
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The evolution of comprehensive strategies for furanoid glycal synthesis and their applications†
Pinki Pal‡
and
Arun K. Shaw
 *
*
Division of Medicinal & Process Chemistry, CSIR- Central Drug Research Institut, Sector 10, Jankipuram Extension, Sitapur Road, Lucknow-226 031, India. E-mail: akshaw55@yahoo.com; Tel: +91-9415403775
First published on 15th May 2017
Abstract
Cyclic enol ether frameworks, especially stereochemically pure furanoid and pyranoid glycals are well known highly functionalized chiral building blocks. Furanoid glycals have been shown to possess great potential, as they have been used as key intermediates for the synthesis of structurally diverse molecules including natural products with various biological activities since their discovery in 1963 by Ness and Fletcher. In this review on furanoid glycals, efforts are made to exhaustively compile the work centered on synthesis and utility of furanoid glycals since the inception to date. This review aims to highlight the importance of furanoid glycals and the strategies developed over the years for their synthesis. Attention has also been focused on the use of these furanoid glycals toward the synthesis of natural and unnatural products including C-and N-nucleosides of biological importance. Apart from that, efforts are also devoted to cover the significant applications of these furanoid glycals for stereoselective synthesis of various cyclic and acyclic key intermediates of significant interest.
 Pinki Pal | Pinki Pal was born in the state of West Bengal, India. She obtained her B.Sc. in Chemistry (Hons.) from The University of Burdwan, India and M.Sc. in Organic Chemistry from University of Kalyani, India in 2002 and 2004 respectively. She received her Ph.D. degree in 2011 jointly from CSIR-Central Drug Research Institute, Lucknow, and Jadavpur University, Kolkata, India, under the supervision of Dr Arun K. Shaw (CSIR-CDRI) and co-supervision of Prof. Gourhari Maiti (Jadavpur University). In her doctoral research, she worked on ‘chiron’ approach to total stereoselective synthesis of bioactive natural product and natural product like molecules starting from commercially available monosaccharides. After working as a Project Associate at the Indian Institute of Science, India, with Prof. N. Jayaraman and Post-doctoral Fellow at National Taiwan University, Taiwan, with Prof. Ya-Ching Shen, she joined as Associate Scientist at GVK Biosciences Pvt. Ltd., Hyderabad, India. Later she worked as a Consultant at Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore in Dr Jayanta Haldar's Research Group. At present, she is Lecturer at Department of Chemistry, Mount Carmel College Autonomous, Bangalore, India. |
 Arun K. Shaw | Arun K. Shaw was born in Raniganj, West Bengal. He received his B.Sc. (Hons.) degree in Chemistry from Scottish Church College, Calcutta University, in 1975, his M.Sc. degree in Chemistry from Calcutta University in 1977, and his Ph.D. in Natural Product Chemistry under the supervision of Professor S. N. Ganguly from Bose Institute, Calcutta University, in 1985. After his doctoral work, he worked with Professors S. K. Talapatra and B. Talapatra in the Department of Pure Chemistry, Calcutta University, during 1985–1986. In 1987 he joined as a Scientist Gr. IV (1) in the Department of Food Chemistry, CSIR-Central Food Technological Research Institute, Mysore. He moved in the same capacity to the Division of Medicinal Chemistry, CSIR-Central Drug Research Institute, Lucknow, in 1991. His research interests centred around isolation, purification characterization of natural products of plant origin, syntheses of bioactive natural product or natural product like molecules using carbohydrates as chiral pool. He is also interested in development of new methodology/synthetic reagents and investigation of new reaction pathways in organic synthesis. He has published over 78 papers in peer reviewed international journals including a review article in Chemical Review. He held the visiting scientist positions at University of Rostock, Germany under CSIR-DAAD Exchange Programme of scientists in April–June, 2000 and at University of Glasgow, Glasgow, Scotland in March, 2013 under INSA, New Delhi International Collaboration/Exchange Programme during 2011–2012. He superannuated in 2015 from Division of Medicinal Chemistry, CSIR-Central Drug Research Institute, Lucknow from the position of Chief Scientist and Professor AcSIR. |
1. Introduction
Carbohydrates represent one of the most privileged classes of naturally occurring versatile building blocks in synthetic organic chemistry due to their wealth of unique functional, conformational, and stereochemical information. The diversity and availability of these relatively cheap chiral compounds has led to their use as starting materials for the design and syntheses of naturally occurring compounds and biologically important molecules.1 The preparation of attractive building blocks from carbohydrates and their use for the synthesis of various biologically active simple or complex natural products has received considerable attention from organic chemists. Therefore, the stereocontrolled synthesis of chiral building blocks (CBBs) is an important objective in organic chemistry. Over the last several years our group has been working toward the synthesis of enantiomerically pure sugar derived building blocks and their utilization to accomplish the total synthesis of target biologically relevant natural products2b,d,g,h and natural product like molecules.2a,c,e,f,i Recently our research group has reviewed glycal derived δ-hydroxy α,β-unsaturated aldehydes (Perlin aldehydes).3aWhile there are many carbohydrate derivatives, monosaccharides occupy a significant place among them as starting materials. Glycals, prepared from hexoses and pentoses, are most important and well known highly functionalized CBBs and find their huge applications in ‘chiron’ approach synthesis.3b,c They are highly reactive due to their enol ether geometry (a double bond between the carbon atoms 1 and 2 of the ring). There are two kinds of glycals: (i) pyranoid glycal I (derived from hexose), (ii) furanoid glycal II (derived from pentose) (Fig. 1).
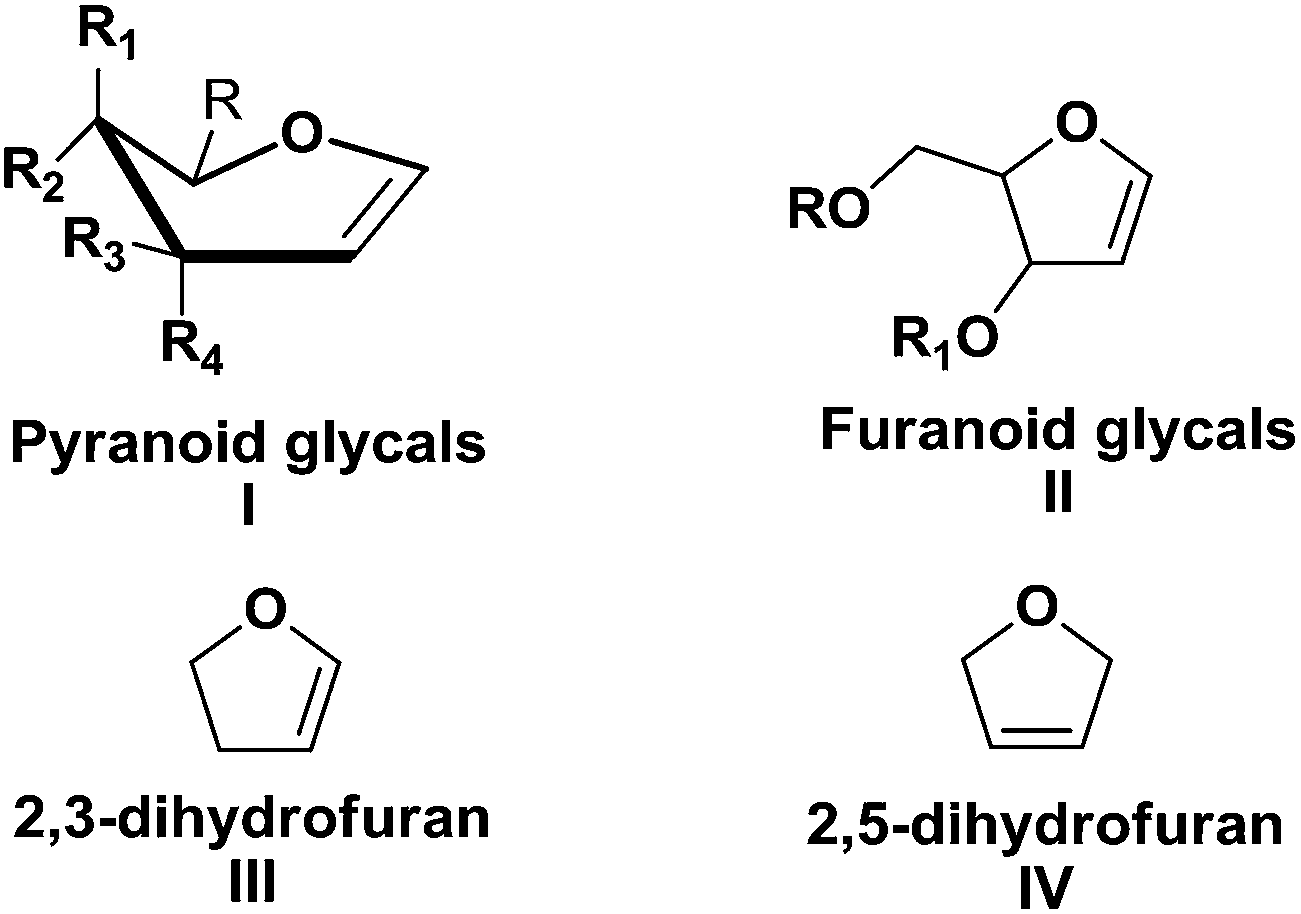 | ||
| Fig. 1 General structures of pyranoid glycals (I), furanoid glycals (II), 2,3-dihydrofuran (III) and 2,5-dihydrofuran (IV). | ||
A considerable effort has been dedicated toward the synthesis of pyranoid glycals and furanoid glycals. In recent years, interest has been devoted to synthesis of furanoid glycals, owing to the fact that they have been used as key intermediates in syntheses of structurally diverse compounds with various biological activities such as polyether antibiotics,4 6-epi-leukotrienes C & D,5 palladium-mediated coupling reaction leading to C-nucleosides,6 antiviral and antitumour C-nucleosides,7 α-arabino nucleosides,8 2′,3′-dideoxynucleosides9 and more recently 2′-deoxynuleosides.10
There are a large number of reports on the synthesis and uses of furanoid glycals. The versatile application of furanoid glycals inspired us to compile the wholesome work centered around their synthesis and applications since the inception to date in the form of a review. This review mainly focuses on various synthetic routes for the preparation of furanoid glycals and their applications toward synthesis of important ‘chiral building blocks’, various kinds of natural and unnatural products of biological importance and C- & N-nucleosides. We have tried to include many recent examples in this review that covers our studies till date, and any omissions on this wide topic are unintentional. It should be noted that, only synthesis and applications of furanoid glycals are described here. Other reactions, in which 2,3- and 2,5-dihydrofurans (III, IV) involve are not described here (Fig. 1).
2. Literature reports on synthesis of furanoid glycals (FGs)
Glycals (pyranoid and furanoid glycals) are important intermediates in the synthesis of a variety of carbohydrate derivative. In 1913, Fischer and Zach11 first synthesized 3,4,6-tri-O-acetyl-1,5-anhydro-2-deoxy-D-arabino-hex-1-enitol. Inspired by the reactivities of glycals and to report the first synthesis of furanoid glycal (FG), Ness and Fletcher tried to synthesise furanoid glycal from tri-O-benzoyl-α-D-arabinofuranosyl bromide after several modification of Fisher and Zach's method.12 But they failed to achieve their goal.However, they successfully synthesised 1,4-anhydro-3,5-di-O-benzoyl-2-deoxy-D-erythro-pent-1-enitol 4 in 1963, known to be the first glycal derivative with a furanose structure, starting from 3,5-di-O-benzoyl-2-O-p-nitrophenylsulfonyl-β-D-ribosyl bromide 3, which was easily prepared from 1,3,5-tri-O-benzoyl-α-D-ribose 1 in two steps.13 The free hydroxyl group in 1 was nosylated with NsCl in the presence of pyridine to obtain nosyl derivative 2. It was brominated at C-1 with HBr in AcOH to afford 3,5-di-O-benzoyl-2-O-p-nitrophenylsulfonyl-β-D-ribosyl bromide 3, which was ultimately treated with NaI in acetone solution at a low temperature to obtain crystalline FG 4 in 72% yield (Scheme 1). While its reaction in DCM with MeOH at room temperature gave the dihydro furan 5, the fufuryl benzoate 6 was obtained when 4 in acetone was allowed to react with water.
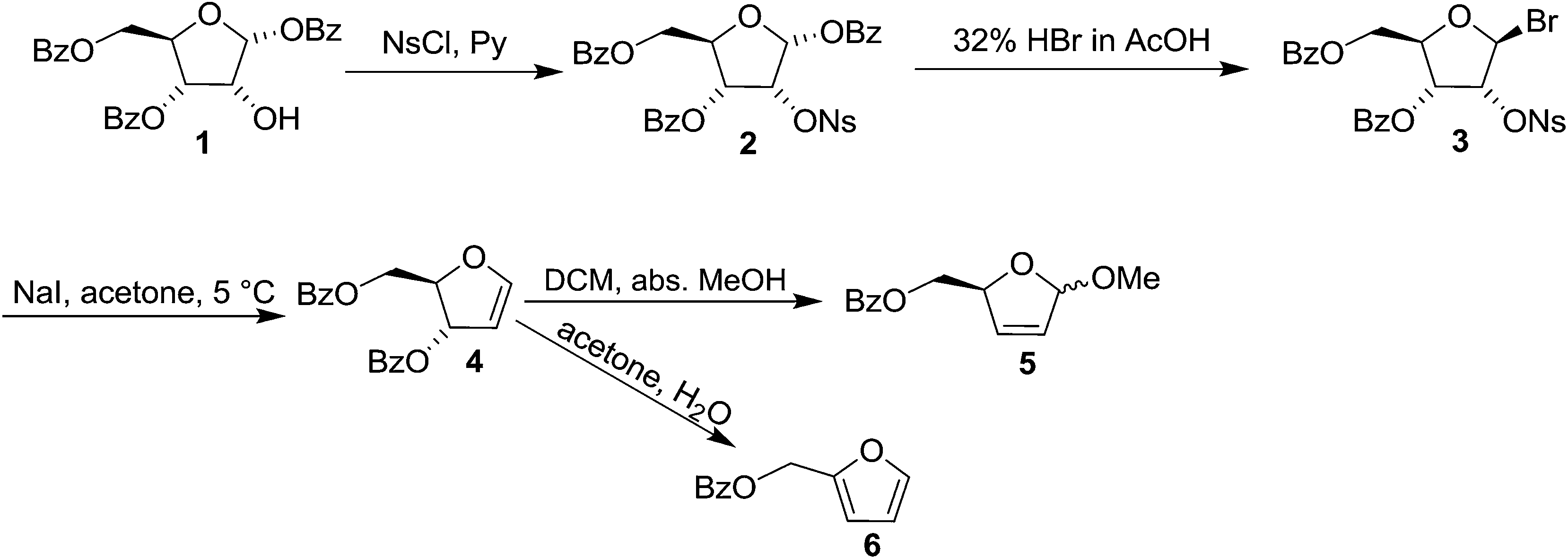 | ||
| Scheme 1 | ||
After the synthesis of labile furanoid glycal 4, Ness et al. synthesised 3,5-di-O-p-anisoyl-1,2-dideoxy-D-erythro-pentofuranos-1-ene 14 and showed that greater the nucleophilicity of the substituent at C-3 of furanoid glycal 14, the lesser would be the tendency to form aromatic products. Its synthesis was started from methyl-β-D-ribofuranoside 7. It was first acylated with p-anisoyl chloride to obtain 8 followed by its bromination with 32% HBr in AcOH to give bromoderivative 9. Its hydrolysis and subsequently crystallization of the resulting product mixture with acetone and water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in DCM yielded isomeric hemiacetals 1,3,5-tri-O-p-anisoyl-α-D-ribofuranose 10 and 2,3,5-tri-O-p-anisoyl-β-D-ribofuranose 11. Compound 10 was nosylated with p-nitrophenyl sulphonyl chloride (NsCl) in pyridine at 0 °C to give 12 in good yield. It was brominated with 32% HBr in acetic acid to furnish 13, which was treated with NaI in acetone solution at 5 °C to obtain crystalline furanoid glycal 14 in 67% yield (Scheme 2).14
:1) in DCM yielded isomeric hemiacetals 1,3,5-tri-O-p-anisoyl-α-D-ribofuranose 10 and 2,3,5-tri-O-p-anisoyl-β-D-ribofuranose 11. Compound 10 was nosylated with p-nitrophenyl sulphonyl chloride (NsCl) in pyridine at 0 °C to give 12 in good yield. It was brominated with 32% HBr in acetic acid to furnish 13, which was treated with NaI in acetone solution at 5 °C to obtain crystalline furanoid glycal 14 in 67% yield (Scheme 2).14
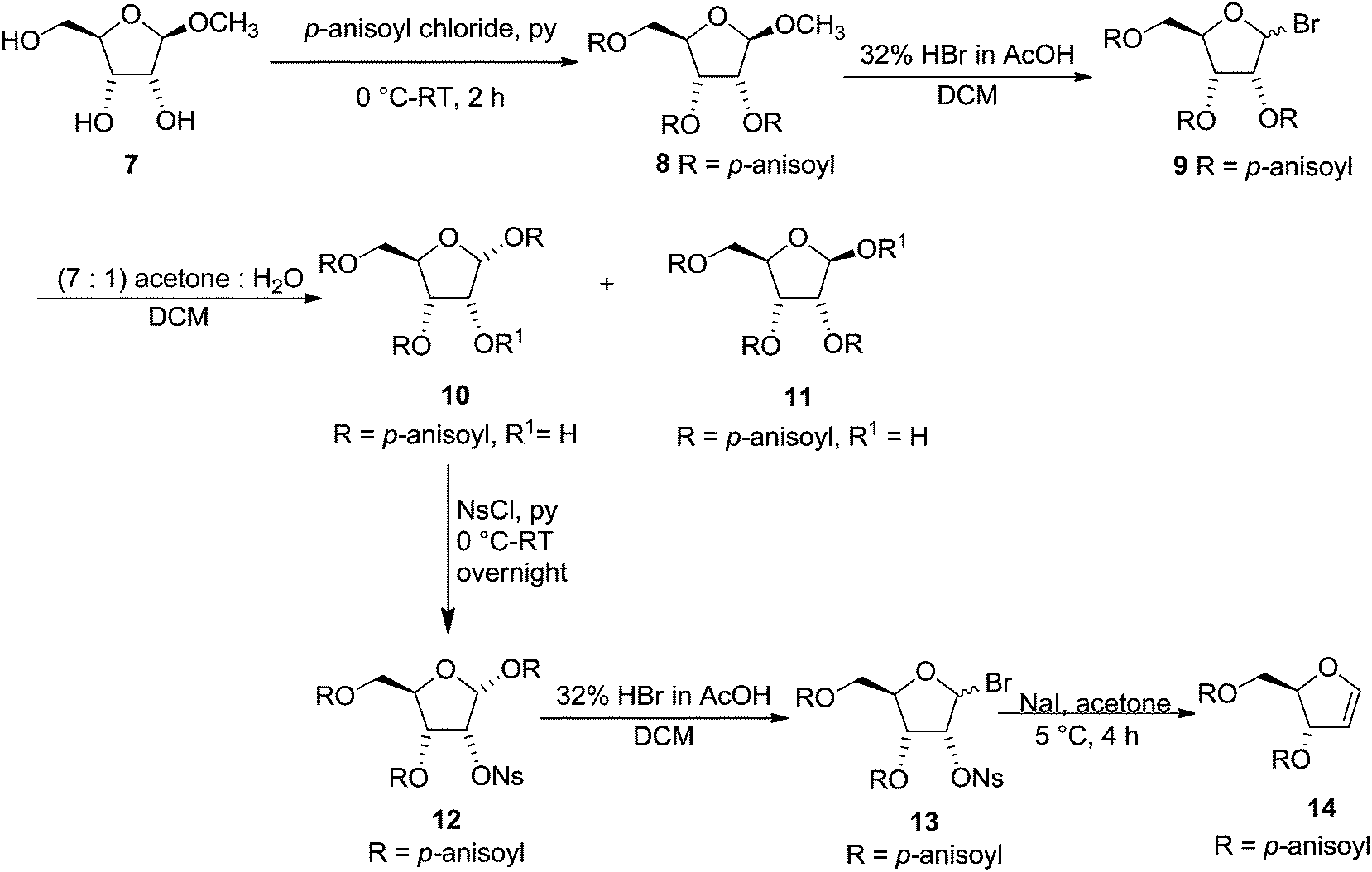 | ||
| Scheme 2 | ||
Bischofberger and Hall attempted to prepare the stable FGs15 by the modification of Fischer and Zach's method in the year 1976.13 However, their method failed to deliver the desired glycals due to the tendency of C-3 substituent to undergo allylic rearrangement. To get rid of this problem, a series of differently substituted furanose derivatives (15a–d) were prepared, where C-3 substituents were less susceptible to allylic rearrangement.15a,b Having these precursors in their hand, they prepared FGs (19a–d) by modifying Fischer and Zach's method for the preparation of glycals (Scheme 3, Table 1).15c The modified method involved the highly reactive cationic species (17a–d), which can either accept two electrons and lose an acetoxyl anion to form the glycals (19a–d), or can combine with hydroxyl or acetoxyl anions to give the free aldoses (18a–d) or starting materials (15a–d). The glycals 19a and 19b were obtained in moderate yields whereas 19c and 19d in low yields. All of them were stable on silica gel and stored under N2 at −5 °C for 6 months. Compounds 20a and 5 were generated from their respective glycals 19a,b by 1,3-sigmatropic shift of the methoxy groups, whereas the furans 21a and 6 were produced by the decomposition of 19a and 19b, respectively (Scheme 3).15d
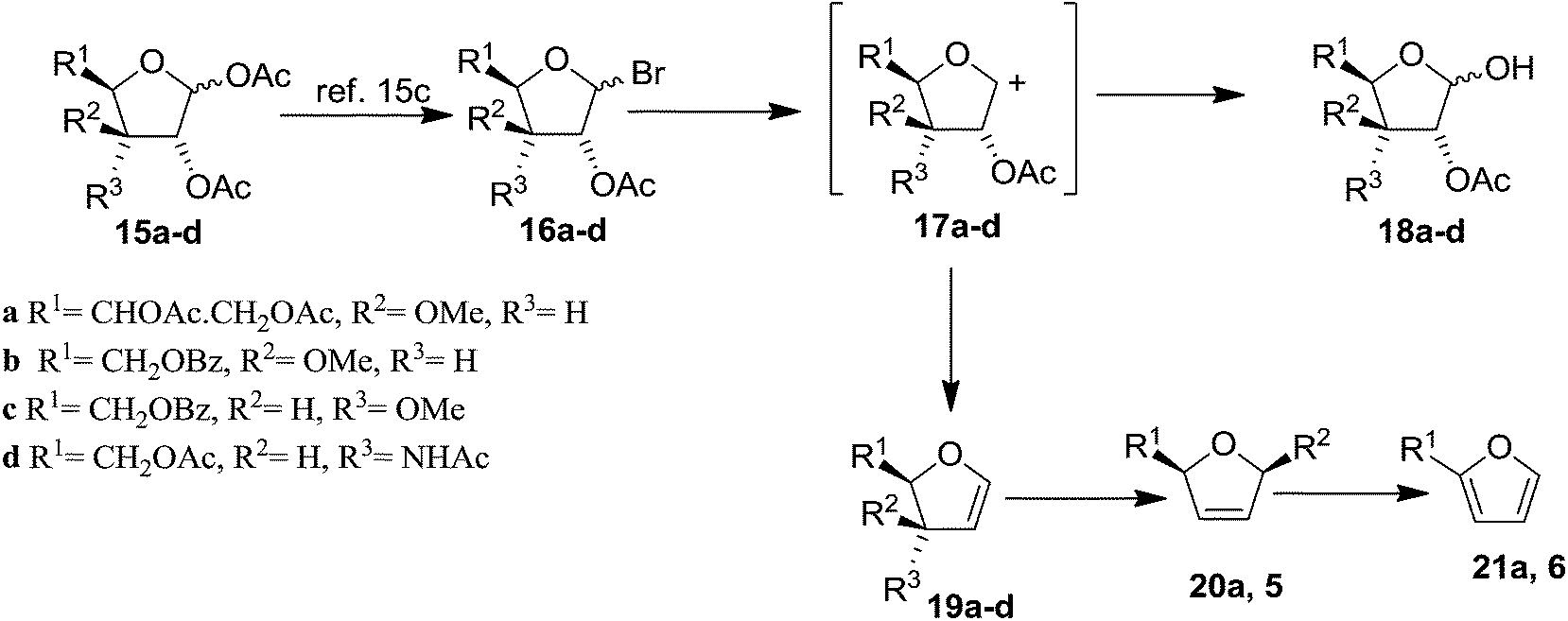 | ||
| Scheme 3 | ||
| Starting material | Yields of products (%) | ||||
|---|---|---|---|---|---|
| Furan (21a, 6) | Glycal (19a–d) | 2,3-Unsaturated compound (20a, 5) | Staring material (15a–d) | Hydroxyl compound (18a–d) | |
| 15a | 1 | 26 | 2 | 3 | 51 |
| 15b | 2 | 20 | 5 | 5 | 48 |
| 15c | 6 | 5 | 83 | ||
| 15d | 3 | 18 | 29 | ||
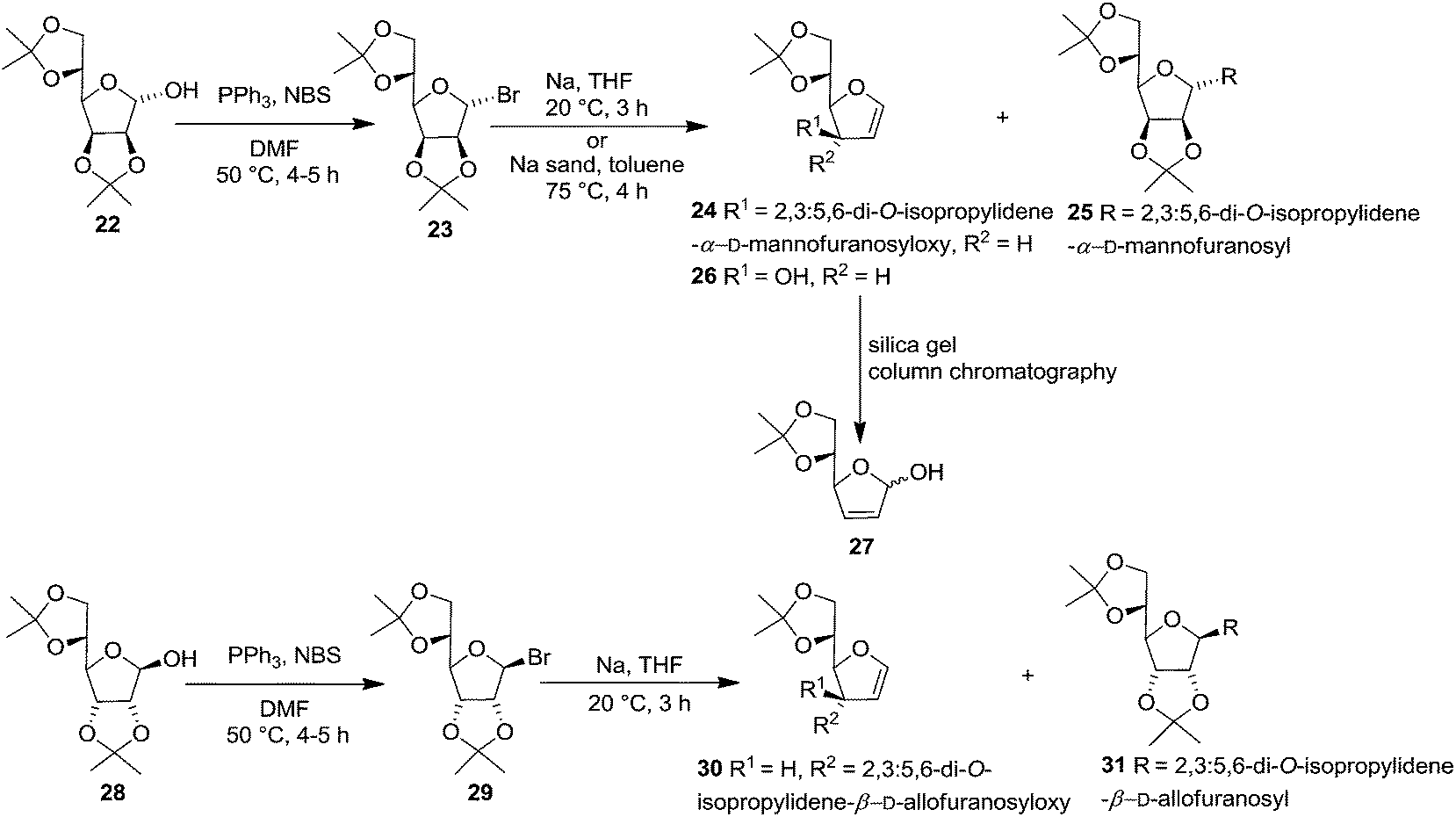
In 1977 Jordaan et al. reported an alternative method for the preparation of furanoid glycals by the reduction of suitably protected furanosyl bromides with sodium or potassium in aprotic solvents.16a 2,3:5,6-di-O-isopropylidene-α-D-mannofuranose 22 derived bromo derivative 23 (ref. 16b) on treatment with an excess of sodium in dry THF at 20 °C for 3 h gave furanoid glycal 24 in 59% yield and a trace of 1,1′-disaccharide 25 (2% yield). Treatment of the mannofuranosyl bromide 23 with sodium sand in toluene at 75 °C for 4 h gave low yields of 24 (9%) as well as the 1,4-anhydrohex-1-enitol 26 (11%). The glycal 26 was unstable and rearranged during chromatography on silica gel to the isomeric compound 27 (Scheme 4).
 | ||
| Scheme 4 | ||
In the similar way, reduction of 2,3:5,6-di-O-isopropylidene-β-D-allofuranosyl bromide 29, derived from 28,16b with sodium in THF (20 °C, 3 h) gave furanoid glycal 30 in 69% yield and 1,1′-disaccharide 31 (2% yield) (Scheme 4).
In 1978, the same group discussed an alternative route for the synthesis of furanoid and pyranoid glycals.17a While treating 2,3:5,6-di-O-isopropylidene-α-D-mannofuranose 22 derived furanosyl chloride 32 (ref. 17b) with sodium naphthalide in THF followed by acetic acid furnished FG 26 in 59% yield, under the identical reaction condition furanosyl chloride 35 (derived from 28) yielded FG 36 in 54% yield (Scheme 5).
 | ||
| Scheme 5 | ||
On the other hand, treatment of furanosyl chloride 32 and furanosyl bromide 23 each with Na or K metal in THF instead of sodium naphthalide gave 3-O-furanosyl furanoid glycal 24 as the major product along with glycoside 33 and disaccharides 25, 34 as by-products. Similarly, the furanosyl chloride 35 (ref. 17c) and the analogous bromide 29, gave only the 3-O-furanosyl glycal 30 (69%) and a trace of the disaccharide 31 (Scheme 5).
Further, warming furanosyl bromide 23 with sodium sand in toluene at 70 °C afforded the glycal 26 in low yield along with its isomeric 2,5-dihydro derivative 27 (Scheme 5).
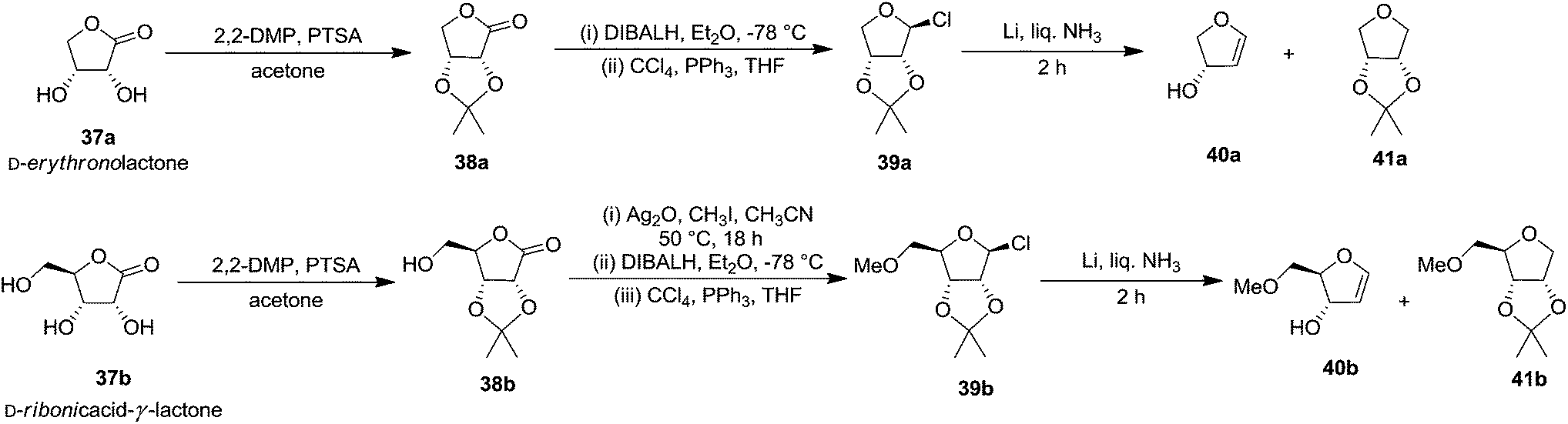
In the same year, Ireland and co-workers developed a general procedure for the synthesis of high yielding 3-hydroxylated FGs, in 4 steps, starting from D-erythronolactone 37a and from D-ribonic acid-γ-lactone 37b in 5 steps, involving the reductive fragmentation of 2,3-O-isopropylidene protected furanosyl chloride with Li in liquid ammonia as a key step (Scheme 6).4,18
 | ||
| Scheme 6 | ||
The starting material 2,3-O-isopropylidene-β-D-erythro-furanosyl chloride 39a was prepared from D-erythronolactone 37a in 79% overall yield by a sequence of reactions that involves acetonide formation of D-erythronolactone 37a followed by partial reduction of the resulting acetonide 38a with DIBALH in Et2O at −78 °C to an intermediate 2,3-O-isopropylidene-D-erythrose, which was immediately treated with CCl4 and Ph3P in THF to obtain furanosyl chloride 39a. Its reduction with 4 equiv. of Li in liquid NH3 in the presence of 6 equiv. of NH4Cl afforded a mixture containing glycal 40a and THF 41a in a ratio of 6:1 (NMR) with 60% yield. They also used D-ribonic-acid-γ-lactone 37b as the starting material in their study to achieve the synthesis of furanoid glycal 40b. The acetonide protection of the lactone was done by adopting the standard process to obtain acetonide 38b. Methylation of its primary hydroxyl group with Ag2O/CH3I/CH3CN at 50 °C for 18 h, followed by reduction of the resulting O-methyl derivative with DIBALH in Et2O at −78 °C for 1 h gave hemiacetal in 90% overall yield, which on treatment with CCl4/PPh3/THF resulted furanosyl chloride 39b in 90% yield. Its reduction with Li in liquid NH3 as described above, afforded a mixture of glycal 40b and the corresponding THF derivative 41b in a ratio of 6:1 (NMR) with 75% yield. It was also reported there that the mixture was not separated by silica gel column chromatography because the furanoid glycal resulted poor recovery after purification (Scheme 6).
