
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of novel F-18-labeled pyrimidine derivatives: potential FAK inhibitors and PET imaging agents for cancer detection†
Dawei Wang ,
Yu Fang,
Hang Wang,
Xingyu Xu,
Jianping Liu and
Huabei Zhang*
,
Yu Fang,
Hang Wang,
Xingyu Xu,
Jianping Liu and
Huabei Zhang*
Key Laboratory of Radiopharmaceuticals of Ministry of Education, College of Chemistry, Beijing Normal University, No. 19 Xinjiekouwai Street, Haidian District, Beijing 100875, People's Republic of China. E-mail: hbzhang@bnu.edu.cn
First published on 24th April 2017
Abstract
Based on computer-assisted drug design, a series of novel pyrimidine derivatives was successfully synthesized and characterized by 1H NMR, 13C HNMR, and MS spectra. All the new compounds were evaluated for their activity against focal adhesion kinase and showed low IC50 values in comparison with control drugs. In particular, for compound 8i, its IC50 value was 0.060 μM, suggesting its advantage as a focal adhesion kinase inhibitor. To evaluate the potentiality of these compounds as PET imaging agents in cancer detection, compounds 8a, 8c, 8h, and 8i were successively labeled with 18F. The four 18F-labeled pyrimidine derivatives showed appropriate log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values and high stability in physiological saline and mouse plasma. Noticeably, compound [18F]-8a with a 4-methoxyl group at the benzene ring exhibited good in vivo biodistribution data in mice bearing the S180 tumor, which promoted a further microPET imaging study of compound [18F]-8a. The microPET image of [18F-8a] administered into the S180 tumor-bearing mice acquired at 60 min post-injection illustrated that the uptake in S180 tumor was obvious. These results suggested that compound [18F]-8a might be a new probe for PET tumor imaging.
P values and high stability in physiological saline and mouse plasma. Noticeably, compound [18F]-8a with a 4-methoxyl group at the benzene ring exhibited good in vivo biodistribution data in mice bearing the S180 tumor, which promoted a further microPET imaging study of compound [18F]-8a. The microPET image of [18F-8a] administered into the S180 tumor-bearing mice acquired at 60 min post-injection illustrated that the uptake in S180 tumor was obvious. These results suggested that compound [18F]-8a might be a new probe for PET tumor imaging.
Introduction
Cancer is now not only the leading cause of death, but also a major public health problem all around the world.1 The increasing growth and aging of the population are the main factors for the occurrence of cancer,2 while in developed countries, it may be induced by a plethora of both external and internal factors. To date, many anticancer drugs have been marketed. The most well-known and widely clinical used type of anticancer agents is nitrogen mustard (nonspecific bifunctional DNA-alkylating agents), which has a serious drawback in that it could cause DNA damage by interrupting DNA biosynthesis. Melphalan, chlorambucil, cyclophosphamide, and bendamustine are revolutionary discoveries in the treatment of cancers. However, many drawbacks exist in these derivatives such as low specificity to tumor cells, eventual loss in activity, high chemical reactivity, and bone-marrow toxicity.3 Therefore, the development of novel anticancer drugs with high specificity, activity retention, and low chemical reactivity as well as no bone-marrow toxicity is of great emergency.Focal adhesion kinase (FAK) is a multi-domain non-receptor tyrosine kinase and scaffold protein localized to focal adhesions,4 which is uniquely positioned at the convergence point of integrins and receptor tyrosine kinase signal transduction pathways, transmitting signals from the extracellular matrix (ECM) to the cell cytoskeleton. A variety of diseases have been identified to be related with FAK, especially cancers, in which FAK is highly expressed or over-expressed at both the transcriptional and translational level. FAK signaling pathways can stimulate tumor progression and metastasis through the regulation of cell migration, invasion, ECM, and angiogenesis.5–7 Thus, this protein has emerged as a promising therapeutic target,8 and efforts to investigate FAK for an anticancer effect are under intense investigation.9 Accordingly, the inhibition against FAK is considered as an effective antineoplastic strategy by inducing apoptosis and sensitizing tumor cells to chemotherapy.10,11
TAE-226, PF-562271, and PF-573228 are the classical FAK inhibitors. TAE-226 is a novel low-molecular-weight ATP-competitive tyrosine kinase inhibitor targeting FAK with potent and selective in vitro activity (IC50 = 5.5 nM). It could inhibit the phosphorylation of FAK, the downstream oncogenic signals, the extracellular signal-related kinase, and the S6 ribosomal protein.12 TAE-226 shows impressive antitumor activity against various cancers, such as neuroblastoma,13 breast cancer,14,15 and ovarian cancer.16 However, the development of TAE-226 has been discontinued in the preclinical stage due to its severe effects on glucose metabolism in animal studies and its inhibition of insulin activity (IC50 = 44 nM).17 PF-562271, developed by Pfizer, is a dual FAK/Pyk2 inhibitor, and is identified as the first-in-class and first-in-human FAK inhibitor (FAK IC50 = 1.5 nM, PYK2 IC50 = 14 nM) being evaluated in the clinic. PF-573228 is another novel small molecule FAK inhibitor that interacts with FAK in the ATP-binding pocket and blocks the catalytic activity of recombinant FAK protein or endogenous FAK. The treatment of cells with PF-573228 blocks FAK phosphorylation on Tyr397 and concomitantly reduces the tyrosine phosphorylation of paxillin. PF-573228 provides an appropriate tool to dissect the role of FAK in the regulation of cell adhesion signaling and adhesion dynamics.18
From the above three FAK inhibitors, it can be seen that the same structural feature is the pyrimidine ring, which attracts our intensive interest in studying pyrimidine derivatives as novel FAK inhibitors related to cancers. As we know, the pyrimidine ring is a cyclic amine with two nitrogen atoms. It is the main component of many heterocyclic compounds and plays important roles in many biological processes, such as the synthesis and function of nucleic acids, several vitamins, co-enzymes, and purines, etc.19,20 Pyrimidines and their derivatives exhibit a broad spectrum of biological activities including significant in vitro activity against DNA and RNA, potential inhibition property against polio herpes viruses, and as antitumor, anti-HIV, antimicrobial, insecticidal, and antiviral agents.21,22
In view of the above observations, a series of novel pyrimidine derivatives as FAK-targeted tumor imaging agents were designed based on the following considerations:
(1) The key framework structure of TAE-226, PF-562271, and PF-573228 should remain as in Fig. 1 to keep the excellent inhibitory activity against FAK. While the strong electron-withdrawing chloro and trifluoromethyl groups are changed into a bromo moiety to explore the effects of electron density at the frame structure on the bioactivity.
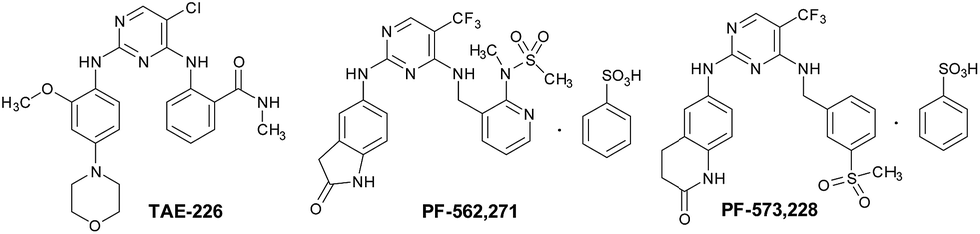 | ||
| Fig. 1 The structures of some classical FAK inhibitors. | ||
(2) The literature has reported that the aliphatic chain is usually considered to be a good substituent to regulate the solubility of target compounds. Given this, aliphatic chains at different lengths are introduced into the phenyl ring at the 2-position of pyrimidine ring in order to investigate the physicochemical characters of the designed compounds on the ability against FAK.
(3) To get relatively comprehensive good structure–activity relationships of the target compounds, another phenyl ring is modified by an electron-withdrawing or electron-donating group at different positions, such as methoxyl and carboxyl moieties (Fig. 2).
 | ||
| Fig. 2 The key framework structure of the designed novel FAK inhibitors. | ||
To make this design more rational, molecular docking between the title compounds and FAK was carried out first (see ESI†). The total score of compound 8a was demonstrated to be 6.04, and it could form two hydrogen bonds with the FAK residues GLU430 and CYS502 through the O and N atoms, which motivates our interest in further investigating its potentiality as a small molecular antitumor-agent targeting FAK (Fig. 3).
 | ||
| Fig. 3 Molecular docking of compound 8a with FAK (PDB code 4c7t): (left) the lowest energy binding modes; (right) hydrogen bond interactions. | ||
The noninvasive imaging of FAK by positron emission tomography (PET) has the ability to not only provide an innovative pharmacological approach for the diagnosis of diseases but can also contribute to a better understanding of the physiological and pathophysiological functions of FAK.23 18F is the most favorable positron emission isotope out of all the commonly used ones, which is mainly due to its longer physical half-life of 110 min, its comparable size to the H atom, and its lower positron energy.24 However, until now, only a few research studies on 18F-labeled FAK imaging agents have been reported. Therefore, there is still a need to develop an 18F-labeled FAK imaging agent with high effect and low toxicity, and for providing useful diagnostic tools to investigate diseases related to FAK, including cancer.25
In this paper, all the newly synthesized compounds were evaluated for their in vitro inhibitory activity against FAK, and some target compounds were radiolabeled by 18F. The radioactive pyrimidine derivatives were further investigated via partition coefficients determination, in vitro stability studies, and in vivo biodistribution studies in S180-bearing mice to better support their potency as PET imaging agents for cancer detection. Importantly, microPET imaging was also studied to develop new F-18 labeled PET imaging agents based on pyrimidine derivatives.
