials were pressed into pellets prior to the XPS analysis. XPS spectra were deconvoluted with CasaXPS 2.3.12 software, using non-linear least squares fitting routine after a Shirley-type background subtraction and the peaks were interpreted using a combination of Gaussian/Lorentzian functions. The adjusted parameters were FWHM, binding energy and peak area. To correct possible deviations caused by electric charge of the samples, the C1s band at 284.6 eV was taken as internal standard. Surface atomic percentages were calculated from the corresponding peak areas upon spectra deconvolution and using the sensitivity factors provided by the manufacturer.Temperature programmed desorption (TPD) was performed in an AMI-200 apparatus, from Altamira Instruments. Typically, samples of 100 mg were heated from room temperature up to 1100 °C, at 5 °C min−1, under a flow of helium (25 cm3 min−1). The amounts of CO (m/z = 28) and CO2 (m/z = 44) released from the samples were monitored with a Dymaxion mass spectrometer (Ametek Process Instruments). Spectra deconvolution was performed by using a multiple Gaussian function and the Levenberg–Marquardt iteration method.
Transmission electron microscopy (TEM) measurements were carried out at Instituto de Biologia Molecular e Celular (IBMC), Porto, Portugal. TEM micrographs were obtained with a JEOL JEM-1400 transmission electron microscope with a CCD camera Gatan, operating at 80 kV. Graphene samples were dispersed in ethanol (99.99%, Fisher) under sonication, after which a carbon-coated 400 mesh copper grid was immersed in the suspension and then air-dried.
The micro-Raman analysis was conducted in the backscattering configuration on a Jobin Yvon HR800 instrument (Horiba, Japan), using a 1800 lines/mm grating and the 532 nm laser line from a Nd:YAG DPSS laser (Ventus, Laser Quantum, U.K.). For the Rayleigh rejection, a pair of edge filters placed in series were used allowing Raman acquisition from 50 cm−1. A 100× objective (spot size ∼ 2 μm, numeric aperture = 0.9, Olympus, Japan) was used to focus the laser light onto the sample and to collect the backscattered Raman radiation to be detected by a Peltier cooled (223 K) CCD sensor. The spectrometer was operated in the confocal mode, setting the iris to 300 μm and the acquisition time to 2 × 10 s. The data treatment of the acquired spectra (background removal and Gauss/Lorentzian curve fitting) was carried out using the native NGS LabSpec software.
Fourier transform infrared spectroscopy (FTIR) spectra were obtained with a Jasco FT/IR-460 Plus spectrophotometer in the range of 400–4000 cm−1, with a resolution of 4 cm−1 and 32 scans. The spectra were collected in KBr pellets containing 0.2% weight of graphene materials.
The UV-visible spectra of graphene dispersions in water (c = 0.1 mg mL−1) were recorded at room temperature on a Shimadzu UV-2550 UV-vis spectrophotometer, in the range of 180–800 nm, with a quartz cuvette (10 mm path length).
Electrochemical studies were carried out using an Autolab PGSTAT 30 potentiostat/galvanostat (controlled by GPES software). A three-electrode electrochemical cell was used with a glassy carbon (GC) electrode (3.0 mm of diameter, BASi) as working electrode, a Pt wire as counter electrode and an Ag/AgCl (1.0 M NaCl) reference electrode (Metrohm). The reference electrode was separated from the working and counter electrodes through a porous glass frit and this compartment containing the reference electrode was filled with supporting electrolyte, which was a 0.1 M KCl solution. All the reported electrochemical potentials are referenced to the Ag/AgCl reference electrode.
Cyclic voltammetry experiments in the presence of 1.0 mM K3[Fe(CN)6] solution were performed by cycling the potential 5 times between 0.9 and −0.5 V, at a scan rate of 0.1 V s−1. For the modification of GC electrode with graphene samples, GF, GF_HNO3, GF_KMnO4, GF_O3_disp, GF_O3_gas and GF_m-CPBA were previously dispersed (0.5 mg mL−1) in DMF for 5 minutes with sonication. The immobilization onto the GC electrode surface was performed by drop-casting: 3 μl of the corresponding material suspension were deposited with a micropipette and allowed to dry; prior to each material immobilization, the GC electrode surface was polished with alumina particles (0.3 μm, Buehler) and thoroughly washed with Milli-Q water. Electron transfer constants,  , were determined according to the Nicholson approach,35 which establishes a relation between the ΔEp–p values and the dimensionless kinetic parameter, ψ, and that recently has been applied to evaluate the heterogeneous electron transfer rates of graphene oxides.36,37 For the calculation of
