DOI:
10.1039/C6RA28865K
(Paper)
RSC Adv., 2017,
7, 11840-11851
The optimization of donor-to-acceptor feed ratios with the aim of obtaining black-to-transmissive switching polymers based on isoindigo as the electron-deficient moiety†
Received
30th December 2016
, Accepted 30th January 2017
First published on 20th February 2017
Abstract
Isoindigo (iI)-containing donor (D)–acceptor (A)-type polymers have rarely been used as electrochromic materials although their applications in the field of organic photoelectric devices have achieved significant progress in only a few years. Three conjugated polymers, namely, P1–P3, were synthesized via the random copolymerization of three units, including thiophene and 3,3-bis-decyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (ProDOT-decyl2) as the D units, and alkyl-substituted isoindigo as the A unit, and the reactions were conducted by the Stille polymerization method. The results showed that the feed ratio of the D units to the A unit had a substantial effect on the electrochemical properties, optical band gap, color presentation, and electrochromic parameters (changes in transmittance, response time, coloration efficiency and persistence of optical contrasts for a long period) of the conjugated polymers. Polymers were easily obtained with the feed ratios (thiophene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :iI:ProDOT) of 2:1:1, 4:1:3, and 5:1:4 for P1, P2 and P3, respectively. P1 exhibits a cyan color in the neutral state and a transmissive grey color in the oxidized state. Both P2 and P3 display a change from black to transmissive on conversion from the neutral to the oxidized state. An increase in the ProDOT content was helpful for enabling the extension of the absorption peak of the polymers across the entire spectrum and also improving the switching property of the polymers. In general, the introduction of iI as the A unit for the construction of D–A-type polymers provides the field of electrochromic devices with verifiable performance and deserves further investigation to obtain more promising electrochromic materials.
:iI:ProDOT) of 2:1:1, 4:1:3, and 5:1:4 for P1, P2 and P3, respectively. P1 exhibits a cyan color in the neutral state and a transmissive grey color in the oxidized state. Both P2 and P3 display a change from black to transmissive on conversion from the neutral to the oxidized state. An increase in the ProDOT content was helpful for enabling the extension of the absorption peak of the polymers across the entire spectrum and also improving the switching property of the polymers. In general, the introduction of iI as the A unit for the construction of D–A-type polymers provides the field of electrochromic devices with verifiable performance and deserves further investigation to obtain more promising electrochromic materials.
1. Introduction
Conjugated polymers have received extensive attention since the discovery of the conductive character of polyacetylene upon doping.1,2 Significant efforts have been made by scientists to design and synthesize later models of conjugated polymers (CPs), which have superior electrical and optical properties.3,4 The modifiable structures of CPs make them fascinating candidates for a variety of applications including sensors,5 supercapacitors,6 photodiodes,7 field-effect transistors,8 photovoltaics,9 and, in particular, electrochromic materials.1–10 Electrochromic CPs are characterized by the reversible switching of the displayed colors upon tuning the difference in the applied voltage. As electrochromic materials, many of their excellent properties have provided them with valuable uses, and the merits of CPs include high solubility in common organic solvents, good film processing ability, strong optical contrasts, fast response times, and fine-tuning of colors via structural modification.11 At present, a large number of polymers have been designed and synthesized, which include polypyrrole and its derivatives,12 polythiophene and its derivatives and polyaniline, etc.13,14 Differences in the electrochromic properties of the polymers may arise from differences in terms of their key physical parameters, e.g., molar absorptivities, energy levels of the frontier molecular orbitals and geometries, which control the essential attributes of CPs such as their redox behavior and the generation of polarons or bipolarons.11
As is generally known, the band gap is the difference in energy between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO),13,15 which determines the intrinsic optical properties of electrochromic polymers. According to a previous report, the band gap of CPs with donor–acceptor configurations can be effectively tuned owing to the occurrence of resonance and intramolecular charge transfer,13 by which the donor unit can elevate its HOMO energy level and the acceptor unit can lower its LUMO energy level, respectively. Thiophene and its derivatives, e.g., 3,4-dialkoxythiophene (DalkOT), 3-alkylthiophene, propylenedioxythiophene (ProDOT), 3,4-ethylenedioxythiophene (EDOT), etc., are usually used as the donor unit.11 Moreover, electron-deficient units are frequently used as the acceptor unit, incorporating benzothiadiazole (BTD), benzimidazole, benzotriazole, quinoxaline or pyridopyrazine as the acceptor.11
CPs with a D–A configuration usually exhibit double distinct absorption bands, which can be tuned by changing the compositions of the moieties in the polymer backbone, in particular, the molar ratios of the donor units to the acceptor units.16 In ideal circumstances, “merging” of the bands could be anticipated, which suggests the possible availability of neutral black polymers (that switch to a transmissive state in the oxidized state), which have remained elusive up to the present, apart from the remarkable studies conducted by Reynolds et al., Xu et al., Kim et al., and Cihaner et al.17–22
Reynolds et al. selected three units as precursors for polymerization, namely, 2,5-dibromo- and 2,5-bis(tributylstannyl)-substituted ProDOT and 4,7-dibromo-2,1,3-benzothiadiazole (BTD), and conducted random polymerization with a series of molecular ratios. Different CPs with variable absorption profiles was obtained by the above procedure, which even included a neutral black polymer with seamless coverage of the entire wavelength range of visible light.16 A newly reported neutral-state black polymer was obtained by the copolymerization of four different units, namely, diketopyrrolopyrrole (DPP), DalkOT, ProDOT and thieno[3,2-b]thiophene units.19 With the condition that suitable monomers are chosen, electrochemical copolymerization has also been shown to be an effective way of preparing neutral-state black polymers.21,22 It is a necessary, but not sufficient, condition that an overlap in the absorption profiles of the homopolymers of the respective monomers could provide saturation coverage of the entire visible spectrum.21,22
A relatively new receptor, which is receiving increasing attention, is isoindigo (iI), which is an isomer of the commonly used dye indigo.15 The related precursor compounds are commercially available in bulk quantities, and existing synthesis facilities can be used for the further development of the materials. In view of these advantages, many D–A-type polymers that employ iI as the acceptor have been synthesized and their applications in the field of organic photovoltaics (OPVs) have been well documented.23 The absorption spectra of D–A-type polymers that contain iI are mainly determined by the structure of the electron donor units and usually display two distinct bands, of which one is located between 400 and 500 nm and the other is located between 550 and 750 nm.23 Both these bands are due to π–π* transitions: the short-wavelength band is directly related to a local excited state and the long-wavelength band can be attributed to intramolecular charge transfer.23 The optical band gaps of polymers containing iI that have been reported to date ranged between 1.87 eV for a fluorene-iI copolymer24 and 1.35 eV for a diketopyrrolopyrrole-iI copolymer.25 Lower optical band gaps are available when the electron donor units are exclusively composed of thiophene rings, including thiophene, bithiophene, terthiophene, and even their fused derivatives such as thienothiophene (TT) or dithienosilole (DTS).23
As far as we know, there are few reports concerning the employment of D–A-type polymers containing iI as electrochromic materials, not to mention the preparation of neutral black polymers based on iI. In this paper, isoindigo was selected as the acceptor unit for the preparation of D–A-type electrochromic materials, as shown in Scheme 1, in which thiophene and 3,3-didecyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (ProDOT-decyl2) were selected as the donor units with the aim of obtaining polymers with a low optical band gap. The feed ratios of the donor units (D) to the acceptor unit were modulated by adjusting the molar ratios of ProDOT to iI. It is suggested that the random copolymerization of three units provided the lowest optical band gaps among D–A-type polymers containing iI with solution-processable properties. Furthermore, the two distinct absorption bands due to π–π* transitions merged into a homogeneous broad band covering the entire visible wavelength range at specific feed ratios. Three candidates for use as polymeric electrochromic (PEC) materials exhibited moderate transmittance (ΔT%) and high coloration efficiency (CE), as well as valuable color changes, and represent fascinating candidates as active materials for the fabrication of near infrared devices (IRD).
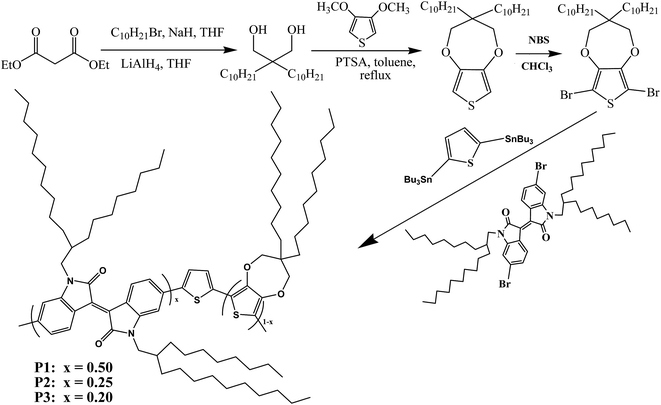 |
| | Scheme 1 Synthetic steps of monomers and polymers P1, P2, and P3. | |
2. Experimental
2.1 Materials
2,2-Didecylpropane-1,3-diol was obtained as described in a previous report.26 3,4-Dimethoxythiophene, 4A sieves, magnesium sulfate, N-bromosuccinimide (NBS, 99%), p-toluenesulfonic acid (PTSA), tetrabutylammonium hexafluorophosphate (TBAPF6, 98%), and bis(triphenylphosphine)dichloropalladium (Pd(PPh3)2Cl2, 99%) were all purchased from Aladdin Industrial Corporation and used without further treatment. Methanol, tetrahydrofuran (THF), chloroform, acetone, diethyl ether, toluene and acetonitrile (ACN) were all of analytical grade and were bought from Laiyang Fine Chemical Factory and used as received. 6,6′-Dibromo-1,1′-bis(2-octyldodecyl)-1H,1′H-[3,3′]biindolylidene-2,2′-dione and 2,5-bis(trimethylstannyl)-thiophene were both bought from Derthon Optoelectronic Materials Science and Technology Co., Ltd. Indium tin oxide (ITO)-coated glass (sheet resistance: <10 Ω sq.−1, purchased from Shenzhen CSG Display Technologies, China) was washed with ethanol, acetone and deionized water sequentially under ultrasound and then dried in a N2 flow before use.