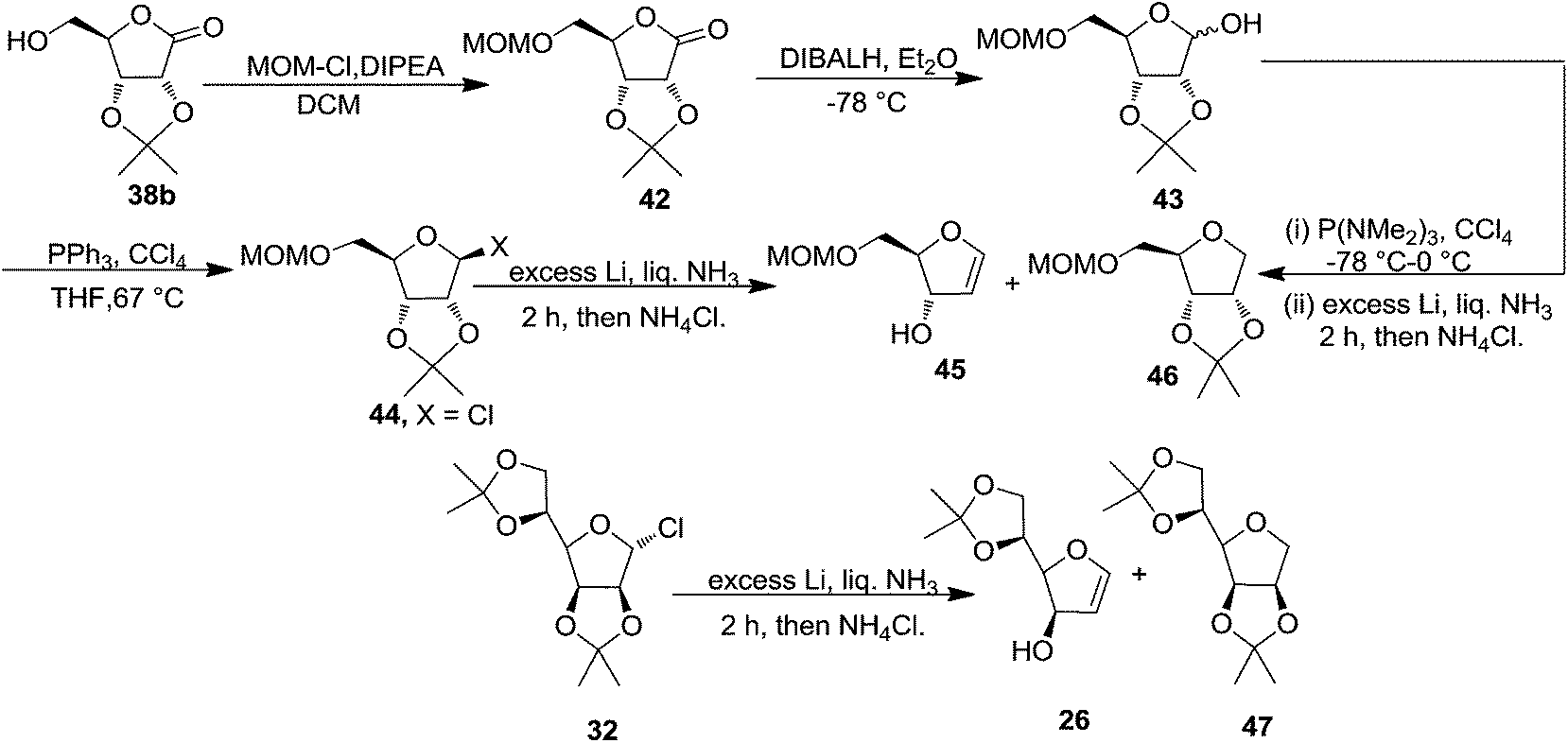
This group again utilized acetonide 38b for the synthesis of furanoid glycal 45.4 The MOM protected lactone 42, prepared from 38b by treating with chloromethyl methyl ether (MOMCl) in the presence of diisopropylethylamine ((iPr)2EtN) in DCM, on reduction with DIBAL-H at −78 °C provided lactol 43. Its chlorination with PPh3 and CCl4 in THF gave the chloride 44. Its reduction with Li in liquid NH3 at −78 °C yielded a mixture of glycal 45 and THF 46 in the ratio 6:1 with 93% overall yield (Scheme 7).
 | ||
| Scheme 7 | ||
Under identical reaction condition furanosyl chlorides 32 yielded furanoid glycal 26 in 75% yield along with THF 47 in 9% yield (Scheme 7).
Due to the instability of the intermediate chlorides formed in this process, this group further examined an alternative method for the transformation of lactols to glycals. Hexamethylphosphorus triamide (tris-(dimethylamino)phosphine, TDAP) reacts at very low temperatures with CCl4 in the presence of alcohols to form adducts of type 48 (Fig. 2).
 | ||
| Fig. 2 Adduct 48. | ||
First TDAP was added to a solution of lactol 43 and CCl4 in THF at −78 °C. The resulting reaction mixture was warmed to 0 °C and then immediately it was added to a solution of excess Li in liquid NH3 to afford a mixture of 45 and 46 in 6:1 with 93% yield after passing the crude product mixture through silica gel to remove HMPA (Scheme 7).
After that, they showed ester enolate Claisen rearrangement on furanoid glycal esters 48 and 50 derived from 45 and 26 respectively. These esters were prepared by the reaction of a lithium alcoholate with the appropriate butanoyl chloride and propanoyl chloride respectively, and the resulting solution of ester was used immediately for the Claisen rearrangement. Silyl ketene acetals 49 and 51, derived from the furanoid glycal esters (48, 50) by deprotonation with LDA in a 23 vol% mixture of hexamethylphosphoric triamide (HMPA) in THF or in 100% THF at −78 °C followed by silylation of enol ether with TMSCl (Scheme 8). Protected furanoid glycal 49 was further converted to natural product Lasalocid A (X537A) 174 after several steps (Schemes 31 and 33).
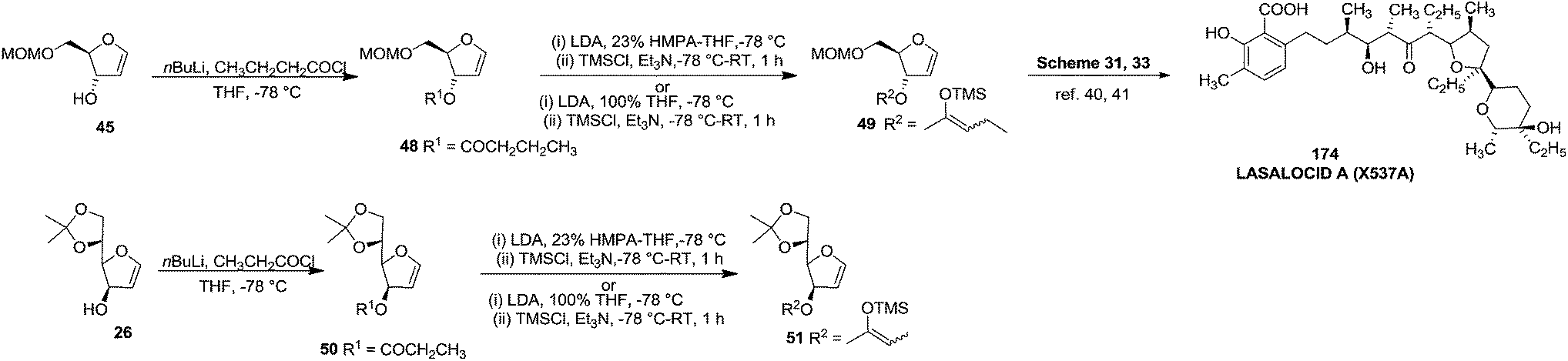 | ||
| Scheme 8 | ||
To obtain high yielding furanoid glycal 26, they further extended their studies on reductive fragmentation of furanosyl chloride 32 with various reductants like metal/NH3 systems, sodium napthalene and lithium di-tert-butylbiphenyl (Table 2) and found that lithium di-tert-butylbiphenyl afforded the furanoid glycal 26 in high yields (Scheme 9, Table 2).19
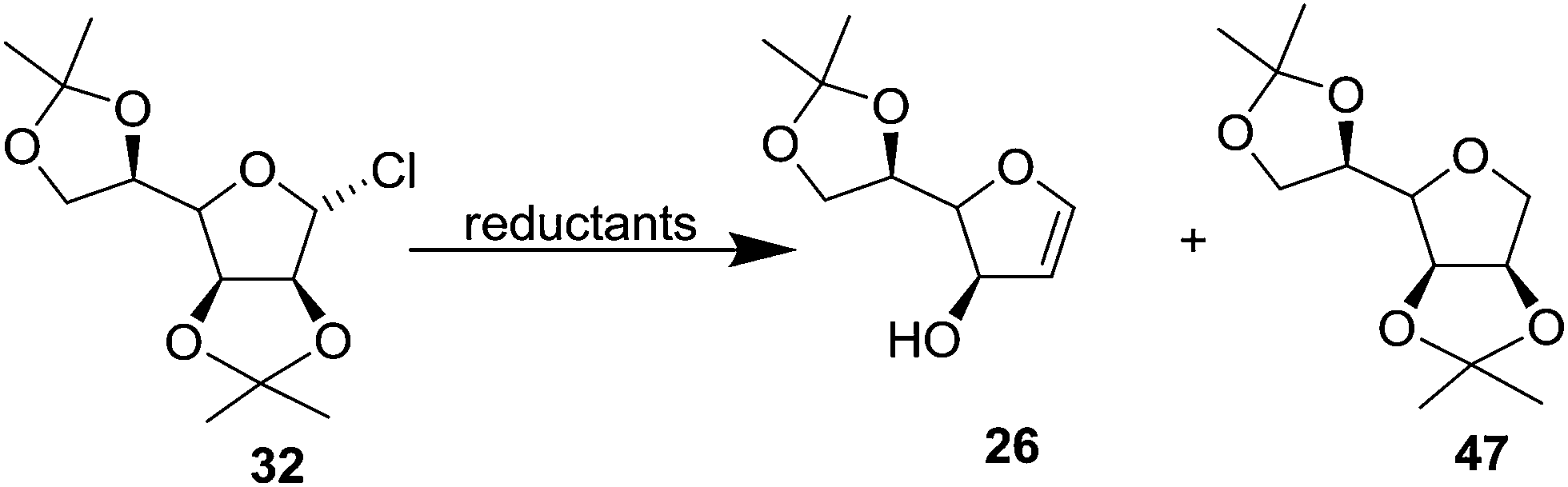 | ||
| Scheme 9 | ||
| Reductants | Yield of 26 (%) | 26:47 |
|---|---|---|
| a 35 equiv. of metal, 0.5 M, 1:10 THF/NH3, −78 °C, 30 min, then NH4Cl.b 2 equiv. 0.07 M THF, 25 °C, 3 h.c 6 equiv. 0.21 M THF, −35 °C, 20 min, then H2O.d 5 equiv. 0.50 M THF, 25 °C.e 5 equiv. 0.25 M THF, 25 °C.f 5 equiv. 0.25 M THF, −20 °C → 0 °C, 1 h, then H2O.g 5 equiv. 0.20 M THF, −78 °C, 15 min, then H2O.h No reaction. |
||
| Li/NH3a | 75% | 7.9:1 |
| Na/NH3a | 77% | 10.7:1 |
| K/NH3a | 79% | 15.0:1 |
| SmI2b | 0% | — |
| Sodium naphthalenec | 82% | >50:1 |
| Lithium benzophenoned | NRh | — |
| Sodium anthracenee | NR | — |
| Sodium trimesitylboranef | 70% | >50:1 |
| Lithium 4,4′-di-tert-butylbiphenylg | 94% | >50:1 |
The same reaction protocol was latter followed by Daves et al. in 1985 to synthesize 3-hydroxy furanoid glycals (45, 53a–c), and also a series of symmetrically (54a, 54f) and differentially protected (54b–e, 54g, 54h) 3,5-bis-O-substituted furanoid glycals from 2,3-isopropylidene protected ribolactone 38b. They also synthesized 1,4-anhydro-2-deoxy-D-erythro-pent-1-enitol 55 and 3-O-derivatized 5-hydroxy glycals 56 from their corresponding starting materials by removal of the silyl protecting groups (Scheme 10).20a
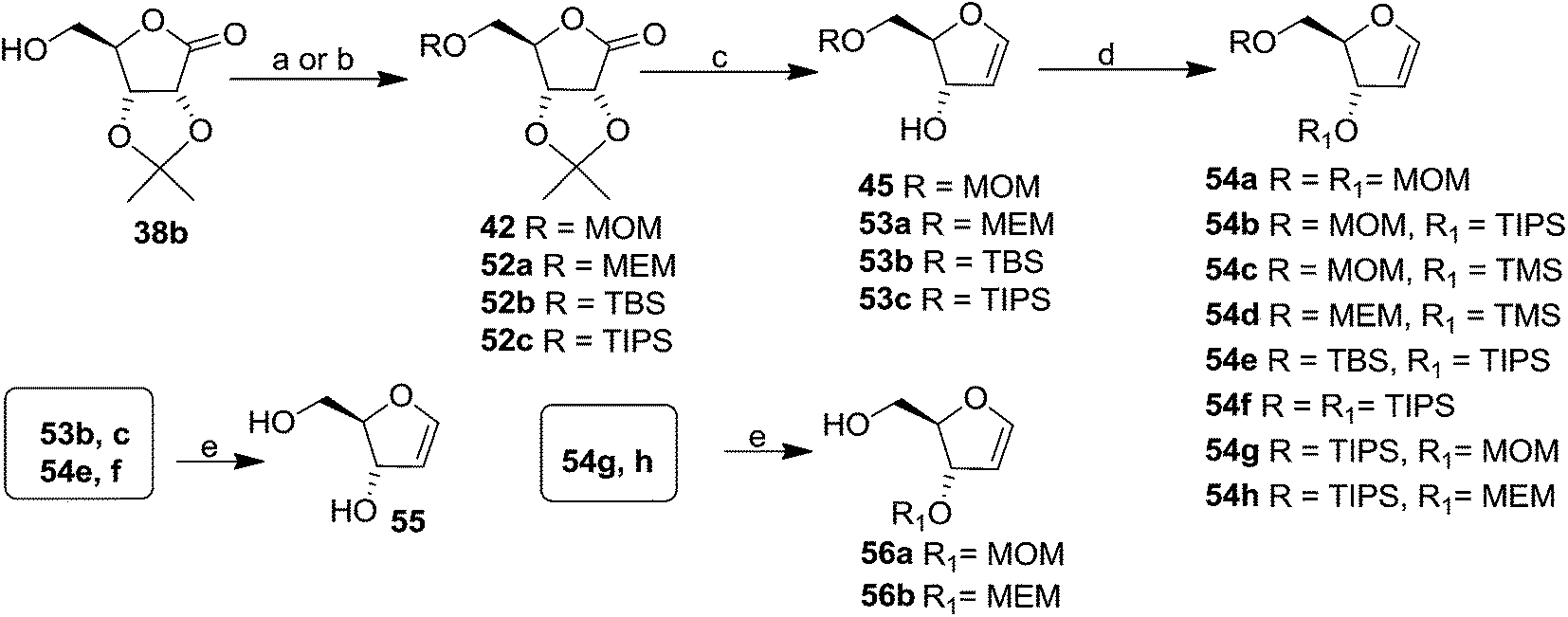 | ||
| Scheme 10 Reagents and conditions: (a) To obtain 42 and 52a: (i) MOMCl, iPr2EtN, DCM; (ii) MEMCl, iPr2EtN, DCM; (b) to obtain 52b and 52c: (i) TBSCl, imidazole, DMF; (ii) TIPSCl, imidazole, DMF; (c) ref. 4, 18 and 19; (d) ref. 20a; (e) ref. 20b. | ||
In the same year, Schlosser and coworkers described the synthesis of furanoid glycal of threo configuration from D-mannose (Scheme 11).21 The free hydroxyl group at anomeric position of D-mannose 57 derived intermediate 1,2:5,6-di-O-isopropylidene-D-mannofuranose was benzylated with BnCl in the presence of NaH in dry DMF to afford benzyl protected compound 58 in good yield. The globally –OH protected compound 58 was treated with HCl in aq. MeOH to form diol 59 in 92% yield, which on treatment with NaIO4 in 3:1 MeOH:H2O mixture gave aldehyde 60 in 70% yield. Its reduction with NaBH4 in EtOH followed by MOMCl protection of the resulting primary alcohol produced 61 in 89% yield. Its debenzylation with Li in liq. NH3 followed by chloride formation with PPh3 and CCl4 in THF afforded 62 in 64% yield. Finally, it was reduced with Li/liq. NH3 and the free 2° alcohol was protected with MOMCl to give threo furanoid glycal 63 in 56% yield (Scheme 11). It was utilized as key intermediate for the synthesis of erythro-(2S,3R)-sphingosine 232 which has been discussed in Scheme 39.
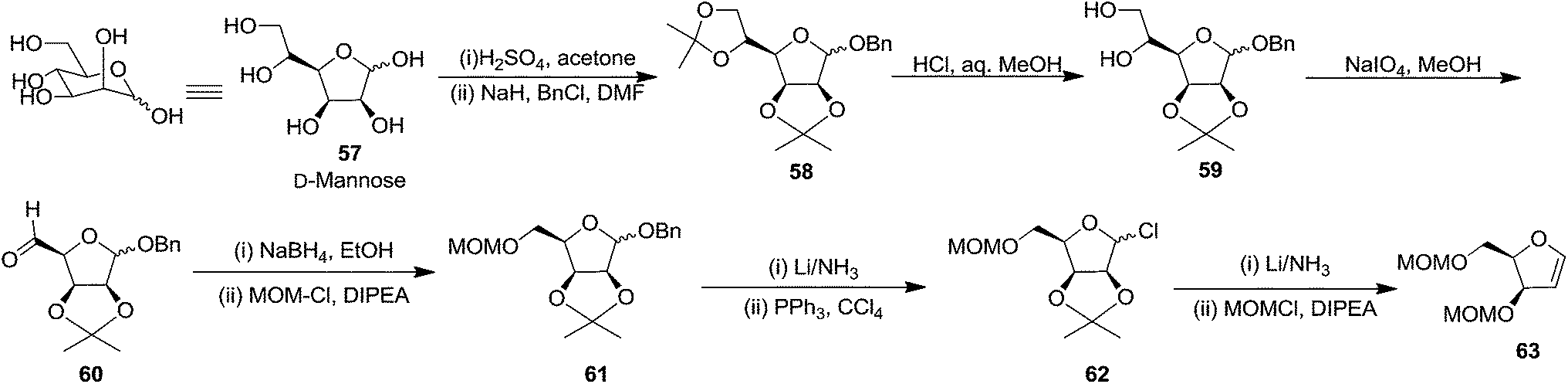 | ||
| Scheme 11 | ||
Pederson et al. reported the synthesis of erythro furanoid glycals from commercially available free thymidine 64a or 5′-O-(tert-butyldiphenylsilyl)thymidine 64b in a single step.22 The nucleosides 64a & 64b were refluxed with 1,1,1,3,3,3-hexamethyldisilazane (HMDS) in the presence of (NH4)2SO4 for 2 h under N2 atmosphere to afford furanoid glycals 65a & 65b in 75–95% yields (Scheme 12).
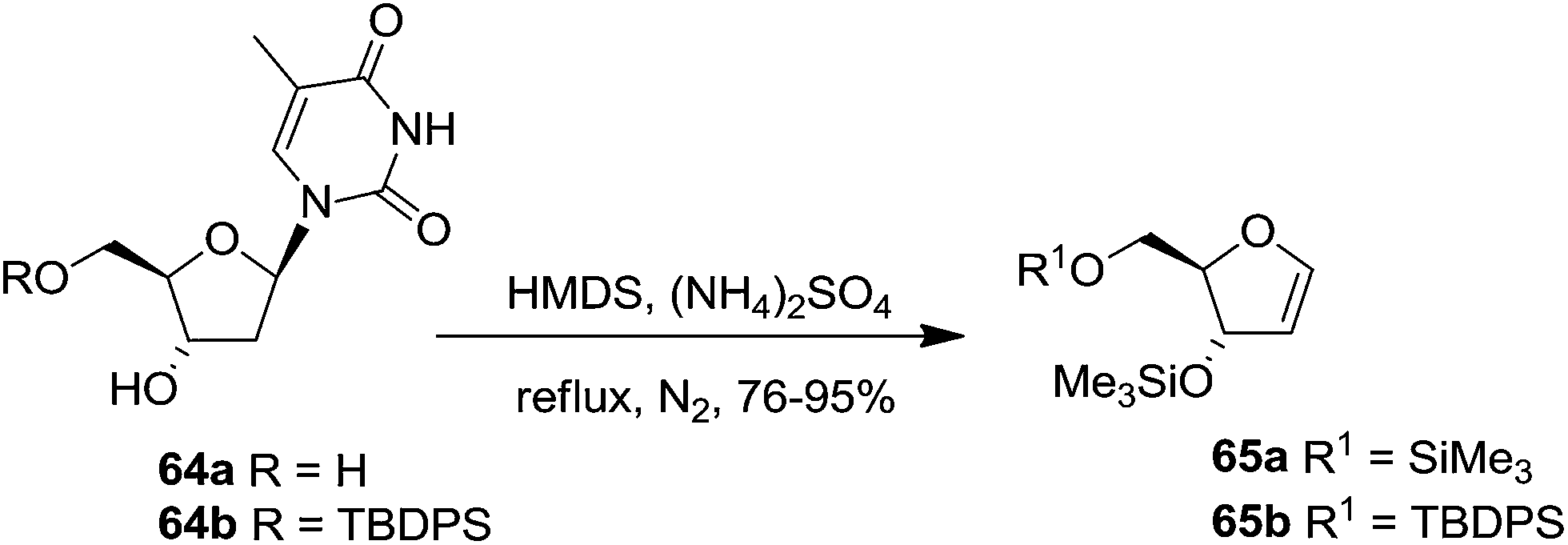 | ||
| Scheme 12 | ||
This approach was further followed by Hammer et al. in 1997 to synthesize gram quantities of furanoid glycals (Scheme 13).23 They synthesized wide range of O-silyl protected furanoid glycals. They also included 5′-ester (toluoyl) protected glycals as well as various combinations of 5′-ester and 3- and 5-TBS and TBDPS protected furanoid glycals (Table 3).
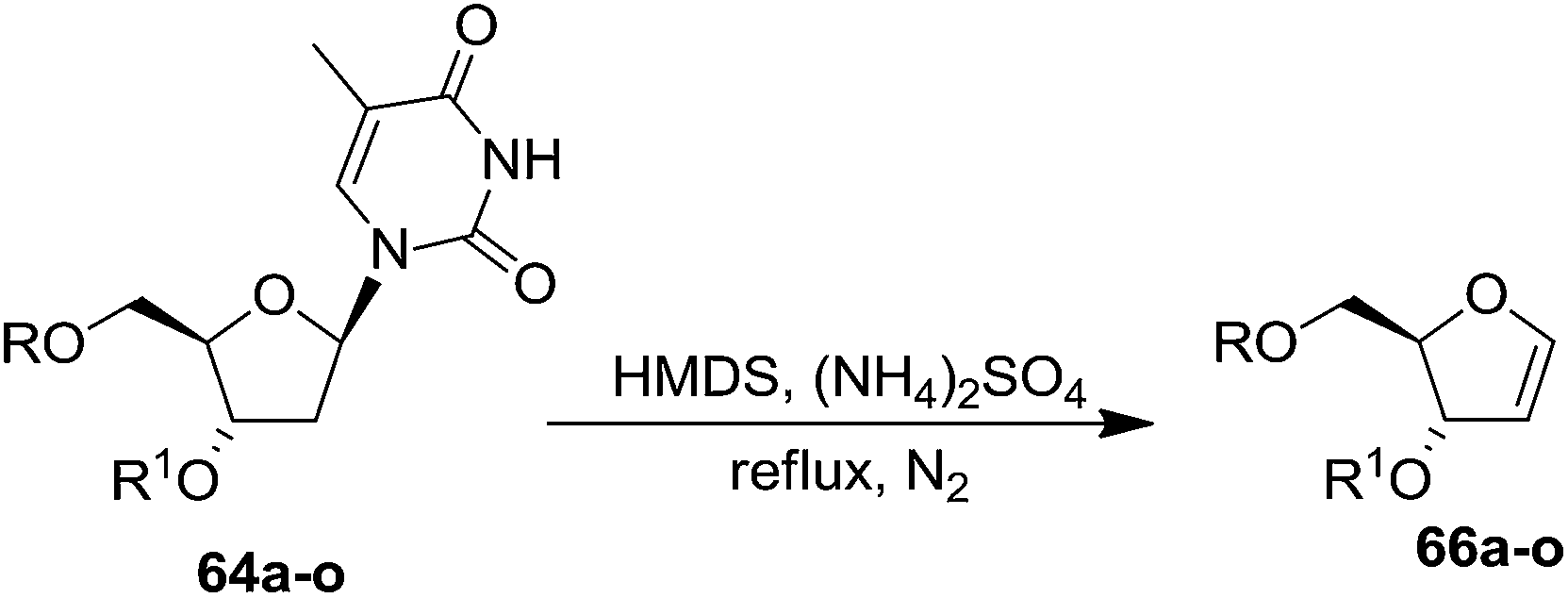 | ||
| Scheme 13 | ||
| Thymidine | Glycal | R | R1 | Yield (%) | Yield (%) from 64aa |
|---|---|---|---|---|---|
| a The number in parentheses indicates the number of steps required to prepare the glycal from commercially available free thymidine 64a. | |||||
| 64a | 66a | H | H | 80 | 80(1) |
| 64b | 66b | TBDPS | H | 91 | 70(2) |
| 64c | 66c | TBS | H | 74 | 53(2) |
| 64d | 66d | Tol | H | 52 | 38(2) |
| 64e | 66e | TBS | TBS | 69 | 47(2) |
| 64f | 66f | TBDPS | TBDPS | 80 | 76(2) |
| 64g | 66g | Tol | Tol | No glycal | No glycal |
| 64h | 66h | TBS | TBDPS | 79 | 55(3) |
| 64i | 66i | TBS | Tol | No glycal | No glycal |
| 64j | 66j | TBDPS | TBS | 59 | 40(3) |
| 64k | 66k | TBDPS | Tol | No glycal | No glycal |
| 64l | 66l | Tol | TBS | 74 | 39(3) |
| 64m | 66m | Tol | TBDPS | 94 | 64(3) |
| 64n | 66n | H | TBS | 36 | 18(4) |
| 64o | 66o | H | TBDPS | 79 | 53(4) |
In 1994 Kassou and Castillón synthesized both erythro and threo furanoid glycals from easily available 2-deoxyribose 67 by using a key selenoxide elimination.24 Methyl furanoside 68 was easily synthesized from 2-deoxyribose 67 by reaction with HCl/MeOH (0.05%), which was benzylated with BnBr/NaH to obtain 69a. Similarly compounds 69b and 72 were prepared by treating methyl furanoside 68 with PivCl and TBDPSCl respectively. The methyl 2-deoxy furanosides 69a, 69b and 72 were treated with PhSeH and BF3/Et2O to afford phenyl-2-deoxy-1-selenofuranosides 70a, 70b and 73 (α/β mixtures) in 85%, 71% and 75% yields respectively. For the synthesis of threo furanoid glycals, inversion of the 3-OH in compound 73 was achieved by its treatment with Tf2O/Py followed by KNO2/18-crown-6/DMF to afford threo derivative 74a in 75% yield for the two steps (Scheme 14). Protection of the secondary alcohol with TBSCl led to compound 74b. Treatment of 74a with nBu4NF led silyl deprotection to give 74c whose phenylseleno derivatives 75a and 75b were easily obtained by usual benzyl and acyl protections respectively. Oxidation followed by thermal elimination of these phenyl-2-deoxy-seleno-furanosides 70a, 70b and 75a, 75b with iPr2EtN/tBuOOH/Ti(O-iPr)4 (1:1:1) system in DCM at 0 °C afforded corresponding furanoid glycals 71a, 71b and 76a, 76b in good yields.
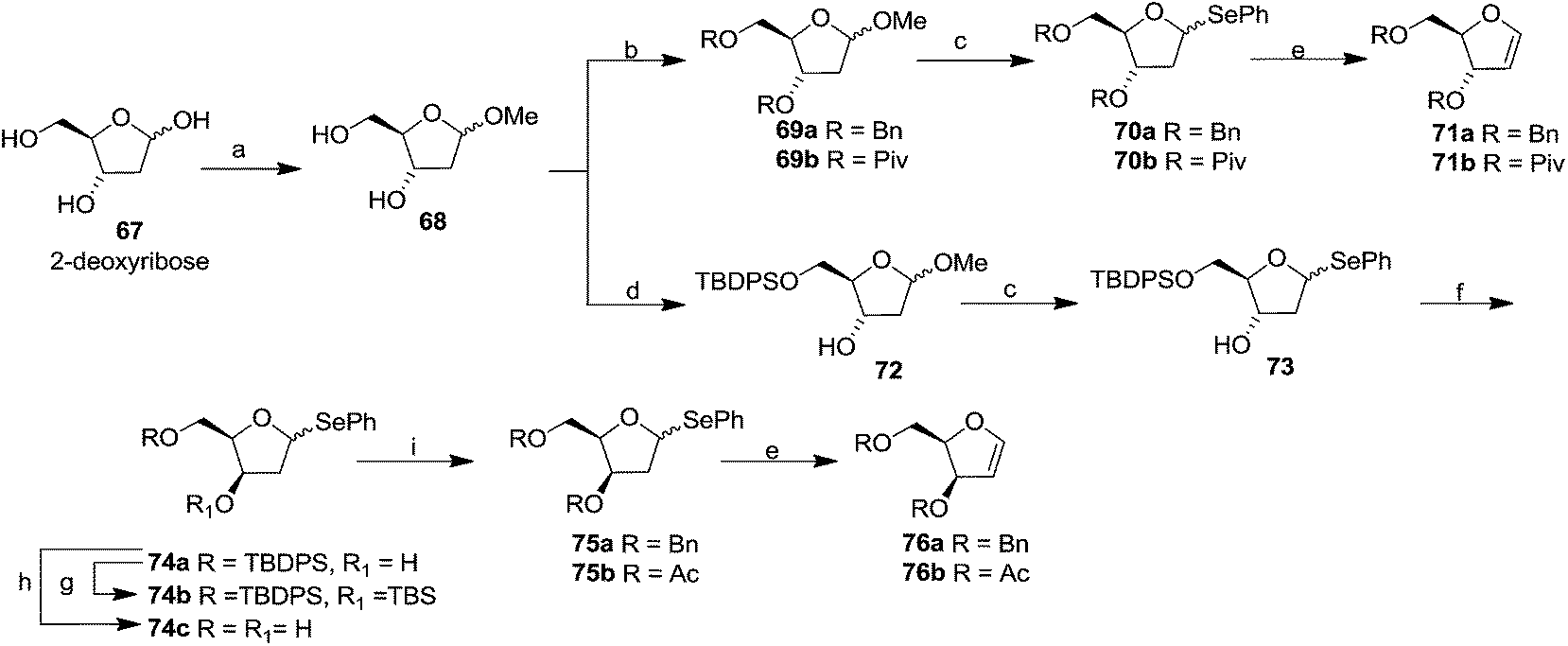 | ||
| Scheme 14 Reagents and conditions: (a) HCl/MeOH (0.05%); (b) (i) BnBr. NaH, THF (69a); (ii) PivCl, Py (69b); (c) PhSeH, BF3/Et2O, DCM, −5 °C (70, 73); (d) TBDPSCl/Im/DMF 72; (e) iPr2EtN/tBuOOH/Ti(O-iPr)4 (1:1:1), DCM, 0 °C; (f) (i) Tf2O, Py, 0 °C. (ii) KNO2, 18-crown-6, DMF; (g) TBSCl, DBU, benzene; (h) nBu4NF, THF; (i) (i) BnBr. NaH, THF (75a), (ii) Ac2O, py (75b). | ||
Their work was further continued to synthesize differently protected erythro and threo furanoid glycals from 4-pentene-1,2,3-triol involving selenium induced 5-endo-trig cyclization followed by selenoxide elimination.25 They utilized inexpensive D-mannitol derived D-glyceraldehyde 77 as the starting material which on treatment with vinylmagnesium chloride in ether/THF furnished the alcohol 78 (1:1 mixture). Benzylation of free hydroxyl group followed by isopropylidene deprotection afforded separable diols 79 and 80. The primary hydroxyl group of compounds 79 and 80 were selectively protected with BnBr via the stannylidene procedure to give alcohols 81 and 83. In another set of experiments, 79 and 80 were selectively protected with TBDPSCl to obtain their respective alcohols 82 and 84 (Scheme 15). The protected alkenic alcohols 81–84 on treatment with N-phenylselenophthalimide (N-PSP) in the presence of CSA in DCM preferencially gave α/β mixture of 4-phenylselenenyl tetrahydrofurans 85–88 in different ratio through unusual 5-endo-trig cyclization. These were oxidised with (tBuOOH, iPr2EtN, Ti(O-iPr)4 in DCM) to obtain the corresponding selenoxides, which on prolong heating in DCM or DCE gave isolated erythro furanoid glycals (89, 90) and threo furanoid glycals (91, 92) respectively in good yields.