Experimental
General methods
All the chemicals and reagents used in the study were of commercial quality and were used as purchased. Proton nuclear magnetic resonance (1H NMR) spectra were acquired using a BRUKER AVANCE® III HD 400 Spectrometer in CDCl3 solutions at room temperature. Chemical shifts were given in chemical shift (δ) as parts per million (ppm) relative to tetramethylsilane (TMS), which was used as an internal standard, and were referenced to the centerline of deuterochloroform (7.27 ppm 1H NMR). The ESI-MS spectra were determined on a Waters Quattro Micro® Quadrupole Mass Spectrometer. Thin layer chromatography was performed on glass plates coated with 60 GF254 silica. Plates were visualized using UV light (254 nm). Flash column chromatography was carried out using an Interchim Puriflash® 4100 medium pressure preparative chromatography apparatus on silica gel (Bonna-Agela® flash silica, 40–60 μm, 60 Å).Truncated human FAK(PTK2) [376-1052(end) amino acids of accession number NP_722560.1] was provided by Carna Biosciences, Inc. The HTRF KinEASE®-TK kit was purchased from Cisbio Biosciences, Inc.
The [18F]fluoride used for the radiosynthesis was produced by the 18O(p, n)18F nuclear reaction by irradiation on 97% enriched H218O at the Nuclear Medicine Department of Peking Cancer Hospital (Beijing, China). QMA light ion-exchange cartridges and C-18 light Sep-Pak® cartridges were obtained from Waters (Milford, MA). We activated the Waters Sep-Pak® Accell™ Plus QMA Plus Light Cartridges (130 mg sorbent per cartridge, 37–55 μm particle size) with 1 N NaHCO3 (10 mL), followed by deionized water (10 mL), and the Waters Sep-Pak® C18 Plus Light Cartridges (130 mg sorbent per cartridge, 55–105 μm particle size) with methanol (10 mL) and deionized water (10 mL) before use. Radiopharmaceuticals were purified and their radiochemical purity were determined on a Shimadzu® LC-20AT HPLC apparatus equipped with a SPD-20A UV detector (λ = 254 nm) and Bioscan® flow count 3200 NaI/PMT γ-radiation scintillation detector. We performed the radiopharmaceutical HPLC separations on an Inertsil® ODS-3 C18 reverse phase semi-preparative column (GL Sciences, Inc. 5 μm, 10 mm × 250 mm), and we carried out the elution with a binary gradient system at a flow rate of 2.0 mL min−1. Their radiochemical purity determinations were achieved on a Kromasil® 100-5C18 reverse phase column (AkzoNobel, 5 μm, 4.6 mm × 250 mm), and elution was carried out with a binary gradient system at a flow rate of 2.0 mL min−1.
S180 ascites sarcoma mice were purchased from Beijing Vitalriver Animal Technology Co., Ltd, and normal ICR mice (20–25 g, female) were provided by Beijing Xinglong Animal Technology Co., Ltd. 18F radioactivity of the tissues and organs of interest was measured on a PerkinElmer® 2480 WIZARD2 automatic gamma counter. The microPET imaging was performed on a SuperArgus PET/CT, which was designed and manufactured by SEDECAL (Sociedad Española de Electromedicina y Calidad, S.A.). All the protocols requiring the use of mice were in accordance with the “guidelines for humane treatment of laboratory animals” promulgated by the National Health and Family Planning Commission of China, and were approved by the Animal Care Committee of Beijing Normal University.
Synthesis of intermediate 2
A mixture of ethane-1,2-diyl bis(4-methylbenzenesulfonate) (13.58 g, 36.66 mmol, 1.5 equiv.), K2CO3 (12.67 g, 91.65 mmol, 3.75 equiv.) and 18-crown-6 (0.32 g, 1.22 mmol, 0.05 equiv.) was added to a solution of 1 (3.40 g, 24.41 mmol) in acetone (30 mL). After stirring at 56 °C for 3–4 h (TLC monitoring showed that the raw material of 1 was completely consumed by that time), the reaction mixture was filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (dichloromethane to dichloromethane/MeOH, 100/1) (Caution! Using petroleum ether/AcOEt as the eluent would cause the crystallization of the product in the column, leading to blocking of the column!) to give relatively pure intermediate 2 as a light yellow solid (2.94 g). (Note: a small part of the superfluous raw material TsO–CH2–CH2–OTs was included and was unable to be completely separated from the intermediate 2, since the polarity of the raw material TsO–CH2–CH2–OTs was close to that of the intermediate 2. Therefore, the 1H NMR analysis was not performed in this step of the reaction. However, the remaining raw material TsO–CH2–CH2–OTs included in the relatively pure intermediate 2 had little effect on the next step of reaction, and the mole of 2 used in the next step of the reaction was calculated as the hypothetical mass of pure 2 divided by the relative molecular mass of 2.)Synthesis of intermediate 3
To a solution of 2 (2.94 g, 8.71 mmol) in dichloromethane (15 mL) and glacial acetic acid (15 mL) was added zinc powder (2.83 g, 43.57 mmol). The mixture was stirred at ambient temperature for 12 h under an inert atmosphere. The reaction mixture was filtered, and the filtrate was concentrated in vacuo. The crude intermediate 3 (2.02 g) was immediately used for the next step of the reaction without further purification since the intermediate 3 was unstable in air. However, according to the immediate ESI-MS analysis of the reaction mixture, we could obviously find the characteristic peak of 3: 308.2 (C15H18NO4S, [M + H]+), 330.1 (C15H17NO4SNa, [M + Na]+). Furthermore, the mole of 3 used in the next step of the reaction was also calculated as the hypothetical mass of pure 3 divided by the relative molecular mass of 3.Synthesis of intermediate 6a
Here, 4-methoxyphenyl amine (150 mmol) was added to a solution of 5-bromo-2,4-dichloropyrimidine 4 (100 mmol) in i-PrOH (200 mL) at ambient temperature. The first batch of the precipitant appeared after a few minutes of stirring at room temperature. After filtration, to the filtrate was added the solution of K2CO3 (300 mmol) in water (200 mL), and the reaction mixture was stirred at room temperature until all of the 5-bromo-2,4-dichloropyrimidine 4 was consumed completely, as shown by TLC monitoring. The resulting second batch of the precipitant was filtered and the filter cake was washed with water and ethyl acetate in sequence. After desiccation, the two batches of precipitant were combined to give the 5-bromo-2-chloro-N-phenylpyrimidin-4-amines 6a as a gray solid (16.17 g, 51.4%). 1H NMR (400 MHz, CDCl3, δ ppm): 8.25 (s, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.16 (br, 1H), 6.93 (d, J = 8.0 Hz, 2H), 3.83 (s, 3H). ESI-MS: calcd for C11H10BrClN3O ([M + H]+) 314.0, found 314.3 ([M + H]+).Synthesis of intermediate 6b
A similar reaction to that described above to prepare 6a was used to obtain 6b as a gray solid (4.9 g, 55%). 1H NMR (400 MHz, DMSO-d6) δ: 3.66 (s, 3H), 3.76 (s, 6H), 7.01 (s, 1H), 8.45 (s, 1H), 9.18 (s, br, 1H). ESI-MS: calcd for C13H14BrClN3O3 ([M + H]+) 374.0, found 373.9 ([M + H]+).Synthesis of intermediate 6c
A similar reaction to that described above to prepare 6a was used to obtain 6c as a white solid (3.5 g, 49%). 1H NMR (400 MHz, DMSO-d6) δ: 7.74 (d, J = 8.4 Hz, 2H), 7.83 (d, J = 8.8 Hz, 2H), 8.55 (s, 1H), 9.55 (s, br, 1H). ESI-MS: calcd for C11H7BrClF3N3 ([M + H]+) 351.9, found 352.1 ([M + H]+).Synthesis of intermediate 6d
