DOI:
10.1039/C6RA28801D
(Paper)
RSC Adv., 2017,
7, 16721-16729
Enhancement of cell uptake and antitumor activity of selenadiazole derivatives through interaction and delivery by serum albumin†
Received
29th December 2016
, Accepted 2nd March 2017
First published on 15th March 2017
Abstract
Serum albumin is an important carrier in the transport of endogenous and exogenous substances across cell membranes. Small molecule drugs could bind to serum albumin to various extents after intravenous injection, which may affect the bioavailability, metabolism, pharmacological and toxicological potency. Organic selenium (Se) compounds exhibit favorable biocompatibility and low toxicity that attracts increased attention from researchers. Herein, a series of selenadiazole derivatives (4a–e) have been synthesized and their cytotoxicity towards various kinds of cells were evaluated. The results suggested that isopropyl benzo[c][1,2,5]selenadiazole-5-carboxylate (4d) exhibited a broad inhibition spectrum on the proliferation of tested cancer cells, with a relatively higher anticancer activity than other derivatives. We found that the differences in their anticancer activities were attributed to the various binding abilities of the Se compounds toward BSA. The bimolecular quenching constant (Kq), apparent quenching constant (KSV), effective binding constant (KA) and binding site number (n) were obtained through fluorescence quenching calculations, which indicated that all compounds could efficiently bind to BSA molecules and the fluorescence quenching mechanism was mainly a static quenching procedure. Moreover, serum albumin was found to interact with selenadiazole derivatives (4a–e), thus promoting the cellular uptake and anticancer activity of the Se compounds. Taken together, this study demonstrated that the synthetic selenadiazole derivatives exhibited high anticancer activity and cellular uptake through delivery by human serum.
Introduction
Although people's living standard has been improved, cancer is still one of the serious diseases threatening human health.1 In the past decades, a large number of agents have been synthesized and investigated for their effects on cancer chemoprevention and chemotherapy. Particularly, selenium (Se), an essential trace element that is fundamentally important to humans,2,3 has been verified as a potential cancer chemopreventive and chemotherapeutic agent by various epidemiological, preclinical and clinical studies.4,5 Comparing with inorganic selenium, organoselenium compounds show several advantages, such as higher bio-absorption, higher anticancer activity and lower cytotoxicity.6,7 Therefore, growing attention has been focused on the application of organoselenium compounds in chemoprevention and chemotherapy. Recently, a large number of potent organoselenium compounds have been designed and synthesized which showed greater chemopreventive efficacy,8 such as selenocyanate,9 selenobetaine and selenium analogues of amino acids.10–13 In addition, the synthesis of heterocyclic compounds with a Se atom in the ring is a fascinating field owing to their pharmacological potential and fascinating chemical properties.5,14–17 For instance, a large amounts of selenazolo derivatives, like ebselen, 6-bromo[1,2,5]selenadiazolo[3,4-b]pyridine, 4-methyl-1,2,3-selenadiazole-5-carboxylic acid amides and 6-phenyl-7(6H)-isoselenazolo[4,3-d]pyrimidone, have been synthesized and found to exhibit antitumor activities in vitro and in vivo.14,18
However, the anticancer mechanisms of the selenium compounds remain elusive, which limits their clinical development. Serum albumin, a globular heart-shaped protein composed of three structurally similar domains (I, II and III), is a 583-amino acid residue monomer with a molecular weight of 66.4 kDa. And each domain consists of two subdomains (A and B) stabilized by 17 pairs of disulfide bridges.19,20 Human serum albumin (HSA)21 is a major constituent of blood plasma, having many physiological functions like transportation facilitation or delivery of several endogenous and exogenous compounds.22,23 It is well known that small molecule drugs are capable to bind with HSA,24 which may have impacts on their bioavailability and antitumor potency. Especially, the binding affinity of drugs toward BSA and HSA affect the rate of the drug release to sites of catabolism or pharmacological action.25 Generally, BSA is chosen as a target protein molecule due to its low cost, ready availability and unique ligand-binding properties.26 The studies on drug's binding to BSA provide the information about structural features that could determine the therapeutic effectiveness of drugs, which become an important research field in chemistry, life science and clinical medicine.
The quantum yield of fluorescence was decreased because the fluorophore was induced by a variety of molecular interactions with a quencher molecule.27 Fluorescence quenching can be dynamic, resulting from collisional encounters between the fluorophore and the quencher, or static, resulting from the formation of a ground-state complex between the fluorophore and the quencher.28 In both cases, molecular contact is required between the fluorophore and the quencher for fluorescence quenching. A number of studies on the interaction of small molecules or drugs with BSA have been recently reported29–31 and elucidation of the molecular aspects of the binding continues to be of great importance in the terms of understanding protein structure–function and designing drug.
In the present study, we have synthesized a series of selenadiazole derivatives (4a–e), and investigated for their underlying anticancer actions, including interactions toward BSA, cellular uptake and anticancer activities. The results showed that, BSA molecules could easily interact with these five selenadiazole, and then quench their intrinsic fluorescence efficiently. Interestingly, all the emission maximum of tyrosine residues have a slight blue shift and the slight red shift of all tryptophan (Trp) residues fluoresce, which indicated that the polarity around the tyrosine (Tyr) residues increased and the hydrophobicity decreased, while the tryptophan residues' hydrophobicity was increased. Among these selenadiazole derivatives, compound 4d with isopropyl group could be easily internalized by cells due to its high binding affinity toward BSA, thus enhancing the anticancer efficacy toward MCF-7 cancer cells. Taken together, this study demonstrates that, the synthetic selenadiazole derivatives could be further developed as effective and safe anticancer agent for cancer chemotherapy.
Results and discussion
Synthesis and in vitro anticancer activities of selenadiazole derivatives (4a–e)
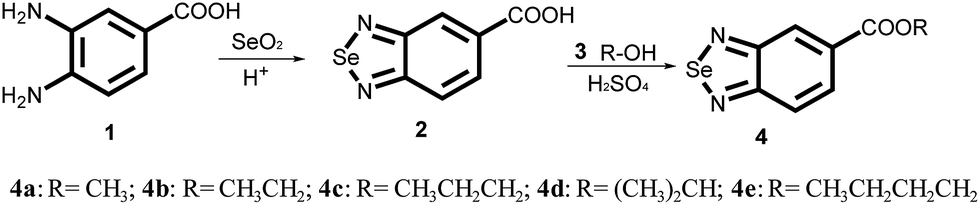
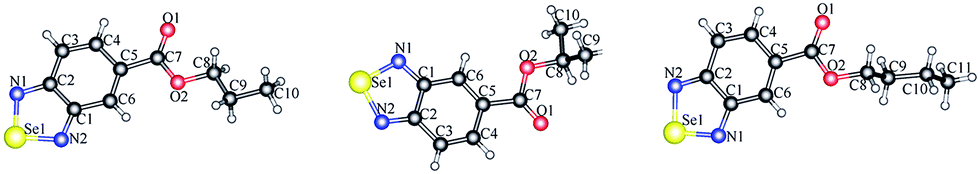
The synthesis of selenadiazole derivatives (4a–e) was outlined in Scheme 1. The crystal structures of 4c, 4d and 4e (Fig. 1) were obtained. Crystal data and structure refinement data of the three compounds were listed in ESI Table S1.† Crystallographic data structures of compound 4c, 4d and 4e have been deposited to the Cambridge Crystallographic Data Centre with CCDC 1516447, 1516438 and 1517017. The in vitro anticancer activities of 4a–e were firstly screened against a panel of five human cancer cell lines, including A549, A375, MCF-7, HepG2, Neuro-2a, and one human normal cells HK-2 by MTT assay32 after 72 h treatment. From the Table 1, we found 4d showed broad inhibition on the tested cancer cells with IC50 values significantly lower than those of its analogues, indicating higher cytotoxic effects of 4d on cancer cells.