2.2 Instrumentation
1H NMR and 13C NMR spectroscopy studies were carried out with a Varian AMX 400 spectrometer, and tetramethylsilane was used as the internal standard and deuterochloroform as the solvent. UV-vis-NIR spectra were recorded using a Varian Cary 5000 spectrophotometer connected to a computer. A self-assembly electrolytic cell was used for spectroelectrochemical analysis and was constructed from a quartz cell (10 mm × 10 mm × 45 mm). A platinum wire (Φ 1 mm) was used as the counter electrode, ITO glass, which was spray-coated with polymer films, was used as the working electrode, and the reference electrode was an Ag wire (Φ 1 mm, 0.02 V vs. SCE). The potential differences applied to the electrodes were controlled by an electrochemical analyzer (CHI 760 C, CHI Co., Ltd, Shanghai, China), which was also used for cyclic voltammetric measurements of the polymers. Spectroelectrochemical and colorimetric measurements were conducted with a Varian Cary 5000 spectrophotometer using the potential differences applied to the electrodes with the help of the electrochemical analyzer. Additional color analysis software was needed for the colorimetric analysis. Digital photographs were taken using a commercially available digital camera with 24 million pixels. The polymers were dissolved in chloroform with a concentration of 5 mg mL−1, the insoluble matter was filtered off, and the polymer solutions were reserved in a dark place for the fabrication of polymer films. The polymer films were coated on the ITO electrode by a spray-coating method using an art spray gun, and the active surface of the polymer films was 0.9 cm × 3.0 cm. The supporting electrolyte was prepared by dissolving 0.1 M TBAPF6 in ACN and was used throughout in spectroelectrochemical and kinetic switching studies.22
2.3 Synthesis procedures
2.3.1 Synthesis of 3,3-didecyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (ProDOT-decyl2). 3,3-Didecyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine was synthesized according to the previous literature,26 and the synthetic route is shown in Scheme 1. A mixed solution consisting of 15 mmol EDOT, 20 mmol diol, 1.8 mmol PTSA and 250 mL toluene was put into a single-neck round-bottom flask. Then, the flask was equipped with a Soxhlet extractor with type 4A molecular sieves in a piece of cotton cloth. The above solution was refluxed under an argon atmosphere for 24 hours. After cooling, the mixed solution was washed with deionized water in a separating funnel. The solvent was removed by distillation under vacuum. The obtained product was purified by column chromatography on silica gel using a mixed solvent (hexanes/DCM, 3:2, v/v). 1H NMR (CDCl3, 400 MHz, ppm): δ = 6.42 (s, 2H), 3.84 (s, 4H), 1.26 (m, 36H), 0.88 (t, 6H). 13C NMR (CDCl3, 101 MHz, ppm): δ = 149.69, 104.63, 77.53, 77.00, 76.68, 43.72, 29.63, 29.33, 22.68, 14.13 (see ESI Fig. S1†).
2.3.2 Synthesis of 6,8-dibromo-3,3-didecyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepine (monomer 1). Monomer 1 was synthesized according to the previous literature.26 NBS (5.34 g, 30 mmol) was dissolved in 100 mL chloroform (solution 1). ProDOT-decyl2 (5.46 g, 12.5 mmol) was dissolved in 150 mL of a mixed solvent of chloroform and acetic acid (1:1, v/v) in a three-neck round-bottom flask (solution 2). Solution 1 was added dropwise to solution 2 with stirring at room temperature. Then, the mixture was maintained at 70 °C for a further 24 hours. After cooling, the mixture was poured into an aqueous solution of sodium thiosulfate, mixed thoroughly, and extracted with chloroform, and the organic layer was rinsed with deionized water three times and dried over magnesium sulfate. The solvent was removed by rotary evaporation under reduced pressure. The crude product was purified by column chromatography with a yield of 87%. 1H NMR (CDCl3, 400 MHz, ppm): δ = 3.90 (s, 4H), 1.26 (m, 36H), 0.88 (t, 6H). 13C NMR (CDCl3, 101 MHz, ppm): δ = 147.14, 90.63, 77.98, 77.00, 76.68, 43.99, 31.65, 29.72, 22.69, 14.10 (Fig. S2 in the ESI†).
2.3.3 Synthesis of P1. Monomers, namely, monomer 1 (0.15 mmol), monomer 2 (0.15 mmol) and monomer 3 (0.30 mmol), were dissolved in 80 mL toluene, and then Pd(PPh3)2Cl2 (2.0 mol%) was added to the solution. The reaction flask was deaerated under reduced pressure, filled with argon three times and then maintained under an argon atmosphere. The reaction mixture was stirred magnetically and heated to 105 °C for 48 hours. After cooling to ambient temperature, the solvent was removed under reduced pressure. The remaining black solid was redissolved in a small amount of chloroform, and was then precipitated in methanol. Subsequently, the polymer was obtained by filtering off the methanol and was purified by Soxhlet extraction. The solvent in the subsequent process was selected from methanol, acetone and chloroform. The chloroform fraction was concentrated and precipitated in methanol. Polymer 1 was obtained by suction filtration as a black solid. P2 and P3 were prepared employing the same procedure, although the feed ratios were different from each other, as shown in Scheme 1.27,28
P1. 1H NMR (main signals) (CDCl3, 400 MHz): δ (ppm) 9.323–8.867 (s, 1H), 7.778–6.211 (m, 11H), 4.312–3.363 (d, 8H), 2.641–2.315 (d, 1H), 2.010–1.733 (s, 1H), 1.642–0.707 (m, 107H), 0.455–0.314 (m, 1H), 0.139–0 (s, 2H) (Fig. S3a†). GPC using PMMA in THF as a standard: (Mn = 21.3k, Mw = 16.4k, PDI = 1.30).
P2. 1H NMR (main signals) (CDCl3, 400 MHz): δ (ppm) 9.310–8.830 (s, 3H), 7.813–6.152 (m, 22H), 4.236–3.423 (d, 16H), 2.546–2.394 (d, 1H), 2.000–1.763 (s, 3H), 1.721–0.585 (m, 220H), 0.487–0.253 (m, 3H), 0.158–0 (s, 4H) (Fig. S3b†). GPC using PMMA in THF as a standard: (Mn = 24.5k, Mw = 15.6k, PDI = 1.57).
P3. 1H NMR (main signals) (CDCl3, 400 MHz): δ (ppm) 9.322–8.836 (s, 2H), 7.864–6.230 (m, 26H), 4.343–3.414 (d, 20H), 2.631–2.377 (d, 1H), 2.013–1.759 (s, 3H), 1.704–0.609 (m, 284H), 0.499–0.257 (m, 4H), 0.123–0 (s, 6H) (Fig. S3c†). GPC using PMMA in THF as a standard: (Mn = 23.6k, Mw = 17.2k, PDI = 1.37).
3. Results and discussion
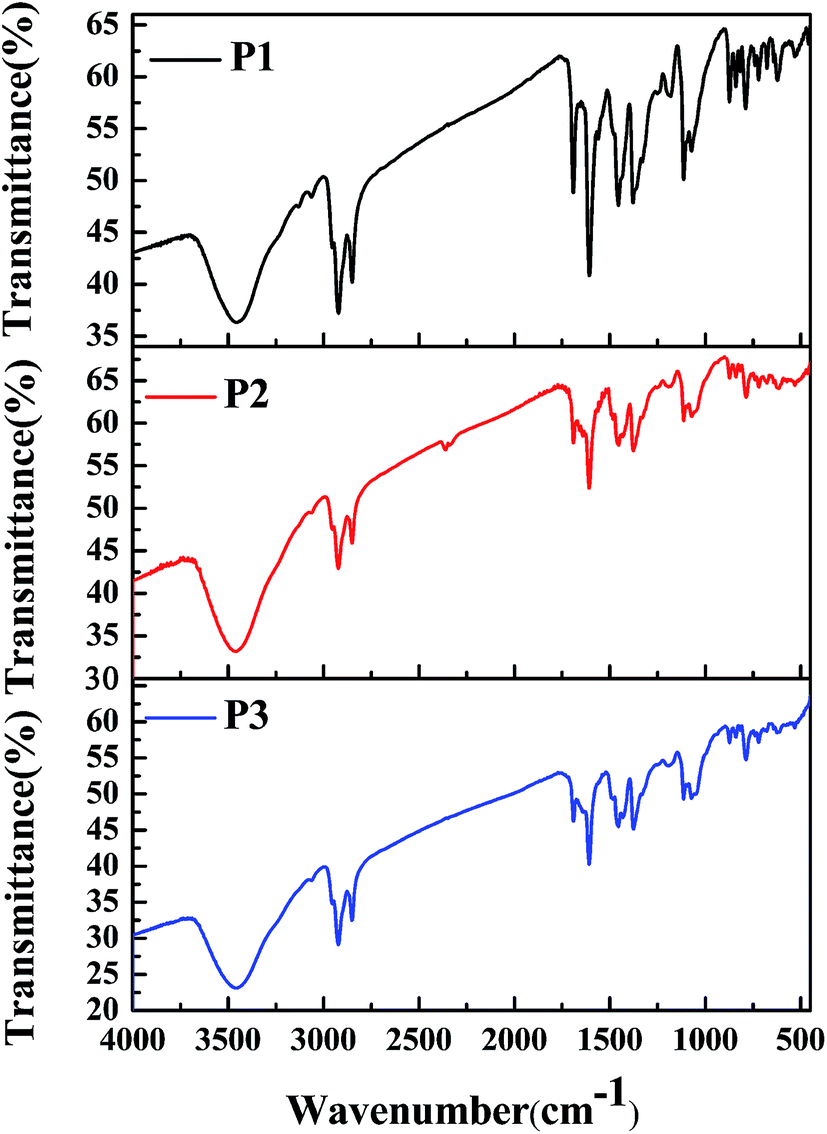
3.1 FT-IR spectra of polymers
To characterize the compositions of the polymers, their FT-IR spectra were recorded, and the results are shown in Fig. 1. The absorption profiles and peak locations of the polymers were nearly the same as each other, although the relative strengths of the specific absorption peaks varied, which was due to the different feed ratios of the monomers in the different polymers. Taking P1 as an example, its infrared absorption peaks were consistent with the molecular composition of the polymer backbone. Specifically, the intense absorption peaks located between 720 cm−1 and 872 cm−1 in the fingerprint region were due to the out-of-plane bending vibrations of C–H bonds on the benzene or thiophene rings. The absorption peaks located at approximately 1072 cm−1, 1112 cm−1, and 1182 cm−1 were due to the stretching vibrations of C–O, C–N, and C–S single bonds, which were present in monomer 1, monomer 2, and monomer 3, respectively.29,30 The absorption peaks located at approximately 1377 cm−1 and 1456 cm−1 were due to the bending vibrations of methyl and methylene groups, which were present in the alkyl side chains of monomer 1 and monomer 2, respectively. The absorption peaks at approximately 1607 cm−1 to 1692 cm−1 were caused by the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or CO bonds or benzene rings, which were present in monomer 2. Furthermore, the absorption peaks at approximately 2851 cm−1 and 2922 cm−1 could be attributed to the stretching vibrations of the saturated C–H bonds in the alkyl side chains in both monomer 1 and monomer 2. Besides, the absorption peak at 3458 cm−1 was due to the stretching vibrations of the unsaturated C–H bonds on the benzene (monomer 2) or thiophene rings (monomer 3). According to the above analysis, it could be concluded that the target polymers were obtained as expected.