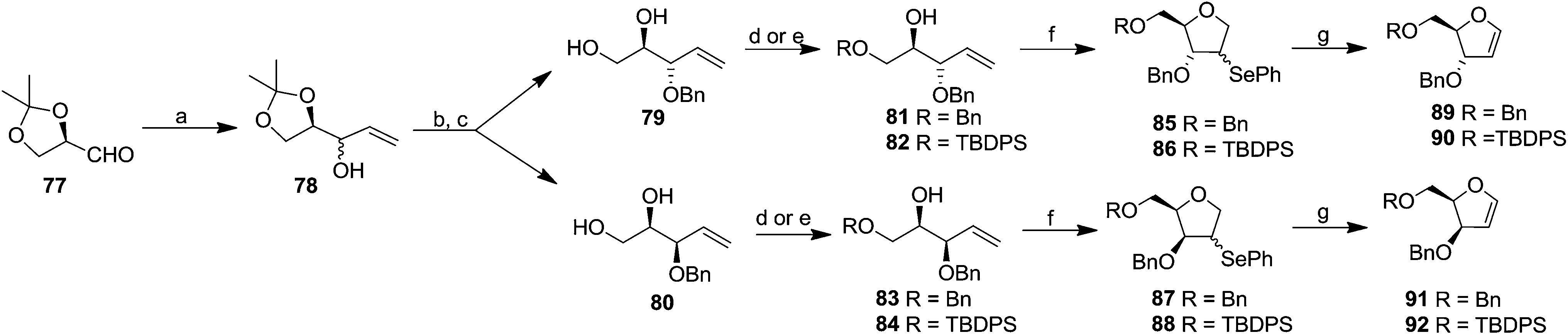 | ||
| Scheme 15 Reagents and conditions: (a) vinylmagnesium chloride, ether/THF; (b) NaH, BnBr; separation of isomers; (c) H +, MeOH; (d) to obtain 81 and 83 (i) Bu2SnO, toluene, 4 Å MS; (ii) BnBr, Bu4NBr; (e) to obtain 82 and 84: TBDPSCI, imidazole, DMF; (f) N-PSP, CSA, DCM; (g) tBuOOH (2.5 equiv.), iPr2EtN (1.7 equiv.) and Ti(O-iPr)4, (1 equiv.) in DCM (48 h) or DCE (4 h), reflux. | ||
They further extended their work to synthesize differently protected erythro and threo furanoid glycals by oxidative elimination of 1-phenylselenenyl furanosides and 2-phenylselenenyl-1,4-anhydroalditol in the presence of tBuOOH, Ti(O-iPr)4 and iPr2EtN system.26
In 1995 Florent and co-workers reported the synthesis of 2′-C-acetoxymethylfuranoid glycal 97a by reductive elemination of glycosyl phenyl sulfone 96 with SmI2-HMPA,27 which was obtained in four steps as discussed below from readily available α-D-isosaccharino-1,4-lactone 93 (Scheme 16). The sequential reduction-acetylation of the lactone 93 afforded peracetyl derivative 94, which on treatment with PhSH/BF3–Et2O/DCM gave thiophenyl glycoside 95. Its oxidation with m-CPBA produced phenyl sulfone 96 whose reductive samariation with SmI2 in THF in the presence of HMPA, followed by elimination of the acetate, afforded the furanoid glycal 97a. It was then converted into 97b via a six membered cyclic rearrangement during purification by silica gel column chromatography. The formation of N-nucleosides 2′,3′-dideoxy-2′-C-methylidene-5-methyl uridine 623 and 3′-deoxy analog of DMDC 626 and 627 from furanoid glycal 97a has been discussed in Scheme 105.
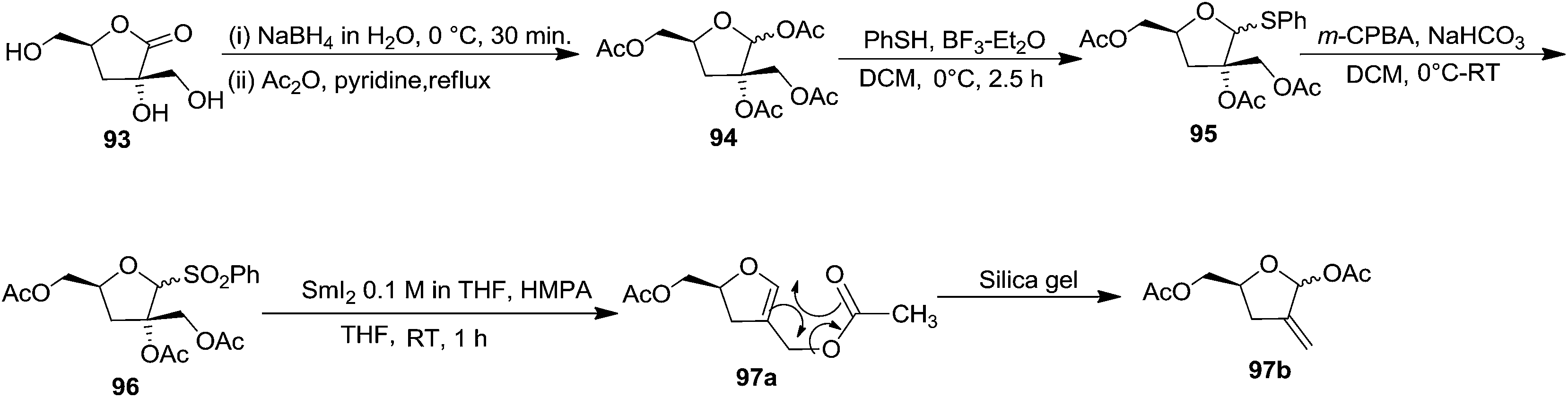 | ||
| Scheme 16 | ||
In 1996 Townsend et al. synthesized ribofuranoid glycals in multigram scale from 2-deoxy-ribose derived 2-deoxy-D-ribono-1,4-lactone 98,28 which was bis-silylated at 3- and 5-positions with different silyl chloride in DMF in the presence of imidazole to give the 3,5-bis-O-silylated products 99a–c. These silyl protected compounds 99a–c were reduced with DIBALH at −78 °C to their corresponding 2-deoxy-D-erythro-pentofuranose derivatives 100a–c (α/β = 1:1). Its mesylation followed by in situ elimination of MsOH furnished ribofuranoid glycals 102a–c (Scheme 17).
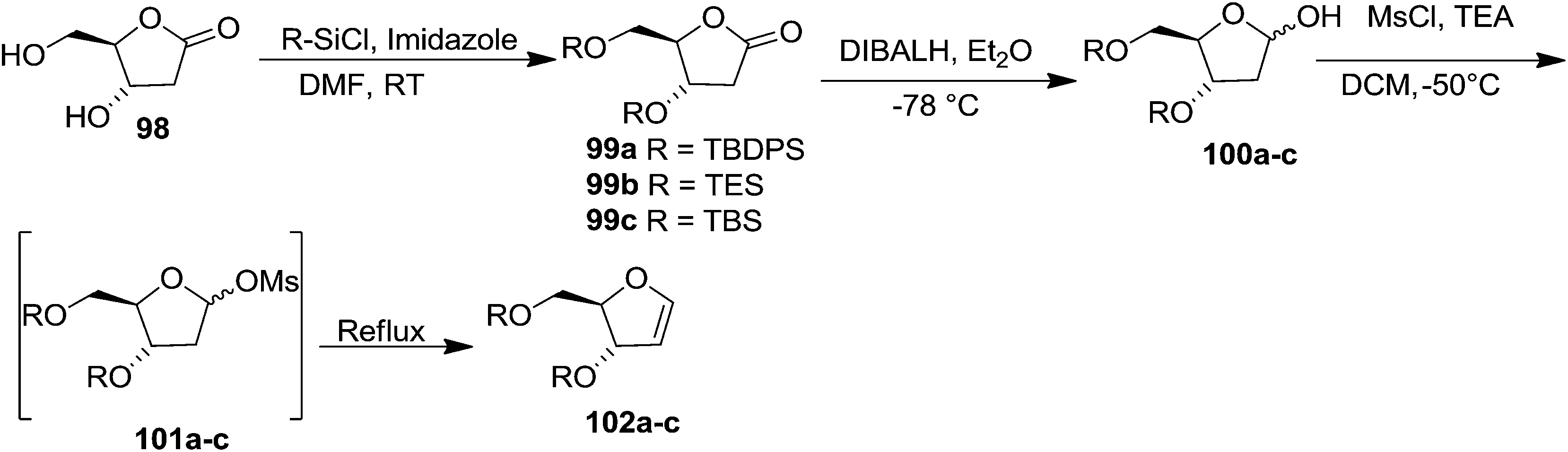 | ||
| Scheme 17 | ||
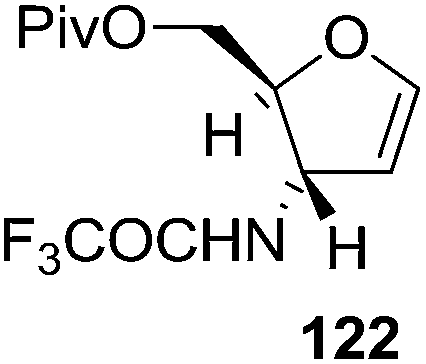
Furanoid glycals were also synthesized by McDonald and Gleason through trialkylamine-molybdenum pentacarbonyl-catalyzed alkynol endo cycloisomerizations,29 which were easily prepared in chiral nonracemic form by short synthetic sequences featuring asymmetric epoxidations of commercially available allylic alcohols (Schemes 18 and 19).
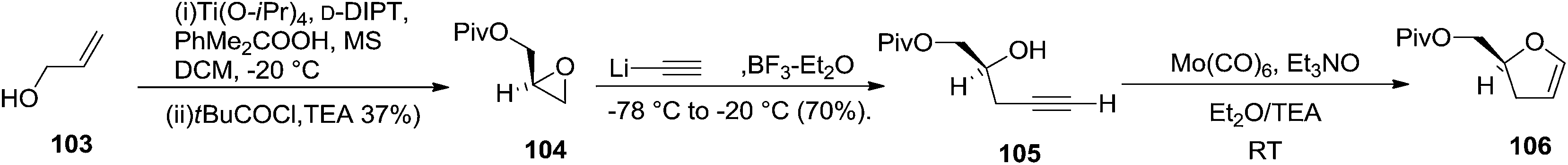 | ||
| Scheme 18 | ||
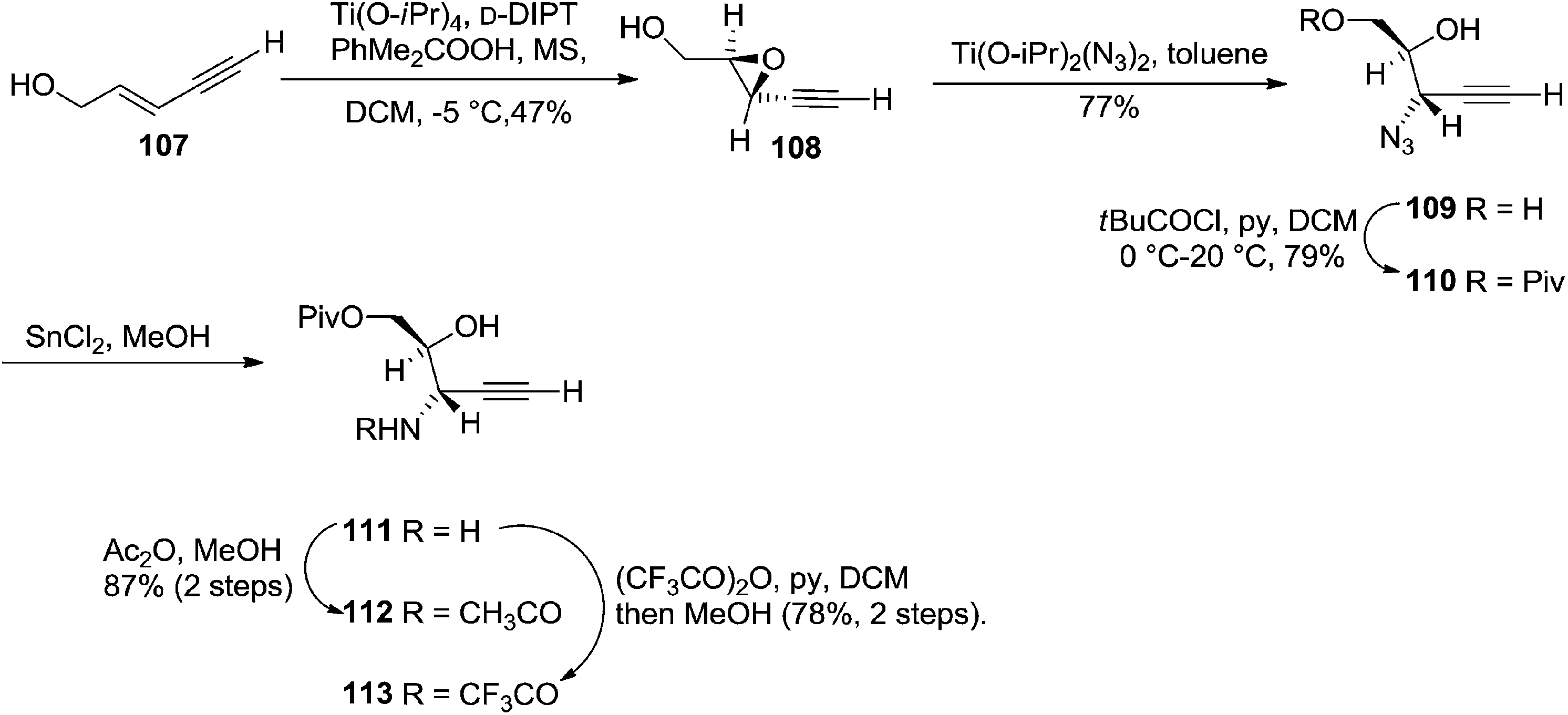 | ||
| Scheme 19 | ||
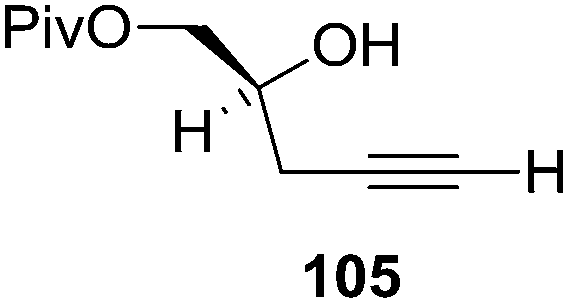
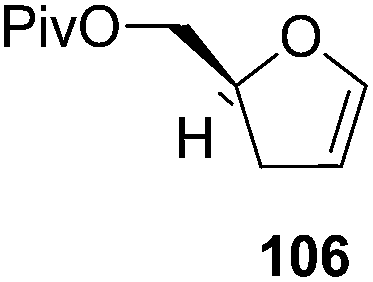
Furanoid glycal 106 was prepared by asymmetric epoxidation of allyl alcohol 103 followed by in situ esterification with pivaloyl chloride to give 104 (Scheme 18). Regioselective addition of LiC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH/BF3·Et2O to 104 at low temperature (−78 °C to −20 °C) provided the homopropargylic secondary alcohol 105. Treatment of 105 with Mo(CO)6 and trimethylamine-N-oxide in ether/Et3N at room temperature gave 106 in 80% isolated yield (Table 4).29
CH/BF3·Et2O to 104 at low temperature (−78 °C to −20 °C) provided the homopropargylic secondary alcohol 105. Treatment of 105 with Mo(CO)6 and trimethylamine-N-oxide in ether/Et3N at room temperature gave 106 in 80% isolated yield (Table 4).29
| Alkynols | Products (isolated yields) |
|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
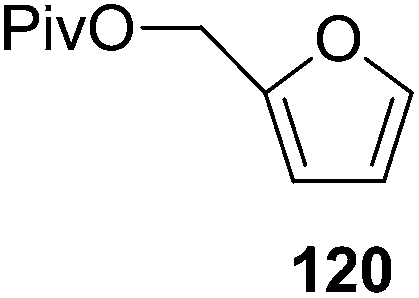 |
 |
 |
 |
 |
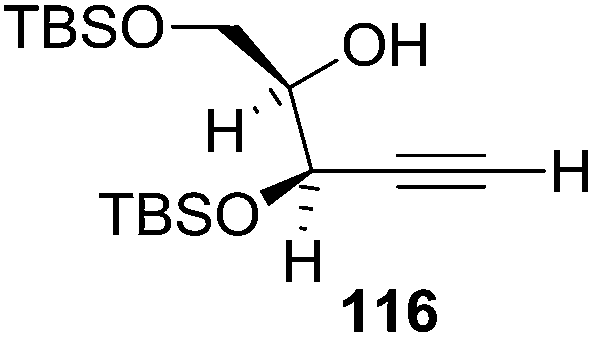
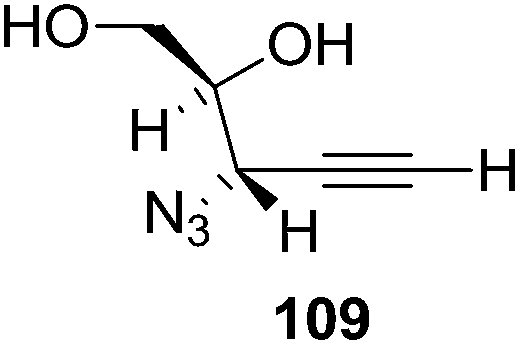
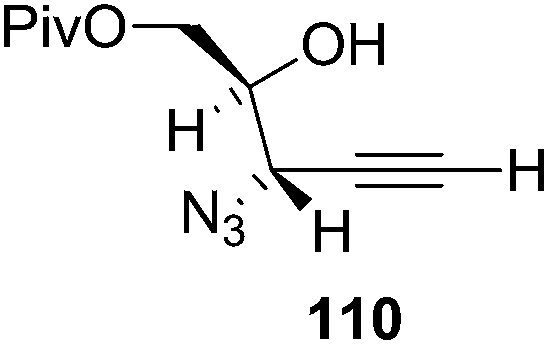
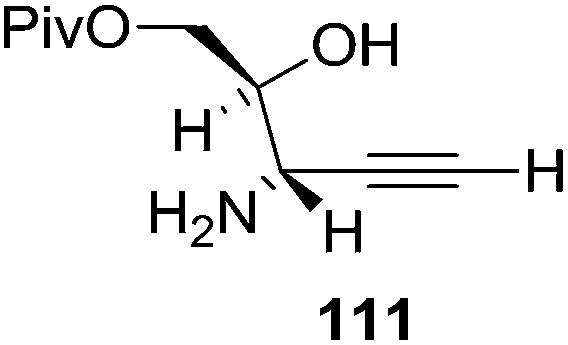
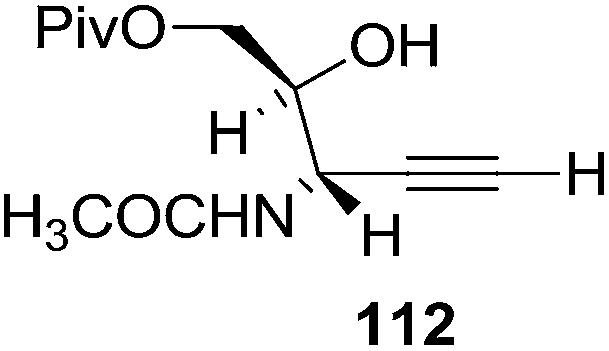
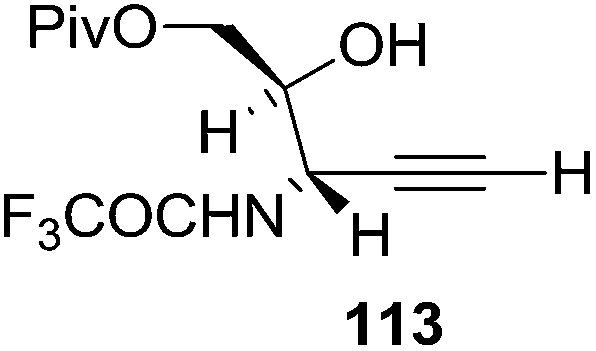
They also reported the asymmetric synthesis of alkynols having propargylic nitrogen substituents. These substrates were prepared from (E)-2-penten-4-yn-1-ol 107 (Scheme 19). Asymmetric epoxidation of 107 furnished 108 which on treatment with Ti(O-iPr)2(N3)2 in toluene followed by the selective protection of the resulting primary alcohol 109 formed the azide 110. Its reduction with tin(II) chloride in MeOH yielded the amine 111 which was acylated with Ac2O and Tf2O to obtain the respective 3-amidoalkynols 112 and 113 respectively.
The various alkynols were subjected to Mo-catalyzed alkynol cyclization to obtain furanoid glycal and THF derivatives as shown in Table 4.
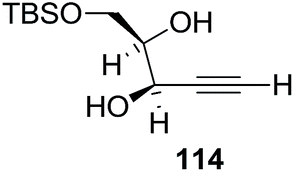
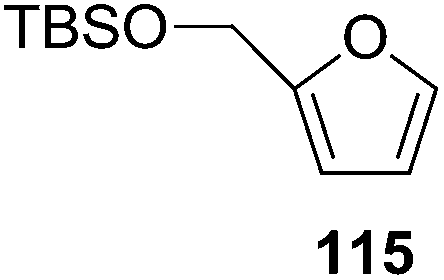
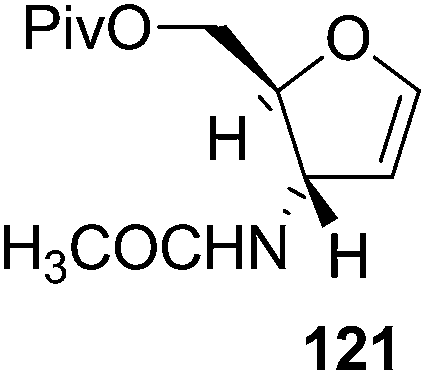
Here it is worth mentioning that the Mo-catalyzed alkynol cyclization method was compatible with alkynol substrates containing propargylic nonbasic, ester and amide functional groups (105, 116, 112, 113) for synthesis of furanoid glycals (106, 117, 121, 122). Whereas, alkynol substrates bearing good leaving groups (i.e., N3 group) and basic groups (i.e. NH2, OR) at the propargylic position (114, 118, 109, 110) underwent cyclization followed by elimination afforded furan derivatives (115, 119, 120). The mechanistic explanations for their formation are delineated below (Schemes 20 and 21). These furanoid glycals further utilized for synthesis of deoxynucleosides (Schemes 106–111).
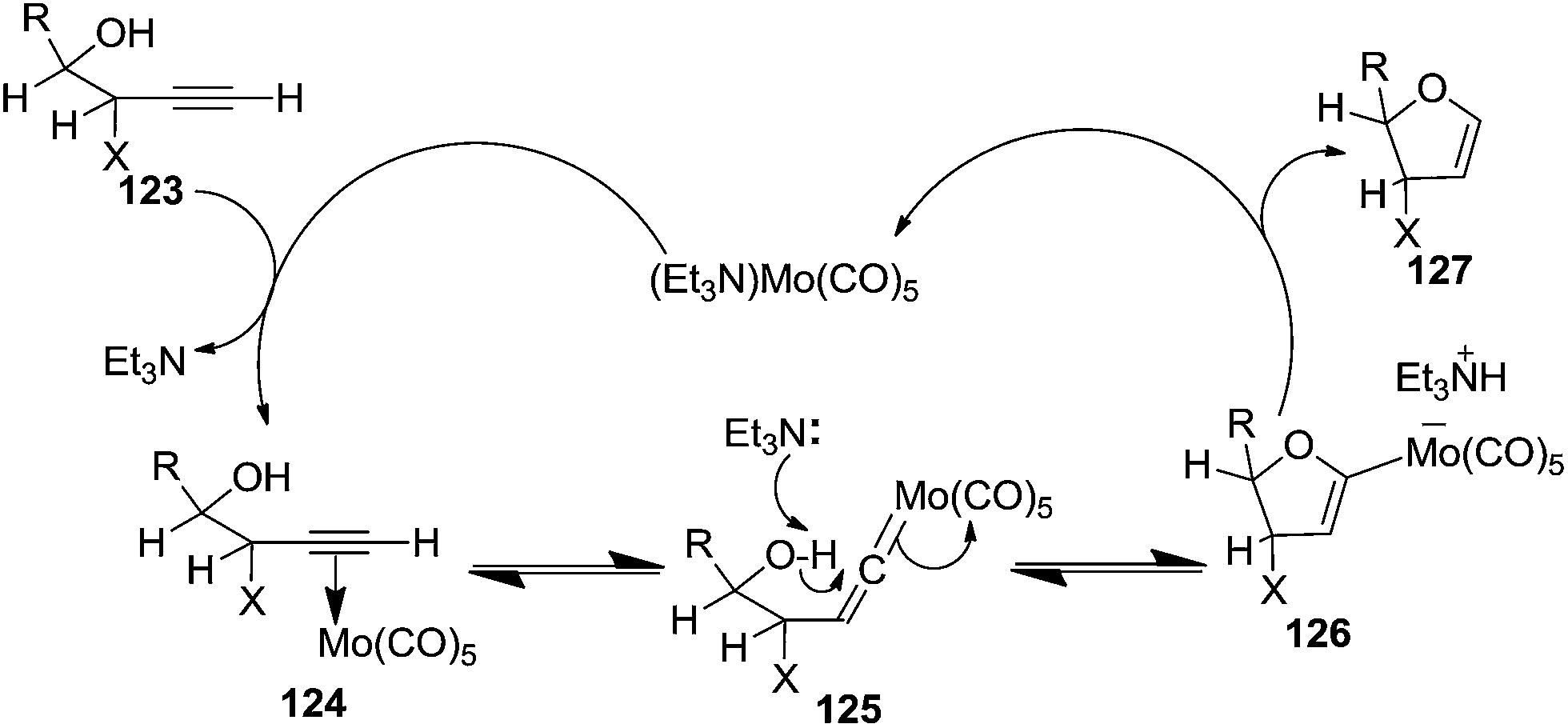 | ||
| Scheme 20 Mechanism for alkynol cycloisomerization (X= non basic group, H, NH(CO)R). | ||
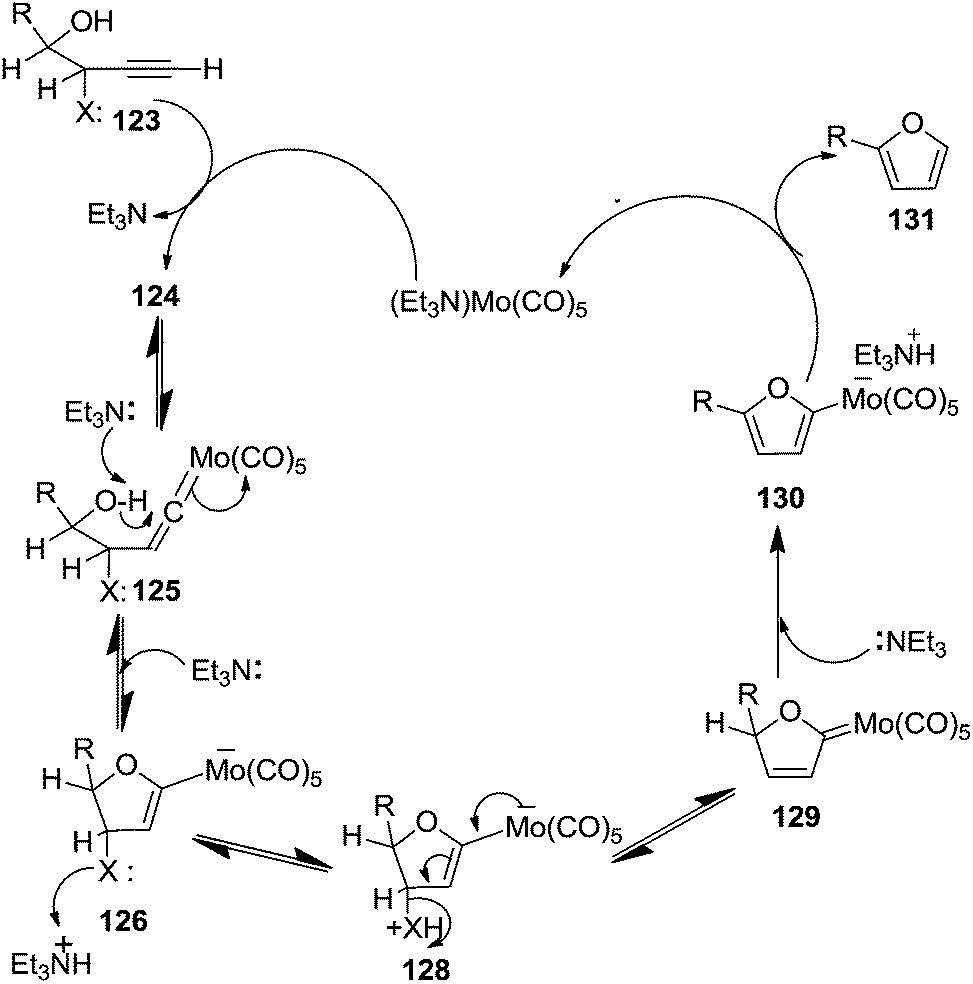 | ||
| Scheme 21 Mechanism for furan formation (X = N3, or basic groups i.e. NH2, OR). | ||
In 1997 Diaz et al. described two synthetic procedures for C-3,4-D-threo and D-erythro furanoid glycals from protected 1,2-dihydroxy pento- and hexo-furanose derivatives with the D-xylo, D-gluco and D-ribo, D-allo configurations as starting materials.30
The first one referred to the Garegg-Samuelsson reaction on vic-diols (132a–138a) in the presence of I2-PPh3-Im caused substitution followed by elimination of unstable vic-diiodide intermediate to the corresponding furanoid glycals (132c–138c) (Scheme 22).
 | ||
| Scheme 22 | ||
The second one was modified Corey's dideoxygenation of 1,2-thiocarbonates (132b–138b) which were derived from the reaction of 1,2-diols (132a–138a) with thiophosgene in alkaline medium in good yields. These 1,2-thiocarbonates (132b–138b) on treatment with 1,3-dimethyl-2-phenyldiazaphospholidine (DMPD) in dry toluene at 70 °C underwent elimination to give furanoid glycals (132c–138c) (Scheme 22).
In 1999 Theodorakis and co-workers disclosed a short and efficient enantioselective synthesis of norrisane side chain 244 from furanoid glycal 139 (Scheme 41) derived from D-mannose 57.31 They followed Ireland's method to synthesize furanoid glycal 139 from D-mannose 57 in good yield. D-Mannose 57 was treated with acetone in presence of iodine as a catalyst to deliver bis-acetonide 22 in 85% yield, after a simple filtration and crystallization. Its treatment with p-TsCl and Et3N afforded glycosyl chloride 32, which upon slow addition to a stirring mixture of sodium naphthalenide in THF gave furanoid glycal 26 in 48% overall yield (for two steps). Due to its labile nature, it was immediately benzylated (BnBr, NaH, TBAI) to produce the glycal 139 in 90% yield (Scheme 23) which was utilized to achieve the synthesis of norrisane side chain 244 in 6 steps discussed in Scheme 41.