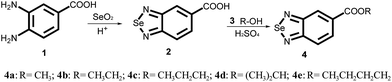 |
| | Scheme 1 Synthetic scheme for the production of selenadiazole derivatives (4a–e). | |
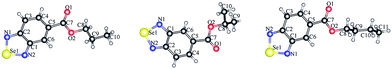 |
| | Fig. 1 Crystal structures of compounds 4c, 4d and 4e. | |
Table 1 Growth inhibition of 4a–e against human cancer and normal cell lines
| Cell linesa |
IC50 (μM) |
| 4a |
4b |
4c |
4d |
4e |
| Cancer cells were exposed to different concentration of SeDs for 72 h and then cell viability was measured by MTT assay. Normal cells. |
| A549 |
125.2 ± 7.36 |
69.14 ± 2.71 |
59.78 ± 2.77 |
27.81 ± 4.33 |
68.54 ± 2.57 |
| A375 |
115.06 ± 4.14 |
109.85 ± 5.00 |
117.63 ± 1.68 |
63.71 ± 1.65 |
118.23 ± 4.7 |
| MCF-7 |
122.17 ± 3.29 |
78.0 ± 8.96 |
46.3 ± 1.61 |
33.2 ± 8.31 |
47.90 ± 7.56 |
| HepG2 |
156.61 ± 5.53 |
141.97 ± 10.19 |
103.28 ± 2.21 |
72.45 ± 9.87 |
76.40 ± 2.39 |
| Neuro-2a |
151.13 ± 3.99 |
143.85 ± 11.51 |
105.83 ± 5.19 |
51.82 ± 9.46 |
69.71 ± 2.78 |
| HK-2b |
217.61 ± 2.71 |
224.63 ± 8.51 |
208.31 ± 3.74 |
136.74 ± 10.77 |
193.61 ± 3.59 |
These results suggest that, the as-synthesized selenadiazole derivatives demonstrate comparable anticancer efficacy with the previously reported compounds. For instance, Xie et al. reported that 4 biphenyl selenadiazole derivatives with IC50 on A375 cells ranging from 23.3 μM to 66.6 μM had a radiation sensitization activity.5 Yang et al. reported 7 biphenylselenadiazole derivatives with IC50 on bladder cancer cells ranging from 1.48 μM to 78.67 μM had activities of inducing apoptosis and inhibition of migration and invasion.17 Liu et al. reported biphenylselenadiazole derivatives with a IC50 at about 30 μM against HepG2 cells had the ability to reverse the drug resistance induced by hyperglycemia.16 Taken together, these studies confirm the anticancer application potential of selenadiazole derivatives.
Binding property of the selenadiazole derivatives (4a–4e) to the serum albumins
Next the binding of 4a–e to BSA was investigated by fluorescence spectroscopy. BSA excited at 296 nm and showed emission at 348 nm, which is mainly attributed to the presence of two tryptophan residues (Trp-134 and Trp-213), that processed intrinsic fluorescence.33,34 Trp-134 in the first domain is located on the surface of the hydrophilic region of the protein while Trp-213 in the second domain is located within a hydrophobic binding pocket.35,36 The absorption spectra of 4a–4e were showed in Fig. S19.† The maximum absorption of 4a–4e were around 320 nm, which will not interfere on the observation the binding property of these compounds to serum albumin by using fluorescence quenching assay. The fluorescence quenching spectra of BSA (1.0 μM) in Tris–HCl buffer with increasing concentration of compounds 4a–e were shown in Fig. 2A–E. The fluorescence intensity was attenuated gradually with the increasing concentration of each compound, which displayed that there was a binding interaction of compounds to BSA and the energy transfer occurred. Generally, the fluorescence quenching can be dynamic or static.35 Firstly, as a hypothetical dynamic quenching process, the data of fluorescence intensities were analyzed by using the Stern–Volmer equation:37| | |
F0/F = 1 + Kqτ0[Q] = 1 + KSV[Q]
| (1) |
where F0 and F are the fluorescence intensities in the absence and presence of the quencher, respectively. Kq is the quenching rate constant. KSV is the dynamic quenching rate constant, τ0 is the average lifetime of the protein without the quencher, which is of the order of 10−8 s (ref. 38) and [Q] is the concentration of compounds. The plot of F0/F versus [Q] was linear (Fig. 2F–J), indicating one type of quenching. The values of KSV could be derived from these plots, Kq could be calculated from the ratio of KSV and τ0 showed value of 1012 order of magnitude. The corresponding plot expressions were shown in Table 2.
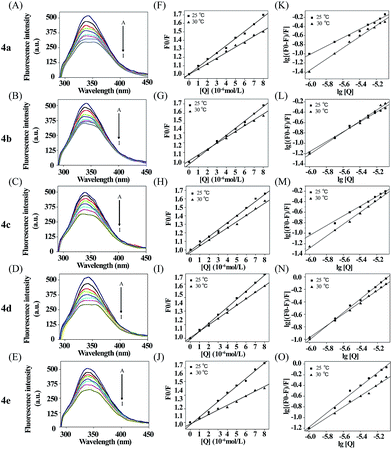 |
| | Fig. 2 (A–E) Fluorescence emission spectra of BSA in the presence of various concentrations of 4a–e, curves from (A to I) correspond to 4a–e concentrations of 0, 1, 2, 3, 4, 5, 6, 7 and 8 μM, respectively (T = 298 K, pH = 7.4 and λex = 296 nm); (F–J) Stern–Volmer plot for the binding of 4a–e with BSA at 298 K and 303 K; (K–O) The Double logarithm plot of 4a–e at 298 K and 303 K. | |
Table 2 Quenching constants (KSV and Kq), binding constants and binding site numbers of the interaction of 4a–e with BSA at two different temperaturea
| System |
Temperature (K) |
Eqn (1) |
Eqn (2) |
| KSV (L mol−1) |
Kq (L mol−1 s−1) |
KA (L mol−1) |
n |
| These values were obtained respectively using Stern–Volmer plots, Double logarithm plots and Van't Hoff plots of the interaction of 4a–e with BSA (compounds concentrations from 0 × 10–6 mol l−1 to 8 × 10–6 mol l−1). |
| BSA + 4a |
298 |
8.42 × 104 |
8.42 × 1012 |
3.87 × 104 |
0.935 |
| 303 |
6.43 × 104 |
6.43 × 1012 |
7.94 × 104 |
1.0209 |
| BSA + 4b |
298 |
7.24 × 104 |
7.24 × 1012 |
3.84 × 104 |
0.9578 |
| 303 |
6.00 × 104 |
6.00 × 1012 |
4.07 × 104 |
0.9707 |
| BSA + 4c |
298 |
8.35 × 104 |
8.35 × 1012 |
3.18 × 104 |
0.9239 |
| 303 |
7.12 × 104 |
7.12 × 1012 |
1.21 × 105 |
1.0335 |
| BSA + 4d |
298 |
11.81 × 104 |
11.81 × 1012 |
3.27 × 105 |
1.0866 |
| 303 |
9.54 × 104 |
9.54 × 1012 |
3.47 × 105 |
1.10 |
| BSA + 4e |
298 |
11.35 × 104 |
11.35 × 1012 |
1.91 × 105 |
1.052 |
| 303 |
6.38 × 104 |
6.38 × 1012 |
1.01 × 105 |
1.0367 |
For dynamic quenching, the maximum value of the scattering collision quenching constant Kdiff is 2.0 × 1010 l mol−1 s−1.39 Since the value of the biomolecular quenching is higher, it can be assumed that the complex formation between the quencher and the protein is a static mechanism. The static quenching mechanism was further confirmed by the temperature dependence of the quenching; KSV values decreased with increasing temperature for static quenching while the reverse is observed for dynamic quenching.40,41 The fluorescence data were analyzed at two different temperatures. Seen from the Table 2, the Stern–Volmer quenching constant KSV and the quenching rate constant Kq values decreased with increasing temperature, suggesting that the quenching mechanism was complex formation rather than dynamic collision. In other words, the fluorescence quenching of BSA induced by these compounds was caused by specific complex formation.