, were determined according to the Nicholson approach,35 which establishes a relation between the ΔEp–p values and the dimensionless kinetic parameter, ψ, and that recently has been applied to evaluate the heterogeneous electron transfer rates of graphene oxides.36,37 For the calculation of  , diffusion coefficient values of Do = 7.39 × 10−5 and Dr = 6.81 × 10−5 cm2 s−1 were used.38
, diffusion coefficient values of Do = 7.39 × 10−5 and Dr = 6.81 × 10−5 cm2 s−1 were used.38
3. Results and discussion
3.1. Composition, textural properties and morphology
Surface composition data of original and oxidized graphene flakes (p- and ox-GF, respectively) was obtained by XPS. The surface atomic percentages of each material are summarized in Table 1. Original GF (p-GF) is composed by 96.2% of carbon, 3.2% of oxygen and a residual amount of nitrogen (0.6%). After the oxidation treatments, the amount of oxygen increased in all the materials, with the percentage varying between the lowest value of 4.5% for GF_HNO3 and the highest value of 16.8% for GF_KMnO4. C/O atomic ratios have considerably decreased, as a consequence of the oxygen percentage increase, for GF_KMnO4, GF_m-CPBA_0.1 M, GF_O3 and GF_m-CPBA_0.01 M (C/O ratios = 4.9, 7.8, 9.8 and 13.0, respectively), when compared with the C/O atomic ratio of p-GF (C/O ratio = 30.1). In the case of GF_HNO3, the XPS results confirmed the occurrence of a milder oxidation since the decrease of C/O ratio is not so pronounced (C/O ratio = 21.2). For comparison, C/O atomic ratios of graphene oxides prepared by the Hummers method are in the range of 2.0–3.0 and, upon its reduction, C/O ratios increase to 6.0–13.5,17,27,39 which are close to the values obtained for the prepared ox-GF. In the case of graphene oxidation by ozone, Z. Xu et al. were able to oxidize thermal-exfoliated graphene nanosheets, achieving a C/O ratio varying from 2.0 to 6.7, although it is worth mentioning that the starting material had already considerably low C/O ratio of 17.7 and thus the comparison should be done cautiously.30| Graphene flakes | % At | C/O ratio | ||
|---|---|---|---|---|
| C1s | N1s | O1s | ||
| GF | 96.2 | 0.6 | 3.2 | 30.1 |
| GF_HNO3 | 95.5 | — | 4.5 | 21.2 |
| GF_O3_disp | 90.4 | 0.4 | 9.2 | 9.8 |
| GF_O3_gas | 90.3 | 0.5 | 9.2 | 9.8 |
| GF_KMnO4 | 82.3 | 0.9 | 16.8 | 4.9 |
| GF_m-CPBA 0.1 M | 88.3 | 0.4 | 11.3 | 7.8 |
| GF_m-CPBA 0.01 M | 92.5 | 0.5 | 7.1 | 13.0 |
The relative atomic percentages of carbon-based functional groups, which were determined based on the deconvolution of C1s high resolution spectra, are summarized in Table 2; deconvoluted C1s and O1s high resolution spectra of the most oxidized material, GF_KMnO4, are presented in Fig. 1, and deconvoluted C1s and O1s high resolution spectra of the other materials are presented in Fig. S1, ESI.†
| Graphene flakes | % C | ||||
|---|---|---|---|---|---|
| 284.6 eV (graphitic structure) | 286.3 eV C–O | 287.4 eV C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
288.9 eV COOH | 290.3 eV π–π* | |
| GF | 71.4 | 5.8 | 5.4 | 1.9 | 15.5 |
| GF_HNO3 | 79.8 | 4.4 | 4.4 | 2.6 | 8.8 |
| GF_O3_disp | 76.4 | 6.4 | 5.3 | 0.5 | 11.4 |
| GF_O3_gas | 67.6 | 13.8 | 6.6 | 1.1 | 10.8 |
| GF_KMnO4 | 64.9 | 19.2 | 10.0 | 1.5 | 4.5 |
| GF_m-CPBA 0.1 M | 76.8 | 6.3 | 4.9 | 0.4 | 11.6 |
| GF_m-CPBA 0.01 M | 75.2 | 6.6 | 5.2 | 0.4 | 12.6 |
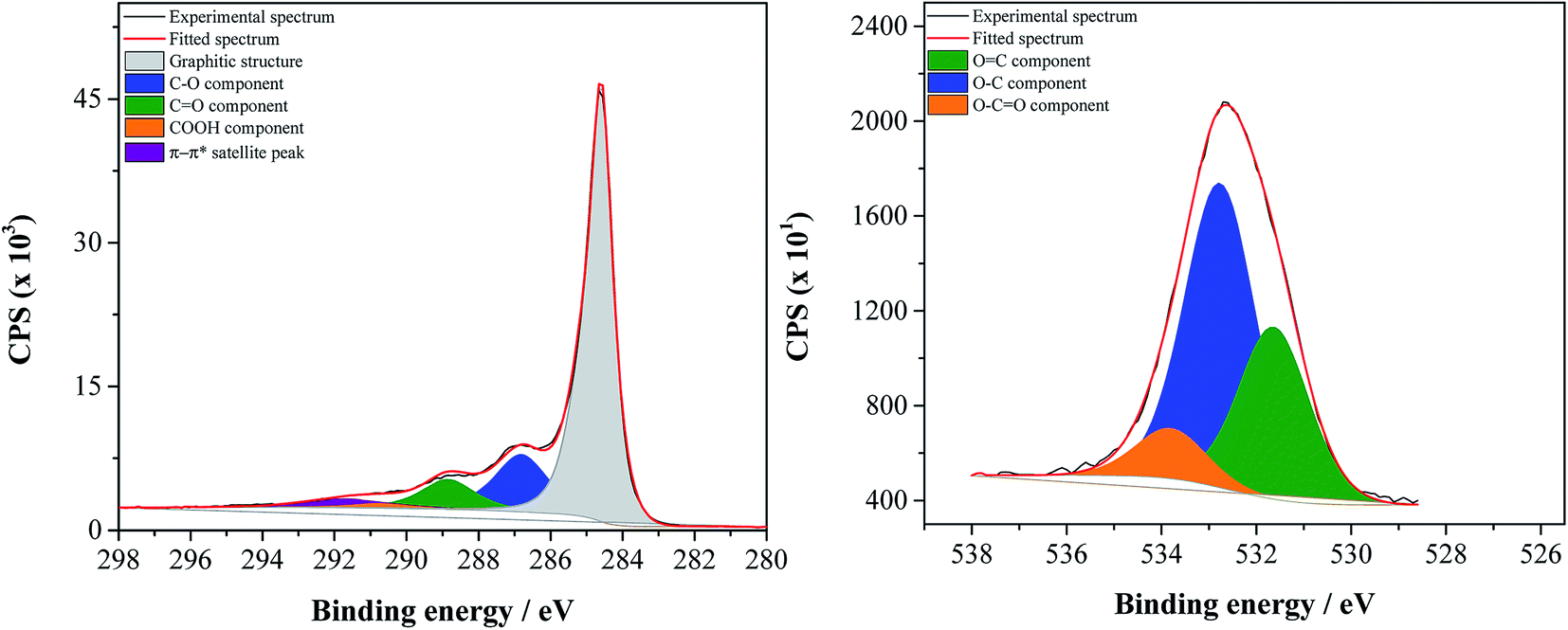 | ||
| Fig. 1 Deconvoluted C1s (on the left) and O1s (on the right) high resolution spectra of GF_KMnO4. | ||
C1s high resolution spectra of all materials were deconvoluted into five peaks: a main peak at 284.6 eV related to the graphitic structure (sp2), a peak at 286.2 eV attributed to C–O in hydroxyls, epoxy groups and phenols, a peak at 287.3 eV associated to CO in ketones, aldehydes and quinones, a peak at 288.9 eV, attributed to O–CO in carboxylic acids, carboxylic anhydrides and esters, and the π–π* shake-up satellite peak from the sp2-hybrized carbon atoms at 290.3 eV.40–45
All spectra of the oxidized graphene flakes present a decrease of the relative intensity of the π–π* satellite peak, corroborating that the graphene oxidation originated a decrease in the electronic delocalization; this decrease is more accentuated for GF_KMnO4, which is the material that presented the highest degree of oxidation. Furthermore, the peaks attributed to carbon atoms bound to oxygen (C–O, CO, O–CO) of ox-GF show some variations in terms of intensity relatively to the corresponding p-GF peaks. For GF_HNO3, the relative atomic percentage of carbon attributed to O–CO in carboxylic acids increased from 1.9 to 2.6%. In the case of GF_O3_gas and GF_KMnO4, it is observed a considerable increase of the C–O peak (from 5.8 to 13.8 and 19.2% for p-GF, GF_O3_gas and GF_KMnO4, respectively).