A similar reaction to that described above to prepare 6a was used to obtain 6d as a light yellow solid (3.8 g, 54%). 1H NMR (400 MHz, DMSO-d6) δ: 9.51 (s, br, 1H), 8.42 (s, 1H), 7.92 (d, J = 8.4 Hz, 2H), 7.67 (d, J = 8.8 Hz, 2H), 4.29 (q, J = 6.8 Hz, 2H), 1.32 (t, J = 6.8 Hz, 3H). ESI-MS: calcd for C13H12BrClN3O2 ([M + H]+) 356.0, found 356.1 ([M + H]+).Synthesis of intermediate 10a
TBAF (36 mL, 1 M in THF) was added to a solution of ethylene di(p-toluenesulfonate), 9a (11.1 g, 30 mmol), and 80 mL THF with stirring. The mixture was refluxed overnight and the solvent was removed under reduced pressure. The residue was partitioned between water and ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and filtered. After concentration, the crude product was purified via silica gel column chromatography by eluting with ethyl acetate/hexane (1/5, v/v) to give 10a as a slightly yellow oil (3.27 g, 50% yield). 1H NMR (300 MHz, CDCl3): δ 7.85 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.2 Hz, 2H), 2.49 (s, 3H), 4.69 (t, J = 4.5 Hz, 1H), 4.53 (t, J = 4.5 Hz, 1H), 4.35 (t, J = 4.5 Hz, 1H), 4.26 (t, J = 4.2 Hz, 1H) (the 1H NMR data of 10a were identical with those in the literature26).Synthesis of intermediate 10b
10b was prepared from 9b (10.25 g, 24.7 mmol) as described above for 10a. The crude product was purified by silica gel column chromatography by eluting with ethyl acetate/hexane (3/10, v/v) to give 10b as a colorless oil (2.66 g, 41% yield). 1H NMR (300 MHz, CDCl3): δ 7.81 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 4.57 (t, J = 4.2 Hz, 1H), 4.41 (t, J = 4.2 Hz, 1H), 4.18 (t, J = 4.8 Hz, 2H), 3.69–3.76 (m, 3H), 3.63 (t, J = 4.2 Hz, 1H), 2.45 (s, 3H) (the 1H NMR data of 10b were identical with those in the literature26).Synthesis of intermediate 14a
Briefly, to a mixture of 13a (124 g, 2 mol) and triethylamine (12 g, 0.12 mol) in dry DCM (100 mL) was added TsCl (19 g, 0.1 mol) in dry DCM (100 mL) dropwise at 0 °C. The solution was stirred at 0 °C for 3 h. Then, the reaction was quenched with water. After extraction with DCM, the organic phase was washed with water two times. After removal of the solvent, the residue was purified by silica gel chromatography to give 14a (12 g, 37.1%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.81 (d, 2H, J = 8.0 Hz), 7.35 (d, 2H, J = 8.0 Hz), 4.15 (t, 2H, J = 4.0 Hz), 3.66–3.71 (m, 2H), 2.45 (s, 3H) (the 1H NMR data of 14a were identical with those in the literature27).Synthesis of intermediate 14b
A similar reaction to that described above to prepare 14a was used to obtain 14b as a colorless oil (15 g, 58%). 1H NMR (400 MHz, CDCl3): δ 7.80 (d, 2H, J = 7.8 Hz), 7.35 (d, 2H, J = 7.8 Hz), 4.20 (t, 2H, J = 4.0 Hz), 3.66–3.71 (m, 4H), 3.52–3.55 (m, 2H), 2.45 (s, 3H) (the 1H NMR data of 14b were identical with those in the literature28).Synthesis of intermediate 15a
A mixture of 14a (12 g, 0.056 mol), 4-nitrophenol (9.3 g, 0.067 mol), and K2CO3 (13.8 g, 0.1 mol) in DMF (50 mL) was heated at 75 °C overnight. The mixture was filtered and the filtrate was concentrated in vacuo. The residue was recrystallized from methanol to give 15a (4.5 g, 14%) as a light yellow solid. 1H NMR (CDCl3): δ 8.10 (d, 2H, J = 8.0 Hz), 7.01–7.06 (m, 2H), 4.09–4.14 (m, 2H), 3.89–3.92 (m, 2H) (the 1H NMR data of 15a were identical with those in the literature29).Synthesis of intermediate 15b
A similar reaction to that described above to prepare 15a was used to obtain 15b as a light yellow solid (10 g, 76%). 1H NMR (CDCl3): δ 8.10 (d, 2H, J = 8.0 Hz), 7.01–7.06 (m, 2H), 4.09–4.14 (m, 2H), 3.89–3.92 (m, 4H), 3.75–3.78 (m, 2H) (the 1H NMR data of 15b were identical with those in the literature30).Synthesis of intermediate 11a
A similar reaction to that described above to prepare 15a was used to obtain 11a as a light yellow solid (9 g, 74%). 1H NMR (400 MHz, CDCl3): δ 8.23 (d, 2H, 3J = 9.0 Hz), 7.00 (d, 2H, 3J = 9.0 Hz), 4.80 (dt, 2H, 3JH–H = 4.4 Hz, 2JH–F = 47.6 Hz), 4.32 (dt, 2H, 3JH–H = 4.0 Hz, 3JH–F = 27.6 Hz) (the 1H NMR data of 11a were identical with those in the literature31).Synthesis of intermediate 12a
A similar reaction to that described above to prepare 15a was used to obtain 12a as a light yellow solid (7.5 g, 68%). 1H NMR (300 MHz, CDCl3): δ 4.141 (dt, J = 1.0 Hz, J = 1.0 Hz, 2H), 4.715 (dt, J = 2.0 Hz, J = 1.0 Hz, 2H), 6.650 (d, J = 2.0 Hz, 2H), 6.771 (d, J = 2.0 Hz, 2H) (the 1H NMR data of 11a were identical with those in the literature32).Synthesis of intermediate 16a
A mixture of 15a (4.5 g, 25 mmol) and 7.5% Pd/C (0.45 g) in ethanol (50 mL) was stirred under a H2 atmosphere for 5 h. The mixture was filtered and the filtrate was concentrated in vacuo. The crude 16a (2.6 g) was used in the next step without further purification since the intermediate 16a was unstable in air. Furthermore, the mole of 16a used in the next step of the reaction was also calculated as the hypothetical mass of pure 16a divided by the relative molecular mass of 16a.Synthesis of intermediate 16b
A similar reaction to that described above to prepare 16a was used to obtain the crude 16b as a red oil (3.2 g), which was then used in the next step without further purification since the intermediate 16b was unstable in air. Furthermore, the mole of 16b used in the next step of the reaction was also calculated as the hypothetical mass of pure 16b divided by the relative molecular mass of 16b.Synthesis of intermediate 12a
A similar reaction to that described above to prepare 16a was used to obtain the crude 12a as a red oil (5.4 g), which was then used in the next step without further purification since the intermediate 12a was unstable in air. Furthermore, the mole of 12a used in the next step of the reaction was also calculated as the hypothetical mass of pure 12a divided by the relative molecular mass of 12a.Synthesis of intermediate 12b
A similar reaction to that described above to prepare 16a was used to obtain the crude 12b as a red oil (4.1 g), which was then used in the next step without further purification since the intermediate 12b was unstable in air. Furthermore, the mole of 12b used in the next step of the reaction was also calculated as the hypothetical mass of pure 12b divided by the relative molecular mass of 12b.Synthesis of precursor 7
A mixture of the crude compound 3 (0.91 g, 3.0 mmol) and intermediate 6a (0.62 g, 2.0 mmol) in 1,4-dioxane (20 mL) using p-toluenesulfonic acid (0.14 g, 0.8 mmol) as the catalyst was stirred under reflux under an inert atmosphere. When intermediate 6a was completely consumed (as monitored by TLC, petroleum ether/ethyl acetate, 3/1, v/v), it was concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography by eluting with petroleum ether/ethyl acetate (6/1–2/1, v/v) to give the pure compound 7 (60 mg) with the yield of 5.2%. 1H NMR (400 MHz, CDCl3, δ ppm): 7.94 (s, 1H), 7.81 (d, J = 8.3 Hz, 4H), 7.30–7.40 (m, 8H), 4.36 (t, J = 4.7 Hz, 2H), 4.13 (t, J = 4.5 Hz, 2H), 3.86 (s, 3H), 2.44 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 158.71, 157.92, 157.24, 153.07, 153.00, 145.52, 135.06, 134.70, 132.79, 130.65, 128.17, 124.87, 121.18, 116.73, 114.77, 92.81, 69.70, 66.12, 55.65, 21.58; HRMS (ESI): calcd for C26H26BrN4O5S ([M + H]+) 585.0729, found 585.0713 ([M + H]+).Synthesis of intermediate 17a