Analysis of binding equilibria
From Fig. 2K–O, the equilibrium constants (KA) and the binding site numbers (n) could be calculated by using the Double logarithm equation:42| |
lg[(F0 − F)/F] = lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KA + nlg[Q] KA + nlg[Q]
| (2) |
Apparently, the KA and n could be measured, respectively, from the intercept and slope obtained through plotting lg[(F0 − F)/F] against lg[Q], shown in Table 2. There was a strong binding force between 4a–e and BSA. In addition, the KA value of these complexes demonstrated that the binding affinity of 4d to BSA was stronger. The values of n were equal to about 1, illustrating that there was one class of binding site for the compounds in the neighborhood of the tryptophan residue.43
Types of interaction force between BSA with selenadiazole derivatives
Following, studies were also carried out to analyse the modes of interaction between BSA and the as-synthesized selenadiazole derivatives. Utilizing KA, the free energy change (ΔG) was calculated from the relationship:| | |
ΔG = −RTlnKA
| (3) |
When the temperature change is not very large, the enthalpy change (ΔH) of a system can be regarded as a constant.43 Under these conditions, both the enthalpy change (ΔH) and entropy change (ΔS) can be evaluated from the following equations:
| | |
ln(K2/K1) = (1/T1 − 1/T2)ΔH/R
| (4) |
In this study, the results (Table 3) showed that the binding of compounds 4a–e to BSA is a spontaneous process, as indicated by the negative ΔG values. From the values of ΔG, it demonstrated that compound 4d bound more easily with BSA molecules. Essentially, there are four main types of noncovalent interactions existing in ligand binding to proteins, which are hydrogen bonds, van der Waals forces, hydrophobic and electrostatic interactions.44,45 According to the principles summarized by Ross and Subramanian, for drug protein interactions, the positive values of ΔH and ΔS indicate that hydrophobic interactions play a major role in the binding process. While, the negative values of ΔH and ΔS suggest that hydrogen bonding and van der Waals force play a crucial role in the binding process. From Table 3, we can deduce that the acting forces of BSA-4a, 4b, 4c system and the BSA-4d, 4e system were mainly hydrophobic interactions and hydrogen bonding, van der Waals force respectively.
Table 3 Thermodynamic parameters of the interaction of 4a–e with BSA at different temperatures
| System |
T (K) |
ΔG° (kJ mol−1) |
ΔH° (kJ mol−1) |
ΔS° (J mol−1) |
| BSA + 4a |
298 |
−26.17 |
543.61 |
1.73 |
| 303 |
−28.42 |
543.61 |
1.73 |
| BSA + 4b |
298 |
−26.15 |
354.44 |
1.28 |
| 303 |
−26.73 |
354.44 |
1.28 |
| BSA + 4c |
298 |
−25.69 |
200.63 |
0.76 |
| 303 |
−23.68 |
200.63 |
0.76 |
| BSA + 4d |
298 |
−31.46 |
−336.80 |
−1.02 |
| 303 |
−32.13 |
−336.80 |
−1.02 |
| BSA + 4e |
298 |
−31.07 |
−498.81 |
−1.55 |
| 303 |
−29.03 |
−498.81 |
−1.55 |
Effect of selenadiazole derivatives (4a–e) on BSA synchronous fluorescence spectra
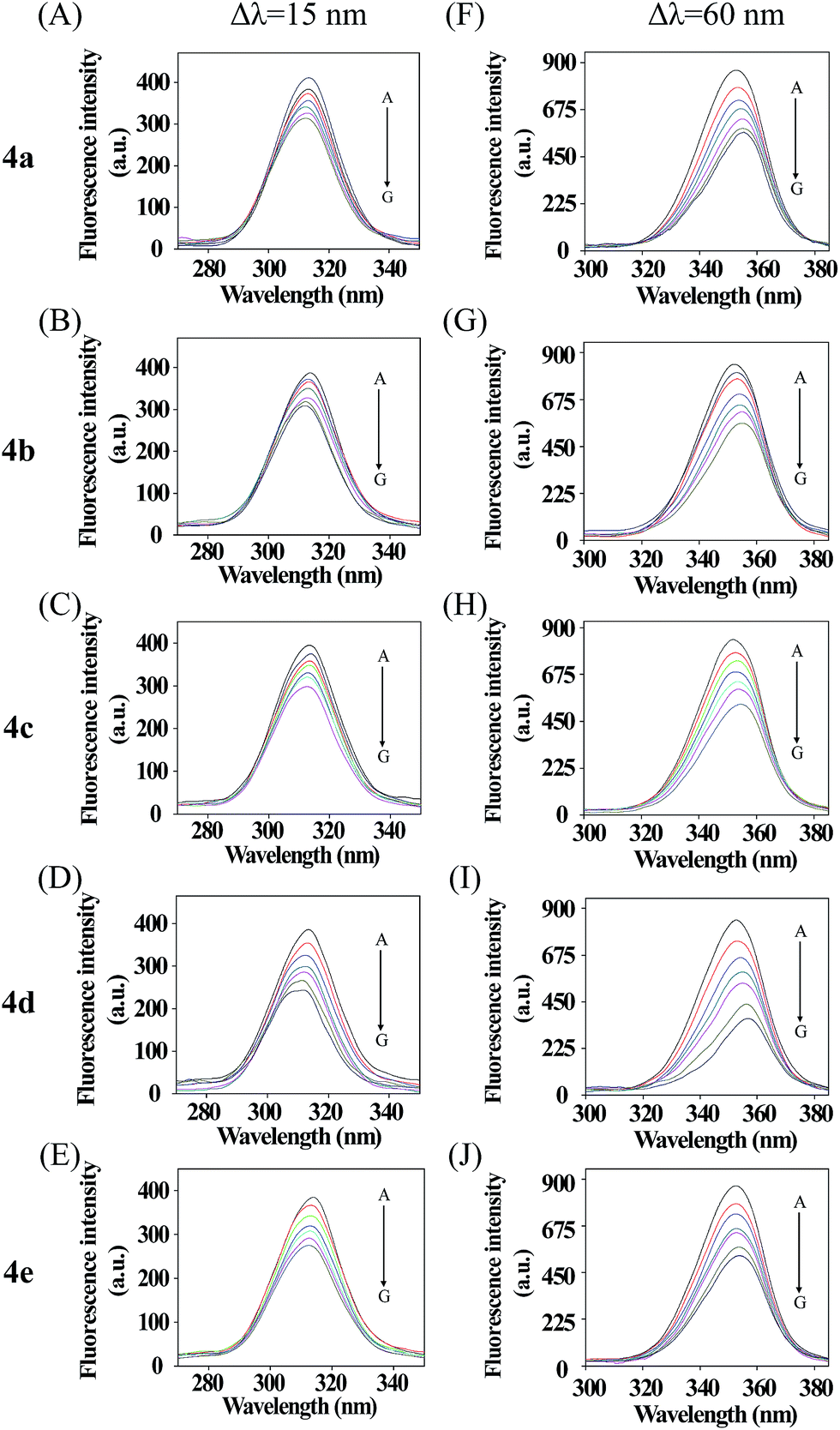