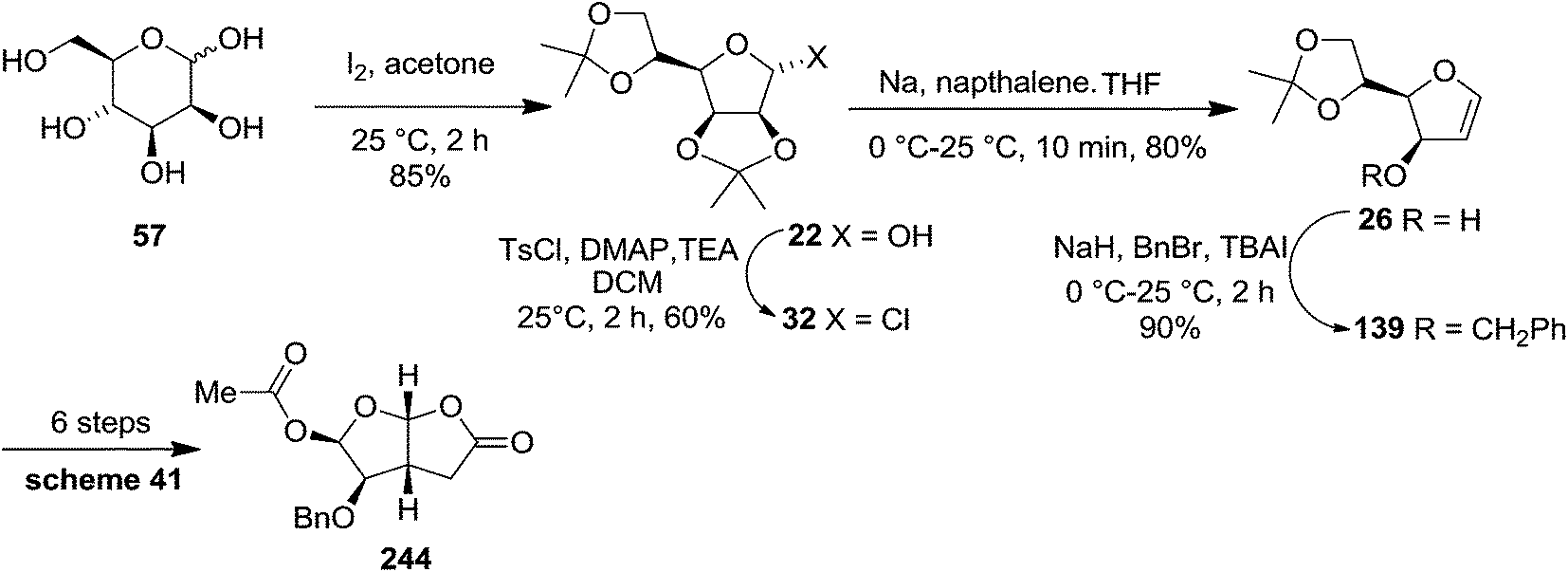 | ||
| Scheme 23 | ||
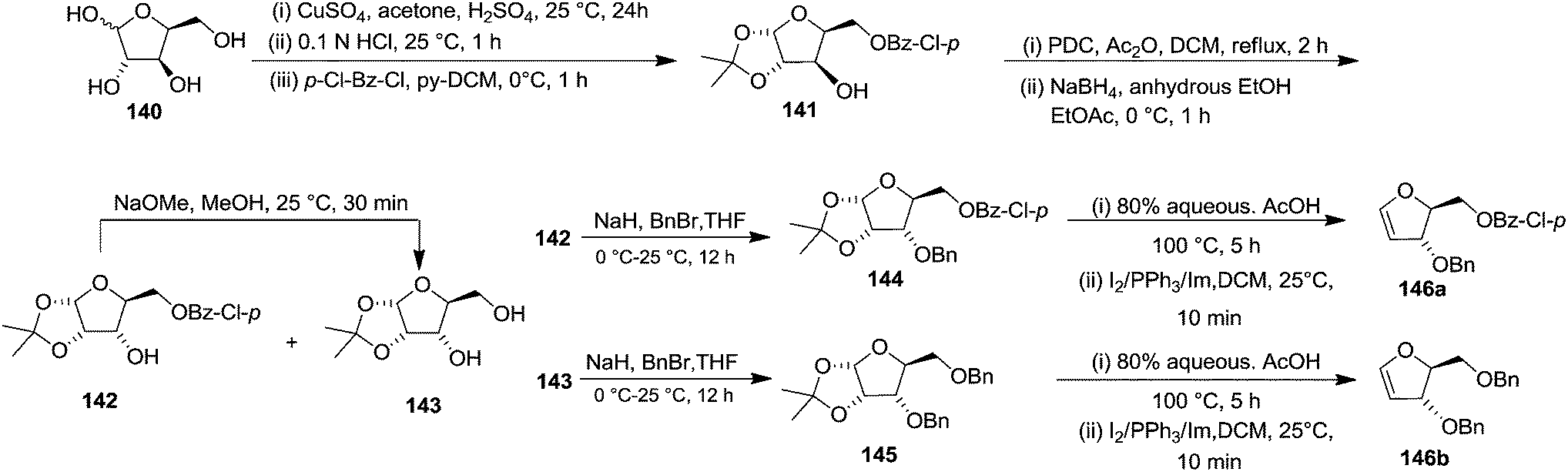
In 2000, Knaus et al. reported the synthesis of analogues of deoxy-β-L-cytidine via furanoid glycal as key intermediates. They followed Garegg Samuelsson reaction for the synthesis of ribo furanoid glycals from vic-diols intermediate, easily derived from L-xylose 140.32 Acetonide protection of L-xylose 140 followed by selective acid hydrolysis with 0.1 N HCl afforded intermediate 1,2-O-isopropylidene-α-L-xylofuranose whose primary hydroxyl group was selectively protected with p-Cl-Bz-Cl in pyridine-DCM at 0 °C to obtain 141. Its free hydroxyl group was oxidised with PDC in DCM, and the resulting ketone was reduced with NaBH4 to afford a mixture of ribose derivatives 142 (68% yield) and 151 (28% yield). The sugar derivative 142 was converted into compound 143 with NaOMe in MeOH. It was benzylated with BnBr in THF in the presence of NaH to afford the 3,5-di-O-benzyl-L-ribose derivative 145 in good yields. The similar protocol was adopted to obtain the 5-O-(p-chlorobenzoyl)-3-benzyl-L-ribose derivative 144 from its precursor 142. The 1,2-O-isopropylidenyl groups in 144 and 145 were readily removed by treating each with 80% aqueous AcOH at 100 °C to give the vic-diols. Their debenzylation with the I2-PPh3-Im system in dry DCM at 25 °C afforded the desired ribofuranoid glycals 146a and 146b in 62% and 45% yield respectively (Scheme 24). These were used as key intermediates for synthesis of unnatural C-aryl 2′-deoxy-β-L-cytidine mimics (529a, b) (Scheme 92).
 | ||
| Scheme 24 | ||
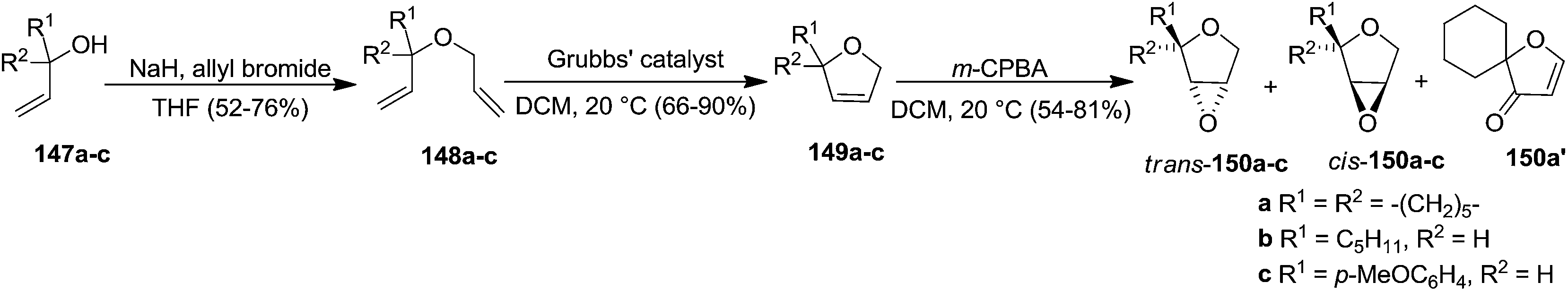
In the same year Schmidt and Wildemann developed the synthesis of 2,3-dihydropyrans or 2,3-dihydrofurans (furanoid glycals) utilizing ring closing methathesis as one of the key reaction steps. The allylic alcohols (147a–c) were subjected to allylation with allyl bromide in the presence of NaH in THF to obtain the diallyl ethers (148a–c). Their ring-closing metathesis yielded 3,4-dihydrofurans (149a–c) followed by their epoxidation with m-CPBA gave dihydrofuran oxides (150a–c). In the case of 150a, spiro dihydrofuranone 150a′ was obtained along with cis, trans-150a (Scheme 25).33
 | ||
| Scheme 25 | ||
The base-induced rearrangement of 150a with LDA in THF at ambient temperature gave a crude product whose NMR spectra identified it as 151a. It was rearranged to hemiacetal 152 during its silica gel column purification. However, this was supressed when free hydroxyl group in 151 was protected as a benzyl ether 153 (Scheme 26).
 | ||
| Scheme 26 | ||
The rearrangement of inseparable diastereoisomeric mixture of dihydrofuran oxide 150b underwent the rearrangement reaction with LDA or LiTMP to give a 4:3:1 mixture of isomers cis-151b, trans-151b and 151b′ which were characterized by NMR spectroscopy of crude reaction mixture.
The dihydrofuran oxides trans- and cis-150c were separable. While the treatment of trans-150c with LiTMP gave three products in a ratio of 9:3:1 which were identified as furanoid glycals 151c′, trans-151c and furan 154c, the cis-150c yielded only one rearrangement product cis-151c.
Recently the furanoid glycal 26 was synthesised by Gómez and López et al. from 155. Its thioglycosylation followed by controlled oxidation of the resulting thioglycoside with m-CPBA afforded furanosyl sulfoxide 156 which on treatment with nBuLi furnished column pure furanoid glycal 26 in good yield (Scheme 27).34
 | ||
| Scheme 27 | ||
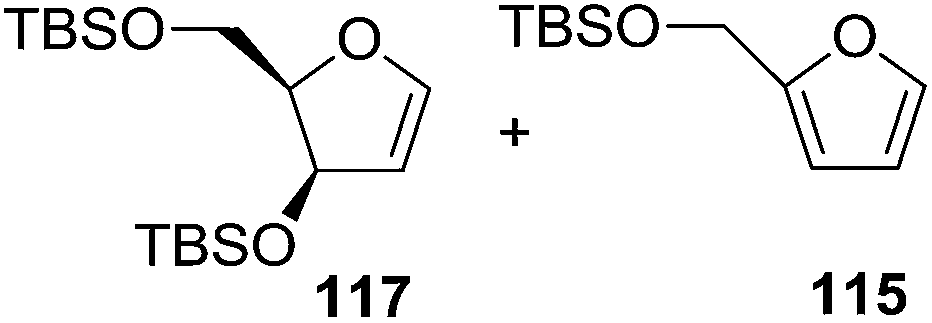
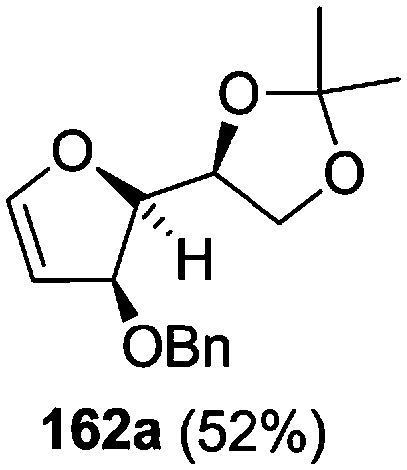
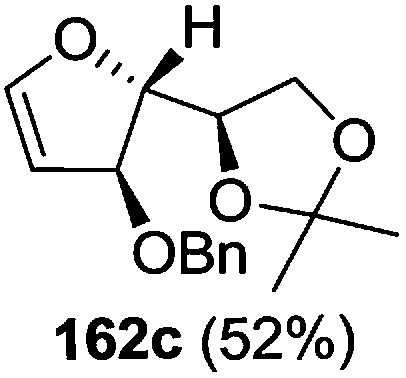
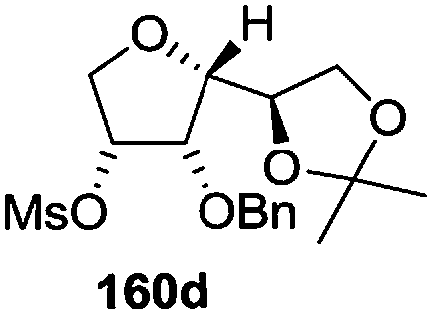
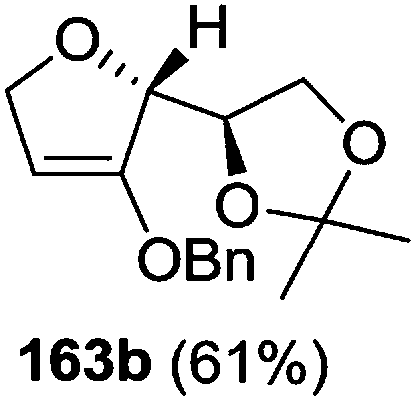
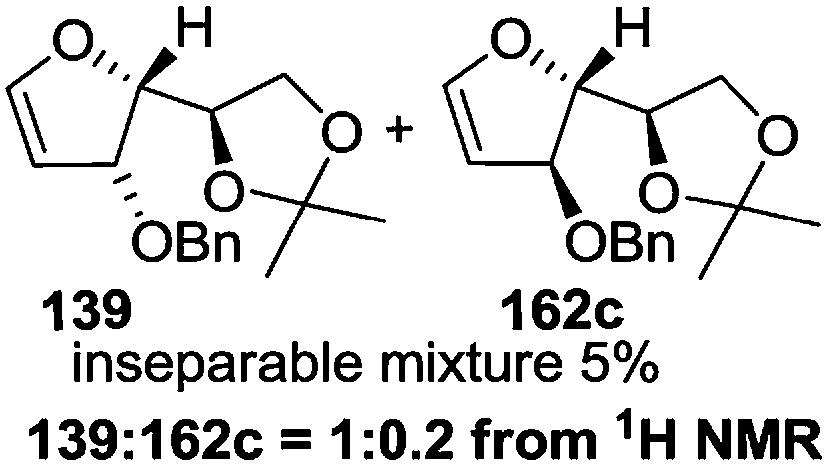
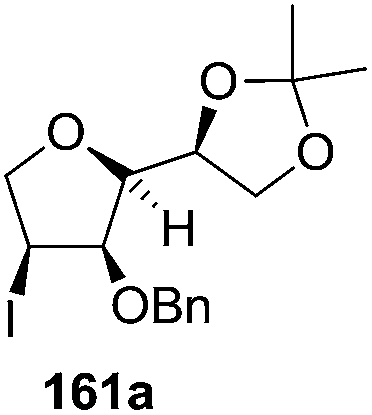
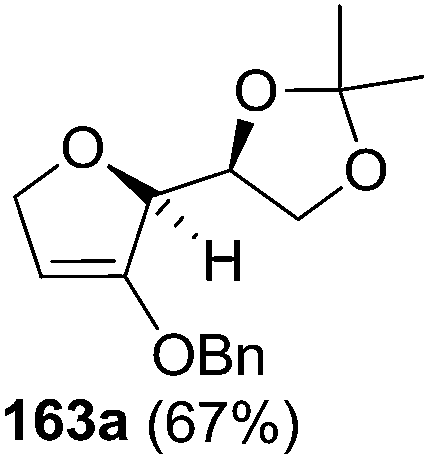
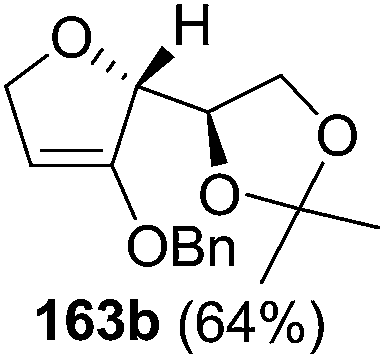
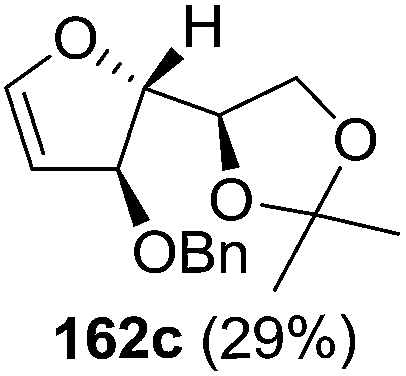
As mentioned above, several methods are available for synthesis of furanoid glycals or 4,5-dihydrofurans but difficulties are generally encountered with the formation of unstable glycals,13 usage of expensive starting materials22,23 and, in some cases, low yield of the desired products and that is why improvements in the existing methods are still desirable. To overcome all these limitations and encouraged by the literature reports on various applications of furanoid glycals in organic synthesis, our research group reported a simple protocol for the synthesis of stereochemically pure different furanoid glycals 162a (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-L-arabino-hex-1-enitol), 162b (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-L-ribo-hex-1-enitol), 162c (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-D-ribo-hex-1-enitol),17a 139 (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-D-arabino-hex-1-enitol)17a,31,36,37 and also highly functionalized 2,5-dihydrofurans (163a, b) (Fig. 3) from easily accessible enantiomerically pure 2,3,4-trisubstituted THF scaffolds (159a–d).35 To the best of our knowledge, examples for the synthesis of enantiomerically pure furanoid glycals (162a, b) and functionalized 2,5-dihydrofurans (163a, b) had not been reported.35a
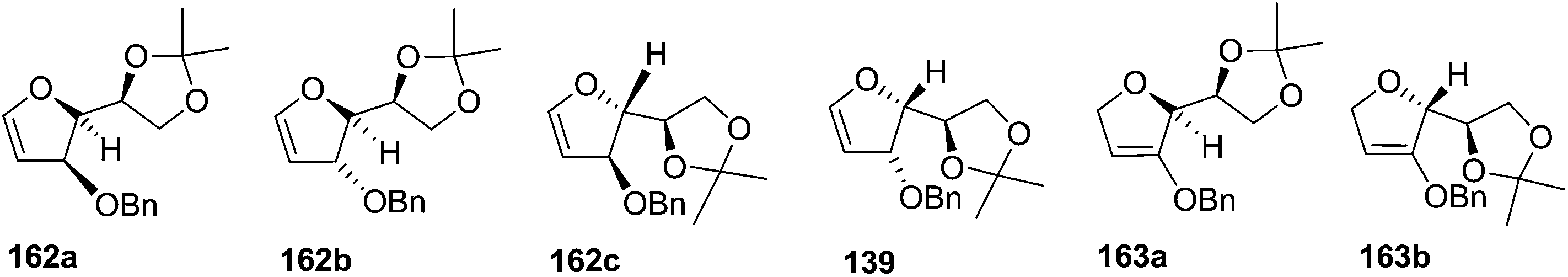 | ||
| Fig. 3 Structures of furanoid glycals (162a–c, 139) and highly functionalized 2,5-dihydrofurans (163a, b). | ||
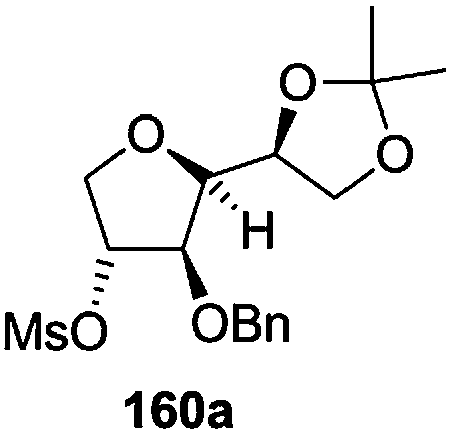
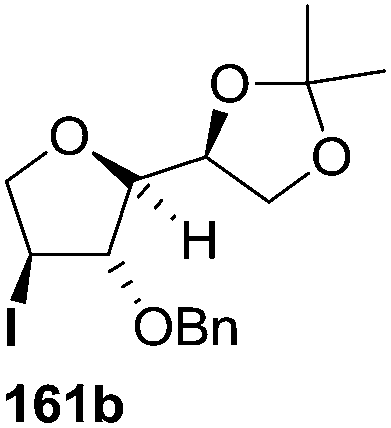
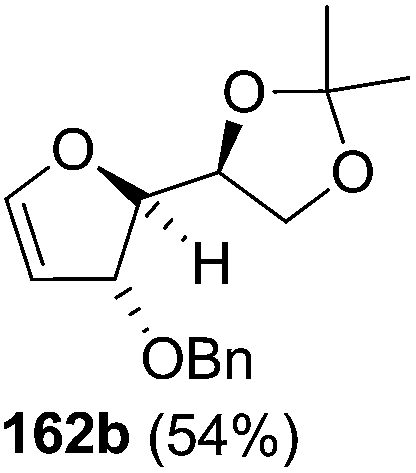
The synthetic protocol to obtain a family of furanoid glycals (162a–c, 139) of different configurations at the 3-, 4- and 5-positions and functionalized 2,5-dihydrofurans (163a, b) of different configurations at the 2- and 1′-positions from enantiopure THF scaffolds (159a–d) is shown in Scheme 28. These THF scaffolds (159a–d) were prepared from Pyranoid glycals (157) (3,4,6-tri-O-benzyl-D-galactal (A) and 3,4,6-tri-O-benzyl-D-glucal (B)) derived allylic alcohols (158) involving the intramolecular asymmetric ring opening (ARO) of the enantiomerically pure 2,3-epoxy alcohols using Sharpless asymmetric epoxidation (SAE) conditions followed by subsequent isopropylidene protection of the diol. The free hydroxyl group in 159 was protected with MsCl in the presence of Et3N to afford the corresponding mesyl derivative 160. The OMs protected THF 160 in dry DMF was subjected to thermal elimination reaction for 8 h in the presence of DBU to furnish the furanoid glycals (162a–c, 139) and 2,5-dihydrofurans (163a, b). These furanoid glycals and 2,5-dihydrofurans were obtained in good yields when the stereochemically inverted iodides 161, prepared by Garegg–Samuelsson reaction (I2/PPh3/imidazole) from 159, were heated at reflux with DBU in dry DMF for 5 h (Scheme 28, Tables 5 and 6).35a
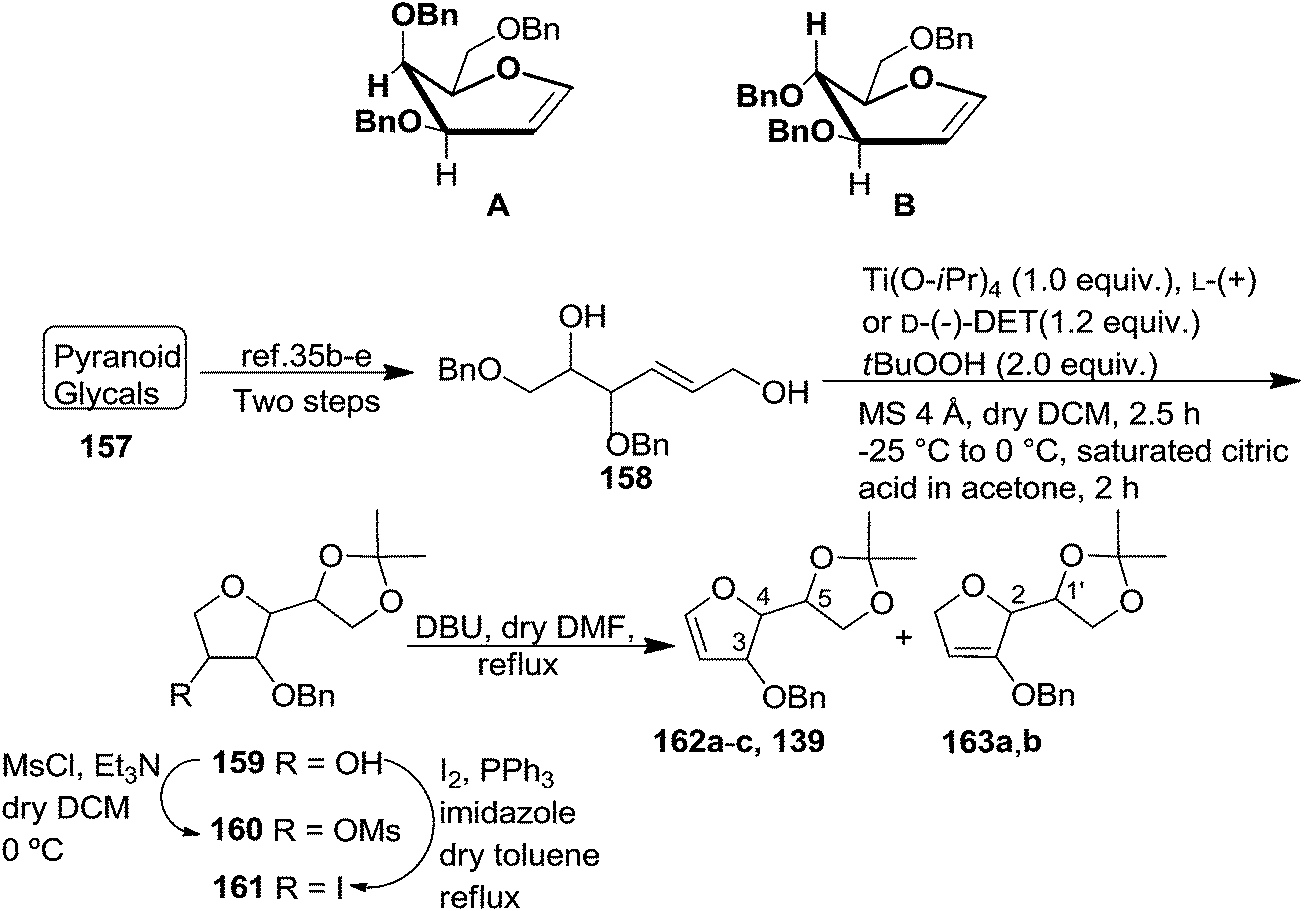 | ||
| Scheme 28 General strategy for the synthesis of furanoid glycals (162a–c, 139) and functionalized 2,5-dihydrofurans (163a, b). | ||
| Entry | THF domains (159a–d) | Mesyl derivatives (160a–d) | Major products | Minor products |
|---|---|---|---|---|
| 1 | 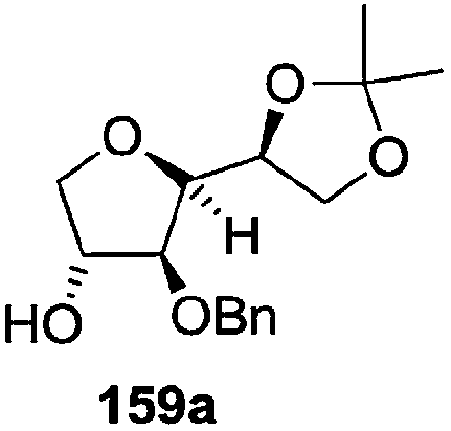 |
 |
 |
— |
| 2 | 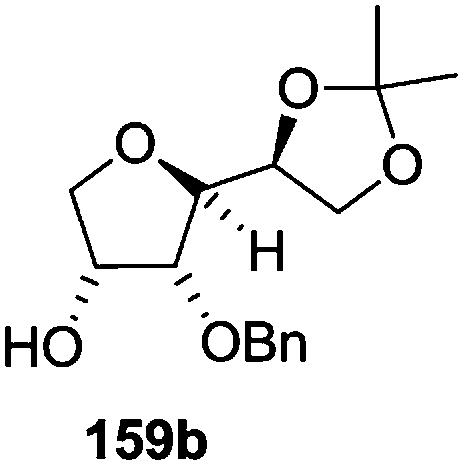 |
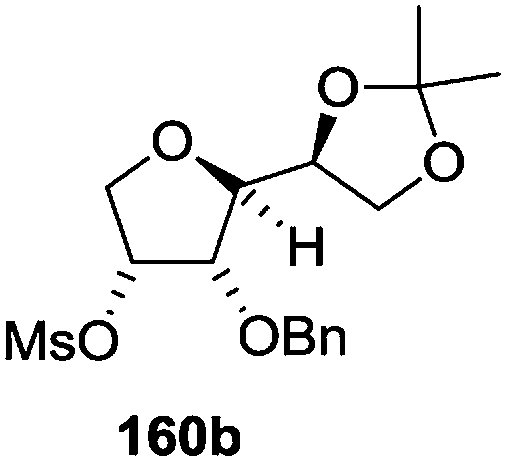 |
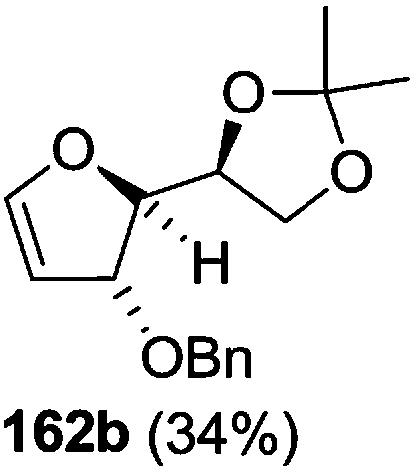 |
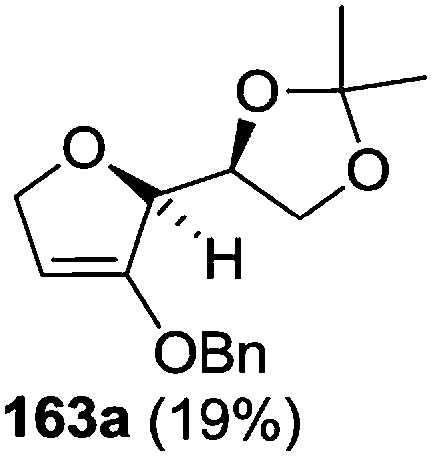 |
| 3 |  |
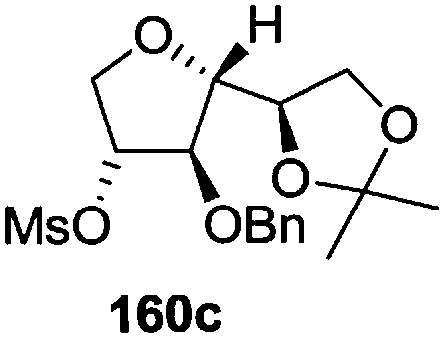 |
 |
— |
| 4 | 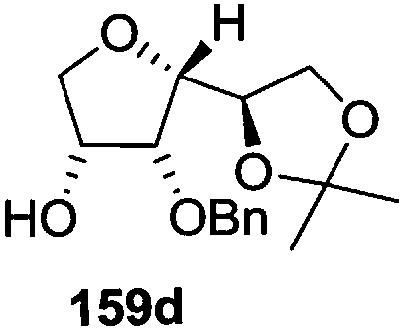 |
 |
 |
 |
| Entry | THF domains (159a–d) | Iodo derivatives (161a–d) | Major products | Minor products |
|---|---|---|---|---|
| 1 | 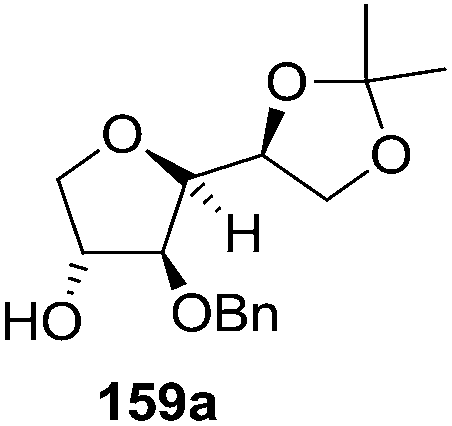 |
 |
 |
— |
| 2 | 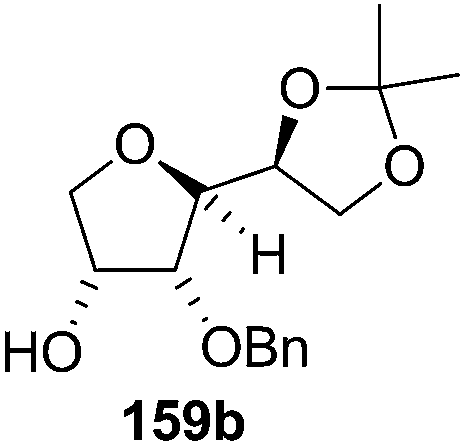 |
 |
 |
— |
| 3 | 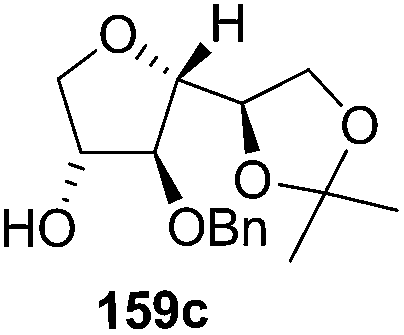 |
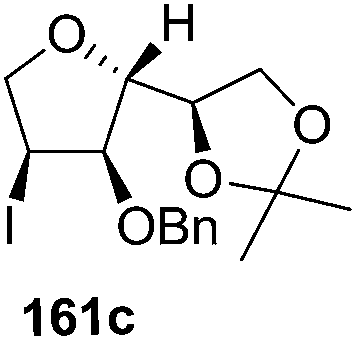 |
 |
 |
| 4 | 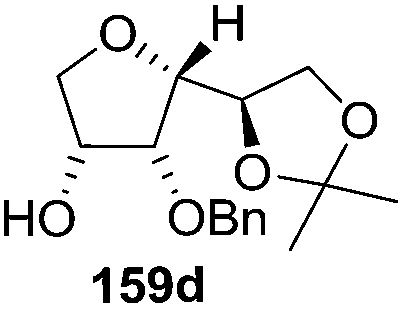 |
 |
 |
— |
The construction of the double bonds was accomplished here by carrying out a base-induced E2 elimination of enantiomerically pure C4 mesylate or iodo THF scaffolds (160 or 161) by utilizing inexpensive reagents with a simple experimental and workup procedure. In the formation of either glycal or olefin, the E2 elimination took place most readily when the hydrogen atom and the leaving group were in an antiperiplanar arrangement. Further, it can be argued that E2 elimination of MsOH from 160b leading to the formation of two products in which furanoid glycal 162b (Hofmann product) was formed predominantly over Saytzeff product 163a (more substituted olefin) may be attributed to the involvement of a conformation in which the leaving group, 4-OMs, and one of the hydrogen atoms at C5 adopted a relatively higher degree of antiperiplanar arrangement relative to the antiperiplanar arrangement of 4-OMs and H3. In contrast, the formation of Saytzeff product 163b as the major product and a mixture of glycals (162c, 139) as the minor product, both from 160d, could be attributed to the comparable torsion angles subtended by the OMs group and the H atom across the C4–C3 and C4–C5 bond, respectively. Similarly, E2 elimination of HI from 161c gave expected Saytzeff product 163b as the major product over furanoid glycal 162c as a result of the same reason described in the case of E2 elimination of MsOH from 160d. We also showed that furanoid glycals 162a and 162c or functionalized 2,5-dihydrofurans 163a and 163b can be synthesized exclusively or in major quantity from the same 2,3,4-trisubstituted THF scaffolds 159a and 159c, respectively, by changing the leaving group at C4. Furthermore, this report provided two pairs of enantiomeric furanoid glycals (4,5-dihydrofurans) (162a, 139) and (162b, 162c) and one pair of enantiomeric 2,5-dihydrofurans (163a, 163b) (Fig. 3).
In 2010, Haraguchi and group described electrophilic glycosidation of erythro-furanoid glycal 164 with nucleobases followed by removal of the substituent X at the 2′-position of the resulting product 165 to give a mixture of β-and α-2′-deoxynucleosides (166, 167) (Scheme 29).38
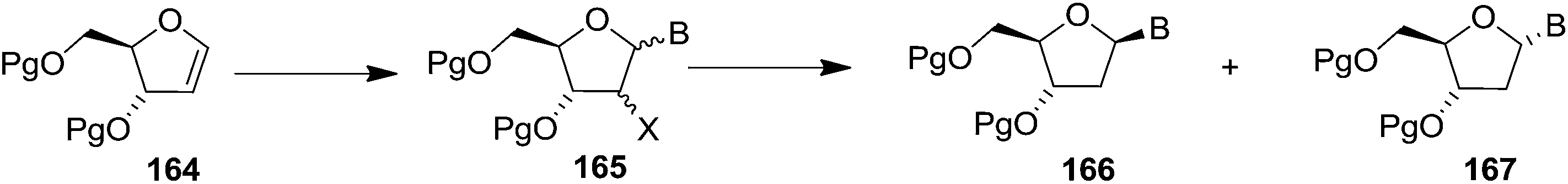 | ||
| Scheme 29 | ||
To improve the β-selectivity, for the first time they reported the synthesis of erythro-furanoid glycals (171, 173) by means of sulfoxide syn-elimination (Scheme 30). 2-Deoxy-D-ribose 67 on treatment with PhSH/H2SO4 in DMF afforded phenyl-2-deoxy-1-thio-D-erythro-pentofuranoside 168 in 97% yield. The free hydroxyl groups of 168 were silylated with TIPDSCl to give 169 in 84% yield. Its oxidation with m-CPBA led to the formation of the sulfoxide 170 in 96% yield. The sulfoxide 170 on treatment with solid NaHCO3 in refluxing xylene underwent sulfoxide syn-elimination to furnish glycal 171 in 84% yield. Likewise, 168 was protected with the DTBS group to give 172 (93% yield), which was then converted to the 3,5-O-DTBS-protected glycal 173 in 83% yield by adopting the reaction sequence similar to that employed for the synthesis of glycal 171. The synthesis of nucleosides utilizing these furanoid glycals (171, 173) has been delineated in Schemes 128 and 129.