A similar reaction to that described above to prepare 7 was used to obtain the crude 17a (0.57 g), which was then used in the next step without further purification since the intermediate 17a was unstable in air. Furthermore, the mole of the 17a used in the next step of the reaction was also calculated as the hypothetical mass of pure 17a divided by the relative molecular mass of 17a.Synthesis of intermediate 17b
A similar reaction to that described above to prepare 7 was used to obtain the crude 17b (0.62 g), which was then used in the next step without further purification since the intermediate 17b was unstable in air. Furthermore, the mole of the 17b used in the next step of the reaction was also calculated as the hypothetical mass of pure 17b divided by the relative molecular mass of 17b.Synthesis of intermediate 17c
A similar reaction to that described above to prepare 7 was used to obtain the crude 17c (0.65 g), which was then used in the next step without further purification since the intermediate 17c was unstable in air. Furthermore, the mole of 17c used in the next step of reaction was also calculated as the hypothetical mass of pure 17c divided by the relative molecular mass of 17c.Synthesis of intermediate 17d
A similar reaction to that described above to prepare 7 was used to obtain the crude 17d (0.64 g), which was then used in the next step without further purification since the intermediate 17d was unstable in air. Furthermore, the mole of 17d used in the next step of the reaction was also calculated as the hypothetical mass of pure 17d divided by the relative molecular mass of 17d.Synthesis of intermediate 17e
A similar reaction to that described above to prepare 7 was used to obtain the crude 17e (0.72 g), which was then used in the next step without further purification since the intermediate 17e was unstable in air. Furthermore, the mole of 17e used in the next step of the reaction was also calculated as the hypothetical mass of pure 17e divided by the relative molecular mass of 17e.Synthesis of intermediate 17f
A similar reaction to that described above to prepare 7 was used to obtain the crude 17f (0.69 g), which was then used in the next step without further purification since the intermediate 17f was unstable in air. Furthermore, the mole of 17f used in the next step of the reaction was also calculated as the hypothetical mass of pure 17f divided by the relative molecular mass of 17f.Synthesis of precursor 18a
Briefly, to a mixture of 17a (570 mg, 1.2 mmol), triethylamine (202 mg, 2 mmol), and 4-dimethylaminepyridine (14 mg, 0.12 mmol) in dry DCM (10 mL) was added TsCl (380 mg, 2 mmol) in dry DCM (10 mL) dropwise at 0 °C. The solution was stirred at 0 °C for 3 h. Then, the reaction was quenched with water. After extraction with DCM, the organic phase was washed with water two times. After removal of the solvent, the residue was purified by silica gel chromatography to give 18a (0.30 g, 45%) as a red solid. 1H NMR (400 MHz, DMSO-d6): δ 9.18 (s, br, 1H), 8.48 (s, br, 1H), 8.16 (s, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.8 Hz, 4H), 6.91 (s, 2H), 6.63 (d, J = 9.2 Hz, 2H), 4.30 (s, 2H), 4.06 (s, 2H), 3.67–3.68 (m, 9H), 2.41 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 158.71, 157.92, 157.24, 153.07, 153.00, 145.52, 135.06, 134.70, 132.79, 130.65, 128.17, 121.18, 116.73, 114.77, 92.81, 69.70, 66.12, 60.65, 56.22, 21.58; HRMS (ESI): calcd for C28H30BrN4O7S ([M + H]+) 645.0940, found 645.1007 ([M + H]+).Synthesis of precursor 18b
A similar reaction to that described above to prepare 18a was used to obtain 18b as a light yellow solid (0.53 g, 66%). 1H NMR (DMSO-d6) δ: 9.26 (s, br, 1H), 8.86 (s, br, 1H), 8.26 (s, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.43–7.47 (m, 4H), 6.71 (d, J = 8.8 Hz, 2H), 4.32 (t, J = 4.0 Hz, 2H), 4.10 (t, J = 4.0 Hz, 2H), 2.40 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 158.73, 156.82, 153.34, 145.47, 143.25, 134.27, 132.79, 130.61, 128.16, 125.87, 124.11, 124.06, 123.85, 123.09, 121.70, 114.83, 69.67, 66.04, 21.55; HRMS (ESI): calcd for C26H23BrF3N4O4S ([M + H]+) 623.0497, found 623.0568 ([M + H]+).Synthesis of precursor 18c
A similar reaction to that described above to prepare 18a was used to obtain 18c as a light yellow solid (0.52 g, 60%). 1H NMR (DMSO-d6) δ: 9.26 (s, br, 1H), 8.80 (s, br, 1H), 8.25 (s, 1H), 7.87–7.90 (m, 4H), 7.79 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 4H), 6.71 (d, J = 8.8 Hz, 2H), 4.29–4.32 (m, 4H), 4.10–4.11 (m, 2H), 2.39 (s, 3H), 1.32 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3, δ ppm): 166.21, 158.61, 157.15, 155.86, 154.43, 145.05, 142.29, 132.94, 132.69, 130.68, 129.97, 128.14, 125.62, 122.62, 119.97, 115.03, 68.26, 65.92, 61.00, 21.55, 14.50; HRMS (ESI): calcd for C28H28BrN4O6S ([M + H]+) 627.0835, found 627.0901 ([M + H]+).Synthesis of precursor 18d
A similar reaction to that described above to prepare 18a was used to obtain 18d as a light yellow solid (0.52 g, 60%). 1H NMR (DMSO-d6) δ: 9.17 (s, br, 1H), 8.46 (s, br, 1H), 8.16 (s, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 9.2 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 6.91 (s, 2H), 6.70 (d, J = 9.2 Hz, 2H), 4.14 (t, J = 4.0 Hz, 2H), 3.92 (t, J = 4.0 Hz, 2H), 3.64–3.68 (m, 13H), 2.38 (s, 3H); 13C NMR (100 MHz, DMSO-d6, δ ppm): 158.73, 157.89, 157.24, 153.75, 152.99, 145.38, 135.07, 134.67, 134.21, 132.93, 130.60, 128.12, 121.30, 114.61, 102.10, 92.72, 70.47, 69.46, 68.53, 67.67, 60.61, 56.21, 55.61, 21.57; HRMS (ESI): calcd for C30H33BrN4O6S ([M + H]+) 689.1202, found 689.1317 ([M + H]+).Synthesis of precursor 18e
A similar reaction to that described above to prepare 18a was used to obtain 18e as a light yellow solid (0.57 g, 61%). 1H NMR (DMSO-d6) δ: 9.25 (s, br, 1H), 8.85 (s, br, 1H), 8.25 (s, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.42–7.47 (m, 4H), 6.78 (d, J = 8.8 Hz, 2H), 4.14 (t, J = 4.0 Hz, 2H), 3.95 (t, J = 4.0 Hz, 2H), 3.64 (q, J = 4.4 Hz, 4H), 2.37 (s, 3H); 13C NMR (100 MHz, CDCl3, δ ppm): 158.92, 157.60, 155.92, 155.15, 144.93, 141.38, 132.99, 132.40, 129.91, 128.08, 126.10, 125.63, 125.41, 125.16, 123.36, 122.87, 120.66, 114.93, 102.10, 93.91, 70.00, 69.35, 68.98, 67.81, 21.72; HRMS (ESI): calcd for C28H27BrF3N4O5S ([M + H]+) 667.0759, found 667.0865 ([M + H]+).Synthesis of precursor 18f
A similar reaction to that described above to prepare 18a was used to obtain 18f as a light yellow solid (0.58 g, 66%). 1H NMR (DMSO-d6) δ: 9.27 (s, br, 1H), 8.81 (s, br, 1H), 8.26 (s, 1H), 7.88–7.90 (m, 4H), 7.78 (d, J = 8.0 Hz, 2H), 7.41–7.49 (m, 4H), 6.80 (d, J = 9.2 Hz, 2H), 4.29 (q, J = 7.2 Hz, 2H), 4.15 (t, J = 4.0 Hz, 2H), 3.96 (t, J = 4.0 Hz, 2H), 3.64–3.67 (m, 4H), 2.36 (s, 3H), 1.31 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3, δ ppm): 166.22, 158.89, 157.44, 155.80, 155.09, 144.91, 142.43, 133.00, 132.43, 130.65, 129.91, 128.07, 125.43, 122.80, 119.87, 114.96, 94.05, 70.00, 69.38, 68.99, 67.81, 60.95, 21.73, 14.49; HRMS (ESI): calcd for C30H32BrN4O7S ([M + H]+) 671.1097, found 671.1213 ([M + H]+).Synthesis of F-19 standard 8a