Synchronous fluorescence spectra can provide much valuable information about the microenvironment around fluorogens in biomolecules.46,47 For protein molecules, when the λ = 15 nm is fixed, the spectrum characteristic of tyrosine residues could be observed, and when λ = 60 nm is done, the spectrum characteristic of tryptophan residues could be obtained.48 In Fig. 3, five courses the fluorescence intensities for λ = 15 nm (upper) and λ = 60 nm (nether) are both quenched more and more severely with increasing compounds concentration, which further demonstrated fluorescence quenching in the binding process. Furthermore, all the emission maximum of tyrosine residues have a slight blue shift and a slight red shift of all tryptophan residues fluoresce, which indicated that the polarity around the tyrosine residues increased and the hydrophobicity decreased,49 while the tryptophan residues' hydrophobicity was increased. In conclusion, these five compounds affect the microenvironment of Tyr and Trp residues through binding to BSA molecules with different accessibility.50
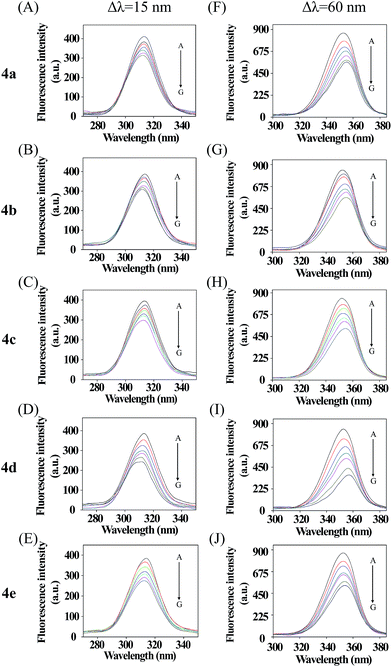 |
| | Fig. 3 Synchronous fluorescence spectra of BSA in the presence of various concentrations of 4a–e, concentrations from 0 to 6 μM. Δλ = 15 nm (A–E); Δλ = 60 nm (F–J) (T = 298 K, pH = 7.4). | |
Selenadiazole derivatives (4a–e) inhibit cancer cell growth by enhanced cellular uptake and induced apoptosis
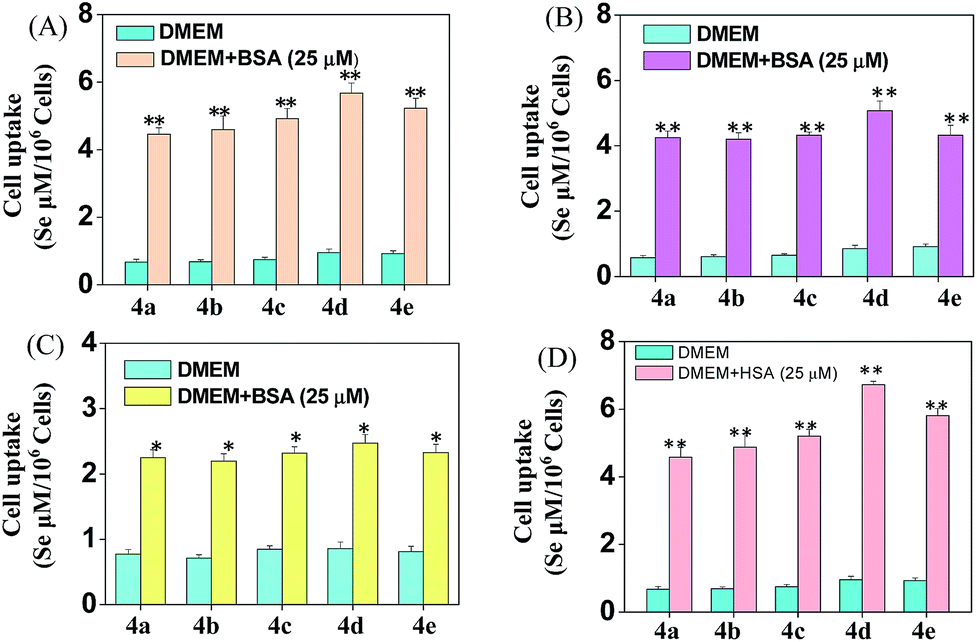
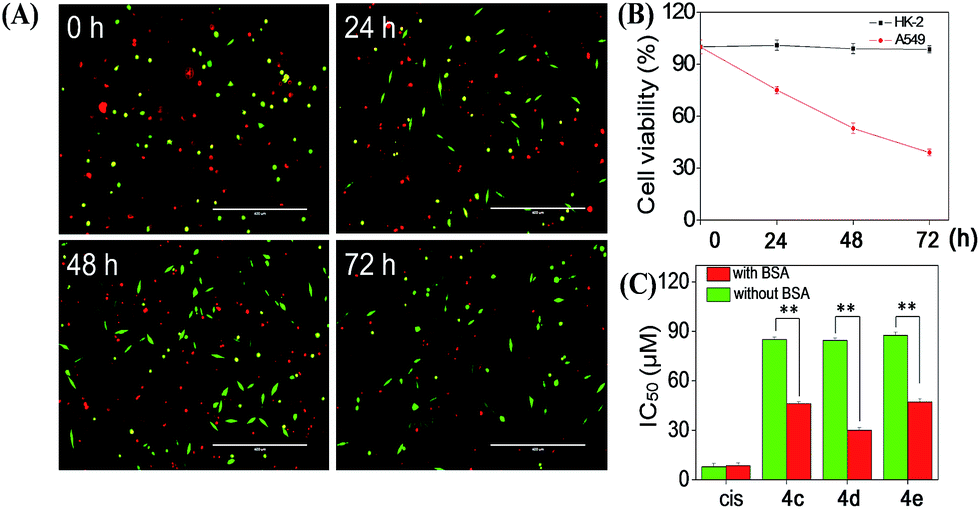
Cellular uptake is a complicated biological process that associates with the interaction between extracellular materials and the plasma membrane or the receptor proteins.51,52 The efficacy of cellular uptake is a critical factor that affects the selenadiazole derivative agent activities and determines their successful clinical application. Therefore, in this study, we have examined the uptake of selenadiazole derivatives (4a–e) in MCF-7 cell in the presence or absence of serum. The stability of BSA in DMEM medium with or without 4a–4b was detected by measuring the size distribution. As shown in Fig. S20,† the size of BSA was stable at around 4 nm in the absent or present of 4a–4b for up to 72 h. These results demonstrate the stability of BSA during drug delivery. As shown in Fig. 5A, the absorption of selenium value of 4d containing an isopropyl substituent in MCF-7 cells was about 5.7 μM/106 cells, which was higher than that of 4a (4.45 μM/106 cells), 4b (4.59 μM/106 cells), 4c (4.91 μM/106 cells), 4e (5.22 μM/106 cells). Consistently, comparing with 4a, 4b, 4c and 4e, 4d showed a higher absorption value and a dramatic enhancement in the anticancer efficacy (Fig. 4A–E). The viabilities of cells treated with different concentrations of the selenadiazole derivatives were more than 80% in basal culture medium without FBS, but the viabilities decreased significantly when BSA (25 μM or 50 μM) was added to the media. These results indicate that BSA as a drug carrier of selenadiazole derivatives effectively promote cell uptake and enhance antitumor activity against cancer cells. Further determination of the cellular uptake of 4d in A549 cells and HK-2 cells shown that uptake of 4d by cancer cells were much higher than that by normal cells (Fig. 5B and C). Moreover, as shown in Fig. 4F–J and 5D, similar results can be gained when BSA was replaced by HSA. To further confirm the selective growth inhibition of 4d on cancer cells, we performed a co-cultured assay. As shown in Fig. 6A and B, the viabilities of A549 cells were significantly decreased when the cells were treated with 4d, while the viabilities of HK-2 cells have no obvious change.