C or CO bonds or benzene rings, which were present in monomer 2. Furthermore, the absorption peaks at approximately 2851 cm−1 and 2922 cm−1 could be attributed to the stretching vibrations of the saturated C–H bonds in the alkyl side chains in both monomer 1 and monomer 2. Besides, the absorption peak at 3458 cm−1 was due to the stretching vibrations of the unsaturated C–H bonds on the benzene (monomer 2) or thiophene rings (monomer 3). According to the above analysis, it could be concluded that the target polymers were obtained as expected.
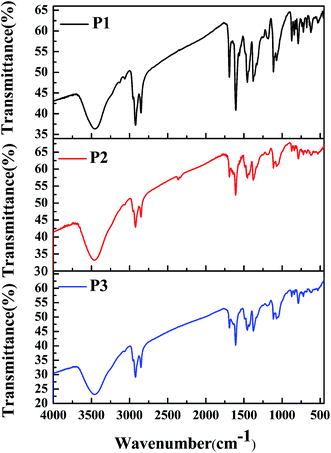 |
| | Fig. 1 FT-IR spectra of P1, P2, and P3. | |
3.2 Electrochemical characterization
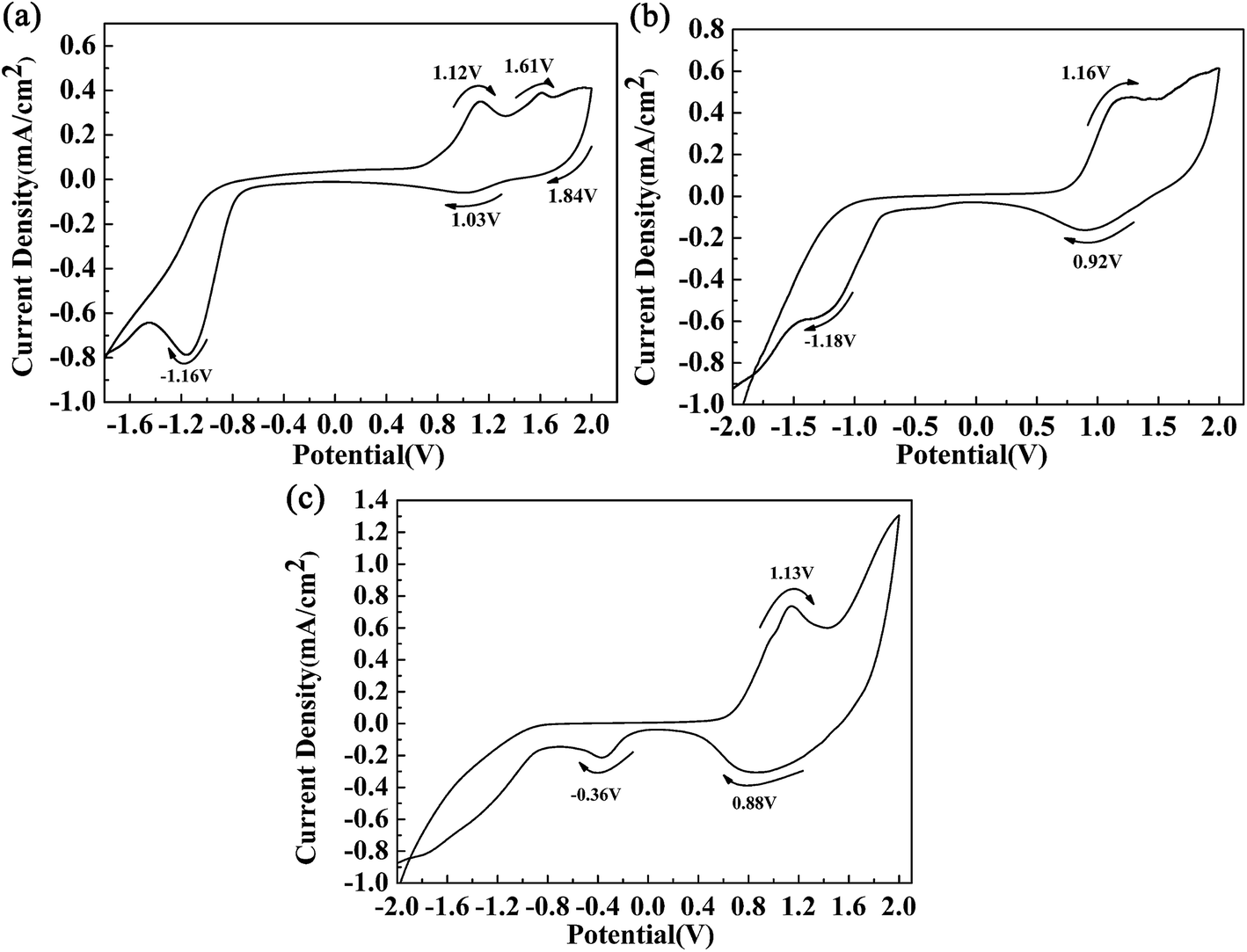
Fig. 2 shows the respective CV curves of the polymers between −2 V and 2 V at a potential scan rate of 100 mV s−1. A polymer-coated ITO electrode was used as the working electrode, and the electrolyte used was 0.2 M TBAPF6 dissolved in ACN. During the p-doping process of P1, two redox peaks were observed at 1.12 V vs. 1.03 V, and an additional oxidation peak was also found at 1.61 V. The peaks located at 1.12 V and 1.61 V were related to the occurrence of polarons and bipolarons, respectively. The peak located at 1.84 V was an irreversible reduction peak corresponding to an oxidation peak at a higher potential, and at this oxidation potential bipolarons were dispersed throughout the polymer backbone. Polymer P1 also had characteristics of n-type doping, which was indicated by the presence of an obvious reduction peak located at −1.16 V. Similar CV profiles were also found for the other polymers. The redox peaks caused by the p-doping process were located at 1.16 V vs. 0.92 V and 1.13 V vs. 0.88 V for P2 and P3, respectively. Moreover, the n-type doping characteristics of the polymers were also confirmed by the presence of reduction peaks located at −1.18 V for P2 and −1.20 V for P3, which were observed in the negative scan. During the positive scan, the films were oxidized gradually owing to the formation of radical cations including polarons and bipolarons.11 In the intermediate potential intervals, for example, −0.75 V to 0.76 V for P1, the polymers were in a neutral state and carried no charge. At a potential of −1.16 V for P1, electrons were transferred from the electrode to the polymer and were then delocalized on the polymer backbone, which caused changes in the energy levels of the polymers. The oxidation onset potentials (Eonset) of the polymers were determined by taking the intersection of the baseline and the tangent to the oxidation peak, and their values were 0.70 V, 0.65 V, and 0.57 V for P1, P2 and P3, respectively.11 It was observed that the donor-to-acceptor feed ratios of the polymers were related to the Eonset values of the polymers. Obviously, the feed ratios of the donor units to the acceptor unit of the polymers were 3, 7 and 9 for P1, P2 and P3, respectively. An increase in the proportion of donor units, in particular the ProDOT-decyl2 unit, had a beneficial effect by improving the electron-donating ability of the donor unit within the polymer.4 Hence, we can see that an increase in the feed ratio of the donor units to the acceptor unit could reduce the Eonset potential of the polymers. According to the donor–acceptor theory, push–pull action between the donor and acceptor units could promote intracellular transfer and increase the π character of double bonds along the backbone of the polymers.31 As a result of orbital hybridization, an electron donor group can increase the energy of the highest occupied molecular orbital (HOMO), which makes oxidation easy to achieve and could account for the reduction in the values of Eonset as the proportion of the donor unit was increased.32
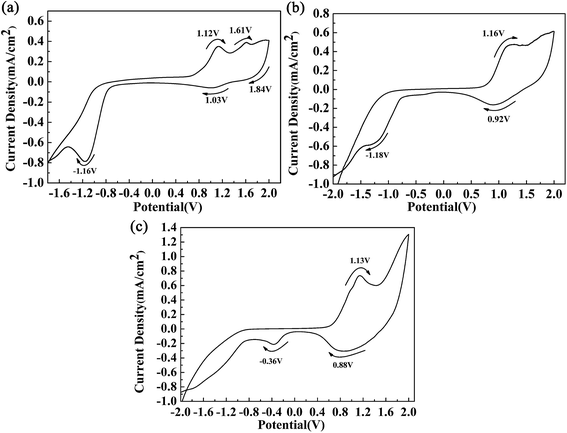 |
| | Fig. 2 CV curves of polymer films of P1 (a), P2 (b) and P3 (c). | |
3.3 Morphology