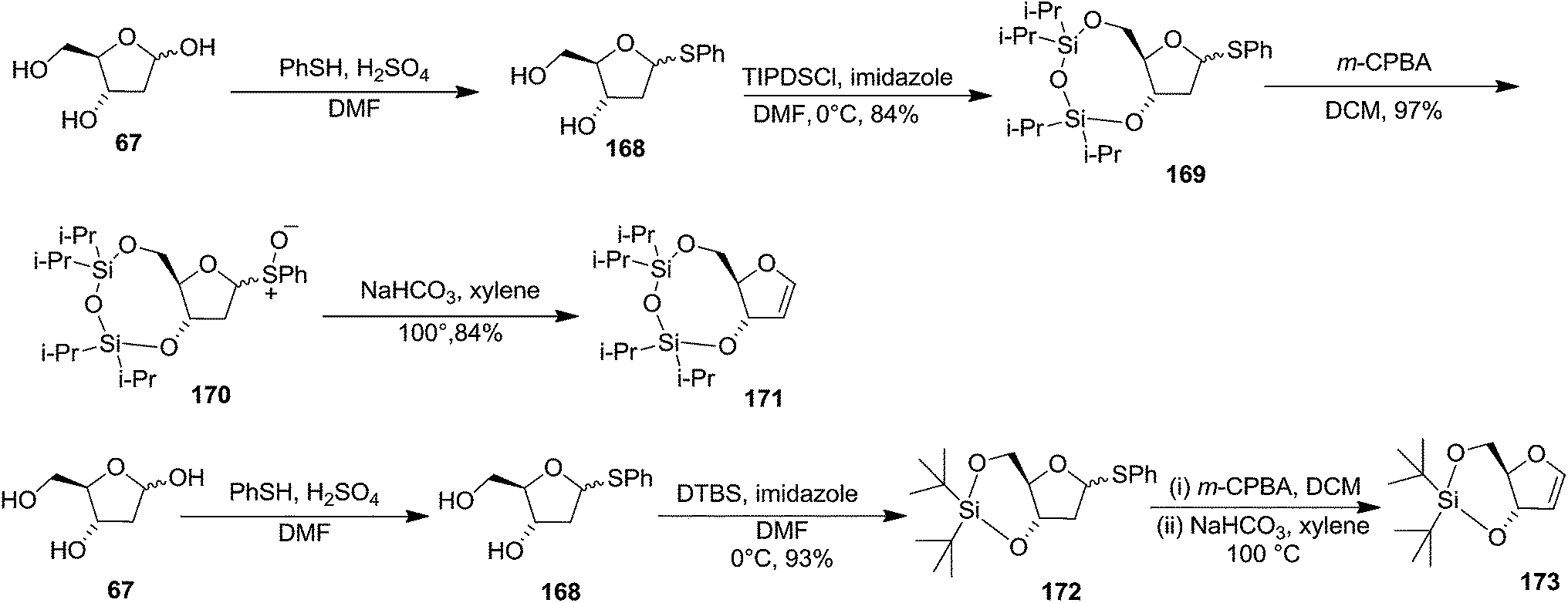 | ||
| Scheme 30 | ||
3. Applications of furanoid glycals
3.1. Synthesis of natural products and related compounds
Nowadays, for the synthesis and utilization of chiral building blocks (CBBs) for the synthesis of target molecules for drug design and enantiomerically pure simple and complex natural products are in great demand. This approach of synthesis utilizing the stereochemistry of the starting material is customarily known as the ‘chiron’ approach synthesis39 and it becomes very cost effective if starting material is derived from inexpensive carbohydrate or amino acid.The furanoid glycals derived from different starting materials have been utilized as chiral pool material by various groups for synthesis of variety of natural products and important target molecules, which are discussed below.
Ireland and group reported the total synthesis of Lasalocid A (X537A) 174 and analogues. For the synthesis of Lasalocid A (X537A)174, they described the construction of both the aldehyde 175 and ketone 176 available from the reverse aldol reaction of lasalocid A (X537A) 174 (Scheme 31).40,41
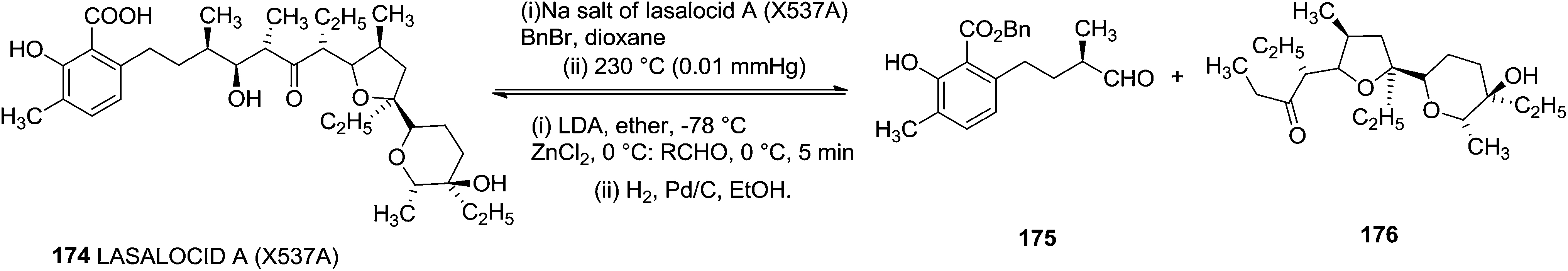 | ||
| Scheme 31 | ||
On the basis of a retrosynthetic strategy using the Ireland-Claisen rearrangement to form the two C-glycosides bearing chiral centres at the C-α positions, they utilized carbohydrate precursors as the source of the furanoid and pyranoid subunits for the construction of ketone 176 (Scheme 32).
 | ||
| Scheme 32 Retro synthetic strategy for synthesis of ketone 176. | ||
First they tried the synthesis of 176 from furanoid glycal 45 which was converted to protected furanoid glycal 49 which is described in Scheme 8,4 from which the isomeric mixture of acid 180 was prepared by [3,3] sigmatropic rearrangement at 35 °C.4 This acid mixture 180 was converted to α-epoxides 182 in 90% overall yield through the intermediate iodolactone 181. Treatment of 182 with lithiated 1,3-dithiane followed by desulfurization of the resulting products provided 183 in moderate yield. It was converted to acid 186 (epimer of 177) via the intermediates 184 and 185 (Scheme 33).
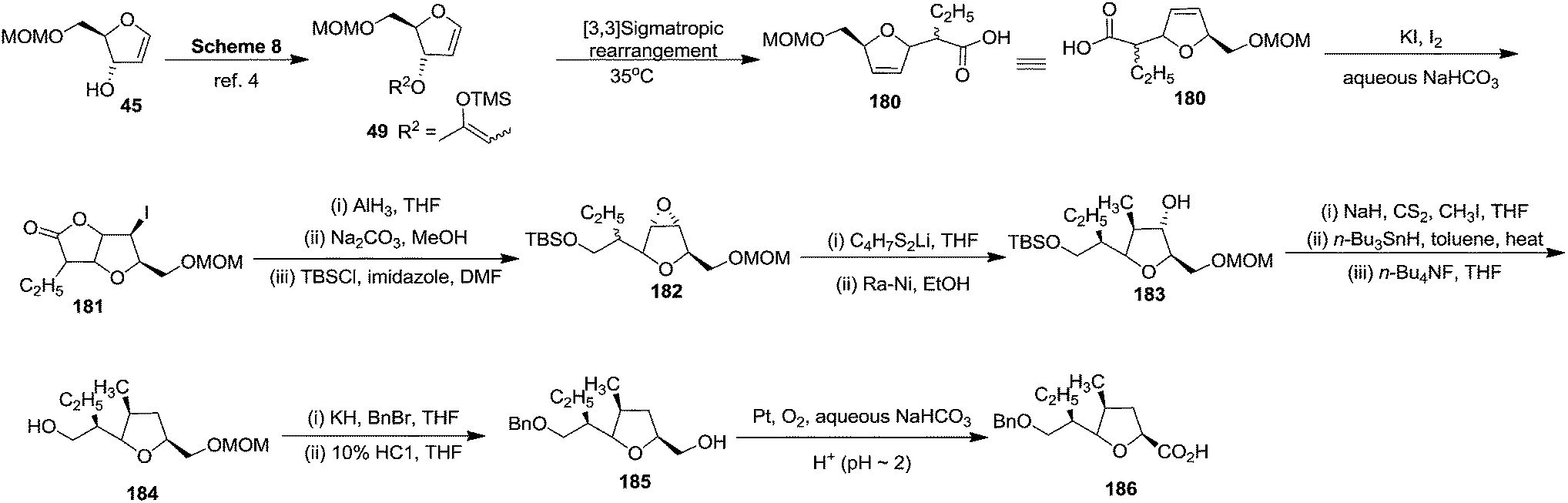 | ||
| Scheme 33 | ||
They chose α-D-glucosaccharino-1,4-lactone 187 as the starting material for the purpose to synthesize 177. It was converted to the mixture of unsaturated isomeric esters 190. Hydrogenation of 190 yielded 191 and 192, which were separated by column chromatography. After LAH reduction of 191 and 192, both the resulting alcohols (193, 194) were readily transformed to the acids 177 and 186 respectively (Scheme 34).
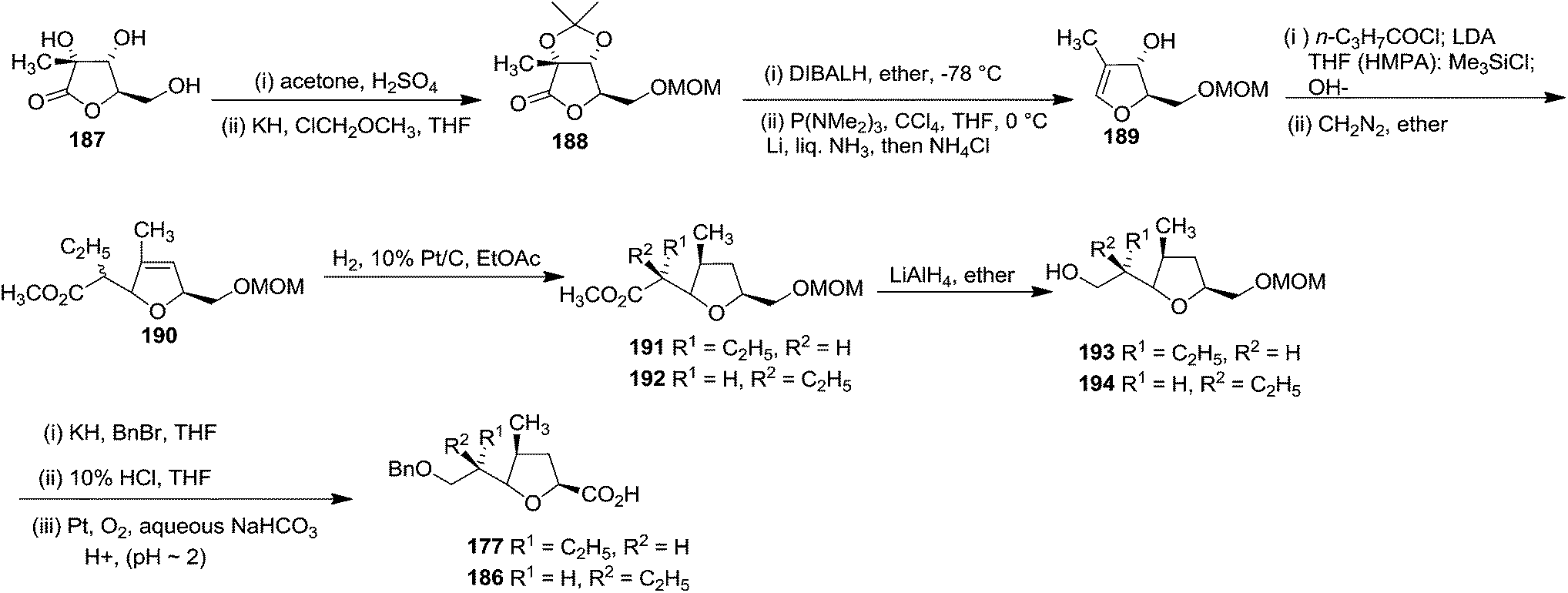 | ||
| Scheme 34 | ||
The construction of Pyranoid glycal 178 started with 6-deoxy-L-gulose 195 as shown in Scheme 35. The hydroxyl groups were differentiated as benzyl glycoside, O-isopropylidene, and methoxymethyl ether in compound 196. Removal of the benzyl ether then led to the lactol 197 which was converted to the desired glycal 178 by their optimised procedure (Scheme 35).
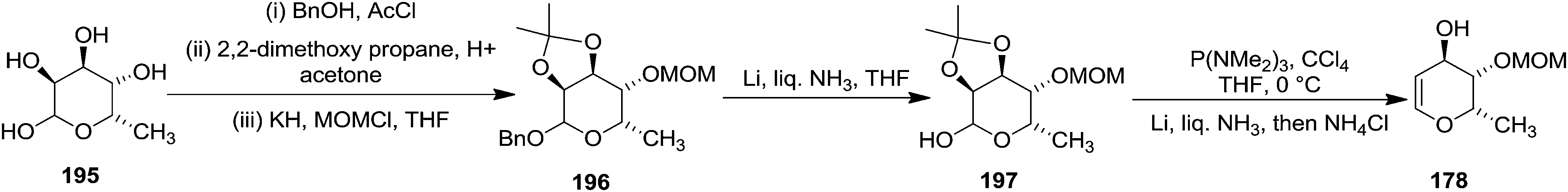 | ||
| Scheme 35 | ||
The connection of the pyranoid subunit 178 to acid 177 resulted in readily separable isomeric esters 198 and 199, while the epimer 186 afforded the isomeric esters 200 and 201. Subsequent transformation of these esters individually to alcohols 206–209 proceeded in excellent yields via 202–205 (Scheme 36).
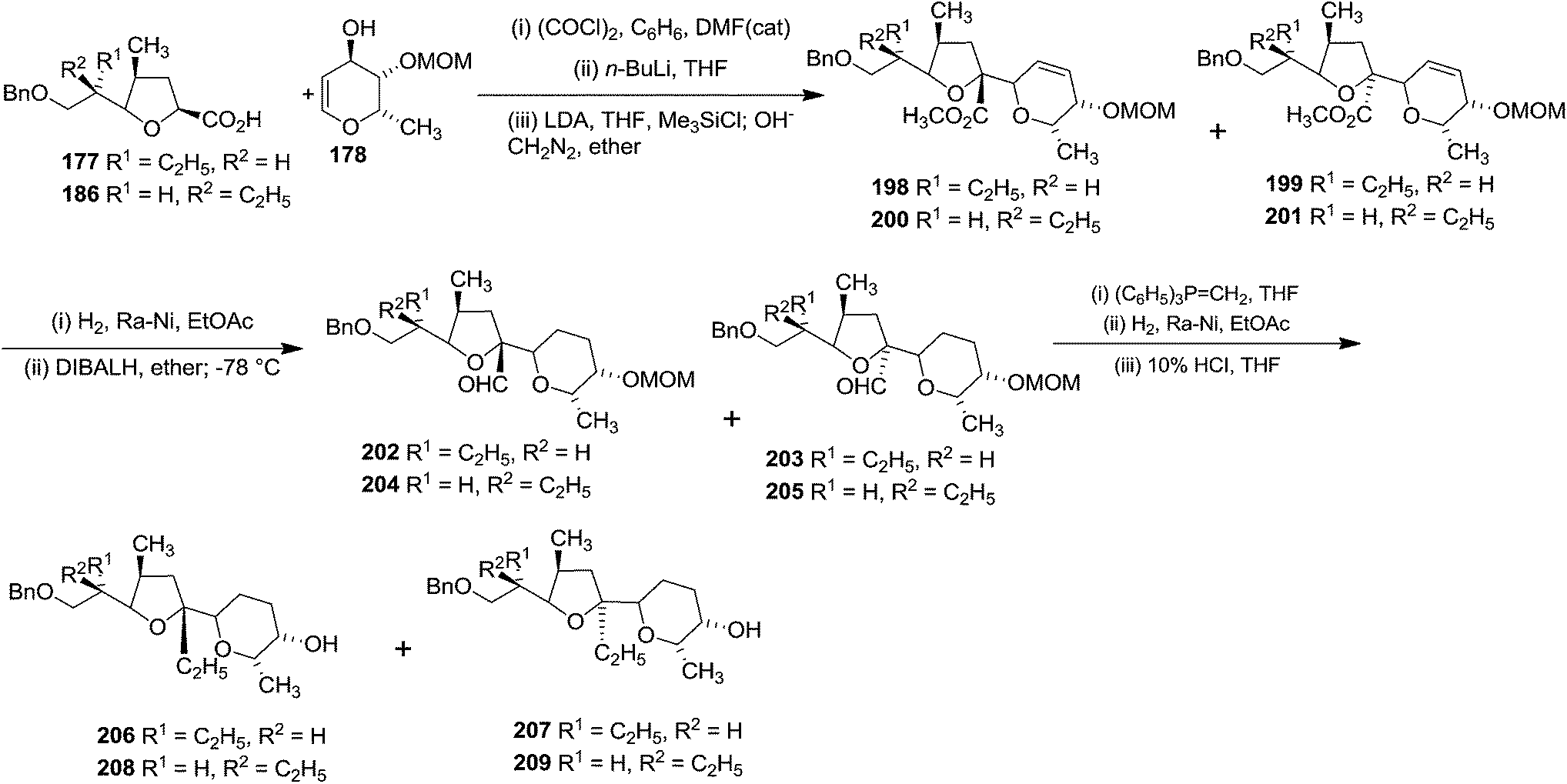 | ||
| Scheme 36 | ||
Alcohol 207 was transformed into ketone 210, which was converted to the exo methylene olefin followed by oxidation with m-CPBA afforded a mixture of epoxides in which the β-epoxide 211 was the major component. Subsequent reductive cleavage of this isomer then led to the tertiary alcohol 212 which was transformed into the aldehyde 213 involving sequence of reactions like debenzylation and oxidation of the resulting primary alcohol. Finally its Grignard reaction furnished the ketone 176 (Scheme 37). The aldol condensation between the aldehyde 175 and the zinc enolate of the ketone 176 completed the total synthesis of Lasalocid A (X537A) 174 (Scheme 31).
 | ||
| Scheme 37 | ||
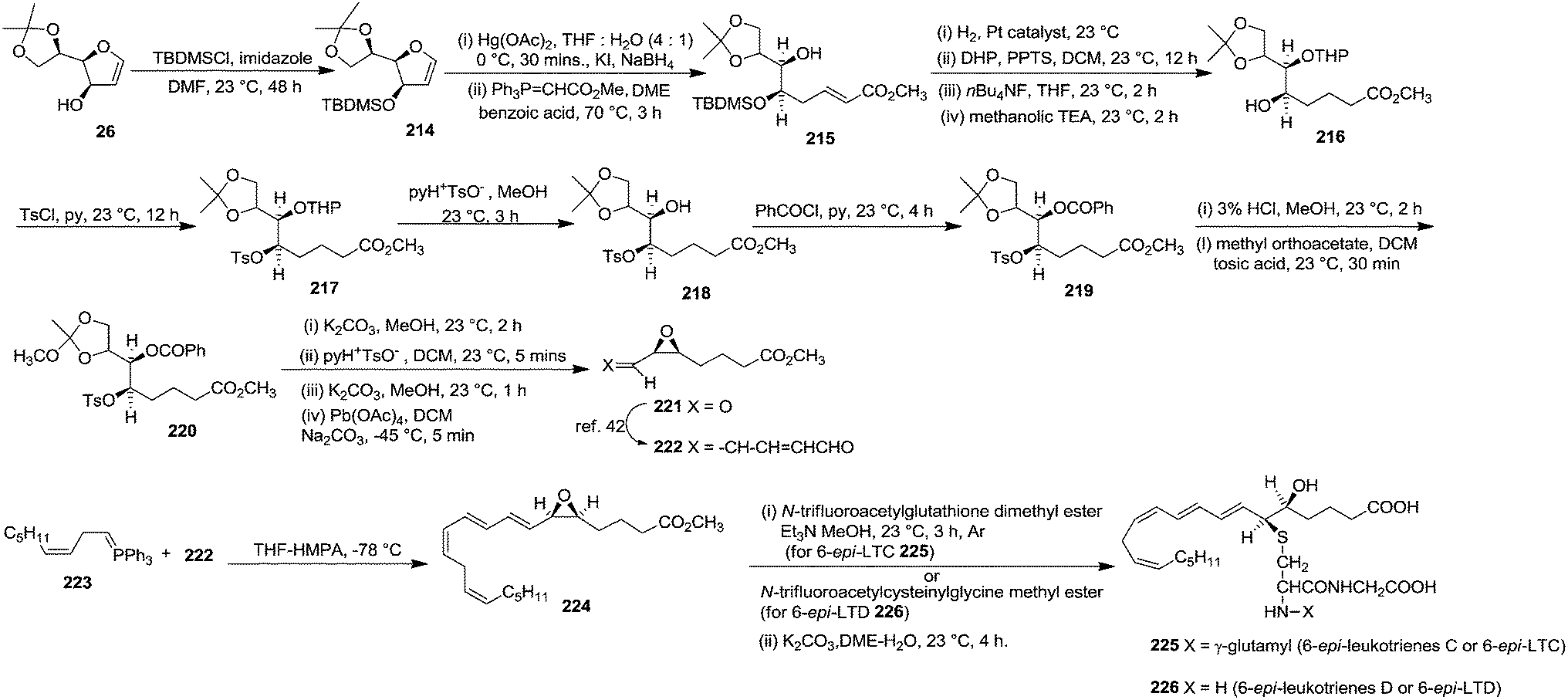
In 1980 Corey et al. synthesized 6-epi-leukotriene C or 6-epi-LTC 225 and 6-epi-leukotriene D or 6-epi-LTD 226 from D-mannose derived furanoid glycal 26. First it was converted to TBDMS ether derivative 214 by treatment with TBDMSCl, imidazole in dry DMF at 23 °C for 48 h (Scheme 38).5,42 Its successive oxymercuration-demercuration reactions followed by esterification of the resulting hemiacetal with methoxycarbonylmethylenetriphenylphosphorane in DME containing a trace of benzoic acid at 70 °C for 3 h gave unsaturated ester 215 in 96% yield. Its sequential hydrogenation followed by tetrahydropyranylation and desilylation formed an intermediate δ-lactone which on treatment with methanolic triethylamine at 23 °C for 2 h provided hydroxy ester 216 (99% yield). Tosylation of 216 gave 217 in 97% yield. Deprenylation of 217 and benzoylation of the resulting 218 (92% yield) gave 219. Acetonide deprotection followed by reaction of the corresponding 1,2-diol with 5 equiv. of methyl orthoacetate in DCM containing a trace of tosic acid (23 °C, 30 min) furnished 220 in 99% yield.
 | ||
| Scheme 38 | ||
The protected benzoate tosylate 220 was transformed into the cis-epoxy aldehyde 221 in 98% yield by the sequential (i) cis-epoxididation with K2CO3 in MeOH (2 h, 23 °C, 96% yield) (ii) conversion of the cyclic orthoacetate to mono acetate by exposure to wet DCM containing a trace of pyH+ TsO− (5 min, 23 °C, 98% yield) to (iii) deacetylation by K2CO3 in MeOH (1 h, 23 °C, 99% yield) to form 1,2-glycol and finally (iv) 1,2-glycol cleavage with 1.05 equiv. of Pb(OAc)4 in DCM containing finely powdered sodium carbonate (−45 °C, 5 min). Its chain extension followed by Wittig reaction of resulting dienal 222 with ylide 223 in THF-HMPA gave epoxy methyl ester 224 (methyl ester of the 5S,6R isomer of leukotriene A).42 It was converted to 6-epi-LTC 225 in two stages: (i) reaction with 2 equiv. of N-trifluoroacetylglutathione dimethyl ester and 3 equiv. of Et3N in a minimum quantity of methanol at 23 °C for 3 h under Ar atmosphere and (ii) deprotection with K2CO3 in 4:1 dimethyoxyethane-water at 23 °C for 4 h to afford 225.
Under the identical reaction condition 6-epi-LTD 226 was prepared from 224 by using N-trifluoroacetylcysteinylglycine methyl ester, and the deprotection step was reported to perform at 23 °C for 18 h (Scheme 38).
Schlosser and coworkers described the synthesis of threo-furanoid glycal 63 (Scheme 11), erythro furanoid glycal 54a (Scheme 10)20a and utilized them as key intermediates for synthesis of erythro-(2S,3R)-sphingosine 232 (Scheme 39) and threo-(2S,3S)-sphingosine 238 (Scheme 40).21
 | ||
| Scheme 39 | ||
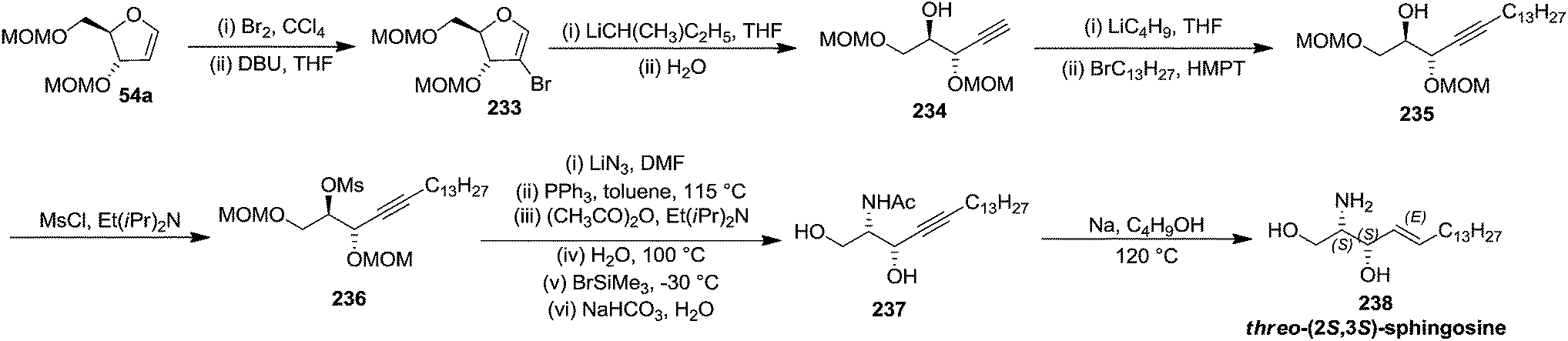 | ||
| Scheme 40 | ||
In 1999, Theodorakis et al. described a short, efficient, and enantioselective synthesis of Norrisane side chain from furanoid glycal 139 as key intermediate (Scheme 23).31 Furanoid glycal 139 was converted into cyclopropanated ester 239 by treatment with ethyl diazoacetate (0.1 M in DCM) and Rh2(OAc)4 at 25 °C which on treatment with dilute ethanolic solution of sulfuric acid afforded 240. After its oxidative cleavage in the presence of NaIO4, the resulting aldehyde was methylated to produce 241 in 63% combined yield. Its Swern oxidation produced ketone 242 in 79% yield, which on MeSO3H mediated cyclization at 0 °C furnished bicycle 243 as a single isomer in 67% yield. Its Baeyer–Villiger oxidation in presence of urea-hydrogen peroxide and trifluoroacetic anhydride yielded 244 in 69% yield (Scheme 41).
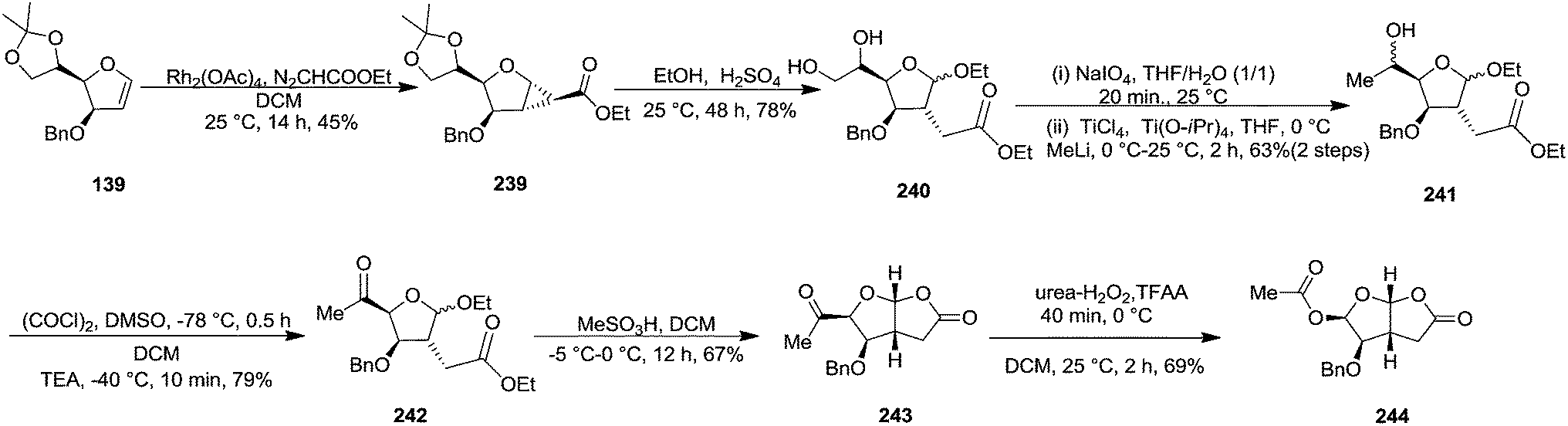 | ||
| Scheme 41 | ||
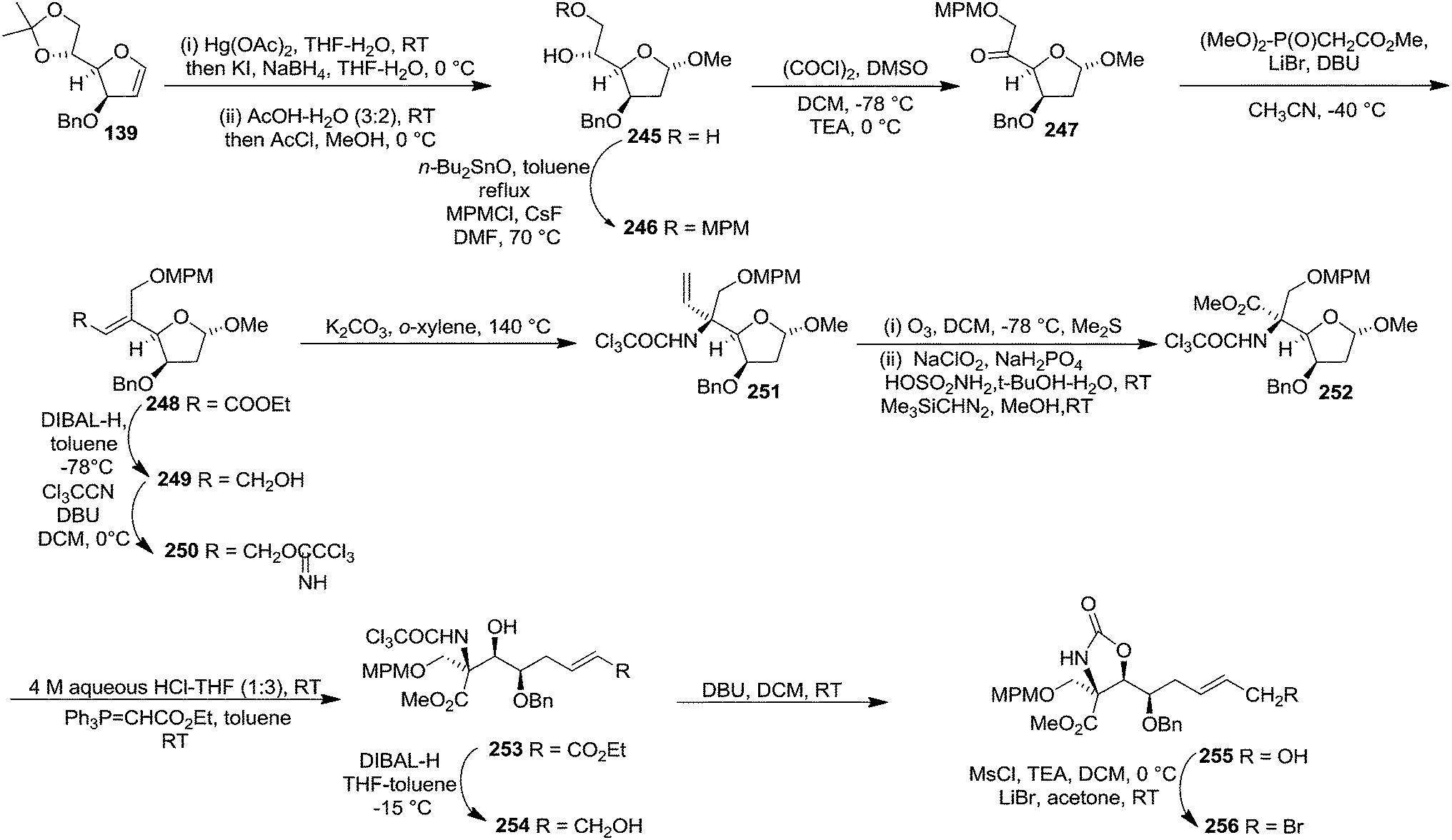
In 2001, Chida and co-worker also utilized furanoid glycal 139 as key intermediate for synthesis of (+)-myriocine 262 (Schemes 42 and 43).37 α-Methyl furanoside 245 was obtained from furanoid glycal 139 in 81% yield by oxymercuration-reduction followed by acid treatment. The primary hydroxyl group in 245 was selectively p-methoxybenzylated to afford 246 (95% yield), which on Swern oxidation generated ketone 247. Its Horner–Emmons reaction provided an inseparable mixture of (E)-alkene 248 and its (Z)-isomer (15:1) in 90% yield. DIBAL-H reduction of the mixture afforded column purified (E)-allyl alcohol 249 and its (Z)-isomer in 93% and 6% isolated yields respectively. The allyl alcohol 249 was converted into trichloroacetimidate 250 which, without further purification, was subjected to Overman rearrangement to yield an inseparable mixture of rearranged products 251 and its epimer in a ratio of 7:1 in 90% yield from 249. Ozonolysis of 251 followed by oxidation and esterification in succession afforded 252 (82% yield). Its acid hydrolysis provided an anomeric mixture of lactol which was then treated with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et to give (E)-alkene 253 in 71% yield. Its DIBAL-H reduction in THF-toluene at 215 °C afforded allyl alcohol 254 (75% yield), which on treatment with DBU in DCM was transformed into cyclic carbamate 255 in 86% yield. The primary hydroxyl group in 255 was converted into corresponding allyl bromide 256 in 92% yield (Scheme 42).