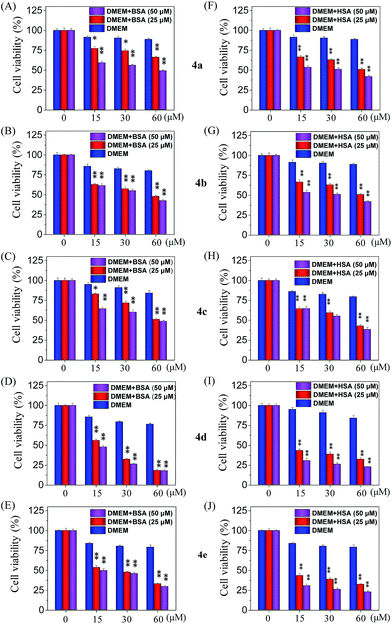 |
| | Fig. 4 Inhibition rate of 4a–e. (A–E) The inhibitory effects of 4a–e on MCF-7 cells after 72 h incubation with or without BSA in DMEM culture medium. (F–J) The inhibitory effects of 4a–e on MCF-7 cells after 72 h incubation with or without HSA in DMEM culture medium. Significant difference between treatment and control groups is indicated at the P < 0.05 (*) or P < 0.01 (**) level. | |
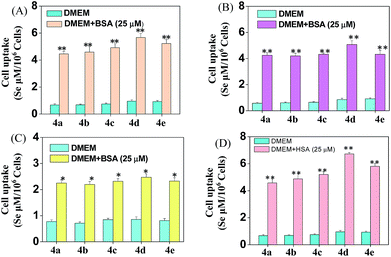 |
| | Fig. 5 Cellular uptake of 4a–e. (A–C) Different cellular uptake of 4a–e (200 μM) on MCF-7, A549 and HK-2 cells treated with or without BSA in DMEM culture medium for 6 h. (D) Different cellular uptake of 4a–e (200 μM) on MCF-7 cells treated with or without HSA in DMEM culture medium for 6 h. Significant difference between treatment and control groups is indicated at the P < 0.05 (*) or P < 0.01 (**) level. | |
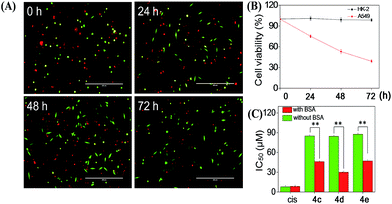 |
| | Fig. 6 (A) The represent images of HK-2 (green) and A549 (red) cells treated with 4d for indicated time. (B) The viabilities of HK-2 and A549 cells treated with 4d in (A) were calculated by normalizing to untreated control group. Bars = 400 μm (C) The IC50 of 4c–e and cisplatin (cis) on MCF-7 cells after 72 h incubation with or without BSA in DMEM culture medium. Significant difference between groups with or without BSA is indicated at the P < 0.05 (*) or P < 0.01 (**) level. | |
To compare the importance of interaction with BSA on the cellular uptake and anti-tumour activity of selenadiazole derivatives and other cancer drugs, the IC50 of a wildly used anti-cancer drug, cisplatin, and 3 selenadiazole derivatives 4c–4e which showed relative higher activity were detected by MTT assay. As shown in Fig. 6C, although cisplatin exhibited lower IC50 then all the selenadiazole derivatives, the values were not affected by the present or absent of BSA. On the contrary, 4c–4e shown significantly reduced IC50 when BSA was added. In addition, the selenadiazole derivatives demonstrate high selectivity between cancer (A549) and normal (HK-2) cells, which is an important advantage for the future clinical application. Taken together, these results fully showed that BSA (or HSA) as a drug carrier could greatly enhance the absorption of selenadiazole derivatives to significantly enhance their anti-tumor activity and selectivity.
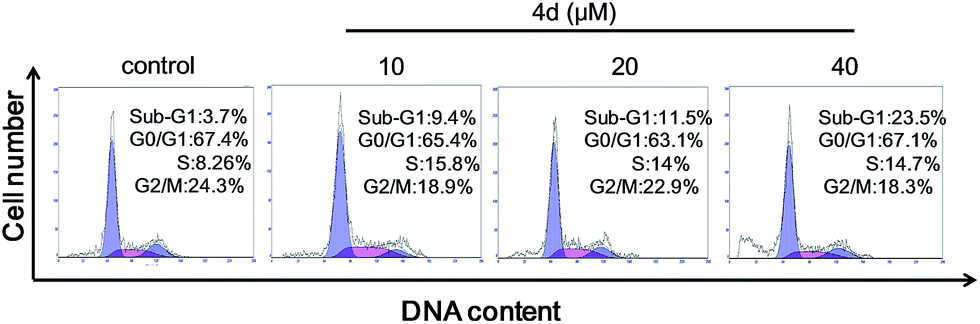
To explore the mechanism of cell death induced by selenadiazole derivatives after uptake by cancer cells, we measured the cell cycle distribution and apoptosis of MCF-7 cells after treated with 4d. The result of PI-staining and flow cytometric analysis (Fig. 7) shown that 4d induced the increase of sub-G1 fraction of MCF-7 cells in a dose-dependent manner, suggesting anti-tumor activity of 4d may be caused by the induction of apoptosis.