In order to describe the surface and bulk morphologies of the polymer films clearly, scanning electron microscopy (SEM) was employed, and the polymer films were also prepared using a spraying method. SEM images of P1, P2, and P3 are shown in Fig. 3. The morphology of P1 is shown in Fig. 3a, which displays a texture like stripes on the surface of the film, which indicates that the particles were isolated from each other. Nanoparticles with flake-like shapes were uniformly distributed on the polymer surface with extremely irregular shapes. The morphologies of the two other polymer films were very similar to that of polymer P1, which might be because the same monomers were used for the preparation of the polymers. There is no definite relationship between the morphology of a polymer film and its electrochromic properties. Nevertheless, the nanostructured smooth morphology of the polymer films would be beneficial for improving their overall electro-optical performance.4
 |
| | Fig. 3 SEM images of films of P1 (a), P2 (b), and P3 (c). | |
3.4 Optical properties of copolymer films and solutions
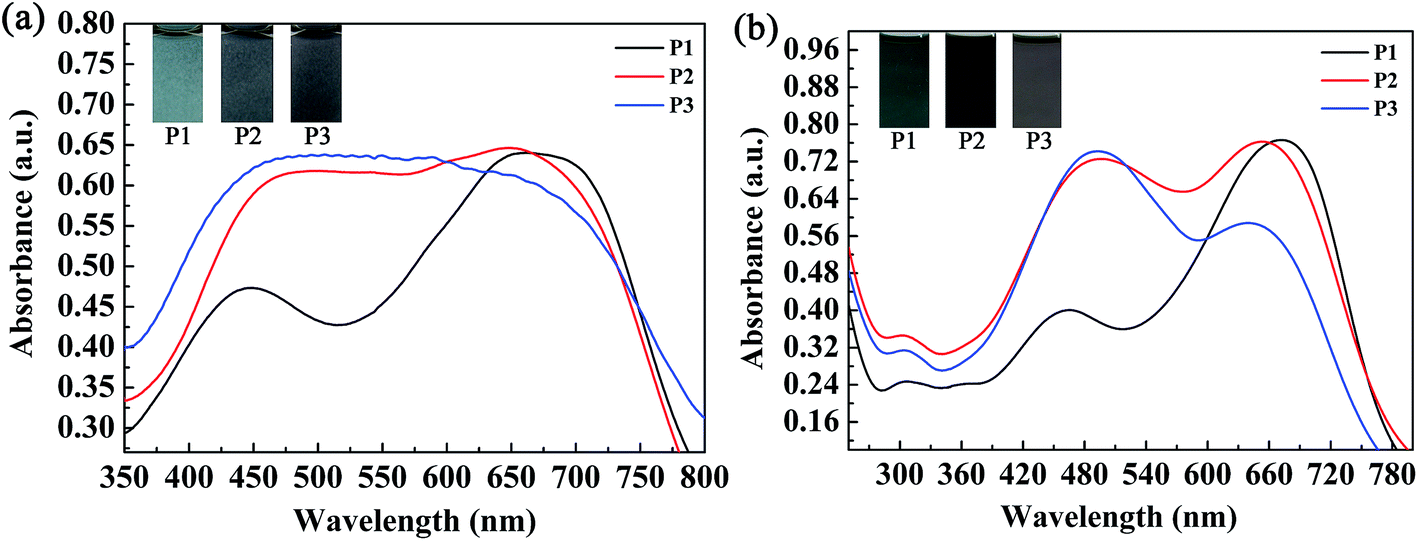
The absorption spectra of the three copolymers in the form of films and solutions were recorded with a spectrophotometer, and the obtained spectral profiles are presented in Fig. 4. The polymer films were obtained by a spraying method, as mentioned above. The polymer solutions were obtained by dissolving the polymers in chloroform with appropriate concentrations. As shown in Fig. 4a, two well-separated well-defined absorption peaks were found at 448 nm and 663 nm for the P1 film, which corresponded to a π–π transition and intramolecular charge transfer, respectively. For P1, the intensity of the high-energy absorption peak (448 nm) was much lower than that of its low-energy absorption peak (663 nm) because its content of the electron-rich ProDOT-decyl2 unit was much lower than in both P2 and P3. Taking into account the reduced absorption between 448 nm and 663 nm, a broad window for the transmission/reflection of violet, blue, cyan, green, yellow and orange light led to the grey-blue color of P1 (Fig. 4a). By increasing the percentage content of the ProDOT-decyl2 unit, the relative intensities of the two bands were reduced. For the polymers P2 and P3, merging of the two absorption bands was observed, which resulted in a homogeneous absorption covering the entire visible spectrum (420–720 nm). Consequently, polymers P2 and P3 exhibited a similar black color owing to their seamless coverage of the entire visible spectrum (Fig. 4a). The absorptions of the three polymers in solution were also recorded, as shown in Fig. 4b. Two distinct absorption bands were observed for the three polymers, which represented short-wavelength and long-wavelength optical transitions, respectively. The absorption peaks were located at 462 nm and 671 nm for P1, 488 nm and 652 nm for P2, and 490 nm and 637 nm for P3, respectively.28 In comparison with the polymer films in the solid state, the feed ratios of the donor units to the acceptor unit had a more profound effect on the relative absorption intensity. It was very interesting to find that there was a regular change regarding the relative absorption intensities of the two distinct absorption bands of the three polymers. For P1, the intensity of the former absorption band was much lower than that of the latter band, and the absorption intensities were comparable to each other for a solution of polymer P2. However, for a solution of polymer P3, the latter absorption band was somewhat weaker than the former band, in contrast to the case for P1. The weak absorption band of the P1 solution between 400 and 550 nm allowed some purple, blue and cyan-colored light to be transmitted through the solution, and therefore the P1 solution exhibited a cyan color because human eyes are more sensitive to cyan than purple and blue light (Fig. 4b). As for the P2 solution, its absorption band uniformly covered the entire visible region between 400 nm and 750 nm, which meant that the P2 solution exhibited a saturated black color. With respect to the P3 solution, there was a weak transmission window between 600 nm and 750 nm, which allowed perceptible red light to be transmitted through the solution, and therefore the solution exhibited a dark gray color.30 There was some difference in UV-vis absorption between the films and the solutions of the polymers, which was mainly found in the relative absorption intensities of the high-energy absorption and low-energy absorption of the polymers. The reason for this difference might arise from the difference in the state of molecular aggregation of the polymers between the film and solution states.