CHCO2Et to give (E)-alkene 253 in 71% yield. Its DIBAL-H reduction in THF-toluene at 215 °C afforded allyl alcohol 254 (75% yield), which on treatment with DBU in DCM was transformed into cyclic carbamate 255 in 86% yield. The primary hydroxyl group in 255 was converted into corresponding allyl bromide 256 in 92% yield (Scheme 42).
 | ||
| Scheme 42 | ||
 | ||
| Scheme 43 | ||
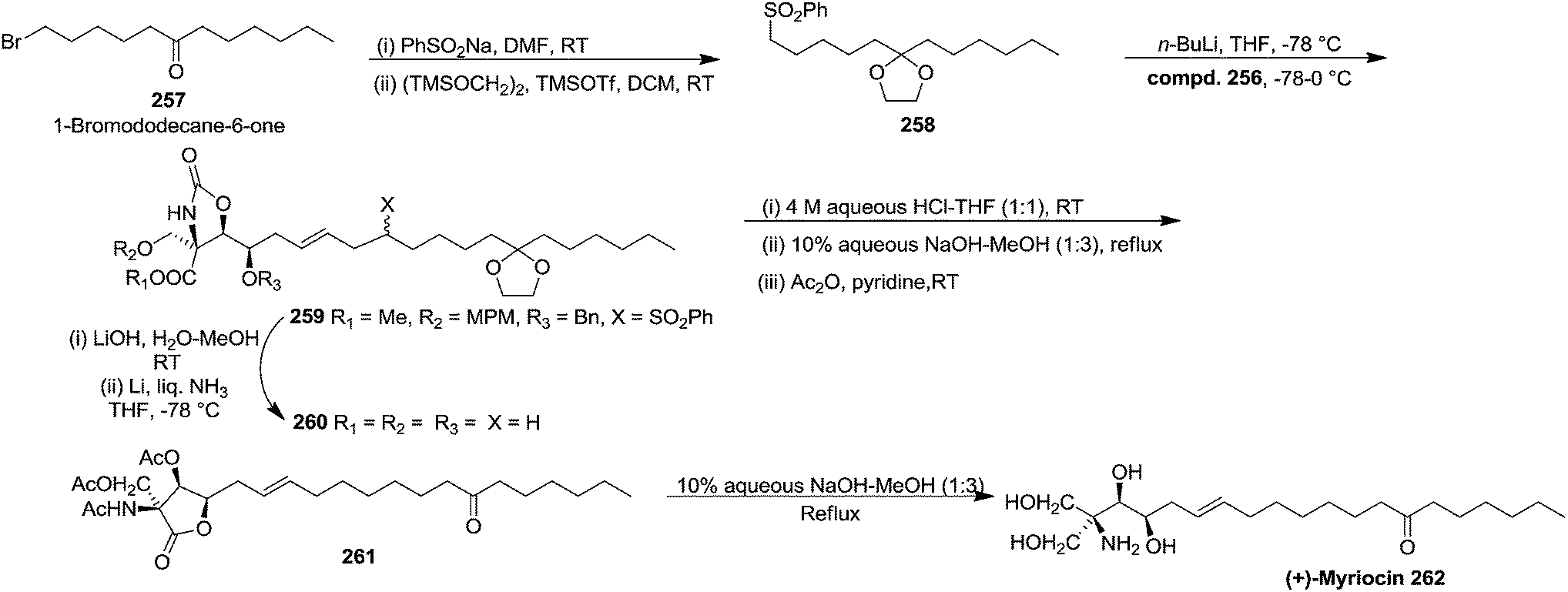
The hydrophobic part of myriocin, sulfone 258, was prepared by treatment of 1-bromododecan-6-one 257 with PhSO2Na, followed by ketalization (82% yield) (Scheme 43). Sulfone 258 on treatment with nBuLi, and allyl bromide 256 afforded the coupling product 259 in 80% yield. Saponification of 259 and subsequent Birch reduction gave crude carboxylic acid 260. Removal of the ketal group and carbamate function in 260 followed by conventional acetylation provided 261. Finally, saponification of 261 followed by neutralization with weak acidic resin (Amberlite IRC-76, H+ form) furnished (+)-myriocin 262 in 82% yield.
In 2007, Chandrasekharan and coworker developed a methodology for the construction of fused perhydrofuro[2,3-b]pyran/furan by using NIS mediated ring opening and cyclization of 1,2-cyclopropanated sugar derivatives, derived from pyranoid and furanoid glycals. They successfully applied this methodology to the synthesis of fused perhydrofuro[2,3-b]pyrano/furano-γ-butyrolactone derivatives using sugar derived 1,2-cyclopropane carboxylic acids.43
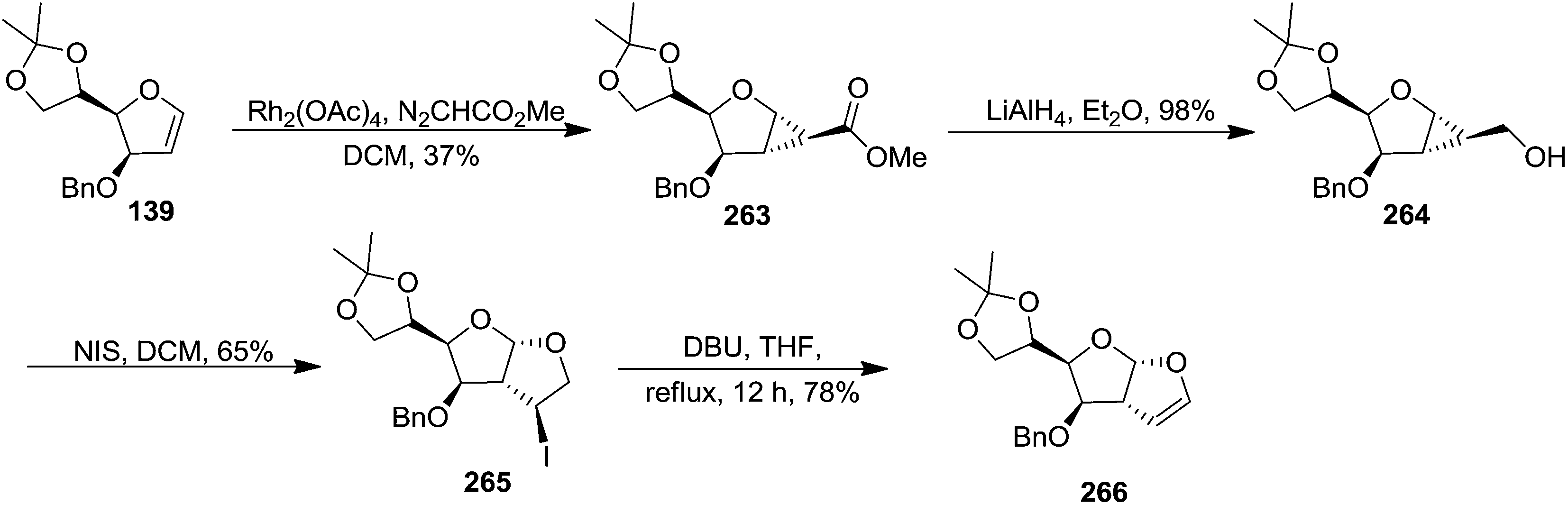
The furanoid glycal 139 on treatment with methyl diazoacetate in the presence of Rh2(OAc)4 gave the cyclopropanated ester 263 as a major product, which on LAH reduction delivered alcohol 264 in 98% yield. It was subjected to NIS mediated ring opening and cyclization reaction to furnish perhydrofuro[2,3-b]furan derivative 265 in 65% yield as a mixture of diastereomers (9:1). The mixture was subjected to dehydrohalogenation with DBU (THF, reflux, 12 h) to obtain the corresponding furofuryl glycal 266 in 78% yield (Scheme 44).
 | ||
| Scheme 44 | ||
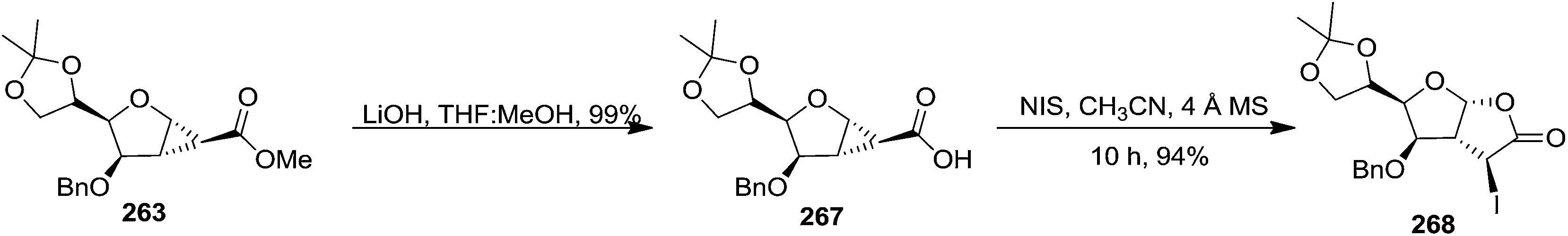
The hydrolysis of the 1,2-cyclopropane carboxylate 263 under the basic medium produced 1,2-cyclopropane carboxylic acid 267 (Scheme 45). Its cyclopropane ring opening with NIS in presence of CH3CN and 4 Å MS for 10 h furnished the corresponding 3-iodoperhydrofuro[2,3-b]furano-γ-butyrolactone derivative 268 in excellent yield (83%) (Scheme 45).
 | ||
| Scheme 45 | ||
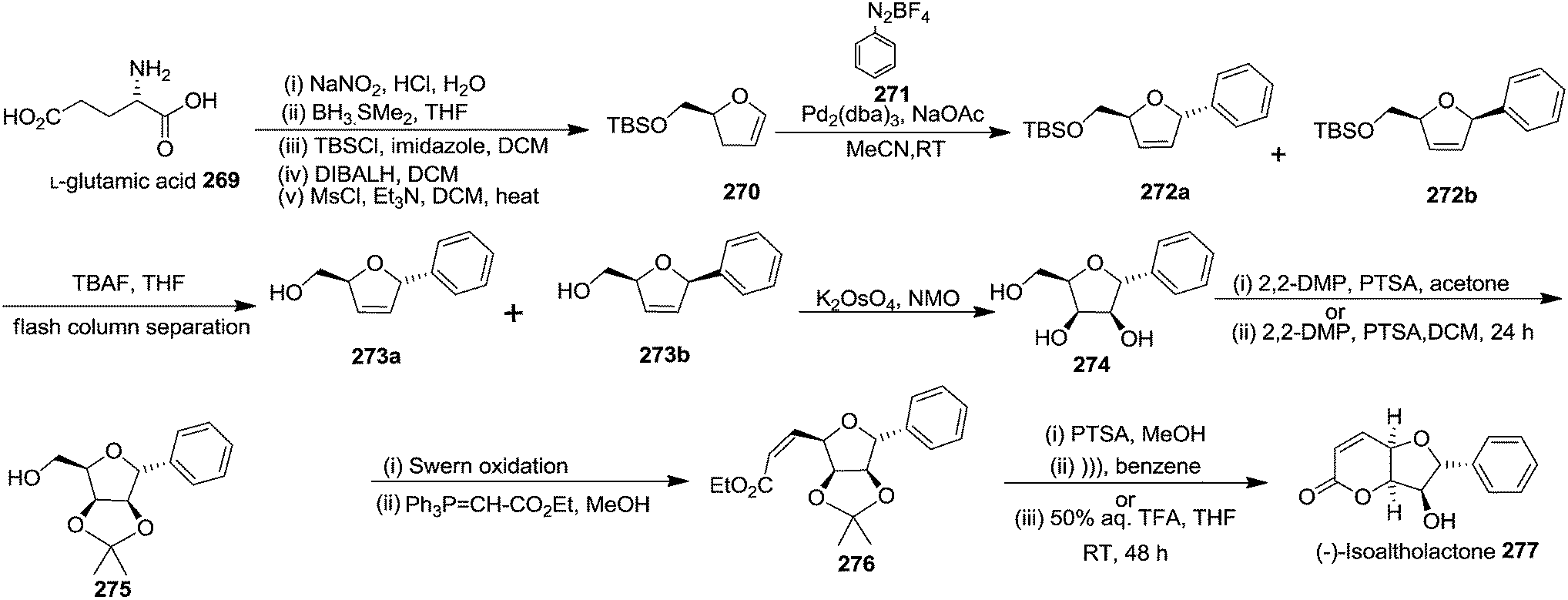
In 2007, Correia and co-workers achieved the synthesis of (−)-isoaltholactone 277 in seven steps with an overall yield of ∼25% from L-glutamic acid 269 derived furanoid glycal 270 (ref. 44) by utilizing Heck–Matsuda arylation with benzenediazonium tetrafluoroborates as the key step.45 After performing several trial and error experiments, they achieved the best condition for Heck–Matsuda arylation of enolether 270 with benzenediazonium tetrafluoroborates 271 in the presence of 4 mol% of Pd2(dba)3, to afford the phenyldihydrofurans 272a and 272b in 90% yield as a 94:06 inseparable diasteromeric mixture. Though the diastereomeric mixture was inseparable by column chromatography, the desilylated Heck adducts were separated. The silyl ether deprotection of 272 followed by column purification resulted 273a and 273b. Treatment of 273a with potassium osmate and N-methylmorpholine N-oxide (NMO) afforded the triol 274 which, without further purification, was treated with 2,2-dimethoxypropane and PTSA to give acetonide 275 in good yields (Scheme 46). Swern oxidation of free hydroxyl group of 275 gave an unstable aldehyde which was immediately subjected to a Wittig olefination with ethoxycarbonylmethylene phosphorane in methanol to furnish the cis-enoate 276 in 75% yield (over 2 steps). It was then treated with catalytic PTSA in methanol followed by sonication to obtain (−)-isoaltholactone 277 in 70% yield (2 steps). However, when cis-enoate 276 was treated with an aqueous solution of trifluoroacetic acid for 48 h at room temperature, the (−)-isoaltholactone 277 was obtained in 80% yield (Scheme 46).
 | ||
| Scheme 46 | ||
After reporting an efficient protocol for the synthesis of stereochemically pure four different furanoid glycals (162a–c, 139) (Fig. 3) from our laboratory, in the year 2011, we were further interested to demonstrate the synthetic utility of these furanoid glycals. In this endeavour, we identified and synthesized four aggregation pheromones brevicomins (285a–d), styryllactones (+)-cardiobutanolide 290a, (−)-cardiobutanolide 290b and (+)-goniofufurone 295a from the above mentioned furanoid glycals (162a–c, 139).2h
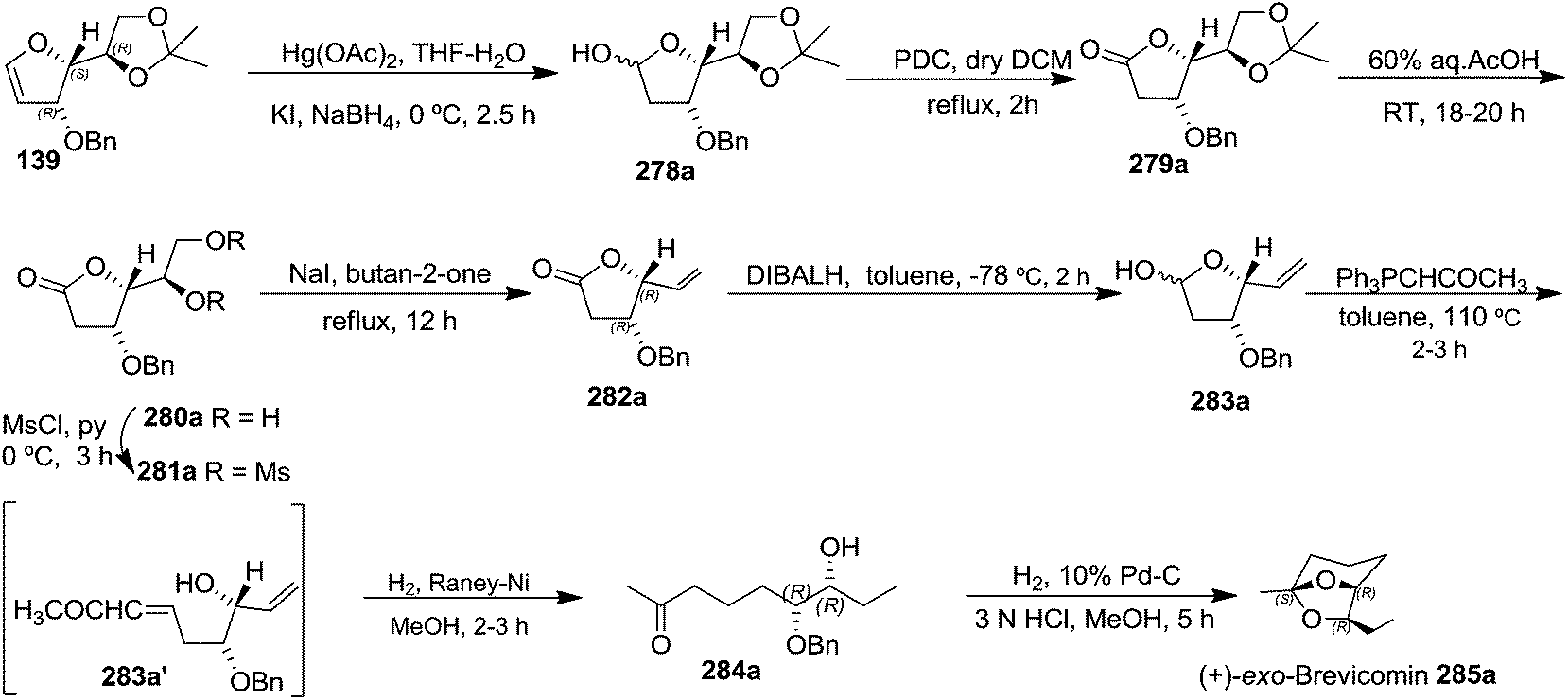
The total synthesis of (+)-exo-brevicomin 285a was initiated from furanoid glycal 139 (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-D-arabino-hex-1-enitol) (Scheme 47), which was converted into 278a by oxymercuration–demercuration sequence in 98% yield. The anomeric OH was oxidized with PDC in dry DCM at refluxing temperature for 2 h to obtain lactone 279a as a white solid in 75% yield. Deprotection of the acetonide in 279a was carried out with 60% aqueous AcOH at room temperature for 18–20 h to give diol 280a as a white solid, which was without further purification, mesylated with MsCl in pyridine at 0 °C for 3 h to afford dimesyl derivative 281a. The reductive elimination of diester 281a with NaI in butan-2-one at reflux temperature for 12 h yielded vinylbutyrolactone derivative 282a in 71% yield for three steps. Its reduction with DIBALH at −78 °C in dry toluene yielded lactol 283a in 86% yield. Its Wittig olefination with Ph3PCHCOCH3 in dry toluene followed by RANEY® hydrogenation of the resulting product 283a' with two double bonds afforded column purified ketone 284a in 52% yield in two steps. Finally, the simultaneous hydrogenolysis of OBn in 284a in presence of Pd/C in MeOH and intramolecular acetalization with a trace of 3 N HCl delivered the target (+)-exo-brevicomin 285a in 44% yield (Scheme 47).
 | ||
| Scheme 47 | ||
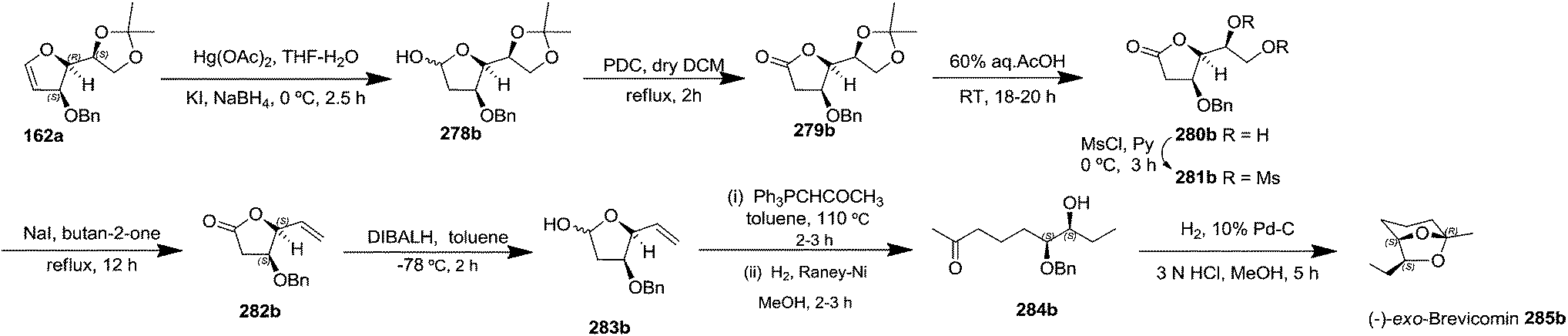
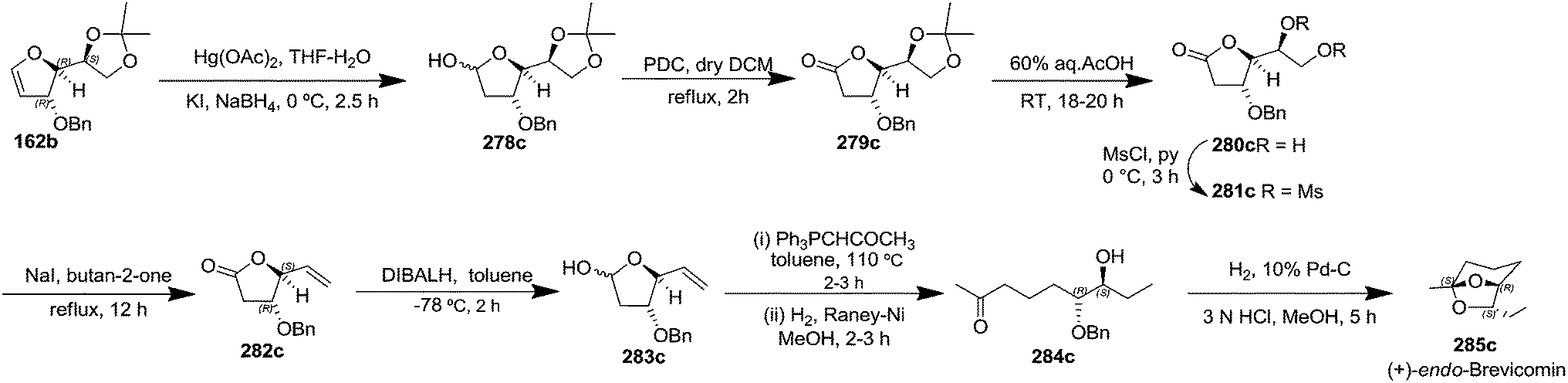
After having completed the total synthesis of (+)-exo-brevicomin 285a from furanoid glycal 139, the similar reaction sequence was successfully followed for the synthesis of (−)-exo-brevicomin 285b from 162a (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-L-arabino-hex-1-enitol) (Scheme 48), (+)-endo-brevicomin 285c from 162b (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-L-ribo-hex-1-enitol) (Scheme 49) and (−)-endo-brevicomin 285d from 162c (1,4-anhydro-2-deoxy-5,6-O-isopropylidene-3-O-benzyl-D-ribo-hex-1-enitol) (Scheme 50).
 | ||
| Scheme 48 | ||
 | ||
| Scheme 49 | ||
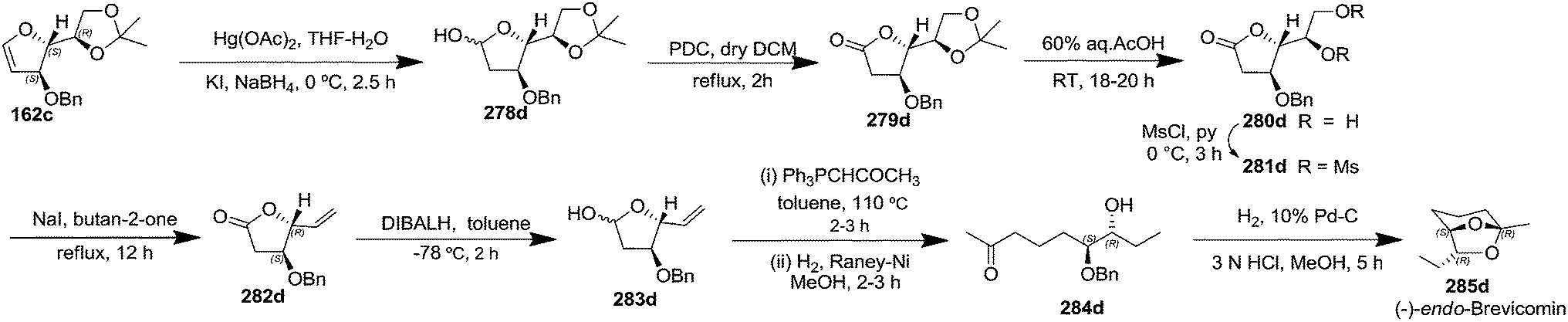 | ||
| Scheme 50 | ||
The key intermediates 282a and 282b (Schemes 47 and 48) were utilized for the synthesis of styryllactones (+)-cardiobutanolide 290a, (−)-cardiobutanolide 290b. The olefin cross metathesis reaction between 282a and (S)-1-phenyl-2-propene-1-ol with Grubb's IInd generation catalyst (2.8 mol%) in refluxing DCM furnished allylic alcohol 286a in 74% yield. It was silylated with TBSCl in dry DCM at 0 °C to afford silyl ether 287a in 93% yield. Its asymmetric dihydroxylation with AD-mix-β in 1:1 tBuOH:H2O afforded 288a in 67% yield which on silyl ether deprotection with amberlyst 15 resin in dry acetonitrile produced 289a in 94% yield. Finally, its O-benzyl deprotection by Pd(OH)2 catalyzed hydrogenolysis in dry MeOH furnished the desired natural product (+)-cardiobutanolide 290a in 67% yield (Scheme 51).
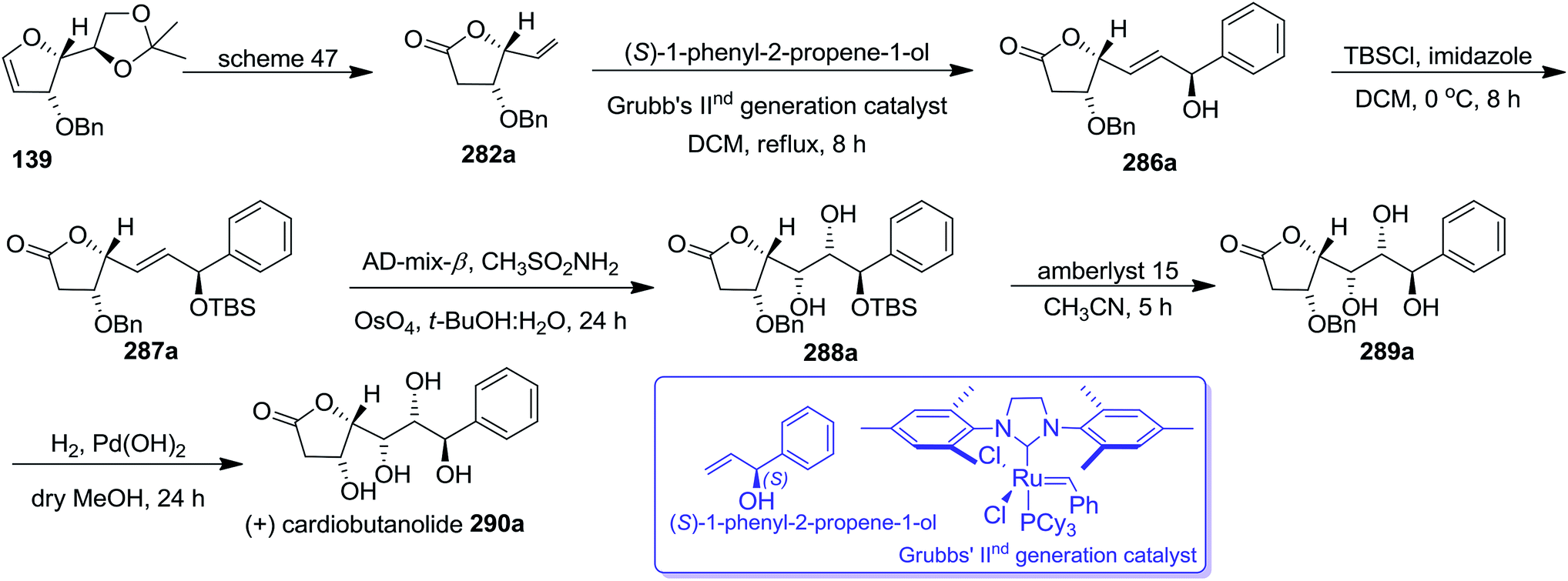 | ||
| Scheme 51 | ||
Similarly, the (−)-cardiobutanolide 290b was synthesized from 162a, an optical antipode of 139 by adopting the reaction sequence similar to that employed for the synthesis of its enantiomer 290a. The olefin cross metathesis reaction between compound 282b derived from 162a (Scheme 48) and (R)-1-phenyl-2-propene-1-ol with Grubb's IInd generation catalyst (2.8 mol%) furnished allylic alcohol 286b in 74% yield. Its silylated derivative 287b on asymmetric dihydroxylation with AD-mix-α in 1:1 tBuOH:H2O afforded 288b. After having 288b in hand, the remaining two synthetic steps similar to that employed for the synthesis of (+)-cardiobutanolide 290a (vide supra) were followed to complete the synthesis of (−)-cardiobutanolide 290b (Scheme 52).
 | ||
| Scheme 52 | ||
The acetonide protection of two free OH in 288a (obtained from furanoid glycal 139, Scheme 51) afforded globally OH protected derivative 291a in 83% yield. Its hydrogenolysis in the presence of Pd(OH)2 in dry EtOAc gave O-benzyl deprotected derivative 292a in 87% yield which on mesylation with MsCl-Et3N at 0 °C in dry DCM for 2 h followed by elimination of MsOH under basic condition furnished the α,β-unsaturated lactone 293a in 92% yield. Its treatment with THF/AcOH/2 N HCl (1:1:1) at room temperature delivered the triol 294a in 56% yield. Finally, it was subjected to DBU catalyzed bicyclic ring formation by the participation of its 6-OH to furnish the title natural product (+)-goniofufurone 295a as a white solid in 64% yield (Scheme 53).