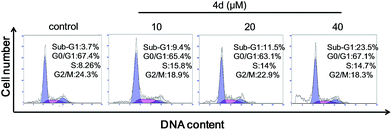 |
| | Fig. 7 Quantitative analysis of cell cycle distribution and apoptotic cells induced by different concentrations of compound 4d by PI-flow cytometric analysis in MCF-7 cells. | |
Conclusions
In conclusion, we have synthesized five selenadiazole derivatives (4a–e), and compared their anticancer activities in vitro. Based on these results, the isopropyl benzo[c][1,2,5]selenadiazole-5-carboxylate (4d) exhibited better anticancer activities than others. The studies of the interaction with BSA suggested that these five compounds can bind to BSA molecules easily, and then quench their intrinsic fluorescence efficiently. The binding parameters further illustrated that all compounds can efficiently bind to BSA molecules. We speculated that a series of complexes were formed between compounds 4a–e and BSA by hydrophobic interactions, hydrogen bond, and van der Waals force. Interestingly, 4d exhibited not only better capacity of binding to BSA, but also higher anticancer activities. The biological significance of this work is that albumin serves as a carrier molecule for selenadiazole derivatives to enhance their antitumor activity, which may have an influence on the fields of pharmacology and clinical medicine.
Materials and methods
Materials
Methanol, ethanol, propanol, isopropyl, n-butanol, chloroform, sodium hydroxide, sodium sulfate anhydrous, hydrochloric acid were purchased from Guangzhou chemical reagent factory and selenium dioxide, 3,4-diaminobenzoic acid, BSA and HSA were purchased from J&K Chemical Company. Thiazolyl blue tetrazolium bromide (MTT), DMSO, DMEM media were purchased from sigma. The solutions of BSA and HSA were prepared in 0.01 M Tris–HCl buffer of pH 7.4 containing 0.01 M NaCl. BSA and HSA solutions were prepared based on their molecular weights of 65000 and 66000, respectively. The water used in all experiments was ultrapure by a Mili-Q water purification system from Milipore.
Synthesis and characterization of selenadiazole derivatives (4a–e)
The synthetic route for SeDs (4a–4e) was elucidated in Scheme 1. The first step, 10 mmol of 3,4-diamino-benzoic acid was dissolved in 250 ml hydrochloric acid solution (HCl:H2O = 1:5) in a 500 ml flask. 10 mmol SeO2 which dissolved in 20 ml of hot distilled water was dropping into the flask through a constant pressure funnel stirred for 2 h. After that, the reacted solution was filtrated and the obtained solids were washed with water for several times until the pH value reached to 7. Then the products were dried at 60 degree to get the pure benzo[1,2,5]selenadiazole-5-carboxylic acid. Yield: 95%. In the second step, 10 mmol of benzo[1,2,5]selenadiazole-5-carboxylic acid dissolved with 50 ml CHCl3 was placed in a 100 ml three-necked flask, which was provided with a stirrer. Then the corresponding alcohol was added slowly and dropwise under stirring and the temperature at 50 °C. And small amounts of sulfuric acid were added as a catalyst. The reaction mixture was stirred for another 4 h. The solvent was removed in vacuum distillation, then, another 300 ml 1 mol/LNaSO4 solution was added to wash the crude product. After that, the crude products were filtrated, and the pure product was obtained through recrystallization from corresponding alcohols. Yield: about 65%.
Benzo[c][1,2,5]selenadiazole-5-carboxylic acid
The compound 2 was prepared according to a general procedure. ESI-MS (in CH3OH): m/z calcd for C7H4N2O2Se (M − H)− 226.9; found 227.0. 1H NMR (500 MHz, DMSO, δ, ppm): 7.96 (d, 1H), 7.94 (d, 1H), 8.42 (s, 1H), 13.18 (s, 1H). 13C NMR (125 MHz, DMSO, δ, ppm): 123.73, 125.86, 128.31, 131.66, 159.63, 161.18, 167.33. Elemental analysis calc. (%) for C7H4N2O2Se: C, 37.03; H, 1.78; N, 12.34; found (%): C, 37.00; H, 1.77; N, 12.35.
Methyl benzo[c][1,2,5]selenadiazole-5-carboxylate
The compound 4a was prepared according to a general procedure. ESI-MS (in CH3OH): m/z calcd for C8H6N2O2Se (M + H)+ 243.0; found 243.2. 1H NMR (500 MHz, DMSO, δ, ppm): 7.95 (d, 1H), 7.96 (d, 1H), 8.45 (s, 1H), 3.93 (s, 3H). 13C NMR (125 MHz, DMSO, δ, ppm): 53.20, 124.02, 126.02, 127.83, 130.46, 159.41, 161.15, 166.30. Elemental analysis calc. (%) for C8H6N2O2Se: C, 39.85; H, 2.51; N, 11.62; found (%): C, 39.82; H, 2.53; N, 11.61.
Ethyl benzo[c][1,2,5]selenadiazole-5-carboxylate
The compound 4b was prepared according to a general procedure. ESI-MS (in CH3OH): m/z calcd for C9H8N2O2Se (M + H)+ 257.0; found 257.2. 1H NMR (500 MHz, DMSO, δ, ppm): 7.95 (d, 1H), 7.96 (d, 1H), 8.45 (s, 1H), 4.37 (d, 2H), 1.37 (t, 3H). 13C NMR (125 MHz, DMSO, δ, ppm): 14.58, 61.92, 123.97, 125.91, 127.83, 130.70, 159.43, 161.16, 165.76. Elemental analysis calc. (%) for C9H8N2O2Se: C, 42.37; H, 3.16; N, 10.98; found (%): C, 42.35; H, 3.17; N, 10.95.
Propyl benzo[c][1,2,5]selenadiazole-5-carboxylate
The compound 4c was prepared according to a general procedure. ESI-MS (in CH3OH): m/z calcd for C10H10N2O2Se (M + H)+ 271.0; found 271.3. 1H NMR (500 MHz, DMSO, δ, ppm): 7.96 (d, 1H), 7.97 (d, 1H), 8.45 (s, 1H), 1.78 (m, 2H), 4.30 (t, 2H), 1.01 (t, 3H). 13C NMR (125 MHz, DMSO, δ, ppm): 10.84, 22.04, 67.28, 124.02, 125.90, 127.83, 130.69, 159.44, 161.17, 165.81. Elemental analysis calc. (%) for C10H10N2O2Se: C, 44.62; H, 3.74; N, 10.41; found (%): C, 44.60; H, 3.77; N, 10.40.