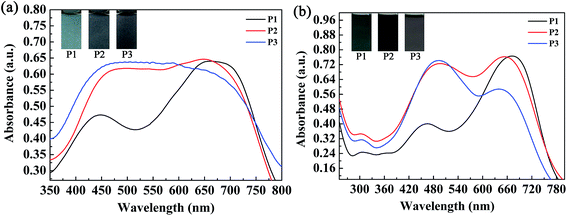 |
| | Fig. 4 Absorption spectra and images of the three copolymers in the form of films (a) and solutions (b). | |
3.5 Spectroelectrochemical properties
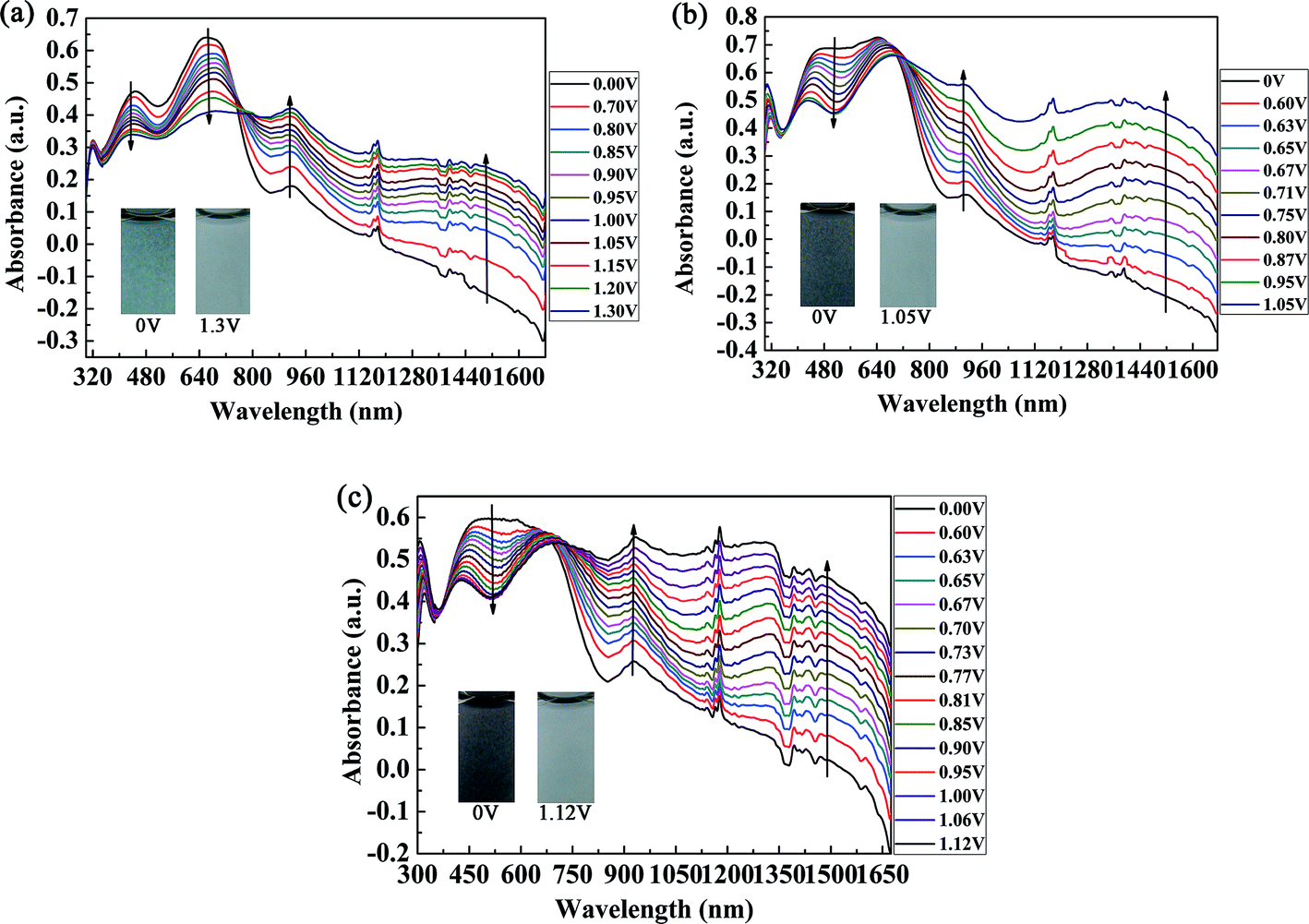
The spectroelectrochemical spectra of the polymers are shown in Fig. 5. Prior to the measurements, repeated CV measurements were conducted for each film until a stable and repeatable curve was obtained. P1 was oxidized from 0 V to 1.30 V in irregular potential steps, and the absorption spectra are shown in Fig. 5a. When the oxidation potential exceeded 0.70 V (Eonset of P1), the double bands in the visible region decreased gradually, and other two peaks due to intramolecular polarization appeared and intensified at 915 nm and 1350 nm in the near infrared region, which were due to the generation of polarons and bipolarons, respectively. Polarons and bipolarons are states of polymers formed during electrochemical oxidation, which are characterized by the presence of radical cations with different contents on the backbones of conjugated polymers; the latter was more stable than the former.30 The absorption peak in the visible region did not completely disappear in the fully oxidized state of P1, and the absorption profiles in the near infrared region also extended into the visible region. In this case, P1 switched between a cyan color and a transmissive grey color. However, for P2 and P3, as a result of the increase in electron donors in the polymer, which led to the complete filling of the low-absorption regions in the short- and long-wavelength transitions of P1, unsurprisingly, the colors of P2 and P3 were very close to black to the human eye (Fig. 5b and c). Owing to the formation of polarons and bipolarons, new absorption peaks were formed in the near infrared region, which were located at 903 nm and 1396 nm for P2 and 931 nm and 1330 nm for P3, respectively. In the fully oxidized states of P2 (1.05 V) and P3 (1.12 V), they both switched to a transmissive light gray color. The isosbestic points of the three polymers were found at 760 nm, 710 nm and 690 nm for P1, P2 and P3, respectively, where the three polymers were successfully interconverted from the neutral state to the radical cation state.31
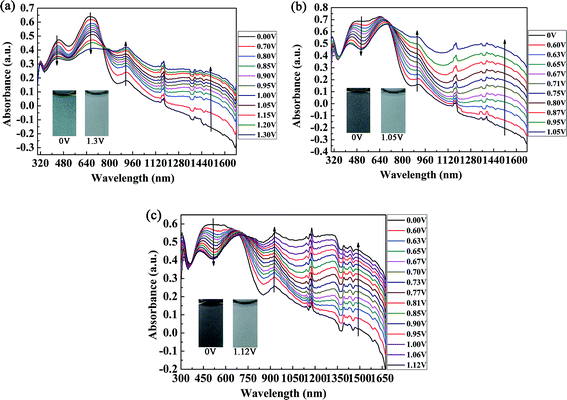 |
| | Fig. 5 Spectroelectrochemical spectra of P1 (a), P2 (b), and P3 (c) with applied potentials of between 0 V and 1.30 V, 0 V and 1.05 V, and 0 V and 1.12 V, respectively. The insets show the various colors of the neutral and oxidized states. | |
Essential parameters regarding the electrochemical and spectroelectrochemical properties of P1, P2, P3 and some donor–acceptor polymers containing isoindigo are summarized in Table 1. The parameters were calculated according to previous reports and include λonset, which was determined by taking the intersection of the baseline and the tangent to the low-energy absorption peak, the absorption peak in solution (λmax,solution) and in the film state (λmax,film), Eonset, the optical band gap (Eg) and the HOMO/LUMO energy levels of both copolymers. Eg, EHOMO and ELUMO were calculated by the formulae Eg = 1241/λonset, EHOMO = −e(Eonset + 4.71) (vs. Ag/Ag+) and ELUMO = EHOMO + Eg, respectively.
Table 1 Essential parameters of the polymers, including P1, P2, P3 and polymers containing isoindigo
| Copolymer |
λonset (film) (nm) |
λmax (solution) (nm) |
λmax (film) (nm) |
Eonset (V) |
Eg (eV) |
HOMO (eV) |
LUMO (eV) |
| Data from ref. 24. Data from ref. 33. Data from ref. 25. Data from ref. 25. |
| P1 |
889 |
671 |
663 |
0.70 |
1.39 |
−5.41 |
−4.02 |
| P2 |
905 |
652 |
646 |
0.65 |
1.37 |
−5.36 |
−3.99 |
| P3 |
979 |
490 |
482 |
0.57 |
1.27 |
−5.28 |
−4.01 |
| P(iI-T)a |
775 |
647 |
604 |
0.71 |
1.60 |
−5.85 |
−3.88 |
| P(iI-ProDOT)a |
800 |
701 |
710 |
0.58 |
1.55 |
−5.68 |
−3.91 |
| P(TT-ID)b |
786 |
684 |
694 |
0.78 |
1.58 |
−5.49 |
−3.91 |
| P(BDT-ID)c |
803 |
680 |
688 |
0.49 |
1.54 |
−5.20 |
−3.66 |
| P(iI-DPP)d |
919 |
— |
— |
0.59 |
1.35 |
−5.30 |
−4.00 |
As shown in Table 1, from P1 to P3 the increase in the feed ratios of the donor units elevated the HOMO levels of the polymers, whereas the LUMO levels of the polymers remained nearly unchanged at the same time. As a result, the Eg values decreased with the increase in donor-to-acceptor feed ratios and P3 had the lowest Eg of 1.27 eV, which is typical of a conjugated polymer with a low optical band gap. Table 1 also lists the parameters of other D–A-type polymers containing iI, including P(iI-T), the copolymer of thiophene and iI;24 P(iI-ProDOT), the copolymer of propylenedioxythiophene (ProDOT) and iI;24 P(TT-ID), the copolymer of thieno[3,2-b]thiophene and iI;33 P(BDT-ID), the copolymer of benzo[1,2-b:4,5-b′]dithiophene derivatives and iI;25 and P(iI-DPP), the copolymer of diketopyrrolopyrrole and iI.25 Importantly, among the polymers listed in Table 1, P3 had the lowest optical band gap of 1.27 eV, which was slightly lower than that of P(iI-DPP), which had been considered to have the lowest optical band gap up to the present. Studies of the electrochromic performance of D–A-type polymers containing iI have not been reported, although their applications in the fields of organic photovoltaics (OPVs) and organic field-effect transistors (OFETs) have made great progress since the first report of a polymer containing iI by Reynolds et al. in 2010.24,34 A low optical band gap is essential for polymers with excellent electrochromic properties. It was therefore necessary to study the electrochromic properties of the iI-containing polymers P1–P3 synthesized in this work.