Fig. 2 shows the relative atomic percentage of oxygen for each contribution of O1s peak components; the corresponding deconvoluted O1s high resolution spectra and the relative atomic percentages of the oxygen-based functional groups are presented in Fig. 1 for GF_KMnO4 and in Fig. S1† for the other materials, and Table S1, ESI,† respectively. For all materials, the broad band in the O1s high resolution spectra was deconvoluted into three peaks:46,47 a peak at 531.3 eV, assigned to OC in ketones, quinones and aldehydes, a peak at 532.7 eV, associated to O–C from phenol and epoxy groups, and a peak at 533.9 eV typically due to O–CO in esters and carboxylic acids.
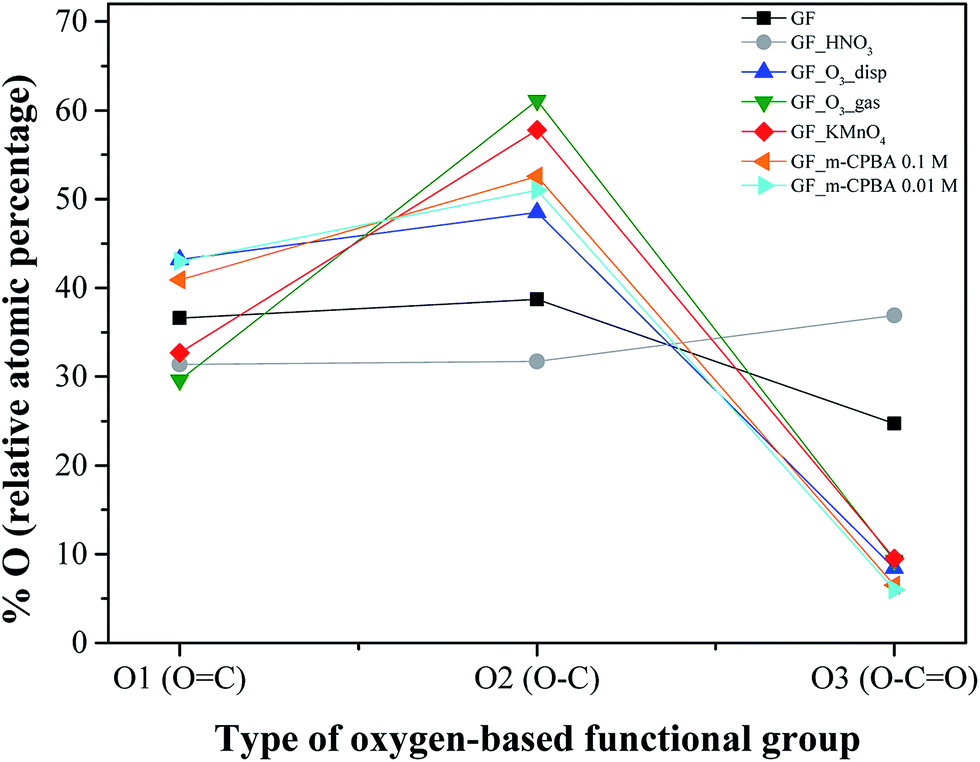 | ||
| Fig. 2 XPS relative atomic percentage of oxygen for each contribution of O1s peak components: O1, O2 and O3 components correspond to oxygen in OC, O–C and O–CO, respectively. | ||
The inspection of relative percentages of each type of oxygen groups, Fig. 2, shows for GF_HNO3 an increase in the percentage of the O3 component (attributed to O–CO), in comparison to O1 and O2 components, revealing the significant introduction of high acidic groups such as carboxylic acids, promoted by the oxidation treatment with HNO3.
In regard to GF_O3_gas and GF_KMnO4, the intensity of the peak at 532.7 eV (O–C) increased considerably relatively to the peak at 531.3 eV (OC), when compared to the intensity of the corresponding peaks of p-GF (for GF_m-CPBA and GF_O3_disp, the same trend is observed, although to a lesser extent), implying that functional groups such as epoxides, hydroxyls and phenols were introduced in a more extensive way than other functional groups such as ketones, quinones or aldehydes. These tendencies indicate that the type and the amount of oxygen groups introduced onto graphene surface are highly dependent on the type of oxidant; furthermore, the preparation form of graphene – dispersed in water or dried powdered – during the oxidation with O3 also has some influence, since the relationship between the two peaks is quite different for GF_O3_disp, in comparison with GF_O3_gas.
The content and the nature of oxygen functional groups were also assessed by TPD; it should be noted that, in the next sections, GF_m-CPBA refers to GF_m-CPBA_0.1 M. During a TPD analysis, all the oxygenated surface groups of carbon materials are thermally decomposed, releasing CO and/or CO2. CO2 evolution spectrum results from the contribution of carboxylic acids and lactones decomposition at low and high temperatures, respectively; carboxylic anhydrides originate both CO and CO2 at intermediate temperatures, and finally phenols and carbonyl/quinone groups decompose in CO at high temperatures.48,49
Fig. 3 shows the TPD spectra of p-GF and ox-GF materials. The amounts of CO2 and CO released obtained by integration of the areas under the respective TPD spectra, together with the total mass percentage of oxygen are presented in Table 3. Globally, a substantial increase in the amounts of released CO2 and CO is observed for oxidized graphene flakes, as expected. For GF_HNO3, the overall profiles of CO2 and CO spectra are similar to the TPD profiles of p-GF, although the CO2 spectrum reveals an increase of CO2 evolution from 200 to 450 °C, a temperature range ascribed to the decomposition of carboxylic acid groups, and from 450 to 650 °C, which is indicative of the presence of carboxylic anhydrides, corroborating the XPS results that revealed a high proportion of carboxylic acids. In the case of GF_KMnO4, GF_m-CPBA and GF_O3, there is a considerable increase in the amount of CO2 – specially for GF_KMnO4 and GF_O3_gas – indicating the introduction of carboxylic acid groups, carboxylic anhydrides and lactones (CO2 released from 550 to 700 °C). However, the TPD spectra of the previous oxidized GF present a distinctive feature – both CO2 and CO spectra present substantial peaks below 210 °C – that may be assigned to the decomposition of more labile groups, such as epoxyl or hydroxyl groups present in the basal plane, which is in agreement with TPD studies performed for graphite and graphene oxides by Dongil,50 Solís-Fernández51 and Kuo52 (although the precise differentiation between the temperature evolution of hydroxyl and epoxyl groups has not been yet clarified), and it is also corroborated by the work of Lerf,53 which demonstrated the elimination of epoxyl and hydroxyl groups upon calcination of graphite oxide, in vacuum at low temperature.