Isopropyl benzo[c][1,2,5]selenadiazole-5-carboxylate
The compound 4d was prepared according to a general procedure. ESI-MS (in CH3OH): m/z calcd for C10H10N2O2Se (M + H)+ 271.0; found 271.2. 1H NMR (500 MHz, DMSO, δ, ppm): 7.94 (d, 1H), 7.95 (d, 1H), 8.45 (s, 1H), 5.91 (m, 1H), 1.37 (d, 6H). 13C NMR (125 MHz, DMSO, δ, ppm): 22.09, 69.53, 123.94, 125.83, 127.85, 131.03, 159.46, 161.16, 165.24. Elemental analysis calc. (%) for C10H10N2O2Se: C, 44.62; H, 3.74; N, 10.41; found (%): C, 44.59; H, 3.77; N, 10.40.
Butyl benzo[c][1,2,5]selenadiazole-5-carboxylate
The compound 4e was prepared according to a general procedure. ESI-MS (in CH3OH): m/z calcd for C11H12N2O2Se (M + H)+ 285.0; found 285.2. 1H NMR (500 MHz, DMSO, δ, ppm): 7.95 (d, 1H), 7.96 (d, 1H), 8.44 (s, 1H), 1.45 (m, 2H), 1.74 (m, 2H), 4.05 (t, 2H), 0.95 (t, 3H). 13C NMR (125 MHz, DMSO, δ, ppm): 14.08, 19.22, 30.63, 65.54, 123.96, 125.86, 127.78, 130.64, 159.41, 161.14, 165.75. Elemental analysis calc. (%) for C11H12N2O2Se: C, 46.65; H, 4.27; N, 9.89; found (%): C, 46.62; H, 4.28; N, 9.88.
Cell line and cell culture
All human cell lines used in this research, including A549 (adenocarcinoma human alveolar basal epithelial cells), MCF-7 (breast adenocarcinoma cells), Neuro-2a (neuroblastoma cells), HepG2 (hepatocellular carcinoma cells), A375 (human melanoma cells), HK-2 (human renal proximal tubule cells), were purchased from American Type Culture Collection (Manassas, VA). All the cell lines were incubated in DMEM media supplemented with penicillin (100 units per ml), streptomycin (50 units per ml) and fetal bovine serum (10%) at 37 °C in a humidified incubator with 5% CO2 atmosphere.
MTT assay
The anticancer activity of selenadiazole derivatives on cells were evaluated by MTT assay as reported.32 Data was expressed as the percentage of MTT reduction relative to the absorbance of control cells. Cell viability assays included the addition of different concentrations BSA (or HSA) in DMEM (the DMEM used in these comparative experiments did not contain fetal bovine serum).
Cellular uptake of selenadiazole derivatives (4a–4e)
MCF-7 cells (8 × 104 cells per ml, 8 ml) were seeded in 10 cm cell culture dishes and pre-cultured for 24 h. After the 10 cm cell culture dishes were washed twice lightly, the cells were then incubated in DMEM with or without BSA (or HSA) containing 200 μM SeDs (4a–4e) for another 6 h, respectively. Cellular uptake was determined by atomic fluorescence spectrometry of intracellular selenium content obtained.
Selenadiazole derivatives (4a–4e)–protein interactions
Based on preliminary experiments, the BSA concentrations were fixed at 1 μM and the drug concentration was varied from 0 to 8 μM for BSA. Fluorescence spectra were recorded at two temperatures (298 and 303 K) in the range of 290–450 nm upon excitation at 296 nm for BSA.
Flow cytometric analysis
Flow cytometric analysis was used to examine the cell cycle distribution of MCF-7 cells. Briefly, cells were seeded into 6 cm culture dishes with or without 4d for 24 h, and then the treated cells were collected and fixed with 70% ethanol overnight at −20 °C.
Next, the cells were stained with propidium iodide (PI, in 1.21 mg ml−1 Tris) for 2 h in darkness and were analysed with Epics XL-MCL flow cytometer (Beckman Coulter, Miami, FL). Cell cycle distribution was analysed using Multi Cycle software (Phoenix Flow Systems, San Diego, CA).
Co-culture assay
To evaluate the selective inhibition of 4d on the growth of cancer cells and normal cells. HK-2 normal cells and A549 cancer cells were stained with CellTracker™ Green CMFDA and CellTracker™ Red CMPTX (Life technology), respectively. Then the cells were mixed at the ratio of 1:1 and plated in 6-well plate. After attachment, 4d and DMSO were added to the tested and control wells. The cells were imaged under the fluorescent microscopy at different time points. The cell viabilities were measured by comparing the number of the corresponding cells in the tested wells to those in control wells.
Statistical analysis
All experiments were carried out in triplicate and data were expressed as mean ± standard deviation. Difference between the control and the experimental groups were analyzed by two-tailed Student's t-test. Differences of P < 0.05 (*) or P < 0.01 (**) were considered statistically significant.
Acknowledgements
This work was supported by Natural Science Foundation of China (21271002, 21371076), Science Foundation for Distinguished Young Scholars of Guangdong Province (S2013050014667), YangFan Innovative & Entepreneurial Research Team Project (201312H05), Guangdong Special Support Program and Guangdong Frontier Key Technological Innovation Special Funds (2014B050505012) and Fundamental Research Funds for the Central Universities.
Notes and references
- H. Shen, G. Zeng, B. Sun, X. Cai, L. Bi, G. Tang and Y. Yang, Tumor Biol., 2015, 36, 4825–4831 CrossRef CAS PubMed
 .
. - M. P. Rayman, S. C. Bath, J. Westaway, P. Williams, J. Mao, J. J. Vanderlelie, A. V. Perkins and C. Redman, Br. J. Nutr., 2015, 9, 1–10 Search PubMed .
- G. Alfthan, M. Eurola, P. Ekholm, E.-R. Venäläinen, T. Root, K. Korkalainen, H. Hartikainen, P. Salminen, V. Hietaniemi and P. Aspila, J. Trace Elem. Med. Biol., 2015, 31, 142–147 CAS .
- R. Sinha and K. El-Bayoumy, Curr. Cancer Drug Targets, 2004, 4, 13–28 CrossRef CAS PubMed .
- Q. Xie, Y. Zhou, G. Lan, L. Yang, W. Zheng, Y. Liang and T. Chen, Biochem. Biophys. Res. Commun., 2014, 449, 88–93 CrossRef CAS PubMed .
- T. Wirth, Angew. Chem., Int. Ed., 2015, 54, 10074–10076 CrossRef CAS PubMed .
- M. A. Rizvi, M. Zaki, M. Afzal, M. Mane, M. Kumar, B. A. Shah, S. Srivastav, S. Srikrishna, G. M. Peerzada and S. Tabassum, Eur. J. Med. Chem., 2015, 90, 876–888 CrossRef CAS PubMed .
- J. Lü, J. Zhang, C. Jiang, Y. Deng, N. Özten and M. C. Bosland, Nutr. Cancer, 2016, 68, 1–17 CrossRef PubMed .
- L. S. Konstantinova, E. A. Knyazeva, A. A. Nefyodov, P. S. Camacho, S. E. Ashbrook, J. D. Woollins, A. V. Zibarev and O. A. Rakitin, Tetrahedron Lett., 2015, 56, 1107–1110 CrossRef CAS .
- H. Tapiero, D. M. Townsend and K. D. Tew, Biomed. Pharmacother., 2003, 57, 134–144 CrossRef CAS .