3.6 Electrochromic switching studies
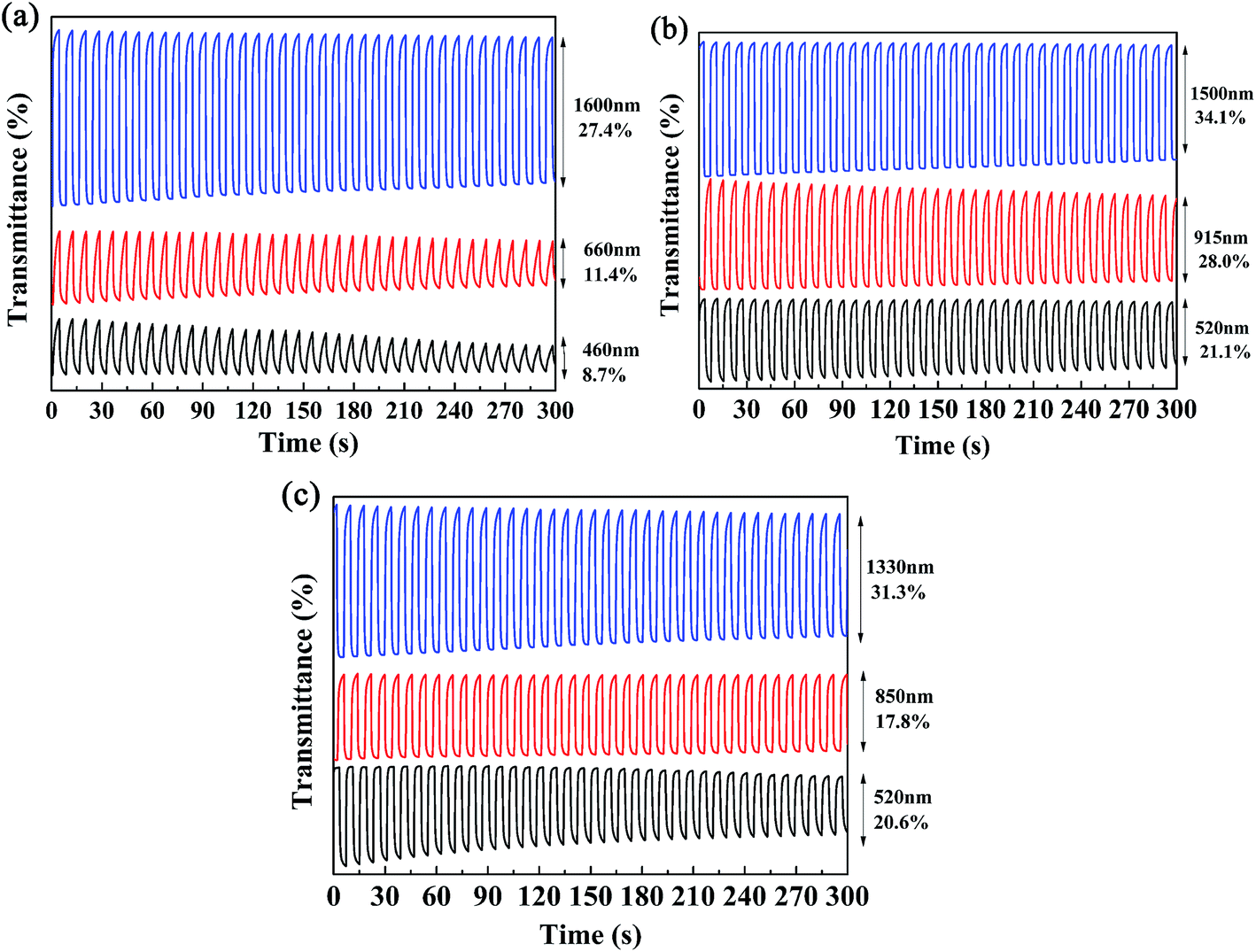
Kinetics studies were carried out by regulating the potentials applied to the polymer films via the multi-potential steps technique performed using an electrochemical analyzer, and at the same time the transmittance of light through the polymer films was monitored with a spectrophotometer.11 Then, the dynamic switching parameters of the polymers were determined by analysis of the data obtained from the kinetics studies, which included the optical contrast (ΔT%), response time (t95%), and coloration efficiency (η) of the polymers. Among these, ΔT% refers to the percentage change in transmittance between redox states at a given wavelength, which is usually recorded at the maximum absorption wavelength. The t95% value was defined as the time necessary for 95% of full optical switching when switching between the neutral and oxidized states.11 For P1, the ΔT% values were 8.7% at 460 nm and 11.4% at 660 nm, which were somewhat lower than those of P2 (21.0% at 520 nm) and P3 (20.6% at 510 nm) (Fig. 6). It is apparent that an increase in the feed ratios of the donor units to the acceptor units was beneficial for improving the ΔT% values in the visible region, which might be due to the intensification of the high-energy absorption peaks, as discussed above. Moreover, all three polymers possessed strong optical contrasts in the near infrared region, and the ΔT% values were 27.4% at 1600 nm for P1, 28.0% at 915 nm and 34.1% at 1500 nm for P2, and 17.8% at 850 nm and 31.3% at 1330 nm for P3 (Fig. 6). With respect to the response times of the polymers, as seen in Table 2 the response times decreased from P1 to P3 at the maximum absorption wavelengths in the visible region. However, the regular pattern was not observed in the NIR region. As reported in previous studies, PProDOTs-type polymers displayed the fastest response times of less than 0.6 s in the visible region. In this case, the decrease in response times from P1 to P3 in the visible region could be attributed to the increase in the percentage content of ProDOT-decyl2 units in the copolymers.35 The coloration efficiency (η) is a key parameter for evaluating the energy utilization efficiency during the switching process driven by electrical power, and is defined as the change in optical density (ΔOD) with respect to the charge consumed per unit area of the electrode (ΔQ). The equations used for calculation are given below:36,37
where Tb and Tc represent the transmittance in the neutral and oxidized states, respectively, which could be obtained from Fig. 6 and were used for the calculation of ΔOD, the change in optical density at a given wavelength. ΔQ is the amount of charge injected per unit area of the sample. Q could be determined via the integration of the electricity consumption during a cycle of performance in the multi-potential step curves, and A refers to the active area of the films. The η values of the three polymers are summarized in Table 2, from which it can be seen that P2 exhibited the best performance in terms of η values at all wavelengths, followed by P3 and P1 in succession. It is also apparent that the η values were generally higher in the NIR region than in the visible region.
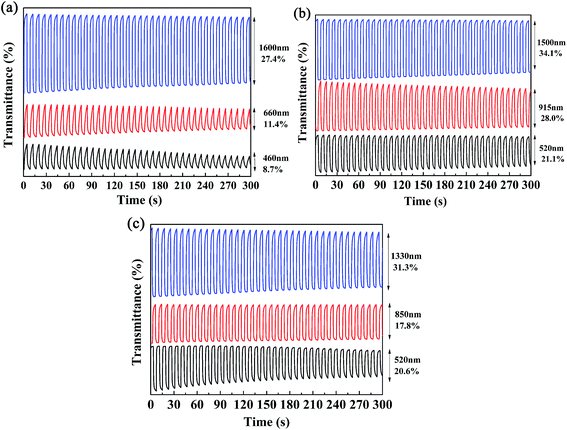 |
| | Fig. 6 Electrochromic switching of P1 (a), P2 (b), and P3 (c) at different wavelengths. | |
Table 2 Optical contrast (ΔT%), response time (t95%) and coloration efficiency (η) of copolymers
| Copolymer |
λ (nm) |
ΔT% (%) |
Response time (t95%) (s) |
Coloration efficiency (cm2 C−1) |
| P1 |
460 |
8.7 |
2.85 |
53.15 |
| 660 |
11.4 |
2.92 |
38.14 |
| 1600 |
27.4 |
0.45 |
162.49 |
| P2 |
520 |
21.0 |
1.90 |
155.97 |
| 915 |
28.0 |
1.02 |
145.61 |
| 1500 |
34.1 |
0.38 |
181.73 |
| P3 |
510 |
20.6 |
0.72 |
167.61 |
| 850 |
17.8 |
0.88 |
50.37 |
| 1330 |
31.3 |
0.77 |
96.88 |
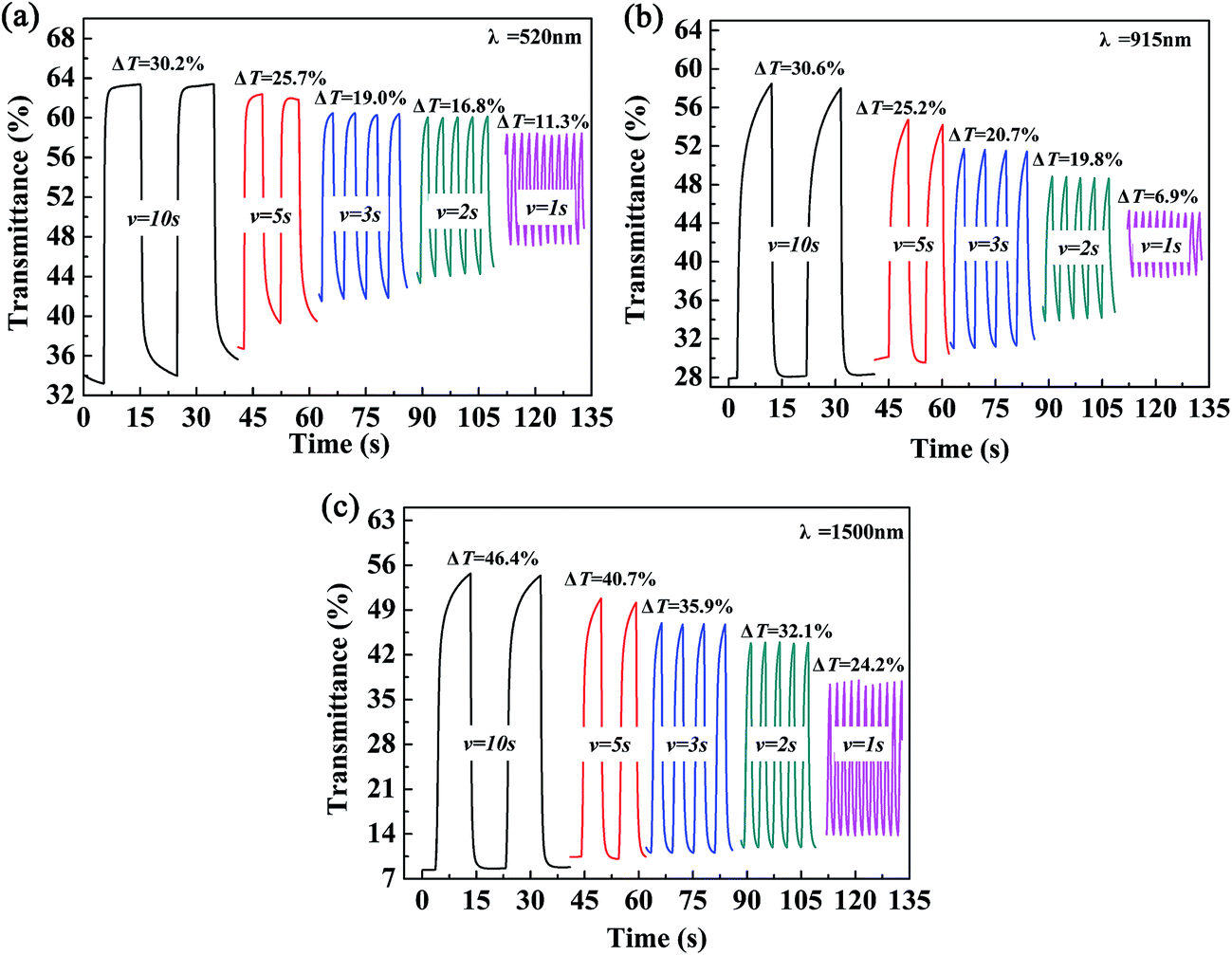
The color-switching speed of EC materials is a very important parameter that determines whether or not they can be employed in the field of display devices. In this case, the changes in ΔT% values were measured at a determined wavelength while changing the retention times in multi-potential switching to periods of 10 s, 5 s, 3 s, 2 s and 1 s. For P2, a ΔT% value of 30.2% was recorded at 520 nm (10 s), which was reduced to 25.7%, 19.0%, 16.8% and 11.3% when the switching time was decreased to 5 s, 3 s, 2 s and 1 s, respectively (Fig. 7a). At a wavelength of 915 nm, the ΔT% value changed from 30.6% to 6.9% as the switching time was decreased from 10 s to 1 s, i.e., a considerable decline of 77.5% in the change in transmittance was observed (Fig. 7b). At a wavelength of 1500 nm, the ΔT% value was reduced from 46.4% (10 s) to 24.2% (1 s), i.e., a reduction of 47.9% was observed (Fig. 7c). In summary, the ΔT% values were greatly dependent on the length of the switching time, and the changes in transmittance in the visible region were more sensitive to the switching time than those in the near infrared region. The other two polymer films, namely, P1 and P3, also followed a similar pattern to that of the P2 film, which suggested that the three polymers reported in the present study are not particularly suitable for use in display-type devices, but would find applications in the field of smart windows and other fields. The data for P1 and P3 are presented in Fig. S4 and S5,† respectively.