 | ||
| Fig. 3 TPD spectra of original and oxidized graphene flakes: (a) CO2 evolution and (b) CO evolution. | ||
| Graphene flakes | [CO2]/μmol g−1 | [CO]/μmol g−1 | % O |
|---|---|---|---|
| GF | 396 | 1608 | 3.8 |
| GF_HNO3 | 984 | 1692 | 5.9 |
| GF_KMnO4 | 3036 | 2208 | 13.2 |
| GF_m-CPBA | 1548 | 1836 | 7.9 |
| GF_O3_disp | 2424 | 1524 | 10.2 |
| GF_O3_gas | 4872 | 4368 | 22.6 |
Deconvoluted CO2 and CO spectra are shown in Fig. S2, ESI† and the detailed information resulting from the deconvolution procedure, such as the temperature of the component peak maximum, TM, and the width at half-height, W, and the integrated area of each peak component, is presented in Tables 4 and 5. For TPD spectra deconvolution, the following assumptions were generally adopted:48,49
| Graphene flake | Peak #1 | Peak #2 | Peak #3 | Peak #4 | A/μmol g−1 s−1 | A/μmol g−1 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TM/°C | W/°C | A/μmol g−1 s−1 | % rel | TM/°C | W/°C | A/μmol g−1 s−1 | % rel | TM/°C | W/°C | A/μmol g−1 s−1 | % rel | TM/°C | W/°C | A/μmol g−1 s−1 | % rel | |||
| GF | 307 ± 25 | 177 ± 37 | 5 ± 2 | 15 | 533 ± 24 | 177 ± 42 | 15 ± 7 | 45 | 702 ± 32 | 177 ± 42 | 13 ± 6 | 40 | 33 | 400 | ||||
| GF_HNO3 | 261 ± 5 | 169 ± 5 | 44 ± 3 | 54 | 444 ± 7 | 169 ± 17 | 23 ± 4 | 28 | 668 ± 7 | 169 ± 10 | 15 ± 1 | 18 | 82 | 978 | ||||
| GF_KMnO4 | 163 ± 4 | 64 ± 4 | 65 ± 9 | 26 | 323 ± 7 | 158 ± 15 | 82 ± 10 | 32 | 517 ± 13 | 158 ± 38 | 36 ± 12 | 14 | 722 ± 31 | 158 ± 42 | 12 ± 5 | 5 | 253 | 3040 |
| 203 ± 1 | 39 ± 1 | 59 ± 8 | 23 | |||||||||||||||
| GF_m-CPBA | 170 ± 0.4 | 74 ± 1 | 29 ± 1 | 23 | 262 ± 7 | 178 ± 7 | 39 ± 3 | 30 | 473 ± 3 | 178 ± 11 | 43 ± 4 | 33 | 702 ± 8 | 178 ± 10 | 17 ± 2 | 14 | 129 | 1546 |
| GF_O3_disp | 191 ± 6 | 96 ± 7 | 84 ± 35 | 42 | 291 ± 22 | 140 ± 48 | 89 ± 47 | 44 | 479 ± 30 | 140 ± 89 | 19 ± 18 | 9 | 652 ± 52 | 140 ± 59 | 9 ± 7 | 5 | 202 | 2421 |
| GF_O3_gas | 171 ± 0.3 | 71 ± 1 | 173 ± 6 | 43 | 246 ± 12 | 138 ± 8 | 116 ± 13 | 28 | 403 ± 13 | 138 ± 42 | 72 ± 42 | 18 | 531 ± 33 | 138 ± 22 | 46 ± 27 | 11 | 406 | 4877 |
| Graphene flakes | Peak #1 | Peak #2 | Peak #3 | Peak #4 | Peak #5 | A/μmol g−1 s−1 | A/μmol g−1 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TM/°C | W/°C | A/μmol g−1 s−1 | % rel | TM/°C | W/°C | A/μmol g−1 s−1 | % rel | TM/°C | W/°C | A/μmol g−1 s−1 | % rel | TM/°C | W/°C | A/μmol g−1 s−1 | % rel | TM/°C | W/°C | A/μmol g−1 s−1 | % rel | |||
| GF | 533 | 177 | 15 | 11 | 681 ± 3 | 159 ± 3 | 86 ± 3 | 64 | 836 ± 7 | 159 ± 7 | 33 ± 3 | 25 | 134 | 1613 | ||||||||
| GF_HNO3 | 444 | 169 | 23 | 16 | 593 ± 29 | 158 ± 22 | 22 ± 10 | 16 | 729 ± 7 | 158 ± 5 | 96 ± 10 | 68 | 141 | 1688 | ||||||||
| GF_KMnO4 | 140 ± 1 | 64 | 15.4 ± 0.3 | 8 | 323 | 88 | 23.6 | 13 | 517 | 157 | 36 | 19 | 687 ± 7 | 195 ± 5 | 77 ± 6 | 42 | 857 ± 46 | 195 ± 37 | 11 ± 6 | 6 | 184 | 2207 |
| 203 | 39 | 20.7 ± 0.2 | 11 | |||||||||||||||||||
| GF_m-CPBA | 170 | 74 | 17.3 ± 0.2 | 11 | 262 | 178 | 7.6 ± 0.5 | 5 | 473 | 178 | 25 ± 0.6 | 17 | 690 ± 2 | 182 ± 3 | 71 ± 2 | 47 | 896 ± 6 | 182 ± 7 | 31 ± 2 | 20 | 153 | 1832 |
| GF_O3_disp | 191 | 96 | 19.7 ± 0.4 | 16 | 291 | 140 | 14.7 ± 0.5 | 12 | 479 | 140 | 19 | 15 | 666 ± 5 | 142 ± 6 | 41 ± 3 | 33 | 817 ± 6 | 142 ± 7 | 32 ± 3 | 25 | 127 | 1520 |
| GF_O3_gas | 171 | 71 | 41.3 ± 0.5 | 11 | 246 | 138 | 28 ± 1 | 8 | 420 | 138 | 62 | 17 | 581 ± 3 | 171 ± 3 | 140 ± 5 | 38 | 767 ± 4 | 171 ± 5 | 93 ± 5 | 25 | 364 | 4366 |
(a) CO2 spectra results from the contribution of peak #2, peak #3 and peak #4, attributed to carboxylic acids, carboxylic anhydrides and lactones, respectively; the same width at half-height (W) was imposed for peaks #2, #3 and #4;
(b) CO spectra are decomposed into four components: peak #2, attributed to the decomposition of carbonyl groups in α-substituted ketones and aldehydes, and that has the same temperature of the component peak maximum (TM) and W as CO2 peak #2; peak #3 that results from the releasing of one CO molecule during carboxylic anhydride decomposition – this peak has the same TM, W and magnitude as the CO2 peak #3; peak #4 and peak #5, ascribed, respectively, to the decomposition of phenols and carbonyl/quinones – the W was taken the same for both peaks.
(c) In this work, and for the specific cases of the samples GF_KMnO4, GF_m-CPBA and GF_O3, an additional assumption was made: it was necessary to include a new CO and CO2 peak (peak #1), at TM < 200 °C. This peak is easily identified in both CO and CO2 spectra and with the same TM and W. The only exception was for GF_KMnO4 sample, whose spectra clearly reveal the existence of two peaks in this region.