- T. Chen, W. Zheng, Y. S. Wong and F. Yang, Biomed. Pharmacother., 2008, 62, 77–84 CrossRef CAS PubMed .
- K. El-Bayoumy, R. Sinha, J. P. Richie Jr, R. Brigelius-Flohe and H. Sies, Diversity of Selenium Functions in Health and Disease, 2015, vol. 38, p. 137 Search PubMed .
- Y.-W. Liang, J. Zheng, X. Li, W. Zheng and T. Chen, Eur. J. Med. Chem., 2014, 84, 335–342 CrossRef CAS PubMed .
- D. Plano, E. Moreno, M. Font, I. Encio, J. A. Palop and C. Sanmartin, Arch. Pharm., 2010, 343, 680–691 CrossRef CAS PubMed .
- L. He, S. Ji, H. Lai and T. Chen, J. Mater. Chem. B, 2015, 3, 8383–8393 RSC .
- Y. Liu, Y. Luo, X. Li, W. Zheng and T. Chen, Chem.–Asian J., 2015, 10, 768 CrossRef .
- Y. Yang, S. Deng, Q. Zeng, W. Hu and T. Chen, Dalton Trans., 2016, 45, 18465–18475 RSC .
- P. Arsenyan, K. Rubina, I. Shestakova and I. Domracheva, Eur. J. Med. Chem., 2007, 42, 635–640 CrossRef CAS PubMed .
- M. Hossain, A. Y. Khan and G. Suresh Kumar, PLoS One, 2011, 6, e18333 CAS .
- S. K. Chaturvedi, E. Ahmad, J. M. Khan, P. Alam, M. Ishtikhar and R. H. Khan, Mol. BioSyst., 2015, 11, 307–316 RSC .
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed .
- U. Kragh-Hansen, Pharmacol. Rev., 1981, 33, 17–53 CAS .
- K. B. Johnsen, J. M. Gudbergsson, M. N. Skov, L. Pilgaard, T. Moos and M. Duroux, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1846, 75–87 CrossRef CAS PubMed .
- T. Hu and Y. Liu, J. Pharm. Biomed. Anal., 2015, 107, 325–332 CrossRef CAS PubMed .
- M. Shahlaei, B. Rahimi, A. Nowroozi, M. R. Ashrafi-Kooshk, K. Sadrjavadi and R. Khodarahmi, Chem.-Biol. Interact., 2015, 242, 235–246 CrossRef CAS PubMed .
- H. Larsericsdotter, S. Oscarsson and J. Buijs, J. Colloid Interface Sci., 2005, 289, 26–35 CrossRef CAS PubMed .
- C.-N. Yan, H.-X. Zhang, P. Mei and Y. Liu, Chin. J. Chem., 2005, 23, 1151–1156 CrossRef CAS .
- B. K. Sahoo, K. S. Ghosh and S. Dasgupta, Biopolymers, 2009, 91, 108–119 CrossRef CAS PubMed .
- J. S. Mandeville and H. A. Tajmir-Riahi, Biomacromolecules, 2010, 11, 465–472 CrossRef CAS PubMed .
- P. Bourassa, C. D. Kanakis, P. Tarantilis, M. G. Pollissiou and H. A. Tajmir-Riahi, J. Phys. Chem. B, 2010, 114, 3348–3354 CrossRef CAS PubMed .
- M. Hossain, A. Y. Khan and G. S. Kumar, PLoS One, 2011, 6, e18333 CAS .
- T. Chen and Y. S. Wong, Cell. Mol. Life Sci., 2008, 65, 2763–2775 CrossRef CAS PubMed .
- N. Tayeh, T. Rungassamy and J. R. Albani, J. Pharm. Biomed. Anal., 2009, 50, 107–116 CrossRef CAS PubMed .
- C. Du, D. Deng, L. Shan, S. Wan, J. Cao, J. Tian, S. Achilefu and Y. Gu, Biomaterials, 2013, 34, 3087–3097 CrossRef CAS PubMed .
- A. Papadopoulou, R. J. Green and R. A. Frazier, J. Agric. Food Chem., 2005, 53, 158–163 CrossRef CAS PubMed .
- S. Hideshima, R. Sato, S. Inoue, S. Kuroiwa and T. Osaka, Sens. Actuators, B, 2012, 161, 146–150 CrossRef CAS .
- M. J. Martinez-Tome, R. Esquembre, R. Mallavia and C. R. Mateo, Biomacromolecules, 2010, 11, 1494–1501 CrossRef CAS PubMed .
- C. Q. Jiang, M. X. Gao and J. X. He, Anal. Chim. Acta, 2002, 452, 185–189 CrossRef CAS .
- S. Tabassum, N. P. Singh and J. Mussarat, Synth. React. Inorg. Met.-Org. Chem., 2003, 33, 509–517 CrossRef CAS .
- P. Schwille and J. Ries, Biophotonics, 2011, 63–85, DOI:10.1007/978-90-481-9977-8_4 .
- I. J. Joye, G. Davidov-Pardo, R. D. Ludescher and D. J. McClements, Food Chem., 2015, 185, 261–267 CrossRef CAS PubMed .
- B. Sandhya, A. H. Hegde, S. S. Kalanur, U. Katrahalli and J. Seetharamappa, J. Pharm. Biomed. Anal., 2011, 54, 1180–1186 CrossRef CAS PubMed .
- S. Z. Hao, S. D. Liu, X. H. Wang, X. J. Cui and L. P. Guo, J. Lumin., 2009, 129, 1320–1325 CrossRef CAS .
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed .
- R. Esfandiary, A. Parupudi, J. Casas-Finet, D. Gadre and H. Sathish, J. Pharm. Sci., 2015, 104, 577–586 CrossRef CAS PubMed .
- J. Q. Gao, Y. W. Guo, J. Wang, Z. Q. Wang, X. D. Jin, C. P. Cheng, Y. Li and K. Li, Spectrochim. Acta, Part A, 2011, 78, 1278–1286 CrossRef PubMed .
- L. Zhang, B. Liu, Z. Li and Y. Guo, Luminescence, 2015, 30, 686–692 CrossRef CAS PubMed .
- M. Hu, X. Wang, H. Wang, Y. Chai, Y. He and G. Song, Luminescence, 2012, 27, 204–210 CrossRef CAS PubMed .
- Y. J. Hu, Y. Liu, W. Jiang, R. M. Zhao and S. S. Qu, J. Photochem. Photobiol., B, 2005, 80, 235–242 CrossRef CAS PubMed .
- H. M. Zhang, Y. Q. Wang, Z. H. Fei, L. Wu and Q. H. Zhou, Dyes Pigm., 2008, 78, 239–247 CrossRef CAS .
- H.-B. Pang, G. B. Braun and E. Ruoslahti, Sci. Adv., 2015, 1, e1500821 Search PubMed .
- B. György, M. E. Hung, X. O. Breakefield and J. N. Leonard, Annu. Rev. Pharmacol. Toxicol., 2015, 55, 439 CrossRef PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1516438, 1516447 and 1517017. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28801d |
| ‡ These authors contributed equally to the work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a