To a stirred solution of compound 7 (30 mg, 0.05 mmol) in dry THF (4 mL), tetrabutylammonium fluoride (0.5 mL, 1 M in THF) was added. The mixture was heated using a digestion high-pressure tank at 100 °C for 6–7 h. After the intermediate 7 was completely consumed (as monitored by TLC, petroleum ether/ethyl acetate, 6/1, v/v), the reaction was quenched with water, and then the mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure, which was further purified by silica gel column chromatography eluted with petroleum ether/ethyl acetate (10/1–4/1, v/v) to give the pure compound 8a (1.4 mg) as a white solid. Yield: 6.3%; 1H NMR (CDCl3, 400 MHz) δ: 7.82 (s, 1H), 7.30 (d, J = 8.8 Hz, 4H), 6.83 (d, J = 8.6 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 4.75 (t, J = 4.1 Hz, 1H), 4.63 (t, J = 3.9 Hz, 1H), 4.16 (t, J = 3.9 Hz, 1H), 4.10 (t, J = 4.0 Hz, 1H), 3.78 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 158.74, 157.93, 157.24, 153.51, 152.99, 135.07, 134.67, 134.47, 122.67, 121.33, 116.71, 114.74, 92.78, 83.58, 81.91, 67.87, 67.68, 55.61; HRMS (ESI): calcd for C19H19BrFN4O2 ([M + H]+) 433.0597, found 433.0621 ([M + H]+).Synthesis of F-19 standard 8b
A similar reaction to that described above to prepare 7 was used to obtain 8b (0.46 g, 68%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ: 9.18 (s, 1H), 8.47 (s, 1H), 8.16 (s, 1H), 7.50 (d, 2H), 6.91 (s, 2H), 6.75 (d, 2H), 4.79–4.75 (m, 1H), 4.67–4.63 (m, 1H), 4.17 (s, 1H), 4.12–4.07 (m, 1H), 3.69–3.65 (m, 10H); 13C NMR (100 MHz, DMSO-d6) δ: 158.74, 157.93, 157.24, 153.51, 152.99, 135.07, 134.67, 134.47, 121.33, 114.74, 102.11, 92.78, 83.58, 81.91, 67.87, 67.68, 60.61, 56.21, 55.61; HRMS (ESI): calcd for C21H23BrFN4O4 ([M + H]+) 493.0808, found 493.0887 ([M + H]+).Synthesis of F-19 standard 8c
A similar reaction to that described above to prepare 7 was used to obtain 8c (0.69 g, 90%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ: 9.26 (s, 1H), 8.85 (s, 1H), 8.25 (s, 1H), 7.89 (d, 2H), 7.66 (d, 2H), 7.47 (d, 2H), 6.83 (d, 2H), 4.80–4.75 (m, 1H), 4.70–4.63 (m, 1H), 4.23–4.17 (m, 1H), 4.15–4.11 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 158.81, 156.82, 153.84, 143.26, 134.14, 125.82, 124.11, 123.84, 123.10, 121.96, 114.80, 83.27, 82.17, 67.78, 67.66; HRMS (ESI): calcd for C19H16BrFN4O4 ([M + H]+) 471.0365, found 471.0440 ([M + H]+).Synthesis of F-19 standard 8d
A similar reaction to that described above to prepare 7 was used to obtain 8d (0.94 g, 78%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ: 9.26 (s, 1H), 8.80 (s, 1H), 8.26 (s, 1H), 7.94–7.80 (m, 4H), 7.49 (d, 2H), 6.85 (d, 2H), 4.82–4.76 (m, 1H), 4.70–4.63 (m, 1H), 4.30 (q, 2H), 4.24–4.19 (m, 1H), 4.17–4.12 (m, 1H), 1.32 (t, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.23, 158.81, 157.33, 155.83, 154.92, 142.35, 132.59, 130.67, 125.55, 122.90, 119.92, 115.08, 94.15, 82.66, 81.52, 67.69, 67.55, 60.97, 14.47; HRMS (ESI): calcd for C21H21BrFN4O3 ([M + H]+) 475.0703, found 475.0784 ([M + H]+).Synthesis of F-19 standard 8e
A similar reaction to that described above to prepare 7 was used to obtain 8e (1.20 g, 82%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ: 9.16 (s, 1H), 8.46 (s, 1H), 8.16 (s, 1H), 7.60–7.40 (m, 3H), 6.91 (s, 3H), 6.77–6.69 (m, 3H), 4.68–4.55 (m, 1H), 4.52–4.44 (m, 1H), 4.04–3.95 (m, 3H), 3.79–3.72 (m, 4H), 3.69–3.64 (m, 13H); 13C NMR (100 MHz, DMSO-d6) δ: 158.76, 157.93, 157.23, 153.80, 152.99, 135.07, 134.67, 134.21, 121.32, 114.63, 102.10, 92.70, 84.39, 82.74, 70.41, 70.22, 69.58, 67.78, 60.61, 56.21, 55.60; HRMS (ESI): calcd for C23H27BrFN4O5 ([M + H]+) 537.1071, found 537.1141 ([M + H]+).Synthesis of F-19 standard 8f
A similar reaction to that described above to prepare 7 was used to obtain 8f (1.08 g, 75%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ: 9.25 (s, 1H), 8.85 (s, 1H), 8.25 (s, 1H), 7.97–7.78 (m, 2H), 7.70–7.62 (m, 2H), 7.50–7.41 (m, 2H), 6.86–6.76 (m, 2H), 4.65–4.56 (m, 1H), 4.52–4.45 (m, 1H), 4.10–3.99 (m, 2H), 3.82–3.72 (m, 3H), 3.72–3.64 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 158.84, 157.43, 155.94, 155.32, 141.33, 132.21, 126.16, 122.93, 120.66, 115.02, 83.85, 82.73, 70.76, 70.63, 70.06, 67.93; HRMS (ESI): calcd for C21H20BrF4N4O2 ([M + H]+) 515.0628, found 515.0704 ([M + H]+).Synthesis of F-19 standard 8g
A similar reaction to that described above to prepare 7 was used to obtain 8g (1.28 g, 88%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ: 9.25 (s, 1H), 8.80 (s, 1H), 8.25 (s, 1H), 7.88 (s, 5H), 7.65–7.36 (m, 2H), 6.95–6.73 (m, 2H), 4.68–4.55 (m, 1H), 4.55–4.43 (m, 1H), 4.37–4.23 (m, 3H), 4.10–4.01 (m, 3H), 3.82–3.72 (m, 4H), 3.70–3.63 (m, 1H), 1.35–1.30 (m, 4H); 13C NMR (100 MHz, CDCl3) δ: 166.24, 158.98, 157.54, 155.77, 155.18, 142.45, 132.42, 130.65, 125.40, 122.84, 119.82, 115.00, 93.98, 83.86, 82.74, 70.76, 70.63, 70.07, 67.91, 60.95, 14.49; HRMS (ESI): calcd for C23H25BrFN4O4 ([M + H]+) 519.0965, found 519.1032 ([M + H]+).Synthesis of F-19 standard 8h
A mixture of compound 8d (0.50 g, 1.1 mmol) and potassium hydroxide (2.00 g, 35.7 mmol) in EtOH/H2O (20 mL, 1/1, v/v) was stirred at 90 °C. When the compound 8d was completely consumed (as monitored by TLC, petroleum ether/ethyl acetate, 6/1, v/v), the mixed solution was concentrated under reduced pressure. The pH of the resulting residue was adjusted to 1–2 by 1 M HCl (aq.), and then filtrated and washed with water to get the pure compound 8h (0.30) as a white solid. Yield: 63%; 1H NMR (400 MHz, DMSO-d6) δ: 9.26 (s, 1H), 8.75 (s, 1H), 8.25 (s, 1H), 7.95–7.78 (m, 4H), 7.49 (d, 2H), 6.84 (d, 2H), 4.82–4.76 (m, 1H), 4.70–4.64 (m, 1H), 4.24–4.19 (m, 1H), 4.17–4.11 (m, 1H) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 167.83, 165.99, 158.81, 158.56, 156.67, 153.80, 144.08, 143.59, 134.14, 130.25, 126.08, 124.63, 121.97, 114.81, 93.35, 83.57, 81.92, 67.84, 67.65, 60.96; HRMS (ESI): calcd for C19H16BrFN4O3 ([M + H]+) 447.0390, found 447.0461 ([M + H]+).Synthesis of F-19 standard 8i
Compound 8i (0.37 g) was obtained as a white solid according to the general procedure described for 8h starting from the pyrimidine derivative 8g (0.50 g, 0.96 mmol) and potassium hydroxide (2.00 g, 35.7 mmol). Yield: 78%; 1H NMR (400 MHz, DMSO-d6) δ: 12.72 (s, 1H), 9.25 (s, 1H), 8.75 (s, 1H), 8.25 (s, 1H), 7.91–7.80 (m, 4H), 7.54–7.42 (m, 2H), 6.88–6.78 (m, 2H), 4.64–4.58 (m, 1H), 4.52–4.46 (m, 1H), 4.06 (t, 2H), 3.80–3.73 (m, 3H), 3.71–3.66 (m, 1H) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 167.61, 165.96, 158.83, 158.59, 156.65, 154.13, 143.76, 133.85, 130.28, 130.06, 125.62, 124.63, 122.03, 114.73, 93.28, 84.40, 82.75, 70.40, 70.22, 69.57, 67.75, 60.95; HRMS (ESI): calcd for C19H16BrFN4O3 ([M + H]+) 491.0652, found 491.0718 ([M + H]+).FAK inhibitory assay
The inhibition tests of the title compounds 8a–8i against FAK were performed using the HTRF® (Homogeneous Time-Resolved Fluorescence Methodologies) kinEASE™ TK kit. The concentration of the enzyme used in the assay, as well as the enzymatic reaction time and the ATP concentration were optimized before the compound testing. Here, 0.11 ng μL−1 enzyme was chosen to be used with 13.8 μM ATP to react for 50 min at room temperature.During the enzymatic reaction step, 4 μL of the compound and 2 μL TK substrate–biotin were incubated with 2 μL kinase. 2 μL ATP was added to start the enzymatic reaction. Then, the enzymatic buffer from the HTRF® kinEASE™ TK kit was added, following by the addition of 5 mM MgCl2, 1 mM DTT, and 25 nM SEB.