Regarding the region below T = 200 °C, TPD spectra of GF_KMnO4, GF_m-CPBA and GF_O3_gas present CO2 and CO peaks at T = 163–171 °C, and GF_O3_disp spectra present a peak at T = 191 °C, which is close to the temperature of the second fitted peak of GF_KMnO4 spectra (T = 203 °C). Based on our results, we ascribe the peaks at the lowest temperature, T = 163–171 °C, to the decomposition of epoxyl groups (which are unstable rings and therefore decompose at lower temperatures) and the peaks at T = 190–203 °C to the decomposition of hydroxyl groups. Thus, TPD spectra of GF_KMnO4 revealed that this material has an equivalent number of epoxyl and hydroxyl groups, since the first two peaks (T = 163 °C and T = 203 °C) correspond to the decomposition of epoxyl and hydroxyl groups and the relative percentages of each peak are similar. These results are in agreement with other reported works: according to the model of graphite oxide presented by Lerf et al. in 1988,53 oxygen functionalities of GO prepared according to the Hummers method (and therefore using KMnO4 as one of the oxidizing agents) are mainly epoxyl and hydroxyl groups and Cai et al. have also demonstrated by 13C solid-state NMR that each hydroxyl is usually accompanied by an epoxyl group.25,54 Moreover, Acik et al. have shown that during thermal annealing of graphene oxide, epoxyl groups are decomposed in the range of 60–175 °C, while hydroxyl groups are only completely removed at higher temperatures, between 175 and 350 °C.55
For graphene flakes treated with 0.1 M m-CPBA solution, CO2 and CO peaks at T = 171 °C corresponding to epoxyl species decomposition were also expected, since m-CPBA is typically used to promote epoxidation of CC in alkenes.33,34
Even though the results obtained by XPS indicate the occurrence of moderate oxidation for GF_O3_gas, in the TPD spectra this material exhibited the highest amount of global released of CO2 and CO, comparable to the values observed for GF_KMnO4 (total amounts of CO2 and CO released correspond to 4872 and 4368 μmol g−1, respectively, for GF_O3_gas, and 3036 and 2208 μmol g−1, respectively, for GF_KMnO4). The majority of the CO2 and CO evolution is observed at T = 171 °C, which can be assigned to epoxyl groups. According to a theoretical study performed by Lee, ozone gas molecules adsorb on graphene basal planes and epoxyl groups can be rapidly formed if the physisorbed molecule chemically react with graphene.56
Concerning GF_O3_disp, we assign the peak at T = 191 °C to the decomposition of hydroxyl groups. Although this material was prepared with ozone, the reaction occurred in the presence of water, which promotes the breakage of epoxyl groups to give two hydroxyl groups (per epoxyl).57 In sum, the analysis of the areas and the relative percentages of each peak of deconvoluted CO2 and CO spectra (Tables 4 and 5) allow us to reach some conclusions: (i) the oxidation of GF with HNO3 exclusively promotes the introduction of carboxylic groups and carbonyl/quinones. Besides the carboxylic acids, the other oxidants also favour the formation of different oxygen functional groups: (ii) m-CPBA is responsible for the introduction of epoxyl groups and carboxylic anhydrides; (iii) GF_O3_disp presents a high content of hydroxyl groups and a moderate content of carbonyl groups in α-substituted ketones and aldehydes; (iv) GF_O3_gas presents predominantly in its structure epoxyl groups, carboxylic anhydrides, phenols, quinones and lactones, and in a lesser extension some carbonyl groups in α-substituted ketones and aldehydes; (v) finally, the oxidation of GF with KMnO4 favours the introduction of epoxyl and hydroxyl groups and also some content of carboxylic anhydrides.
The morphology of ox_GF materials was analyzed by TEM and some examples of TEM micrographs of p- and ox-GF materials are shown in Fig. 4. The panel 4a corresponds to a micrograph of original graphene flakes, while panels 4b, 4c and 4d show images of GF_HNO3, GF_KMnO4 and GF_O3_gas, respectively. TEM images of the other oxidized graphene flakes are shown in Fig. S3, ESI.† The acquired images suggest that p-GF is composed by wrinkled few-layer graphene sheets, whose sizes are in the range of hundreds of nanometres to one micrometre. In the case of the ox-GF – specially for GF_KMnO4 and GF_O3_gas – there are some regions where graphene sheets are defective (marked with a red arrow), probably due to the disorder induced by oxidation, coexisting with large and ordered flat sheets that are wrapped in some of the borders, as already reported.58–60 These wrapped sections may be caused by hydrogen bonding that may be formed between terminal groups, such as carboxyl, ketone and phenol groups within groups present in the basal planes, such as epoxy and hydroxyl groups.61
 | ||
| Fig. 4 TEM micrographs of (a) original and oxidized graphene flakes: (b) GF_HNO3; (c) GF_KMnO4 and (d) GF_O3_gas. Scale bars are 100 nm for a, b and d and 0.2 μm for c. Defective regions are marked with a red arrow. | ||
3.2. Spectroscopic characterization
Raman spectra of p- and ox-GF materials are shown in Fig. 5a. For all the materials, the corresponding spectrum displays a prominent G peak at ∼1584 cm−1, which is common to all sp2 carbon systems and corresponds to the first-order scattering of the E2g mode, and a peak at ∼1345 cm−1, ascribed to the D mode that is induced by structural disorder. In the second-order spectra of original and oxidized graphene flakes, there is a strong peak at ∼2700 cm−1, assigned as the 2D mode and that is the overtone of the D peak. At ∼1620 cm−1 it is also possible to identify a weak shoulder that corresponds to the D′ mode. The intensity ratio of the D and G bands is often used to estimate the disorder degree of graphitic materials.27,39,62,63 Fig. 5b presents the ID/IG ratio for each material. Although the ID/IG ratio remains practically constant for GF, GF_HNO3 and GF_m-CPBA, it increased considerably for GF_O3_gas, GF_KMnO4 and GF_O3_disp, revealing the disorder effect of the introduction of oxygen functionalities on the structure of graphene. In fact, since the ID/IG ratio is much higher, it can also reflect the occurrence of cracks and other structural defects during the oxidation treatment that originated this specific material. | ||
| Fig. 5 (a) Raman spectra of original (a) and oxidized graphene flakes: GF_HNO3 (b); GF_KMnO4 (c); GF_m-CPBA (d); GF_O3_gas (e); GF_O3_disp (f). (b) Calculated ID/IG ratio for original and oxidized graphene flakes. | ||
The I2D/IG ratio is shown in Fig. S4, ESI.† The 2D mode is usually related to the crystalline structure and the stacking order, and also to the number of layers of graphene.64,65 The I2D/IG ratios of GF_HNO3 and GF_KMnO4 materials are slightly higher than those of p-GF and the other oxidized materials; since HNO3 and KMnO4/H2SO4 are strong oxidants, an attack of the outer graphene layers may have occurred, leaving the oxidized graphene flakes only with the inner and more organized layers, and thus promoting an increase of the I2D/IG ratio.