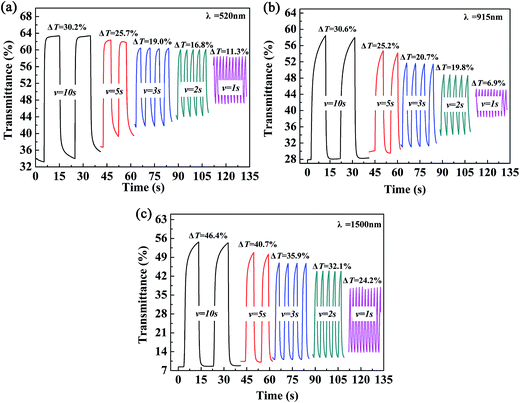 |
| | Fig. 7 Electrochromic switching of P2 (a–c) with intervals of 10 s, 5 s, 3 s, 2 s, and 1 s. | |
3.7 Colorimetry
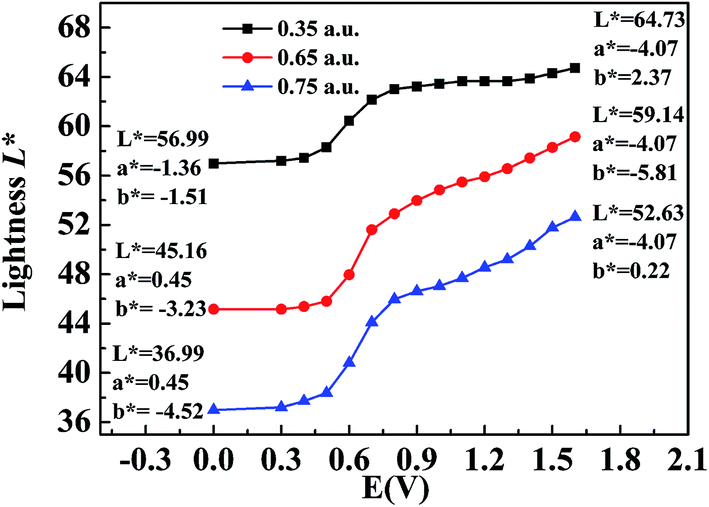
The CIE 1976 L*a*b* color space was used for identifying the color changes at various potentials and three different film thicknesses, and the results are shown in Fig. 8. L* refers to the brightness of the color on a scale from black to white, where the corresponding data range from 0 to 100, a* refers to the contrast between red and green, where a positive value means that the color tends to be red and a negative value means that the color tends to be green, and b* represents the contrast between yellow and blue, where a positive value means that the color tends to be yellow and a negative value means that the color tends to be blue.38 Fig. 8 shows the measured values of CIE 1976 L* as a function of the potential difference, which gives an indication of the relative brightness of the film as it was oxidized when illuminated by a standard D50 light source. At a potential of 0 V (vs. Ag wire), the polymer (P2) was in the neutral state and the L* values were 36.99, 45.16 and 56.99 for polymer films with absorption maxima of 0.75 a.u., 0.65 a.u., and 0.35 a.u., respectively, which ranged from the thickest film to the thinnest film (Fig. 8). The P2 film with an absorption intensity of 0.75 a.u. exhibited a saturated black color with a* and b* values of 0.45 and −4.52, respectively (Fig. 8). This phenomenon was in accordance with the absorption spectra of P2 film, which covered the entire range of the visible spectrum and no colored light was transmitted through the polymer film. With an increase in the potential difference, the L* values of the polymer increased and varied from 52.63 to 64.73 in the fully oxidized state of polymer films with different thicknesses, whereas a* and b* remained at very low values at the same time (Fig. 8). Collectively, the polymers could attain a highly transmissive state as defined by the L*a*b* color coordinates. The color changes of P1 and P3 followed a similar tendency to that of P2, and the related data are presented in Fig. S6a and b,† respectively, in the ESI.
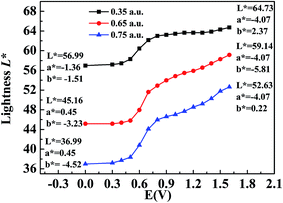 |
| | Fig. 8 L*a*b* values of P2 at applied voltages ranging from 0 V to 1.5 V. | |
3.8 Thermogravimetric analysis
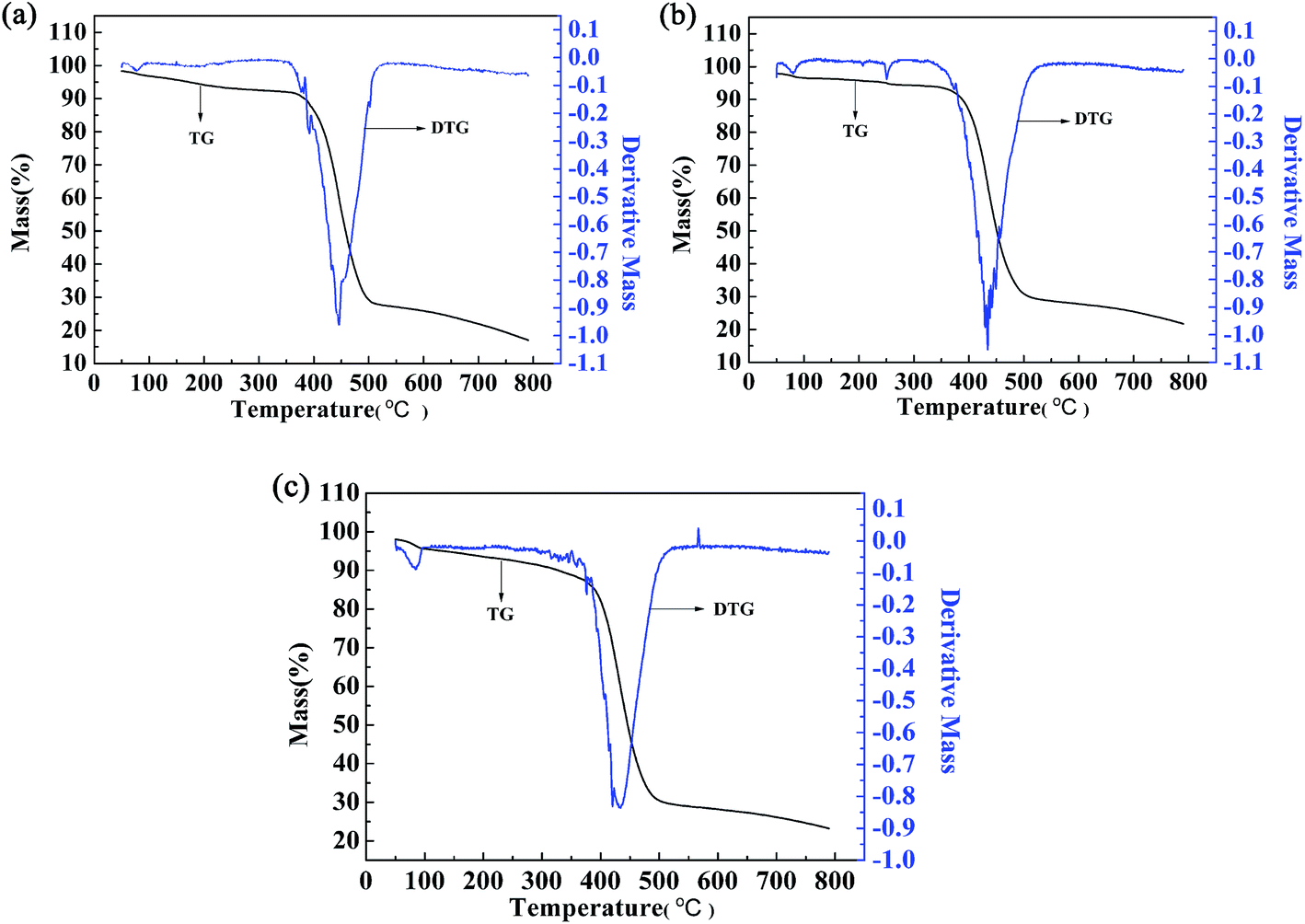
The thermal stability of the polymers was also investigated by thermogravimetric (TG) and differential thermogravimetric (DTG) methods. For polymer P2, the weight loss was only 15.61% up to 410 °C, which might be caused by evaporation of the solvent and water contained in the polymer (Fig. 9b). Significant and rapid weight loss was observed for P2 between 405 °C and 500 °C, and the maximum degradation rate occurred at 433.7 °C, as indicated by the DTG curve. The gross weight was rapidly reduced to 30.57% at 500 °C (Fig. 9b), and then the weight loss became less obvious as the gross weight remained at 22.7% up to 780 °C (Fig. 9b). From the above analysis, it is obvious that the thermal stability of P2 is sufficient to meet the requirements of the operating environment of electrochromic materials.13 The two other polymers had similar thermal stabilities to that of P2, and the data are shown in Fig. 9a and c.