The detection reagents (5 μL Eu3+-cryptate labeled TK-antibody and 5 μL Steptavidin-XL665) dissolved in the detection buffer (in the presence of EDTA) were mixed and added to the reaction system. The TR-FRET signal was proportional to the phosphorylation level and was detected by a plate reader (BMG FS) after incubation for 1 h. For each compound, the IC50 value was determined from a sigmoid dose–response curve using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Partition coefficient determination
The determination of the partition coefficients of the radiotracers [18F]-8a, [18F]-8c, [18F]-8h and [18F]-8i was carried out according to the following procedure: first, the same volume of n-octanol and PBS (phosphate buffer saline, 0.05 M, pH 7.4) was mixed, and each phase was presaturated with the opposite phase overnight before use. Then, to a centrifuge tube was added a solution of 18F-labeled tracer (370 kBq, 10 μCi) in 0.1 mL normal saline, 0.9 mL PBS (presaturated with the opposite phase overnight), and 1.0 mL n-octanol (presaturated with the opposite phase overnight) in sequence. The centrifuge tube was vortexed for 0.5–1.0 min at room temperature, followed by centrifugation for 5 min at 5000 rpm. After standing for a while, two samples were transferred from the n-octanol (100 μL) and PBS (100 μL) layers to the counting tubes and were measured in a gamma counter. The partition coefficient was expressed as the logarithm of the ratio of the radioactivity from the n-octanol phase versus the PBS phase. The measurement was done in triplicate and repeated three times.In vitro stability studies
[18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i (370 kBq, 10 μCi) after HPLC purification were dissolved in 100 μL of normal saline, and then mixed with 500 μL of normal saline at 37 °C for 1 h and 2 h, respectively. About 100 μL of the solution was analyzed by radio-HPLC.The in vitro stability of the 18F-labeled tracers in mouse plasma was determined using the following procedure: (1) first, incubate 370 kBq of the HPLC-purified radiotracers in 100 μL of normal saline with 500 μL of murine plasma at 37 °C for 1 h and 2 h, respectively; (2) after centrifuging the plasma proteins at 5000 rpm for 5 min at 4 °C, they were precipitated by adding 200 μL of acetonitrile. The supernatant was collected. Approximately 100 μL of the supernatant solution was loaded for the HPLC analysis.
In vivo biodistribution studies in S180-bearing mice
An S180 ascites sarcoma mouse was sacrificed and the ascites was immediately drawn off from the carcass and made into an S180 sarcoma cell suspension (diluted four times by normal saline). Then, the normal ICR mice were inoculated subcutaneously into the right front flank with the diluted ascites sarcoma cell suspension (100 μL per mouse, containing about 5 × 106 tumor cells by cell count).The in vivo biodistribution studies could be conducted until the tumors reached a size of 0.5–0.8 cm in diameter (i.e., for about one week). A normal saline solution containing the 18F-labeled tracers [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i (370 kBq per 100 μL) was injected via the tail vein for each of the S180-tumor-bearing mice. The mice (n = 5 for each time point) were sacrificed at 5, 15, 30, 60, and 120 min post-injection. The tissues and organs of interest were anatomized and weighed, and the radioactivity was determined with an automatic γ-counter. The percent dose per gram of wet tissue was calculated by a comparison of the tissue counts to suitably diluted aliquots of the injected material. The values were expressed as the mean ± SD (n = 5).
MicroPET imaging
The S180-tumor-bearing mice were injected intravenously with approximately 3.7 MBq of [18F]-8a in 300 μL normal saline, anesthetized with 1.5% isoflurane in air (about 1.5 mL min−1), and fixed near the center of the microPET scanner (SuperArgus microPET/CT). Static scans (for 30 min and 60 min, 10 min scans for each time point) were obtained on a SuperArgus microPET/CT. Then, the images were reconstructed with the SEDECAL reconstruction software and the images were exported by MMWKS SUPERARGUS software.Results and discussion
Chemistry
The synthetic route used to prepare the pyrimidine derivative 8a is shown in Scheme 1. The starting material 4-nitrophenol 1 was treated with ethane-1,2-diyl bis(4-methylbenzenesulfonate) to give compound 2, which was reduced by Zn to obtain compound 3. Intermediate 6a was obtained by the reaction of 5-bromo-2,4-dichloropyrimidine 4 and 4-methoxyaniline 5a in i-PrOH using K2CO3 as a base at room temperature with a yield of 78%. Compound 7 could be prepared by compounds 3 and 6a in 1,4-dioxane at 100 °C using p-toluene sulfonic acid as the catalyst, which was further refluxed in THF in the presence of TBAF to provide the target compound 8a. | ||
| Scheme 1 Reagents and conditions: (i) ethane-1,2-diyl bis(4-methylbenzenesulfonate), K2CO3, 18-crown-6, 56 °C; (ii) Zn, acetic acid, room temperature, 12 h; (iii) i-PrOH, K2CO3, rt; (iv) TsOH·H2O, 1,4-dioxane, 100 °C; (v) TBAF, THF, reflux, 6–7 h. | ||
The synthetic route used to prepare the pyrimidine derivatives 8b–g is shown in Scheme 2. The fluorinated products 10a–b were obtained starting from the commercially available materials 9a–b, followed by etherification and hydrogenation to give the key intermediates 12a–b in excellent yields. Another important kind of intermediate 6b–d could be easily prepared by compounds 4 and 5b–d, whereby the former further reacted with compounds 12a–b in 1,4-dioxane at 100 °C to provide the target pyrimidine derivatives 8b–g using p-toluene sulfonic acid as the catalyst with 68–90% yields.
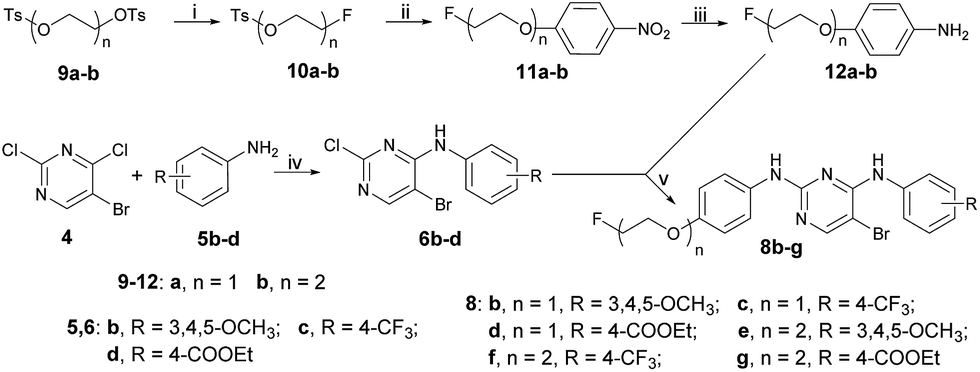 | ||
| Scheme 2 Reagents and conditions: (i) TBAF, THF, N2, 80 °C, 20 h; (ii) p-nitrophenol, K2CO3, DMF, N2, 80 °C, 20 h; (iii) CH3OH–THF, 7.5% Pd–C, H2, rt, 4 h; (iv) i-PrOH, K2CO3 rt; (v) TsOH·H2O, 1,4-dioxane, 100 °C, 3 h. | ||
The synthetic route used to prepare the pyrimidine derivatives 8h and 8i is shown in Scheme 3. The target compounds 8h and 8i with moderate yields were easily synthesized by pyrimidine derivatives 8d and 8g in the mixed solution of ethanol and water at 90 °C in the presence of KOH.
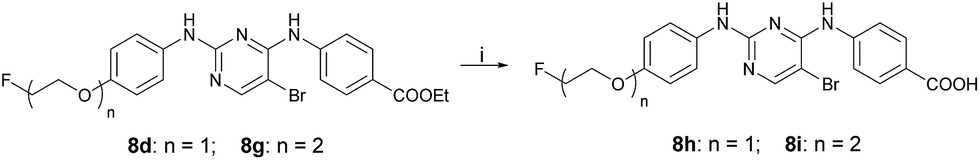 | ||
| Scheme 3 Reagents and conditions: (i) (a) KOH, EtOH–H2O (1:1), 90 °C; (b) 1 M HCl. | ||
All the newly synthesized target pyrimidine derivatives were characterized by 1H NMR, 13C NMR, and MS spectra. The spectral data were in accordance with the assigned structures, and all the spectral data are listed in the Experimental section.
FAK inhibitory assay
The prepared compounds 8a–i were evaluated for their in vitro inhibitory activities against FAK using 16-OTS-1 as the positive control. The obtained results are shown in Table 1.
|
FAK biochemical assay | ||
|---|---|---|---|
| Compds | n | R | IC50/μM |
| 8a | 1 | 4-OCH3 | 1.085 |
| 8b | 1 | 3,4,5-OCH3 | 1.730 |
| 8c | 1 | 4-CF3 | 3.258 |
| 8d | 1 | 4-COOEt | 9.423 |
| 8e | 2 | 3,4,5-OCH3 | 0.768 |
| 8f | 2 | 4-CF3 | 0.722 |
| 8g | 2 | 4-COOEt | 4.419 |
| 8h | 1 | 4-COOH | 0.285 |
| 8i | 2 | 4-COOH | 0.060 |
| 18c | 33.290 | ||

The results in Table 1 demonstrated that all the target compounds showed superior inhibition against FAK compared to the reference 18c. The most active compound 8i displayed an IC50 value of 0.060 μM against FAK, which was more than 500-fold lower than 18c (IC50 = 33.290 μM). When n = 1, compound 8h with a carboxyl group exhibited strong inhibitory activity with an IC50 value of 0.285 μM, while when n = 2, the carboxyl-group-modified derivative 8i was still the most active one. These outcomes suggested that the carboxyl group was favorable to exert FAK inhibition for this type of pyrimidine derivatives. However, with the change of the carboxyl group into a carboxylic ester moiety, the inhibitory activity is reduced a lot. Comparing compounds with n = 1 (8a–d, 8h) to n = 2 (8e–g, 8i), the pyrimidine derivatives 8e–g and 8i generally showed stronger activity, which indicated that the length of the aliphatic chain has an important effect on the FAK inhibitory activity.