Compared to the p-GF Raman spectrum, the oxidized graphene flakes, GF_O3_gas, GF_O3_disp, GF_m-CPBA and GF_KMnO4, show the Raman bands shifted to lower frequencies, being this variation more accentuated for the last two materials. As observed in Fig. 6, the shift is larger for 2D band. This shift can be caused by the existence of tension in the structure of graphene, whose origin may be related to the introduction of epoxyl and hydroxyl groups onto the basal planes of graphene. Furthermore, Ruoff et al. had firstly observed a similar relationship between the degree of oxidation of graphene oxide films and the variation of 2D peak position.66
 | ||
| Fig. 6 Raman shift of D, G and 2D peak positions for oxidized graphene flakes, in relation to the Raman shift of the corresponding peaks of original graphene flakes. | ||
FTIR spectra were also obtained to confirm the presence of oxygen functional groups in the ox-GF materials, Fig. 7. The FTIR spectrum of p-GF shows two bands: a band at 1580 cm−1, assigned to the CC stretching mode of skeletal vibrations from non-oxidized domains, and a broad band at 1220 cm−1 that results from the contribution of phenolic groups vibration modes.31 The FTIR spectra of the oxidized graphene flakes present more resolved vibration bands, since the oxidation introduces some disruption of the electronic conjugation and leads to the rupture of some π–π interactions. Besides the two vibrational bands stated above, these spectra also show typical vibrational bands of OH groups at 3430 cm−1 and 1340 cm−1, due to O–H stretching and bending modes,31,67 respectively. The spectra of GF_KMnO4 and GF_O3_disp materials present two bands at 2935 cm−1 and 2850 cm−1, assigned – respectively – to the C–H bending and stretching modes that can be due to the existence of defects in the sp2 hybridized domains. The FTIR spectra of all ox-GF materials also reveal a vibrational band at 1740 cm−1 due to the CO stretching mode of carboxylic acid, carbonyl, ketone and quinone groups.68,69 It is also worth mentioning that the band at 1220 cm−1 is considerably more intense in the spectra of the oxidized graphene flakes, when compared with the band of the parent material, which suggests the introduction of phenol/hydroxyl groups. A broad and prominent band at 1050 cm−1 also appears in the FTIR spectra of GF_m-CPBA, GF_O3_gas, GF_O3_disp and GF_KMnO4 materials that can be assigned to C–O–C and C–O stretching modes and C–O–H bending mode (in this spectral region typically occurs the overlap of several C–O vibration modes,70 and thus the attribution of C–O and C–OH is still ambiguous31,68,69,71,72).
 | ||
| Fig. 7 FTIR spectra of (a) original and oxidized graphene flakes: (b) GF_HNO3; (c) GF_m-CPBA; (d) GF_O3_gas; (e) GF_O3_disp; (f) GF_KMnO4. | ||
UV-visible absorption spectra of p-GF and ox-GF materials are shown in Fig. 8. When compared with the spectrum of p-GF, the spectra of ox-GF present a prominent peak at λ ∼ 270–280 nm that can be assigned to n → π* transition of CO bonds.73 The intensity of this band is higher for GF_KMnO4 and GF_O3_gas, which is indicative of a higher degree of oxidation and, hence, corroborates our previous results. For GF_m-CPBA, an additional band at λ = 200 nm and a shoulder at λ = 230 nm is also observed; in addition, the spectrum of GF_O3_disp shows a band at λ = 210 nm, the same region in which there is a shoulder in the spectrum of GF_HNO3. No assignment could be made to these observed bands.
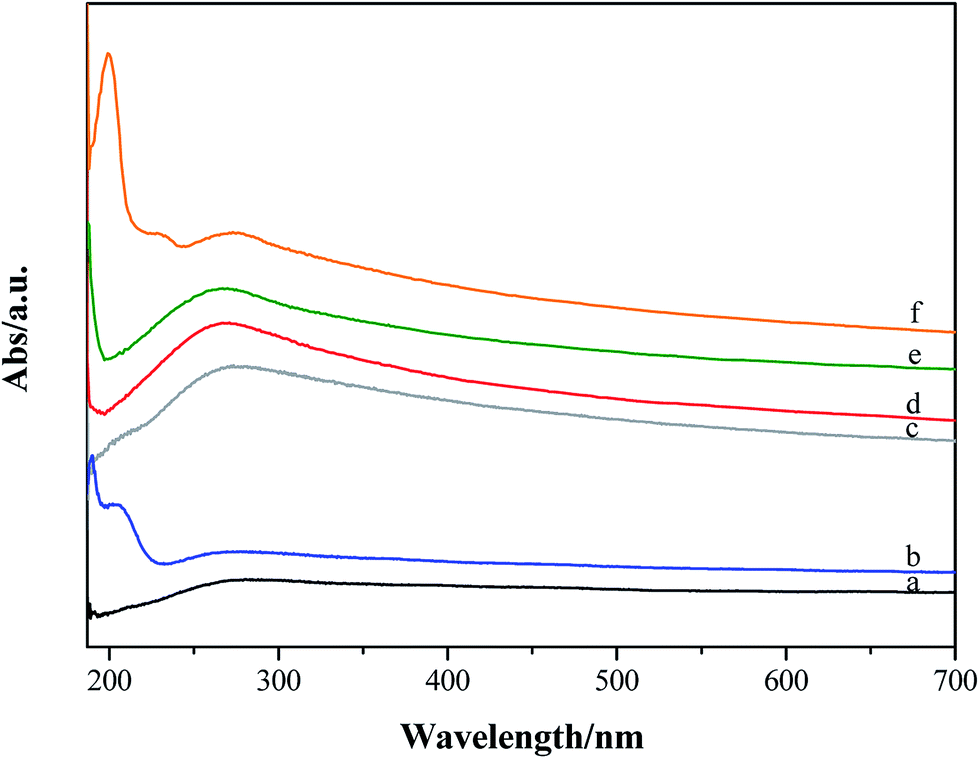 | ||
| Fig. 8 UV-visible absorption spectra of (a) original and oxidized graphene flakes: (b) GF_O3_disp; (c) GF_HNO3; (d) GF_KMnO4; (e) GF_O3_gas; (f) GF_m-CPBA. | ||
3.3. Electrochemical characterization
Voltammetric experiments in the presence of the [Fe(CN)6]3−/4− redox probe were performed. This is a standard probe which has been used to assess the influence of surface oxygen and other functional groups of several carbon materials, such as pyrolytic graphite, carbon nanotubes and graphene oxide, on the electrode kinetics.37,74,75Cyclic voltammograms of bare and modified glassy carbon electrode with original and oxidized graphene flakes in 1.0 mM K3[Fe(CN)6] solution (0.1 M KCl as supporting electrolyte) are shown in Fig. 9a. The cyclic voltammogram of the bare electrode shows the oxidation and the reduction peaks of [Fe(CN)6]3−/4− at Epa = 0.216 V and Epc = 0.105 V, respectively. In the CVs of the p- and ox-GF-modified electrodes, the intensity of the oxidation and reduction peaks (ipa and ipc, respectively) is higher than the intensity of the correspondent peaks for the bare GC electrode. Furthermore, the modification of the GC electrode surface with original and oxidized graphene flakes results in a decrease of peak-to-peak separation (ΔEp–p), as it is presented in Fig. 9b. Consequently, the decrease of the ΔEp–p is indicative of a faster electron transfer that can be quantified by the faster electron transfer constant, 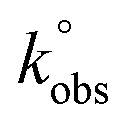 : the
: the  values, determined by the Nicholson methodology, for bare GC electrode is 4.26 × 10−5 cm s−1, while the
values, determined by the Nicholson methodology, for bare GC electrode is 4.26 × 10−5 cm s−1, while the  values for GF, GF_HNO3, GF_O3_disp are respectively: 1.04 × 10−4, 1.37 × 10−4 and 1.04 × 10−4 cm s−1, and for GF_m-CPBA, GF_KMnO4, and GF_O3_gas is 8.74 × 10−5 cm s−1.