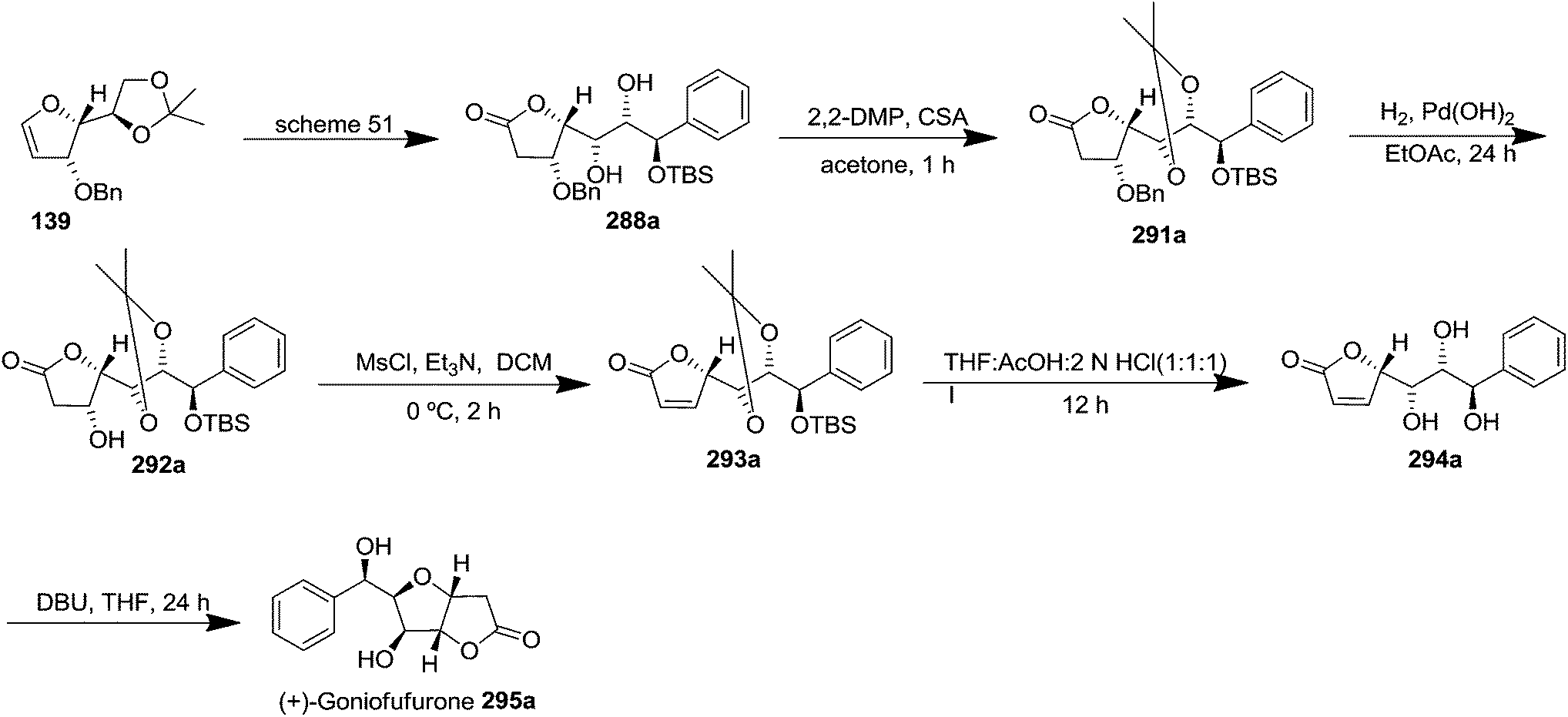 | ||
| Scheme 53 | ||
4. Some reactions of furanoid glycals
4.1. Peroxidation, osmylation, mercuration and bromination
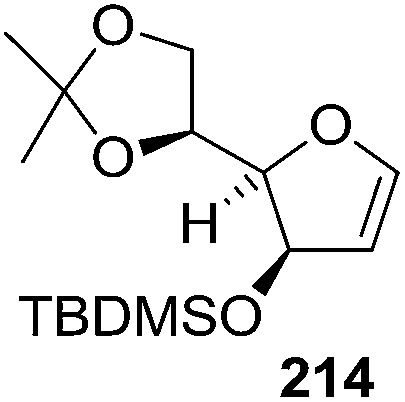
After describing the synthesis of furanoid glycal 24, 26 and 30 (Scheme 4),16a Bischofberger et al. in 1979 discussed some important reactions on furanoid glycals (1,4-anhydro-2-deoxy-3-O-(2,3:5,6-di-O-isopropylidene-α-D-mannofuranosyl)-5,6-O-isopropylidene-D-arabino-hex-1-enitol 24 and 1,4-anhydro-2-deoxy-5,6-O-isopropylidene-D-arabino-hex-1-enitol 26.46 Oxidation of 26 with m-CPBA in absolute ethanol gave hex-2-enofuranosides 296 (26% yield), and also two ethyl furanosides 297 (7% yield) and 298 (15% yield) by trans-ring opening of 1,2-epoxide intermediates. Under the identical reaction condition, furanoid glycal 24 afforded β-D-glucoside 299 as major product in 62% yield. Here the oxidant preferentially attacked from the α-side to form 1,2-epoxide intermediate due to the β-C-3 bulky group (Scheme 54).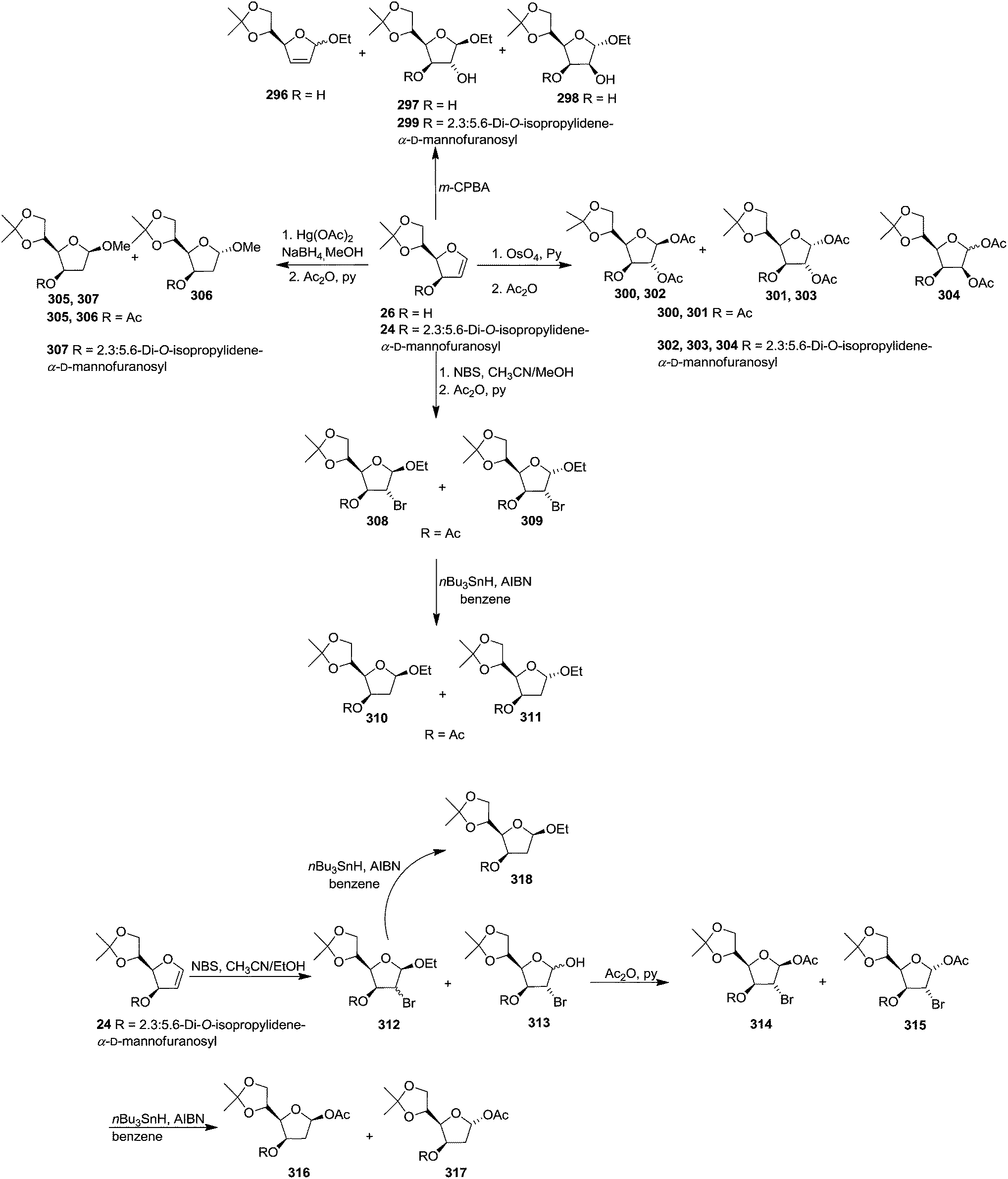 | ||
| Scheme 54 | ||
They further described that osmylation of 26 with OsO4 in pyridine, followed by cleavage of osmate ester and acetylation gave column purified β-D- and α-D-glucofuranoses, 300 (30% yield) and 301 (17% yield), respectively, indicating that attack by the oxidant took place mainly from the α-face. Similarly, osmylation reaction of the glycal 24, followed the same reaction sequence to form isolated anomeric gluco-furanoses 302 (14% yield) and 303 (26% yield) and a mixture of manno-furanoses 304 (gluco:manno, 7:1).
Methoxymercuration of 26 with Hg(OAc)2 in dry methanol, followed by demercuration with NaBH4 and acetylation yielded 2-deoxy derivatives 305 and 306 respectively. Methoxymercuration-demercuration of 24 furnished only one product 307 having the anomeric methoxy group in β-configuration.
Ethoxybromination of 26 with N-bromosuccinimide (NBS) in acetonitrile–ethanol, followed by acetylation produced mainly the ethyl-β-D-glucoside 308 (28% yield), with the α-D-glucoside 309 (3% yield) with other minor side products (6% yield). Debromination of 308 and 309 with nBu3SnH in the presence of AIBN in benzene afforded 2-deoxy derivatives 310 and 311 respectively. Ethoxybromination of 24 furnished a mixture of ethyl-2-bromo-2-deoxy-furanosides 312 (19% yield) and an anomeric mixture of a 2-bromo-2-deoxy-furanose 313. Acetylation of 313 gave separable anomeric acetates having β-D-gluco configuration 314 and α-D-gluco configuration 315 respectively, whose debromination formed 316 and 317 respectively and debromination of 312 yielded 318 (Scheme 54).
In 1987, Dax et al. demonstrated the reaction of acetyl hypofluorite (CH3CO2F) with pyranoid and furanoid glycals and they observed that more stereospecific reactions took place with furanoid glycals. Treatment of 1,4-anhydro-2-deoxy-5,6-O-isopropylidene-D-arabino-hex-1-enitol16,19 26 or 1,4-anhydro-2-deoxy-5-O-methoxymethyl-D-erythro-pent-1-enitol4 45 with gaseous acetyl hypofluorite in DCM-hexane at room temperature gave a complex mixture of compounds. Among them 1-O-acetyl-2-deoxy-2-fluoro-5,6-O-isopropylidene-β-D-mannofuranose 319 was obtained from 26 in 47% yield and 1-O-acetyl-2-deoxy-2-fluoro-5-O-methoxymethyl-α-D-ribofuranose 322 from 45 in 30% yield as major products (Scheme 55).47
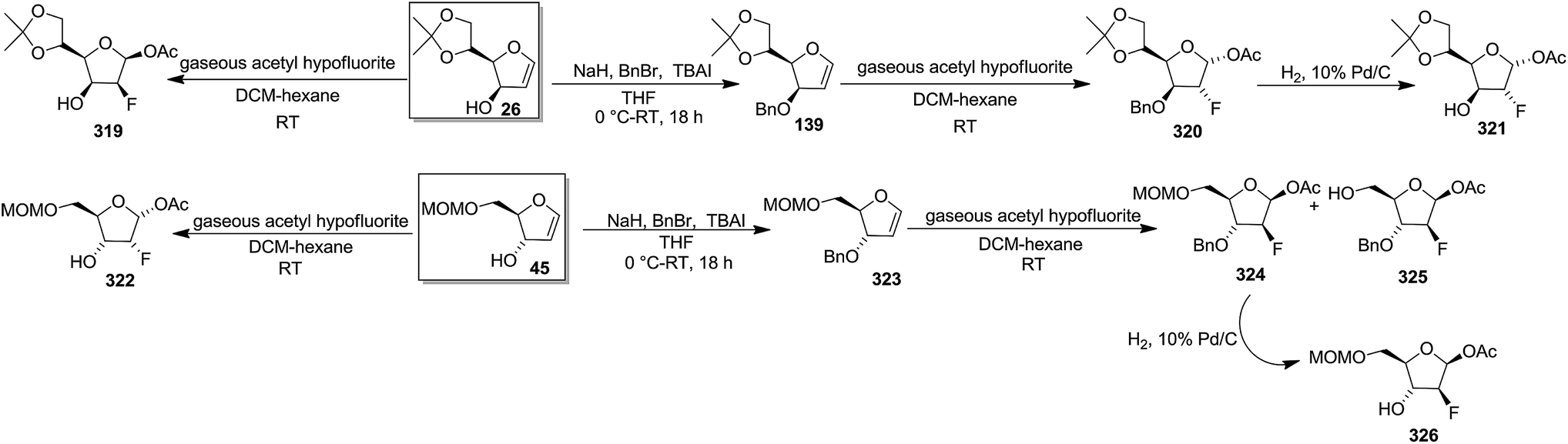 | ||
| Scheme 55 | ||
On the other hand, while 3-O-benzyl derivative 139 (ref. 31 and 35–37) (derived from glycal 26), on treatment with gaseous acetyl hypofluorite led to the formation of only 1-O-acetyl-3-O-benzyl-2-deoxy-2-fluoro-5,6-O-isopropylidene-α-D-glucofuranose 320, and 323 from the glycal 45, under the identical reaction conditions formed two fluorinated products viz. 1-O-acetyl-3-O-benzyl-2-deoxy-2-fluoro-5-O-methoxymethyl-β-D-arabinofuranose 324 and its analogue 325. Compounds 320 and 324 were debenzylated to afford 1-O-acetyl-2-deoxy-2-fluoro-5,6-O-isopropylidene-α-D-glucofuranose 321 and 1-O-acetyl-2-deoxy-2-fluoro-5-O-methoxymethyl-β-D-arabinofuranose 326, respectively (Scheme 55). They concluded from this study that free 3-OH of 26 and 45 induced the exclusive syn addition of acetyl hypofluorite across the double bond to afford their respective compounds 319 and 322, whereas benzyloxy derivatives 139 and 323 caused attack from the less-hindered opposite face of the double bond to afford 320 and 324 respectively.
4.2. Cycloaddition reaction
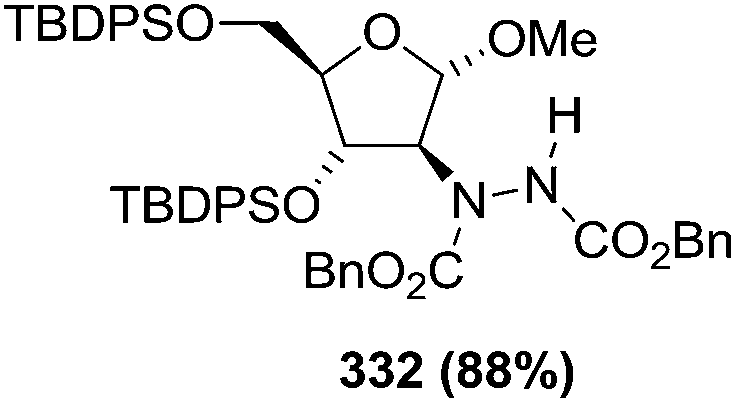
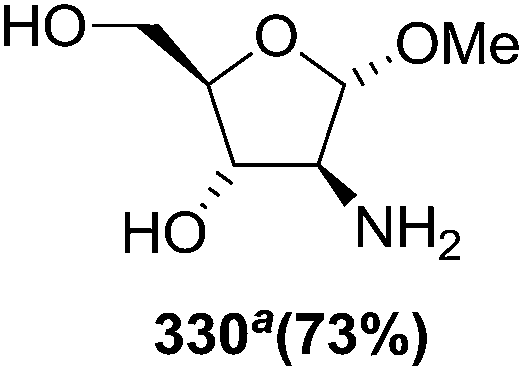
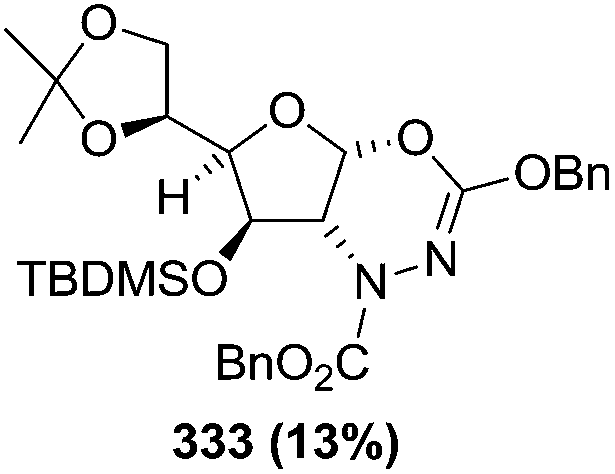
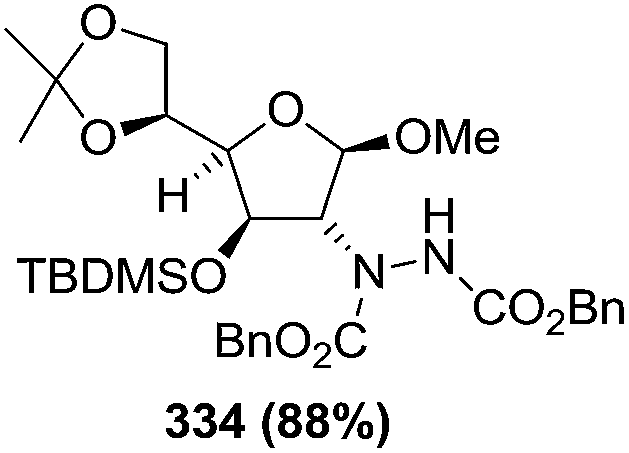
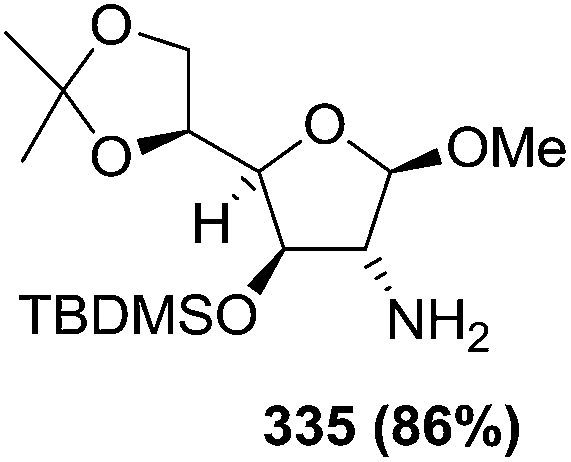
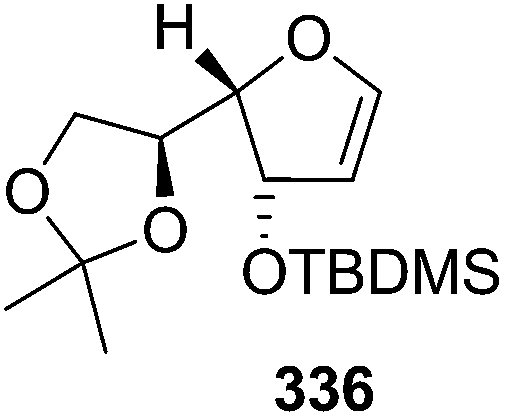
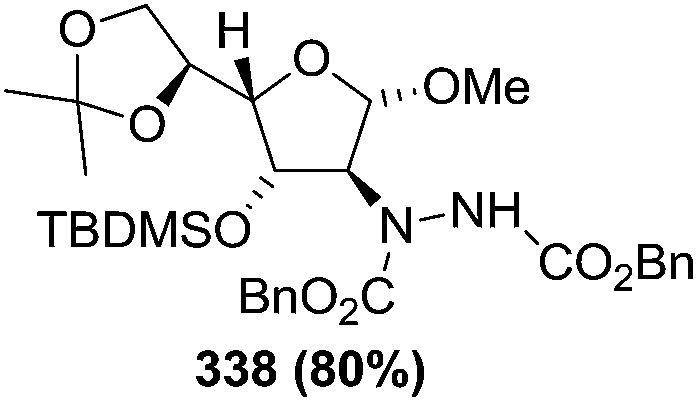
N–CO2Bn) on the appropriate glycals (Scheme 56, Table 7).48 Irradiation of glycals (327, 66f, 214 and 336) with dibenzyl azodicarboxylate (DBAD) in cyclohexane at 350 nm for 18 h gave single [4+2] cycloadduct (328, 331, 333 and 337) respectively. Treatment of these cyclo adducts with a catalytic amount of PTSA in MeOH led to opening at C-1 with inversion of stereochemistry to afford the corresponding methyl glycosides (329, 332, 334, 338) in quantitative yield. Hydrogenolysis of the protected hydrazines gave the 2-amino glycosides (330, 335, 339) in high yields (Scheme 56).
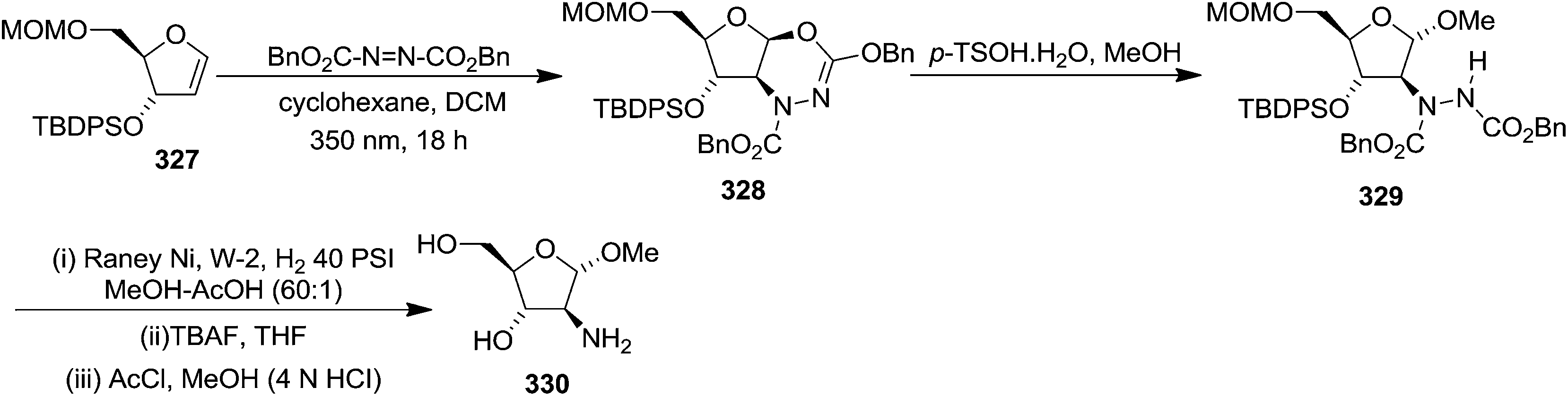 | ||
| Scheme 56 | ||
| Glycal | [4+2]adduct | Glycoside | Free amine |
|---|---|---|---|
| a The hydrogenolysis was performed on the free diol obtained by desilylation of compound 332 (n-Bu4NF 10 equiv, AcOH 3 equiv, THF, 90%). | |||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
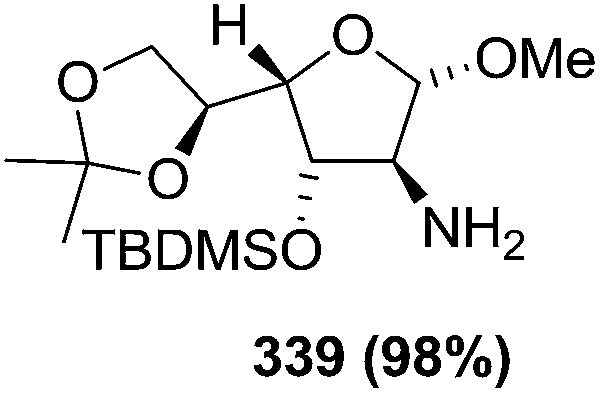 |
Since amino sugars present in natural products are usually in the pyranoside form, that's why they also showed the conversion of furanoside 342 into pyranoside 343 (Scheme 57).48 Later they reported similar reactions with several pyranoid glycals to obtain 2-amino pyranosides.49
 | ||
| Scheme 57 | ||
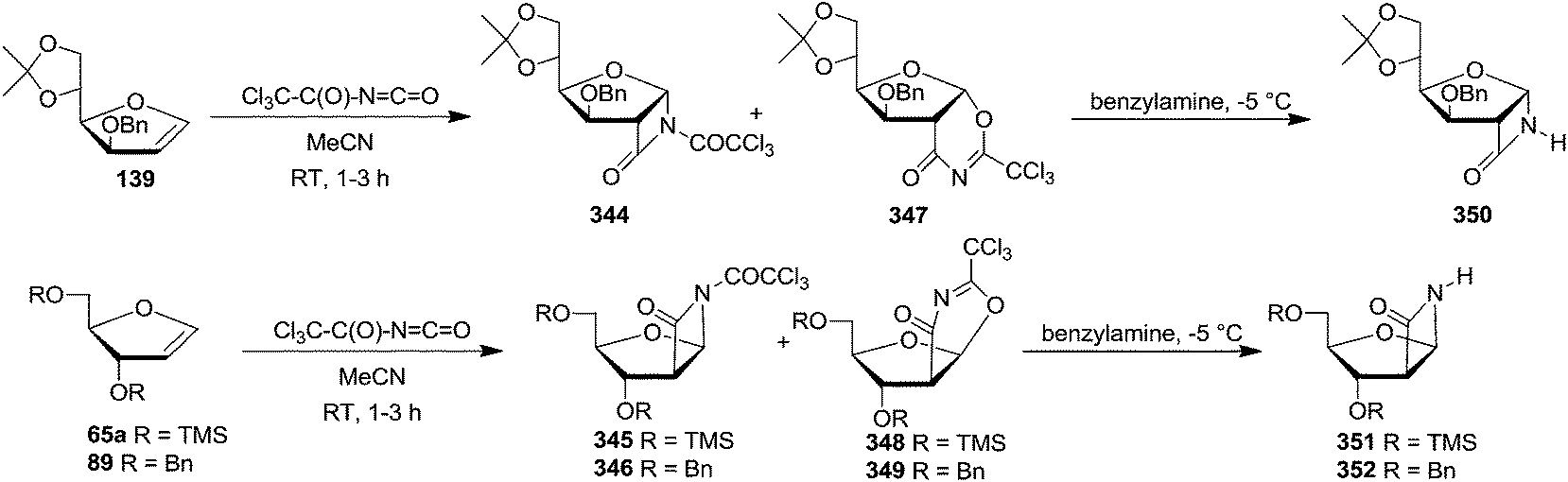
In 1993, Chmielewski et al. showed [2+2] cycloaddition of trichloroacetyl isocyanate to furanoid glycals. Here, the preferential attacked of the reagent to the substrate was governed by the stereochemistry of the C-3 substituent. They selected furanoid glycals 139, 65a and 89 which on treatment with trichloroacetyl isocyanate in acetonitrile at room temperature afforded a mixture of [2+2] cycloadducts (344–346) and [4+2] cycloadducts (347–349) in a 1:1 ratio in each case. Deprotection of the nitrogen atom in (344–346) produced stable bicyclic β-lactams (350–352). The α-D-gluco configuration for 350 and β-D-arabino configuration for 351 and 352 confirmed cycloaddition proceeded exclusively anti to the C-3 substituent (Scheme 58).50
 | ||
| Scheme 58 | ||
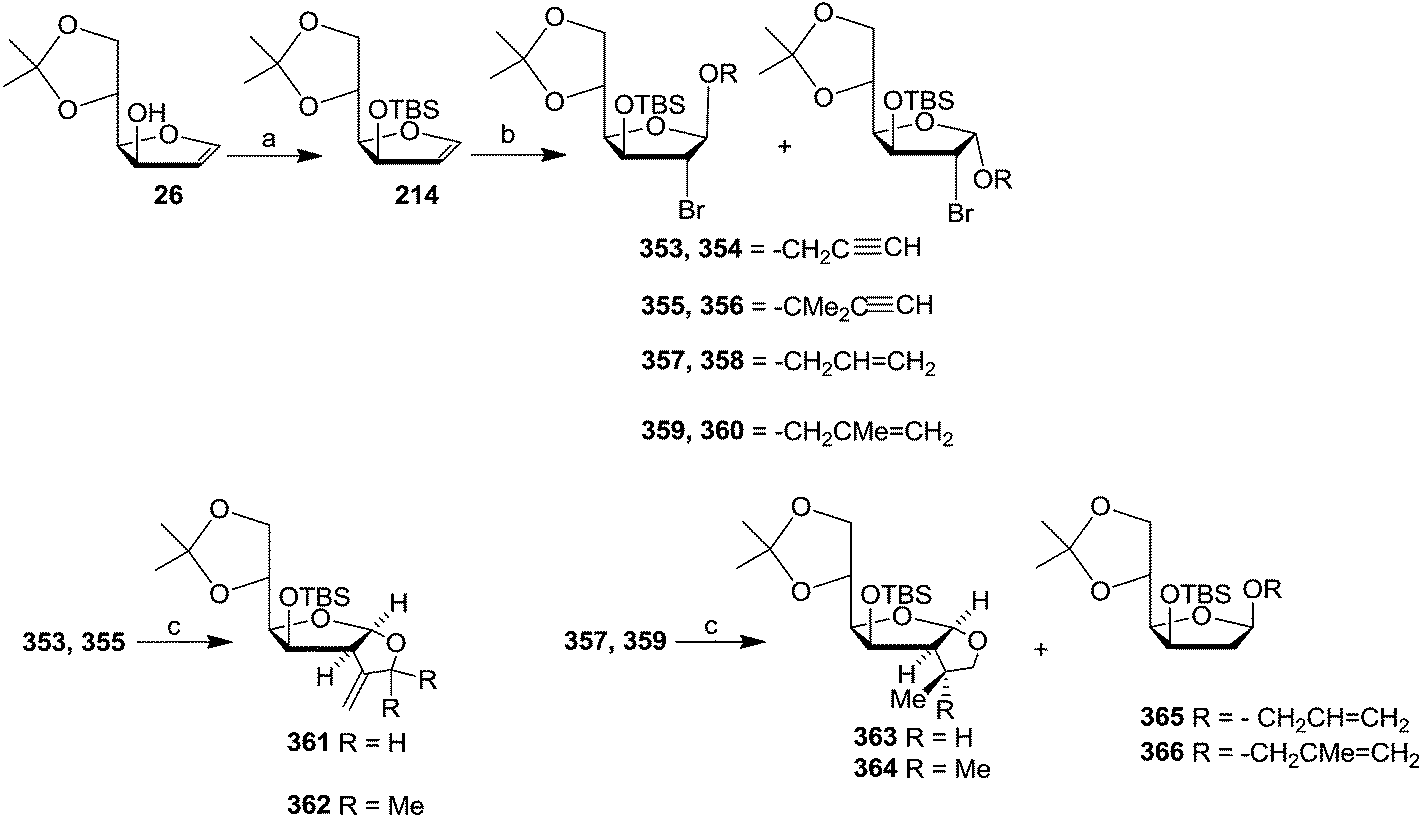 | ||
| Scheme 59 Reagents and conditions: (a) TBSCl, imidazole, DMF, 83%; (b) NBS, 10 equiv. 2-propyn-1-ol (for 353, 354), 2-methyl-3-butyn-2-ol (for 355, 356), 2-propen-1-ol (for 357, 358), and 2-methyl-2-propen-1-ol (for 359, 360) respectively; (c) nBu3SnCl, AIBN, NaBH3CN, tBuOH, reflux. | ||
4.3. Wittig rearrangement
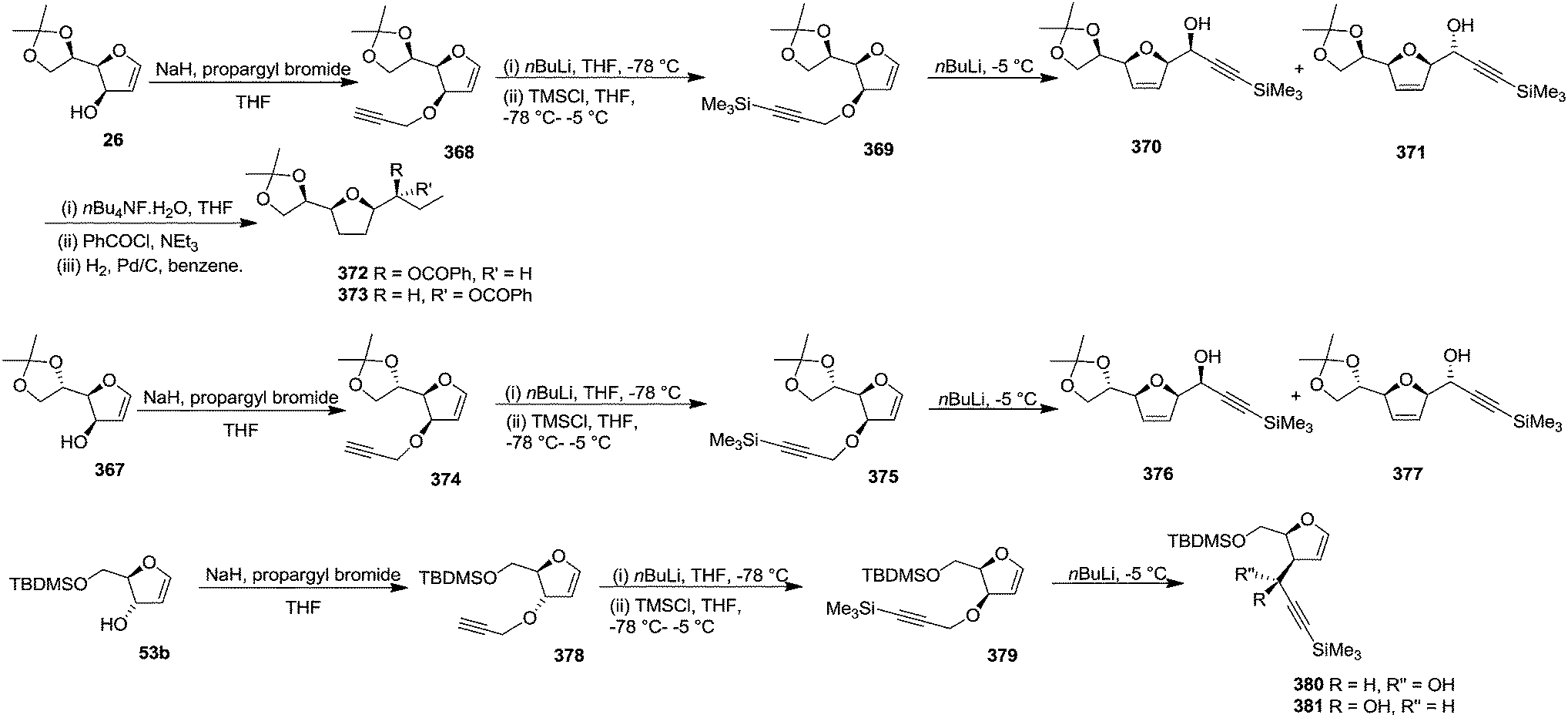
Gesson et al. studied51 the [2,3]-Wittig rearrangement of 369, 375, 379 derived from furanoid glycals 26, 367, 53b which were easily derived from D-mannose, L-gulonic-γ-lactone and D-ribonic-γ-lactone respectively.19,20a The C-3 free hydroxyl group of these furanoid glycals (26, 367, 53b) was alkylated with propargyl bromide in the presence of NaH in THF to form 368, 374, 378 which on treatment with nBuLi (1.1 eq.) in THF at −78 °C followed by addition of TMSCl (1.1 eq., −78 °C to −5 °C) afforded 369, 375, 379 respectively. These trimethylsilylpropargyl ethers of furanoid glycals 369, 375, 379 were used in a one-pot [2,3]-Wittig rearrangement by addition of a further 1.1 eq. of nBuLi at −5 °C. The glycal derivative 369, under the identical reaction condition produced easily separable mixture of 370 and 371 in a 7:3 ratio with 61% overall isolated yield from 26. The formation of the major isomer with erythro configuration was confirmed by converting 370 and 371 into 372 and 373 respectively through sequence of reactions involving desilylation, benzoylation followed by hydrogenation. The rearrangement of 375 afforded 376 and 377 with higher selectivity in 9:1 ratio but in lower overall yield (40%) from 367. Under the same conditions, 379 gave an inseparable mixture of 380 and 381 in a 2:1 ratio (Scheme 60).