Radiolabeling
Encouraged by the good inhibitory activity and docking results of most of the newly prepared pyrimidine derivatives against FAK, a radiofluorination method for the preparation of [18F]-8 was developed with different reactions.Scheme 4 describes the synthesis of compound [18F]-8a, which could be easily obtained with the yield of 20% by the reaction of the tosylate precursor 7 with [18F]fluoride/potassium carbonate and Kryptofix 2.2.2. in anhydrous acetonitrile using a digestion high-pressure tank at 100 °C for 15 min.
 | ||
| Scheme 4 Reagents and conditions: (i) anhydrous KF, Kryptofix 2.2.2., anhydrous acetonitrile, 100 °C, 15 min. | ||
The synthetic route for the preparation of the radioactive derivatives [18F]-8c, [18F]-8d, [18F]-8g, [18F]-8h, and [18F]-8i is outlined in Scheme 5. The starting materials 13a–b were followed by sulfonylation, etherification, and hydrogenation to give the intermediates 16a–b, which further reacted with 6c and 6d to provide the pyrimidine derivatives 17a–c. Then the sulfonylated products 18a–c were treated with [18F]fluoride/potassium carbonate and Kryptofix 2.2.2. in anhydrous acetonitrile using a digestion high-pressure tank at 160 °C for 15 min to give the radioactive derivatives [18F]-8c (16%), [18F]-8d, and [18F]-8g. Compounds [18F]-8h (7%) and [18F]-8i (9%) were obtained by further treating [18F]-8d and [18F]-8g with 1 M NaOH in EtOH at 85 °C for 10 min.
 | ||
| Scheme 5 Reagents and conditions: (i) TsCl, TEA, DCM, 0–5 °C, 3 h; (ii) p-nitrophenol, K2CO3, DMF, 75 °C; (iii) EtOH, 7.5% Pd–C, H2, rt, 4 h; (iv) 6c, 6d, TsOHS·H2O, 1,4-dioxane, 100 °C, 5 h; (v) TsCl, TEA, DMAP, DCM, room temperature, 3 h; (vi) anhydrous KF, Kryptofix 2.2.2., anhydrous acetonitrile, 160 °C, 15 min; (vii) 1 M NaOH, EtOH, 85 °C, 10 min. | ||
The radiochemical purity of the radiotracers [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i was greater than 98% after purification by high performance liquid chromatography (HPLC). The identity of the 18F-labeled tracers was verified by a comparison of their retention times with those of the corresponding nonradioactive F-19 standards 8a, 8c, 8h, and 8i via co-injection. The retention times of [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i were 14.576, 19.197, 6.696, and 6.668 min, respectively, which matched well with the corresponding F-19 standards 8a (14.301 min), 8c (18.899 min), 8h (6.357 min), and 8i (6.313 min) within admissible error, respectively (Fig. 4).
 | ||
| Fig. 4 HPLC chromatogram of the 18F-labeled radiotracers and the corresponding 19F standards. | ||
Partition coefficient determination
Selecting potential candidates as FAK targeting tumor imaging agents for further development requires measuring a range of physiochemical properties.33 The partition coefficient (logP) value governs various biological processes, such as the transportation, distribution, metabolism, and secretion of biomolecules, which is essential to predict their transportation and activity. Therefore, the selected compounds [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i were measured for their logP values.
Table 2 shows that the logP values of the four measured compounds were all over 0. The results indicated that the four compounds were all liposoluble. Notably, the logP value of compound [18F]-8a (4.51) was higher than the others. The difference might result from the substituents, such as trifluoromethyl and the carboxyl groups on compounds [18F]-8c, [18F]-8h, and [18F]-8i, which increased their hydrophilicity.
P values of compounds [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i
| Compds | [18F]-8a | [18F]-8c | [18F]-8h | [18F]-8i |
| logP |
4.51 ± 0.04 | 1.63 ± 0.04 | 1.22 ± 0.06 | 1.35 ± 0.05 |
In vitro stability studies
The in vitro stability of [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i were determined in physiological saline and mouse plasma. Radio-HPLC analysis of the physiological saline and mouse plasma samples revealed that compounds [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i remained sufficiently stable (>95%) during incubation at 37 °C for 2 h, demonstrating a high in vitro stability of these radio-compounds (Fig. 5). | ||
| Fig. 5 (A) HPLC chromatograms of [18F]-8a in normal saline and murine plasma at 37 °C after 1 h and 2 h, respectively. (B) HPLC chromatograms of [18F]-8i in normal saline and murine plasma at 37 °C after 1 h and 2 h, respectively. | ||
In vivo biodistribution studies in S180-bearing mice
In vivo biodistribution data in mice bearing the S180 tumor for [18F]-8a at 5, 15, 30, 60, and 120 min are shown in Table 1, while for compounds [18F]-8c, [18F]-8h, and [18F]-8i the date are shown in the ESI.† The results in Table 1 display that [18F]-8a was accumulated in the tumor with retention values between 3.11 ± 0.22 and 3.71 ± 0.43. At 60 min post-injection, [18F]-8a had the highest uptake in the tumor (3.71 ± 0.43 ID% g−1). The uptake by the tumor of [18F]-8a was found to be at least 2 times more than the uptake in the liver (1.41 ± 0.08 ID% g−1) and in the kidney (1.49 ± 0.13 ID% g−1). Also, a good to moderate uptake was observed in other major organs or tissues, such as the intestine (3.51 ± 0.14 ID% g−1), heart (3.12 ± 0.18 ID% g−1), lung (2.26 ± 0.12 ID% g−1), and muscle (1.97 ± 0.25 ID% g−1) at 60 min post-injection. Since most PET-based radiotracer studies are generally performed within 60 min of the radiotracer administration,34 thus [18F]-8a seems suitable for early tumor imaging (Table 3).| ID% (g−1) | 5 min | 15 min | 30 min | 60 min | 120 min |
|---|---|---|---|---|---|
| a Expressed as % injected dose per gram (% ID g−1) unless otherwise indicated. Data are the average for five mice ± standard deviation.b Expressed as % injected dose per organ (% ID), intest = intestine, stom = stomach.c Tu/Mu = tumor/muscle, Tu/Bo = tumor/bone, Tu/Bl = tumor/blood. | |||||
| Blood | 1.95 ± 0.32 | 1.99 ± 0.09 | 2.19 ± 0.01 | 1.9 ± 0.10 | 0.81 ± 0.01 |
| Brain | 1.20 ± 0.12 | 0.78 ± 0.03 | 1.19 ± 0.18 | 1.51 ± 0.12 | 0.91 ± 0.03 |
| Heart | 1.23 ± 0.15 | 2.44 ± 0.22 | 2.45 ± 0.20 | 3.12 ± 0.18 | 1.72 ± 0.08 |
| Liver | 3.29 ± 0.02 | 1.99 ± 0.23 | 1.87 ± 0.19 | 1.41 ± 0.08 | 0.50 ± 0.06 |
| Spleen | 1.98 ± 0.03 | 1.65 ± 0.11 | 2.28 ± 0.12 | 1.74 ± 0.19 | 0.75 ± 0.23 |
| Lung | 6.98 ± 0.09 | 8.01 ± 0.17 | 6.30 ± 0.18 | 2.26 ± 0.12 | 1.25 ± 0.06 |
| Kidney | 0.70 ± 0.09 | 1.51 ± 0.10 | 1.80 ± 0.03 | 1.49 ± 0.13 | 1.33 ± 0.13 |
| Muscle | 1.38 ± 0.21 | 1.47 ± 0.19 | 1.73 ± 0.20 | 1.97 ± 0.25 | 1.59 ± 0.27 |
| Intestb | 1.18 ± 0.02 | 1.59 ± 0.03 | 3.46 ± 0.26 | 3.51 ± 0.14 | 3.03 ± 0.19 |
| Stomb | 1.14 ± 0.21 | 1.33 ± 0.06 | 1.49 ± 0.08 | 1.61 ± 0.04 | 1.35 ± 0.05 |
| Tumor | 3.69 ± 0.51 | 3.39 ± 0.25 | 3.11 ± 0.22 | 3.71 ± 0.43 | 3.23 ± 0.14 |
| Tu/Muc | 2.67 | 2.31 | 1.80 | 1.88 | 2.03 |
| Tu/Boc | 1.96 | 1.64 | 0.90 | 0.95 | 0.65 |
| Tu/Blc | 1.89 | 1.70 | 1.42 | 1.92 | 3.99 |
MicroPET imaging
Compound [18F]-8a was selected as the microPET imaging agent in the four F-18-labeled pyrimidine derivatives for its good in vivo biodistribution in mice bearing the S180 tumor. In the microPET images, it could be seen that the diffusion of the [18F]-8a in vivo needed a period of time, and with the time increasing, the accumulation of [18F]-8a increased close to the tissue of the tumor. It is worth noting that after 60 min of caudal intravenous injection for [18F]-8a, the intensity of radioactivity in the urinary bladders of the mice could be clearly observed, which might be responsible for the metabolism of [18F]-8a in mice kidney. These results were in accordance with the data for the in vivo biodistribution (Fig. 6). | ||
| Fig. 6 MicroPET images of [18F]-8a at 60 min (left) and 120 min (right) of S180-bearing nude mice. | ||
Conclusions
In conclusion, a series of novel pyrimidine derivatives were successfully synthesized and characterized by 1H NMR, 13C HNMR, and MS spectra. All the new compounds were evaluated for their activity against FAK, and showed low IC50 values in comparison with control drugs. Especially for compound 8i, its IC50 value was 0.060 μM, suggesting its advantage as an FAK inhibitor. To evaluate the potentiality of these compounds as PET imaging agents in cancer detection, compounds 8a, 8c, 8h, and 8i were successively labeled with 18F. The four 18F-labeled pyrimidine derivatives [18F]-8a, [18F]-8c, [18F]-8h, and [18F]-8i showed appropriate logP values and high stability in physiological saline and mouse plasma. Noticeably, compound [18F]-8a with a 4-methoxyl group at the benzene ring exhibited good in vivo biodistribution data in mice bearing the S180 tumor, which promoted its further microPET imaging study. MicroPET image of [18F]-8a administered into S180-tumor-bearing mice acquired at 60 min post-injection illustrated that the uptake in the S180 tumor was obvious. These results suggested that compound [18F]-8a might be a promising PET tracer candidate for tumor detection. On the other hand, on-going efforts to optimize the structure of [18F]-8a aimed at enhancing the tumor-to-nontarget ratios in vivo are under way. Furthermore, in order to enhance the uptake of the F-18-labeled tracer in tumor and its target/nontarget ratios, the interaction between the corresponding F-19 standards and the FAK should be further increased and the logP should be further properly lowered in future designs.