values for GF, GF_HNO3, GF_O3_disp are respectively: 1.04 × 10−4, 1.37 × 10−4 and 1.04 × 10−4 cm s−1, and for GF_m-CPBA, GF_KMnO4, and GF_O3_gas is 8.74 × 10−5 cm s−1.
 | ||
| Fig. 9 (a) Cyclic voltammetry of original and oxidized graphene flakes-modified glassy carbon (GC) electrode in 1.0 mM K3[Fe(CN)6] solution (0.1 M KCl as supporting electrolyte); (b) peak-to-peak separation of [Fe(CN)6]3−/4− redox couple for original and oxidized graphene flakes modified-glassy carbon (GC) electrode. | ||
Interestingly, the modification of the GC electrode with original and oxidized graphene flakes leads to similar results. The existence of oxygen functional groups in graphene can be responsible for the decrease of the electron transfer of [Fe(CN)6]3−/4− redox couple due to electrostatic repulsion between [Fe(CN)6]3−/4− and the functionalized graphene surface,76 so the same trend was not expected for both original and oxidized graphene flakes. Hence, the obtained results suggest that the applied mild oxidation protocols do not affect the electrochemical behaviour of original graphene flakes.
4. Conclusions
The selective oxidation of graphene flakes was successfully achieved through the implementation of four different one-step protocols based on distinct oxidative systems. XPS results confirmed the occurrence of graphene oxidation in different degrees, with C/O atomic ratios ranging from 21.2 to 4.9. TPD and FTIR characterization also revealed that it was possible to tune the surface chemistry of graphene flakes, according to the type of oxidant: the introduction of carboxylic acid groups was predominant in the case of the oxidation reaction with HNO3, while more labile groups such as epoxyl and hydroxyl groups could be introduced in the basal planes when the oxidations were performed with KMnO4, m-CPBA and O3. Besides, the oxidation with O3 revealed that the initial state of graphene flakes is determinant since GF_O3_disp presented a high amount of hydroxyl groups and a moderate content of carbonyl groups in α-substituted ketones and aldehydes, while GF_O3_gas presented predominantly in its structure epoxyl groups, carboxylic anhydrides, phenols, quinones and lactones, and in a lesser extension carbonyl groups in α-substituted ketones and aldehydes. Moreover, this work highlighted the potentiality of TPD analysis in distinguishing epoxyl and hydroxyl groups on graphene flakes surface.The selective one-step oxidation strategies presented in this work contribute to a step forward in the rational design of graphene-based composites with potential applications in electronics and energy storage and conversion, without compromising the disruption of the electronic π delocalization of graphene and avoiding multiple, exhaustive and drastic steps involved in the preparation of GO and rGO based materials.
Acknowledgements
The work was funded by Fundação para a Ciência e a Tecnologia (FCT)/MEC under FEDER under Program PT2020 (projects UID/QUI/50006/2013-POCI/01/0145/FEDER/007265 and UID/EQU/50020/2013-POCI-01-0145-FEDER-006984) and Program FCT – UT Austin (project ref UTAP-ICDT/CTM-NAN/0025/2014). MA thanks FCT for her PhD grant (SFRH/BD/89156/2012) and support from Fundação Calouste Gulbenkian through Programa de Estímulo à Investigação 2013, OSGPS thanks FCT for the BPD grant (SFRH/BPD/97689/2013).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- M. Inagaki, Y. A. Kim and M. Endo, J. Mater. Chem., 2011, 21, 3280–3294 RSC.
- R. Mas-Ballesté, C. Gómez-Navarro, J. Gómez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20–30 RSC.
- Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541–4583 RSC.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- S. Guo and S. Dong, Chem. Soc. Rev., 2011, 40, 2644–2672 RSC.
- N. Yang, G. M. Swain and X. Jiang, Electroanalysis, 2015, 27–34 Search PubMed.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 CAS.
- H.-X. Wang, Q. Wang, K.-G. Zhou and H.-L. Zhang, Small, 2013, 9, 1266–1283 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- X. An and J. C. Yu, RSC Adv., 2011, 1, 1426–1434 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1957, 208, 1939 Search PubMed.
- B. C. Brodie, Ann. Chim. Phys., 1860, 59, 466–472 Search PubMed.
- J.-A. Yan and M. Y. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125403–125410 CrossRef.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- U. Hofmann and R. Holst, Ber. Dtsch. Chem. Ges., 1939, 72, 754–771 CrossRef.
- G. Reuss, Monatsh. Chem., 1946, 76, 381–417 CrossRef.
- W. Scholz and H. P. Boehm, Z. Anorg. Allg. Chem., 1969, 369, 327–340 CrossRef CAS.
- T. Nakajima, A. Mabuchi and R. Hagiwara, Carbon, 1988, 26, 357–361 CrossRef CAS.
- H. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- A. Ciesielski and P. Samorì, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- Z. Xu, M. Yue, L. Chen, B. Zhou, M. Shan, J. Niu, B. Li and X. Qian, Chem. Eng. J., 2014, 240, 187–194 CrossRef CAS.