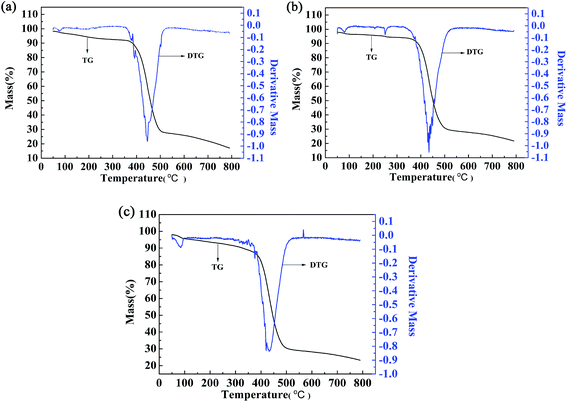 |
| | Fig. 9 Thermogravimetric analysis of P1 (a), P2 (b), and P3 (c). | |
4. Conclusions
In summary, three newly designed donor–acceptor-type conjugated polymers were prepared by the random copolymerization of three units, namely, thiophene and ProDOT-decyl2 as the donor units and isoindigo (iI) as the acceptor unit. The D–A-type polymers containing iI were used as electrochromic materials, and their related properties, including electrochemical and spectroelectrochemical properties and switching abilities, were recorded in detail. The optimization of the feed ratio of the donor units to the acceptor unit provided a powerful and effective means for the precise modulation of the absorption profiles and colorimetric characteristics of the obtained polymers in the dedoped state, which made it possible to obtain neutral black and oxidized transmissive electrochromic polymers. At a low content of ProDOT-decyl2, the obtained polymer P1 exhibited a neutral cyan color, whereas both P2 and P3 exhibited a neutral black color at a relatively high content of ProDOT-decyl2. In the oxidized state, all three polymers switched to a transmissive gray color, which means that valuable color switches are available in the form of the resulting polymers obtained in the present study. Furthermore, P3 has the lowest optical band gap among D–A-type polymers containing iI reported up to the present, in combination with other desirable properties for switches, including moderate optical contrast and a rapid color change rate, which make the polymers reported in this study outstanding candidates for use in a wide range of smart windows.
Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (51473074, 31400044) and the Natural Science Foundation of Shandong Province (ZR2013BQ021).
References
- J. A. Kerszulis, R. H. Bulloch, N. B. Teran, R. M. Wolfe and J. R. Reynolds, Macromolecules, 2016, 49(17), 6350–6359 CrossRef CAS.
- H. Yu, S. Shao, L. Yan, H. Meng, Y. He, C. Yao, P. Xu, X. Zhang, W. Huc and W. Huang, J. Mater. Chem. C, 2016, 4(12), 2269–2273 RSC.
- P. A. Ledin, J. W. Jeon, J. A. Geldmeier, J. F. Ponder, M. A. Mahmoud, M. A. El-Sayed, J. R. Reynolds and V. V. Tsukruk, ACS Appl. Mater. Interfaces, 2016, 8(20), 13064–13075 CAS.
- R. J. Mortimer, K. R. Graham, C. R. Grenier and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2009, 1(10), 2269–2276 CAS.
- D. M. Kim, S. J. Cho, C. H. Cho, K. B. Kim, M. Y. Kim and Y. B. Shim, Biosens. Bioelectron., 2016, 79, 165–172 CrossRef CAS PubMed.
- Y. Shi and G. Yu, Chem. Mater., 2016, 28(8), 2466–2477 CrossRef CAS.
- A. Falco, A. M. Zaidi, P. Lugli and A. Abdellah, Org. Electron., 2015, 23, 186–192 CrossRef CAS.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133(8), 2605–2612 CrossRef CAS PubMed.
- F. Lombeck, A. Sepe, R. Thomann, R. H. Friend and M. Sommer, ACS Nano, 2016, 10(8), 8087–8096 CrossRef CAS PubMed.
- C. G. Granqvist, Thin Solid Films, 2014, 564, 1–38 CrossRef CAS.
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23(3), 397–415 CrossRef CAS.
- L. Beverina, G. A. Pagani and M. Sassi, Chem. Commun., 2014, 50(41), 5413–5430 RSC.
- D. Zhang, M. Wang, X. Liu and J. Zhao, RSC Adv., 2016, 6(96), 94014–94023 RSC.
- G. Gunbas and L. Toppare, Chem. Commun., 2012, 48(8), 1083–1101 RSC.
- P. Deng and Q. Zhang, Polym. Chem., 2014, 5(10), 3298–3305 RSC.
- P. Shi, C. M. Amb, E. P. Knott, E. J. Thompson, D. Y. Liu, J. Mei, A. L. Dyer and J. R. Reynolds, Adv. Mater., 2010, 22(44), 4949–4953 CrossRef CAS PubMed.
- H. Shin, Y. Kim, T. Bhuvana, J. Lee, X. Yang, C. Park and E. Kim, ACS Appl. Mater. Interfaces, 2012, 4(1), 185–191 CAS.
- P. M. Beaujuge, S. Ellinger and J. R. Reynolds, Nat. Mater., 2008, 7(10), 795–799 CrossRef CAS PubMed.
- W. T. Neo, C. M. Cho, Z. Shi, S. J. Chua and J. Xu, J. Mater. Chem. C, 2016, 4(1), 28–32 RSC.
- K. R. Lee and G. A. Sotzing, Chem. Commun., 2013, 49(45), 5192–5194 RSC.
- M. Içli, M. Pamuk, F. Algı, A.
M. Önal and A. Cihaner, Org. Electron., 2010, 11(7), 1255–1260 CrossRef.
- C. Wang, M. Wang, Y. Zhang, J. Zhao and C. Fu, RSC Adv., 2016, 6(83), 80002–80010 RSC.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2013, 26(1), 664–678 CrossRef.
- R. Stalder, J. Mei and J. R. Reynolds, Macromolecules, 2010, 43(20), 8348–8352 CrossRef CAS.
- F. Grenier, P. Berrouard, J. R. Pouliot, H. R. Tseng, A. J. Heeger and M. Leclerc, Polym. Chem., 2013, 4(6), 1836–1841 RSC.
- B. D. Reeves, C. R. G. Grenier, A. A. Argun, A. Cirpan, T. D. McCarley and J. R. Reynolds, Macromolecules, 2004, 37(20), 7559–7569 CrossRef CAS.
- G. Zhang, Y. Fu, Z. Xie and Q. Zhang, Macromolecules, 2011, 44(6), 1414–1420 CrossRef CAS.
- S. V. Vasilyeva, P. M. Beaujuge, S. Wang, J. E. Babiarz, V. W. Ballarotto and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3(4), 1022–1032 CAS.
- P. W. Sadler, Spectrochim. Acta, 1960, 16(9), 1094–1099 CrossRef.
- L. Xu, J. Zhao, C. Cui, R. Liu, J. Liu and H. Wang, Electrochim. Acta, 2011, 56(7), 2815–2822 CrossRef CAS.
- S. Mi, J. Wu, J. Liu, Z. Xu, X. Wu, G. Luo, J. Zheng and C. Xu, ACS Appl. Mater. Interfaces, 2015, 7(21), 11387–11392 Search PubMed.
- J. A. Kerszulis, K. E. Johnson, M. Kuepfert, D. Khoshabo, A. L. Dyer and J. R. Reynolds, J. Mater. Chem. C, 2015, 3(13), 3211–3218 RSC.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24(10), 1762–1770 CrossRef CAS.
- T. lei, Y. Cao, Y. Fan, C. Liu, S. C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133(16), 6099–6101 CrossRef CAS PubMed.
- S. P. Mishra, K. Krishnamoorthy, R. Sahoo and A. Kuma, J. Polym. Sci., Part A: Polym. Chem., 2005, 43(2), 419–428 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Adv. Mater., 2010, 22(47), 5383–5387 CrossRef CAS PubMed.
- A. L. Dyer, C. R. G. Grenier and J. R. Reynolds, Adv. Funct. Mater., 2007, 17(9), 1480–1486 CrossRef CAS.
- B. C. Thompson, P. Schottland, K. Zong and J. R. Reynolds, Chem. Mater., 2000, 12(6), 1563–1571 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28865k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a