Compliance with ethical standards
All protocols requiring the use of mice were approved by the Animal Care Committee of Beijing Normal University.Acknowledgements
This work was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” (Grant No. 2014ZX09507007-001 and 2014ZX09507007-003); the National Science and Technology Support Program (Grant No. 2014BAA03B03) and the National Natural Science Foundation of China (Grant No. 21371026). We also thank the Nuclear Medicine Department of Peking Cancer Hospital (Beijing, China) for providing the fluoride-18 nuclide and the use of the Micro PET of Peking Union Medical College Hospital (Beijing, China).Notes and references
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87 CrossRef PubMed.
- Y. Y. Wang, Y. He, L. F. Yang, S. H. Peng, X. L. He, J. H. Wang, F. Lv, Y. Hao, M. Y. Liu, Z. F. Yi and W. W. Qiu, Eur. J. Med. Chem., 2016, 120, 13 CrossRef CAS PubMed.
- M. X. Zhao, H. Y. Ren, J. Chang, D. Q. Zhang, Y. T. Yang, Y. He, C. M. Qi and H. B. Zhang, Eur. J. Med. Chem., 2016, 119, 183 CrossRef CAS PubMed.
- J. R. Beck, X. Q. Zhou, G. R. Casey and C. I. Stains, Anal. Chim. Acta, 2015, 897, 62 CrossRef CAS PubMed.
- P. Dao, N. Smith, C. Tomkiewicz-Raulet, E. Yen-Pon, M. Camacho-Artacho, D. Lietha, J. P. Herbeuval, X. Coumoul, C. Garbay and H. X. Chen, J. Med. Chem., 2015, 58, 237 CrossRef CAS PubMed.
- J. Zhang and S. N. Hochwald, Pharmacol. Ther., 2014, 142, 154 CrossRef CAS PubMed.
- T. Lechertier and K. Hodivala-Dilke, J. Pathol., 2012, 226, 404 CrossRef CAS PubMed.
- M. A. Cabrita, L. M. Jones, J. L. Q. Z. Lu, A. Sabourin, B. C. McKay and C. L. Addison, Mol. Oncol., 2011, 5, 517 CrossRef CAS PubMed.
- W. W. Ma, Anti-Cancer Agents Med. Chem., 2011, 11, 638 CrossRef CAS PubMed.
- T. Heinrich, J. Seenisamy, L. Emmanuvel, S. S. Kulkarni, J. Bomke, F. Rohdich, H. Greiner, C. Esdar, M. Krier, U. Grädler and D. Musil, J. Med. Chem., 2013, 56, 1160 CrossRef CAS PubMed.
- U. Grädler, J. Bomke, D. Musil, V. Dresing, M. Lehmann, G. Hölzemann, H. Greiner, C. Esdar, M. Krier and T. Heinrich, Bioorg. Med. Chem. Lett., 2013, 23, 5401 CrossRef PubMed.
- Q. Shi, A. B. Hjelmeland, S. T. Keir, L. H. Song, S. Wickman, D. Jackson, O. Ohmori, D. D. Bigner, H. S. Friedman and J. N. Rich, Mol. Carcinog., 2007, 46, 488 CrossRef CAS PubMed.
- E. A. Beierle, A. Trujillo, A. Nagaram, V. M. Golubovskaya, W. G. Cance and E. V. Kurenova, Cancer Invest., 2008, 26, 145 CrossRef CAS PubMed.
- V. M. Golubovskaya, C. Virnig and W. G. Cance, Mol. Carcinog., 2008, 47, 222 CrossRef CAS PubMed.
- N. Kurio, T. Shimo, T. Fukazawa, M. Takaoka, T. Okui, N. M. Hassan, T. Honami, S. Hatakeyama, M. Ikeda, Y. Naomoto and A. Sasaki, Exp. Cell Res., 2011, 317, 1134 CrossRef CAS PubMed.
- J. Halder, Y. G. Lin, W. M. Merritt, W. A. Spannuth, A. M. Nick, T. Honda, A. A. Kamat, L. Y. Han, T. J. Kim, C. H. Lu, A. M. Tari, W. Bornmann, A. Fernandez, G. Lopez-Berestein and A. K. Sood, Cancer Res., 2007, 67, 15 CrossRef PubMed.
- A. Schultze and W. Fiedler, Anti-Cancer Agents Med. Chem., 2011, 11, 593 CrossRef CAS PubMed.
- J. K. Slack-Davis, K. H. Martin, R. W. Tilghman, M. Lwanicki, E. J. Ung, C. Autry, M. J. Luzzio, B. Cooper, J. C. Kath, W. G. Roberts and J. T. Parsons, J. Biol. Chem., 2007, 282, 14845 CrossRef CAS PubMed.
- N. Malik, P. Dhiman, P. K. Verma and A. Khatkar, Res. Chem. Intermed., 2015, 41, 7981 CrossRef CAS.
- A. A. Abu-Hashema and F. A. Badria, J. Chin. Chem. Soc., 2015, 62, 506 CrossRef.
- O. S. Reddy, Ch. V. Suryanarayana, K. J. P. Narayana, V. Anuradha and B. H. Babu, Med. Chem. Res., 2014, 24, 1777 CrossRef.
- H. J. Ma, J. H. Zhang, X. D. Xia, J. Kang and J. H. Li, Pest Manage. Sci., 2015, 71, 1189 CrossRef CAS PubMed.
- P. Brust, W. Deuther-Conrad, K. Lehmkuhl, H. Jia and B. Wunsch, Curr. Med. Chem., 2014, 21, 35 CrossRef CAS PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS PubMed.
- Y. Y. Chen, X. Wang, J. M. Zhang, W. Deuther-Conrad, X. J. Zhang, Y. Y. Huang, Y. Li, J. J. Ye, M. C. Cui, J. Steinbach, P. Brust, B. L. Liu and H. M. Jia, Bioorg. Med. Chem., 2014, 22, 5270 CrossRef CAS PubMed.
- S. Bai, S. G. Li, J. Xu, X. Peng, K. Sai, W. Chu, Z. Tu, C. Zeng and R. H. Mach, J. Med. Chem., 2014, 57, 4239 CrossRef CAS PubMed.
- M. W. Moon and R. A. Wade, J. Org. Chem., 1984, 49, 2663 CrossRef CAS.
- K. Niikura, N. Iyo, T. Higuchi, T. Nishio, H. Jinnai, N. Fujitani and K. Ijiro, J. Am. Chem. Soc., 2012, 134, 7632 CrossRef CAS PubMed.
- L. Ding, M. Xu, J. Wang, Y. Liao and J. Qiu, Polymer, 2014, 55, 1681 CrossRef CAS.
- E. V. Malykhin and V. D. Shteingarts, Russ. J. Appl. Chem., 2012, 85, 1232 CrossRef CAS.
- T. M. Rangarajan, R. Singh, R. Brahma, K. Devi, R. P. Singh, R. P. Singh and A. K. Prasad, Chem.–Eur. J., 2014, 20, 14218 CrossRef CAS PubMed.
- H. Lee, D. L. Chen, J. M. Rothfuss, M. J. Welch, R. J. Gropler and R. H. Mach, Nucl. Med. Biol., 2012, 39, 77 CrossRef CAS PubMed.
- L. Gilfillan, A. Blair, B. J. Morris, J. A. Pratt, L. Schweiger, S. Pimlott and A. Sutherland, Med. Chem. Commun., 2013, 4, 1118 RSC.
- I. AlJammaz, B. Al-Otaibi, H. AlHindas and S. M. Okarvi, Nucl. Med. Biol., 2015, 42, 804 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28851k |
| This journal is © The Royal Society of Chemistry 2017 |