- W. Gao, G. Wu, M. T. Janicke, D. A. Cullen, R. Mukundan, J. K. Baldwin, E. L. Brosha, C. Galande, P. M. Ajayan, K. L. More, A. M. Dattelbaum and P. Zelenay, Angew. Chem., Int. Ed. Engl., 2014, 53, 3588–3593 CrossRef CAS PubMed.
- B. Lesiak, L. Stobinski, A. Malolepszy, M. Mazurkiewicz, L. Kövér and J. Tóth, J. Electron Spectrosc. Relat. Phenom., 2014, 193, 92–99 CrossRef CAS.
- D. Ogrin, J. Chattopadhyay, A. K. Sadana, W. E. Billups and A. R. Barron, J. Am. Chem. Soc., 2006, 128, 11322–11323 CrossRef CAS PubMed.
- J. Chattopadhyay, A. Mukherjee, C. E. Hamilton, J. Kang, S. Chakraborty, W. Guo, K. F. Kelly, A. R. Barron and W. E. Billups, J. Am. Chem. Soc., 2008, 130, 5414–5415 CrossRef CAS PubMed.
- R. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- C. H. A. Wong, A. Ambrosi and M. Pumera, Nanoscale, 2012, 4, 4972–4977 RSC.
- H. L. Poh, F. Šaněk, A. Ambrosi, G. Zhao, Z. Sofer and M. Pumera, Nanoscale, 2012, 4, 3515–3522 RSC.
- S. J. Konopka and B. McDuffie, Anal. Chem., 1970, 42, 1741–1746 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2009, 25, 5957–5968 CrossRef CAS PubMed.
- H. Estrade-szwarckopf, Carbon, 2004, 42, 1713–1721 CrossRef CAS.
- C. Hsieh, B. Yang and Y. Chen, Diamond Relat. Mater., 2012, 27–28, 68–75 CrossRef CAS.
- A. V. Ellis, A. Al-deen, H. Dalal and G. G. Andersson, J. Phys. Chem. C, 2013, 117, 21312–21319 CAS.
- Y. Sun, Z. Y. Wu, X. Wang, C. Ding, W. Cheng, S. H. Yu and X. Wang, Environ. Sci. Technol., 2016, 50, 4459–4467 CrossRef CAS PubMed.
- Y. Sun, C. Ding, W. Cheng and X. Wang, J. Hazard. Mater., 2014, 280, 399–408 CrossRef CAS PubMed.
- Y. Sun, X. Wang, W. Song, S. Lu, C. Chen and X. Wang, Environ. Sci.: Nano, 2017, 4, 222–232 RSC.
- A. B. Dongil, B. Bachiller-Baeza, A. Guerrero-Ruiz and I. Rodríguez-Ramos, J. Catal., 2011, 282, 299–309 CrossRef CAS.
- Y. Sun, S. Yang, Y. Chen, C. Ding, W. Cheng and X. Wang, Environ. Sci. Technol., 2015, 49, 4255–4262 CrossRef CAS PubMed.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. a. Freitas and J. J. M. Órfão, Ind. Eng. Chem. Res., 2007, 46, 4110–4115 CrossRef CAS.
- A. B. Dongil, B. Bachiller-Baeza, A. Guerrero-Ruiz, I. Rodríguez-Ramos, A. Martínez-Alonso and J. M. D. Tascón, J. Colloid Interface Sci., 2011, 355, 179–189 CrossRef CAS PubMed.
- P. Solís-Fernández, R. Rozada, J. I. Paredes, S. Villar-Rodil, M. J. Fernández-Merino, L. Guardia, A. Martínez-Alonso and J. M. D. Tascón, J. Alloys Compd., 2012, 536, S532–S537 CrossRef.
- S.-L. Kuo, W.-R. Liu, C.-P. Kuo, N.-L. Wu and H.-C. Wu, J. Power Sources, 2013, 244, 552–556 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 5647, 4477–4482 CrossRef.
- L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 5672–5676 CrossRef CAS PubMed.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- G. Lee, B. Lee, J. Kim and K. Cho, J. Phys. Chem. C, 2009, 113, 14225–14229 CAS.
- F. A. Carey, Organic Chemistry, McGraw-Hill, University of Virginia, 4th edn, 2000 Search PubMed.
- Y. Sun, Q. Wang, C. Chen, X. Tan and X. Wang, Environ. Sci. Technol., 2012, 46, 6020–6027 CrossRef CAS PubMed.
- Y. Sun, S. Yang, C. Ding, Z. Jin and W. Cheng, RSC Adv., 2015, 5, 24886–24892 RSC.
- Y. Sun, X. Wang, Y. Ai, Z. Yu, W. Huang, C. Chen, T. Hayat, A. Alsaedi and X. Wang, Chem. Eng. J., 2017, 310, 292–299 CrossRef CAS.
- J. Guerrero-Contreras and F. Caballero-Briones, Mater. Chem. Phys., 2015, 153, 209–220 CrossRef CAS.
- H. Kang, A. Kulkarni, S. Stankovich, R. S. Ruoff and S. Baik, Carbon, 2009, 47, 1520–1525 CrossRef CAS.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS PubMed.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and a. K. Geim, Phys. Rev. Lett., 2006, 97, 1–4 Search PubMed.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- F. Lorestani, Z. Shahnavaz, P. Mn, Y. Alias and N. S. A. Manan, Sens. Actuators, B, 2015, 208, 389–398 CrossRef CAS.
- C. K. Chua and M. Pumera, Chemistry, 2014, 20, 1871–1877 CrossRef CAS PubMed.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- E. Fuente, J. A. Menéndez, M. A. Díez, D. Suárez and M. A. Montes-Morán, J. Phys. Chem. B, 2003, 107, 6350–6359 CrossRef CAS.
- G. Eda, J. Ball, C. Mattevi, M. Acik, L. Artiglia, G. Granozzi, Y. Chabal, T. D. Anthopoulos and M. Chhowalla, J. Mater. Chem., 2011, 21, 11217–11223 RSC.
- V. H. Pham, T. V. Cuong, S. H. Hur, E. Oh, E. J. Kim, E. W. Shin and J. S. Chung, J. Mater. Chem., 2011, 21, 3371–3377 RSC.
- Z. Luo, Y. Lu, L. A. Somers and A. T. C. Johnson, J. Am. Chem. Soc., 2009, 131, 898–899 CrossRef CAS PubMed.
- X. Ji, C. E. Banks, A. Crossley and R. G. Compton, ChemPhysChem, 2006, 7, 1337–1344 CrossRef CAS PubMed.
- A. Chou, T. Bocking, N. K. Singh and J. J. Gooding, Chem. Commun., 2005, 842–844 RSC.
- S. M. Tan, A. Ambrosi, C. K. Chua and M. Pumera, J. Mater. Chem. A, 2014, 2, 10668–10675 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28868e |
| This journal is © The Royal Society of Chemistry 2017 |