 | ||
| Scheme 60 | ||
4.4. Reverse polarity strategy
Parker and Su, utilized “Reverse Polarity” strategy for the synthesis of C-aryl furanosides from furanoid glycals.52 They started with furanoid glycal 66e for the synthesis of C-1 aryl arabino-furanosides (385a–c). Lithiation of 66e with tBuLi and addition of the resulting reagent to quinone ketal 382 gave quinol ketal 383, which without further purification was treated with sodium dithionite to afford C-aryl glycal. But instead of that, quinol 384a, the hydrolysis product, was obtained in 53% yield. However, the reductive aromatization and anti-Markovnikov hydration was made possible by treating crude 383 with borane-THF followed by stirring with NaOH/H2O2 and acylation to obtain 2′-acetate 385a in an 40% overall yield from glycal 66e (Scheme 61).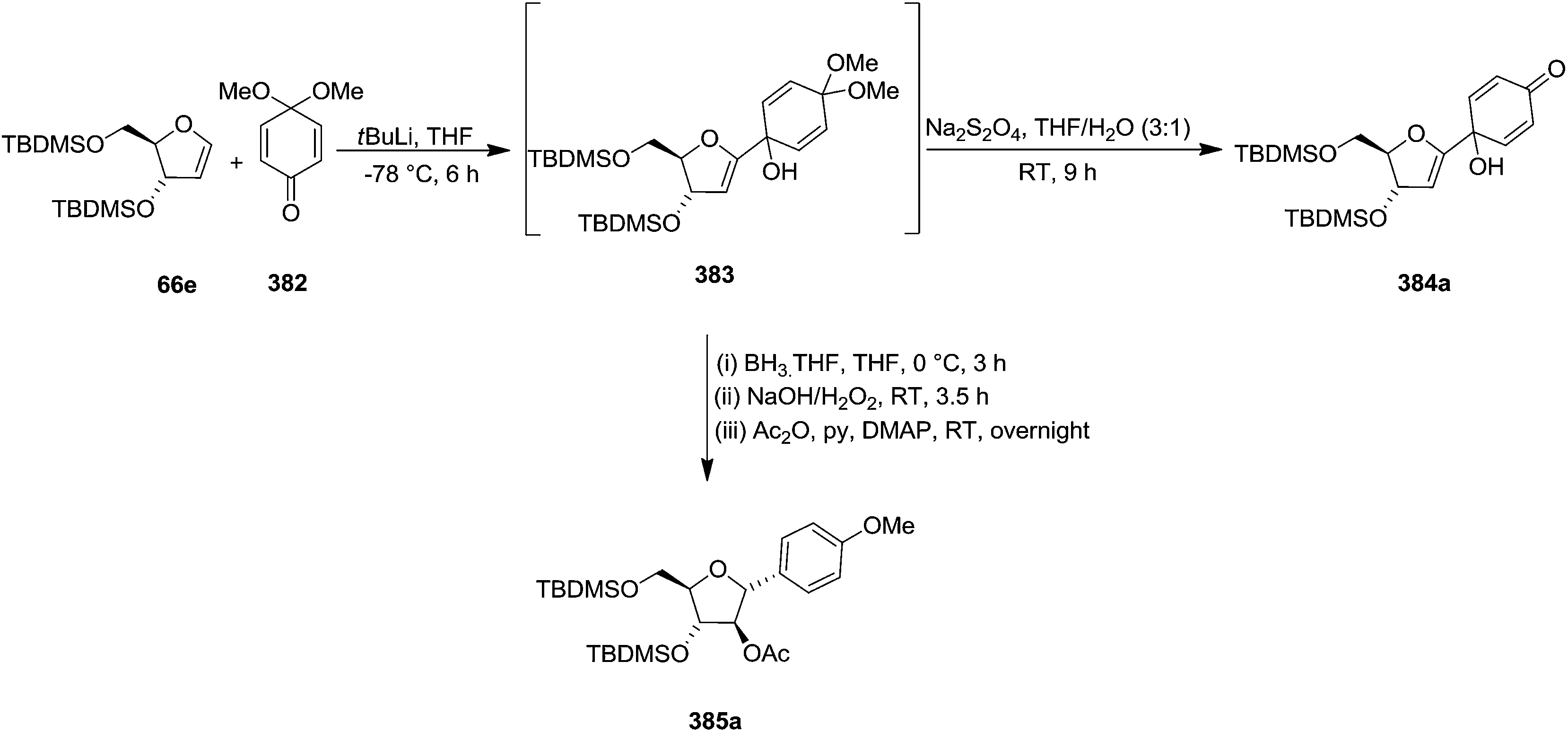 | ||
| Scheme 61 | ||
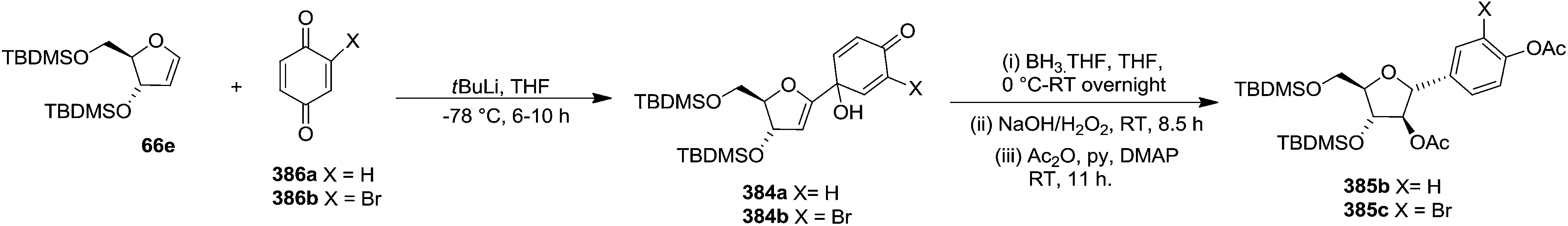
By utilizing same reaction sequence, they synthesized C-aryl furanosides 385b and 385c in good yields starting from furanoid glycal 66e, via intermediates (384a–b) (Scheme 62).
 | ||
| Scheme 62 | ||
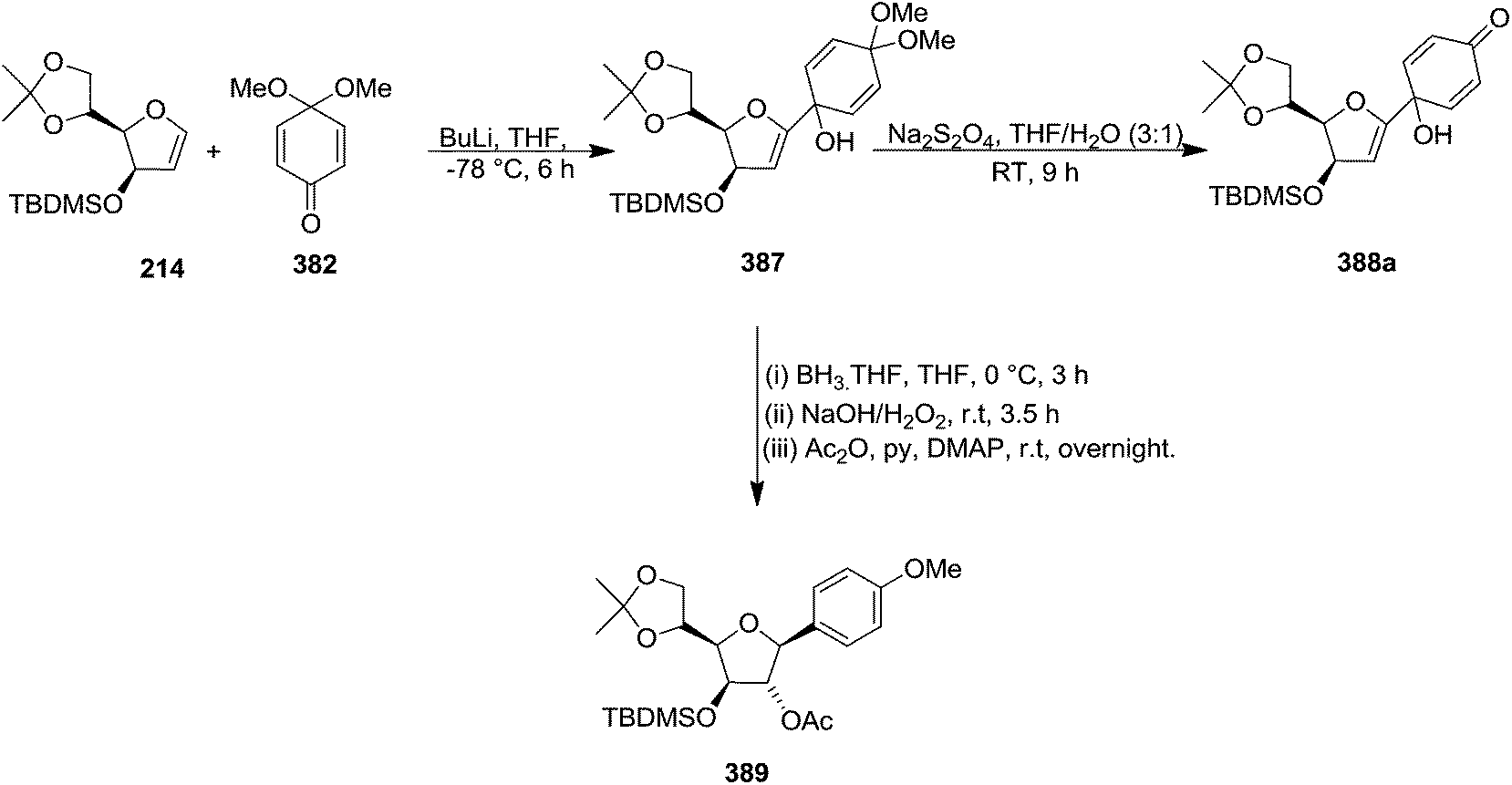
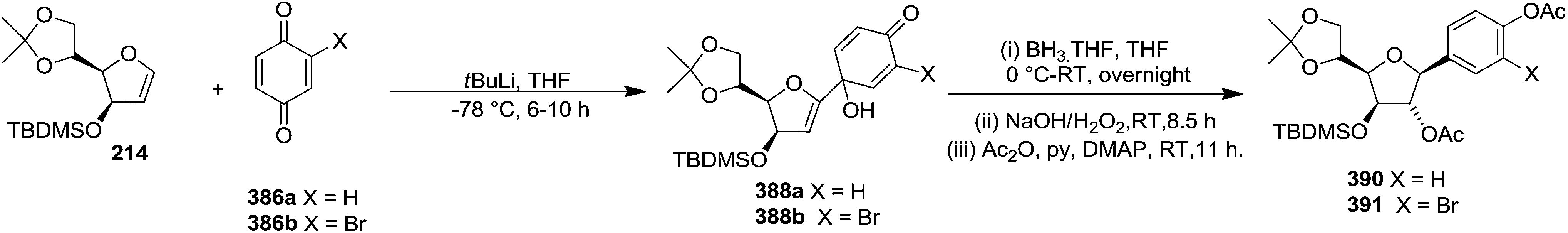
They have also reported the synthesis of C-1 aryl gluco-furanosides 389, 390, 391 in good yields from furanoid glycal 214, by utilizing same reaction sequence (Schemes 63 and 64).
 | ||
| Scheme 63 | ||
 | ||
| Scheme 64 | ||
4.5. Metal mediated amination
Carrier and group have synthesized stereoselectively 2-amino saccharides through metal-mediated amination of glycal substrates. They studied this reaction on differently protected pyranoid and furanoid glycals by utilizing (saltmen)Mn(N) and TFAA to transfer CF3CON unit to electron rich silyl enol ethers. The oxazoline 393 was isolated on treatment of furanoid glycal 392 with (saltmen)Mn(N) and TFAA, which, under mild acidic conditions furnished the N-protected amino alcohol 394. Its structure was confirmed by converting it back to the oxazoline 393 upon treatment with MsCl, Et3N, DCM (Scheme 65).53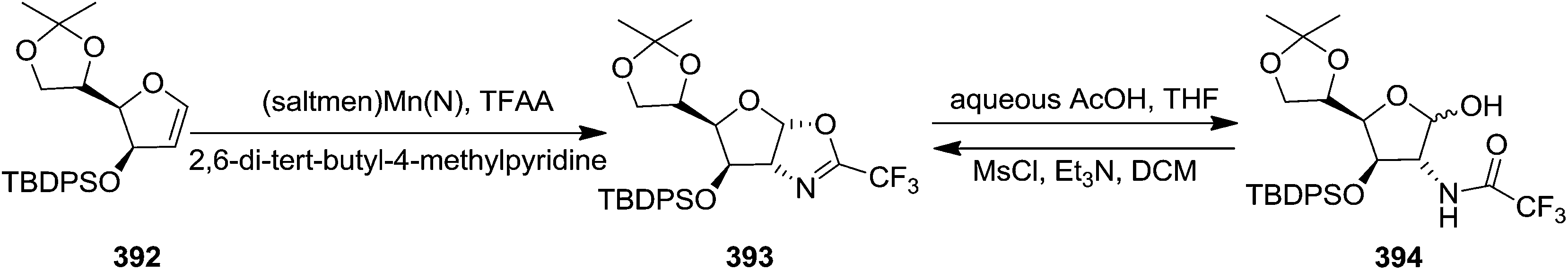 | ||
| Scheme 65 | ||
4.6. Ring expansion
In 2000 Totchtermann et al. reported the formation of heptano-bridged pyranosides by ring enlargement of the glycal rac-396,54 which was prepared from heptano bridge methyl furanoside rac-395 by treatment with BF3·Et2O in DCM in 95% isolated yield. Treatment of rac-396 with 50% aq. KOH in the presence of catalytic amounts of nBu4NBr in CHCl3 for 22 h at 0 °C afforded dichloro intermediate derivative rac-397, which was without further purification refluxed in anhydrous methanol with a large excess of K2CO3 to give chloro-2H-pyran rac-398 in 60% isolated yield over two steps. Oxidation of rac-398 with RuO4 yielded rac-399 in 67%. The stereoselective reduction of rac-399 with LTBAH provided the methyl pyranoside rac-400 in 71% isolated yield, which was transformed into corresponding acetate rac-401 in 90% yield (Scheme 66).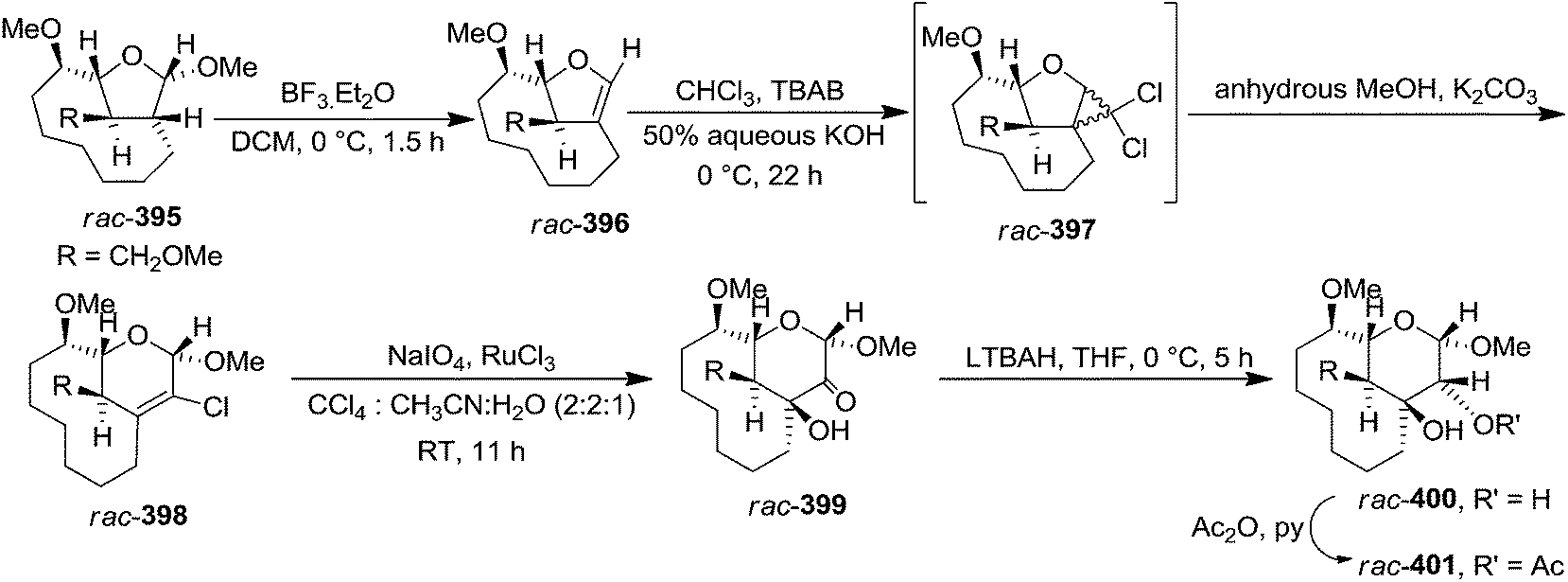 | ||
| Scheme 66 | ||
5. Synthesis of C-nucleosides
The synthesis of nucleoside analogues having modified sugar and/or nucleobases moieties has received much attention because of their general biological activities55 and potential use as antiviral56,57 and antineoplastic58 therapeutic agents. Over the last few decades, deoxynucleosides have attracted the attention of many research groups due to their antiviral and antitumor activities. They are also important components of antisense oligonucleotides.The 2′-deoxynucleosides such as 3′-azido-2′,3′-dideoxythymidine (AZT), 2′,3′-dideoxyinosine (ddI), 2′,3′-didehydro-2′,3′-dideoxythymidine (d4T) and related analogues have shown potent antiviral activity, particularly against human immunodeficiency virus (HIV) (Fig. 4), which is the causative agent for acquired immune deficiency syndrome (AIDS).59
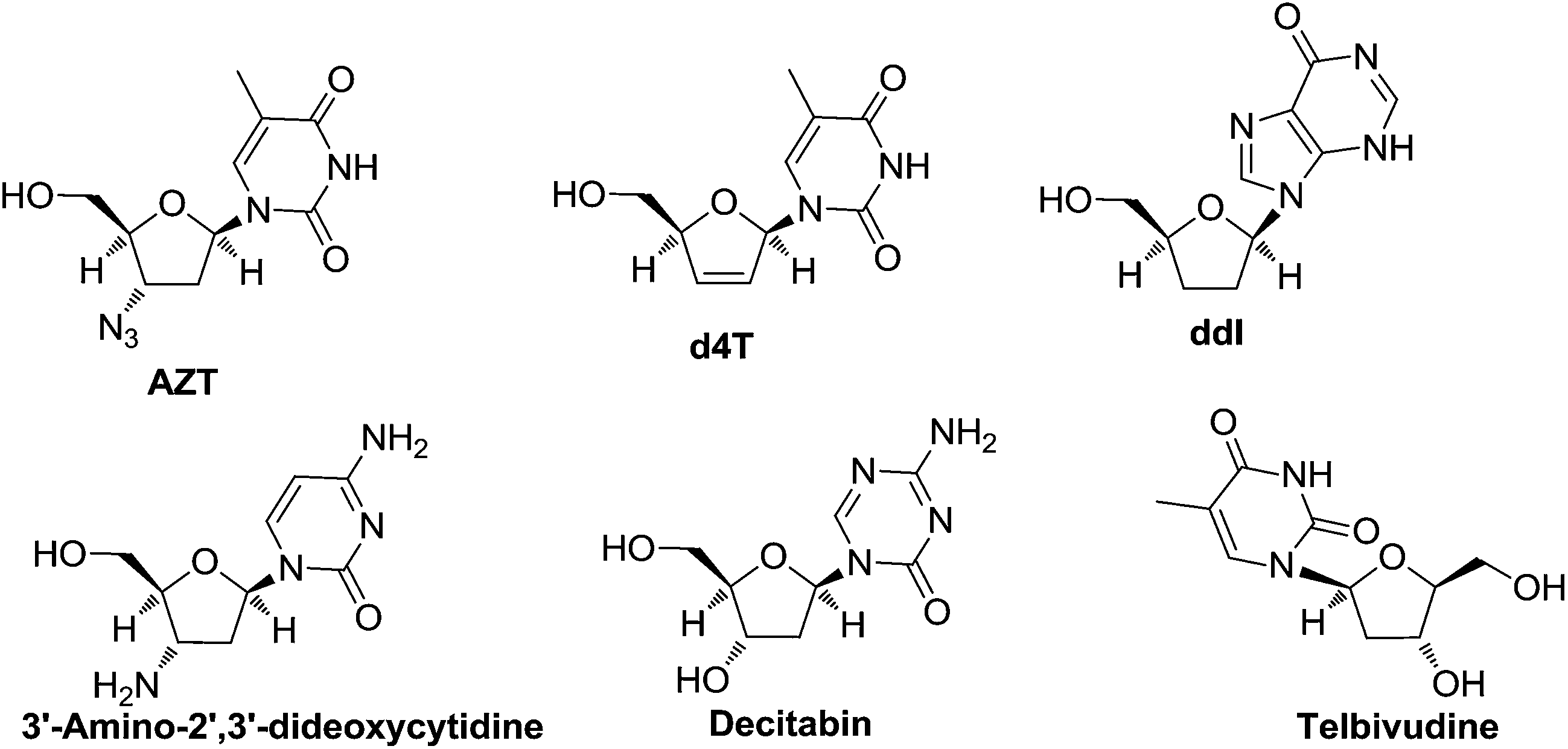 | ||
| Fig. 4 Structures of some 2′-deoxynucleosides have potent antiviral activity, particularly against human immunodeficiency virus (HIV). | ||
Nucleosides are generally considered to be compounds which contain a heterocyclic aglycon and a carbohydrate moiety that are joined together by a carbon–nitrogen bond. However, C-nucleosides differ from the more common nucleosides in that the sugar and heterocyclic aglycon are connected by a C–C, rather than a C–N, bond.60 From the last few years synthesis of C or N-nucleosides and nucleoside analogues by utilizing furanoid glycals as the key intermediates has also received much attention in research. In this overview we have discussed as one of the important application of furanoid glycals for synthesis of C and N-nucleosides.
To synthesize different C-nucleosides Daves Jr and his group contributed a lot in the field of nucleoside chemistry by utilizing differently substituted furanoid glycal.
In 1983 Daves Jr and Hacksell showed Heck coupling reactions of furanoid glycals 404–407, 45, 54a, 54b with (1,3-dimethyl-2,4-dioxo-1,2,3,4-tetrahydrimidin-5-yl)mercuric acetate 402 in the presence of a stoichiometric quantity of Pd(OAc)2 resulted in regio- and stereospecific formation of α or β-C-nucleosides (408–414) through an initial transmetalation leading to an organopalladium reagent which subsequently adds to the olefinic double bond in a syn fashion to form adducts (409A–414A) (Scheme 67).6 These are reasonably stable and decomposed to α-or β-C-nucleosides (409–414) via syn elimination of hydropalladium. The more stable adduct 412A among them on treatment with hydrogen for 8 h was resulted in 2′-deoxy C-nucleoside 412B. In this report, they demonstrated the direction of addition of organopalladium reagent to trans-substituted furanoid glycals (45, 54a, 54b) which was depended on the steric bulk of C3 and C4 substituents of the corresponding trans-furanoid glycals to form C-nucleosides (412–414). Organopalladium reagent always attacked from less sterically hindered site of the furanoid glycals. With cis-substituted glycals (405–407), the attack occured on the unsubstituted face of the ring to form C-nucleosides (409–411).
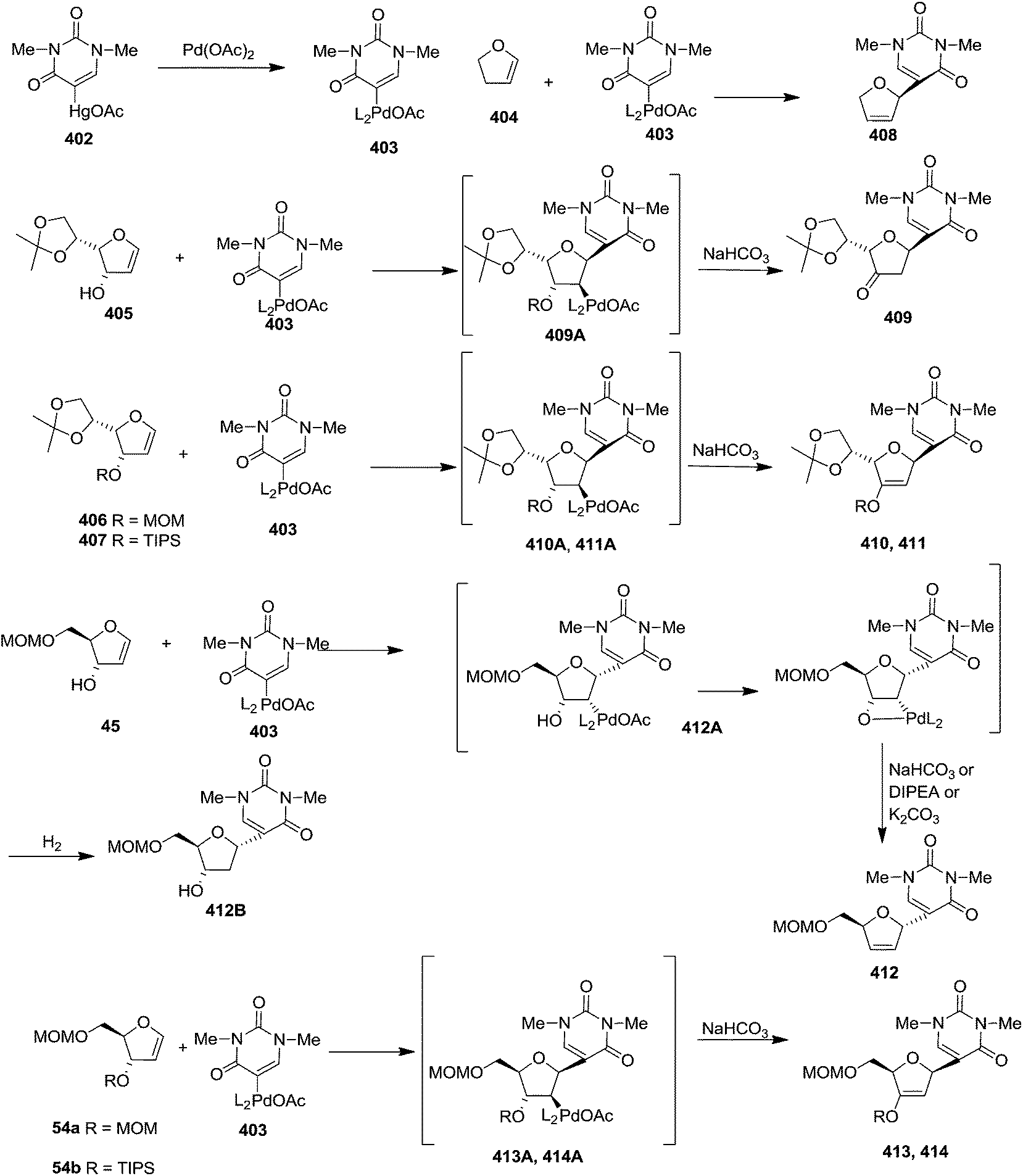 | ||
| Scheme 67 | ||
This research group after reported the synthesis of differently substituted furanoid glycals20a and α-or β-C-nucleosides, they further extended their study to show that when both the 3- and 5-hydroxyls are identically substituted e.g. 54a and 54f, the organopalladium reagent attacked from β-face to form 413 and 416 respectively. Thus, it indicated that the reaction was more sensitive to the steric bulk of the substituent at the allylic position (C3) than to that at position C5 of the furanoid glycal. Use of very bulky group at C3 for 54b, 54e also yielded β-C-nucleoside 414, 415 respectively. The furanoid glycals 56a and 56b whose 3-OH was substituted and 5-OH was free led only to β-C-nucleosides 417 and 418 respectively (Scheme 68).61
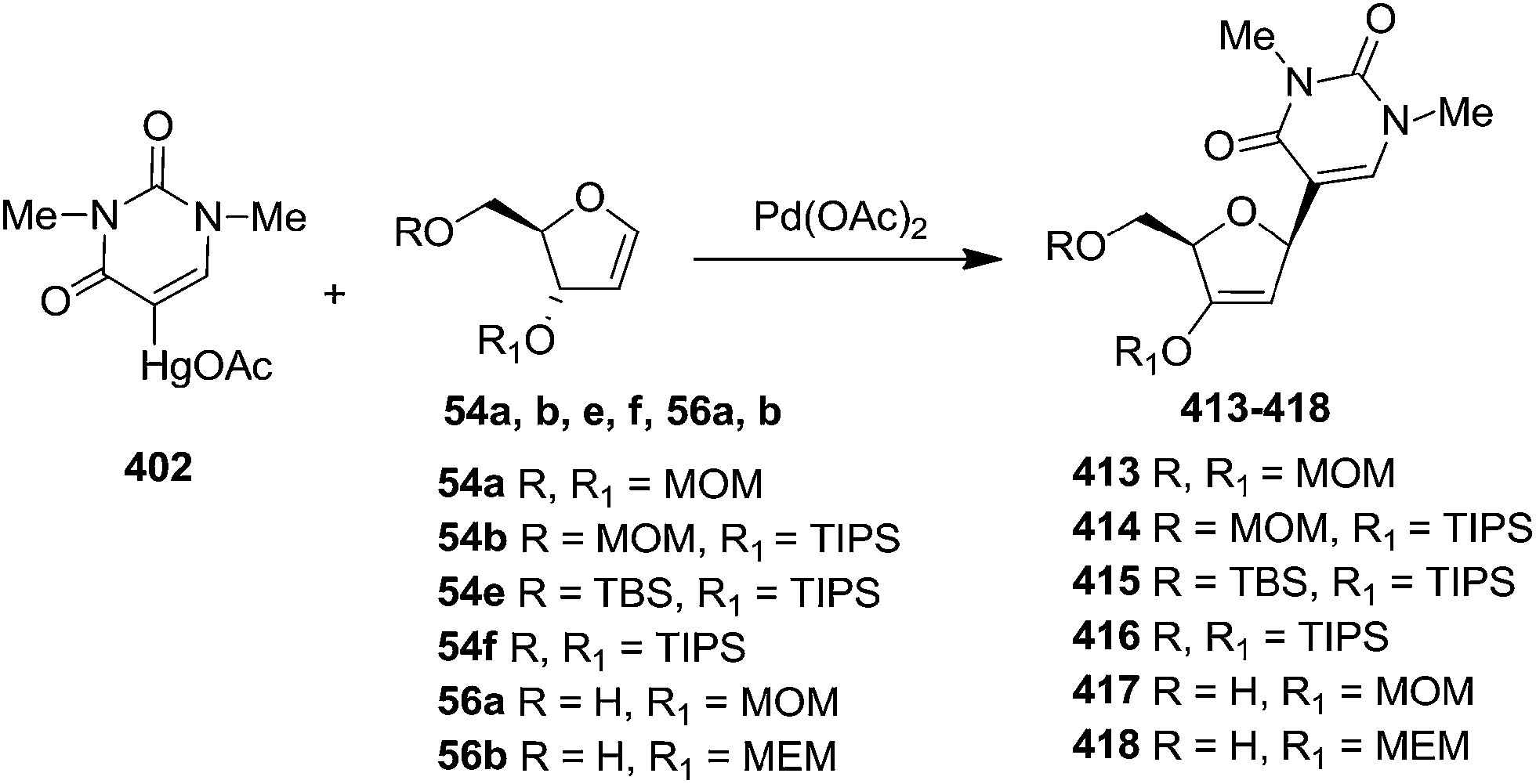 | ||
| Scheme 68 | ||
The methoxymethyl group of C-nucleoside of 413 was removed under acidic condition to form 419a and/or 420. O-trialkylsilyl β-C-nucleosides 414 and 416 on treatment with TBAF in the presence of AcOH in THF at −78 °C yielded 419a and 419b and/or 421 respectively. The removal of (β-methoxyethoxy)methyl group in 418 by using zinc bromide14 resulted in migration of the (β-methoxyethoxy)methyl group from the 3′-O to the 5′-O to form 419c along with 421. The borohydride reduction of (419a–c, 421) gave separable α-3′-hydroxyl (422a–d) and β-3′-hydroxyl derivatives (423a–d) (Scheme 69, Table 8).
 | ||
| Scheme 69 | ||
| Entry | 3′-Keto-C-nucleoside | R | Reducing agents | Temperature °C | Yielda% | 3′-OHα/3′-OHβb | Products |
|---|---|---|---|---|---|---|---|
| a Isolated yield.b Determined by high-pressure liquid chromatography. | |||||||
| 1 | 419a | MOM | NaBH4 | 0 °C | 73 | 1:3 |
422a/423a |
| 2 | 419b | TIPS | NaBH4 | 0 °C | 88 | 1:2 |
422b/423b |
| 3 | 419c | MEM | NaBH4 | −78 °C | 55 | 1:2 |
422c/423c |
| 4 | 421 | H | LiBH4 | −78 °C | 60 | 1:2 |
422d/423d |
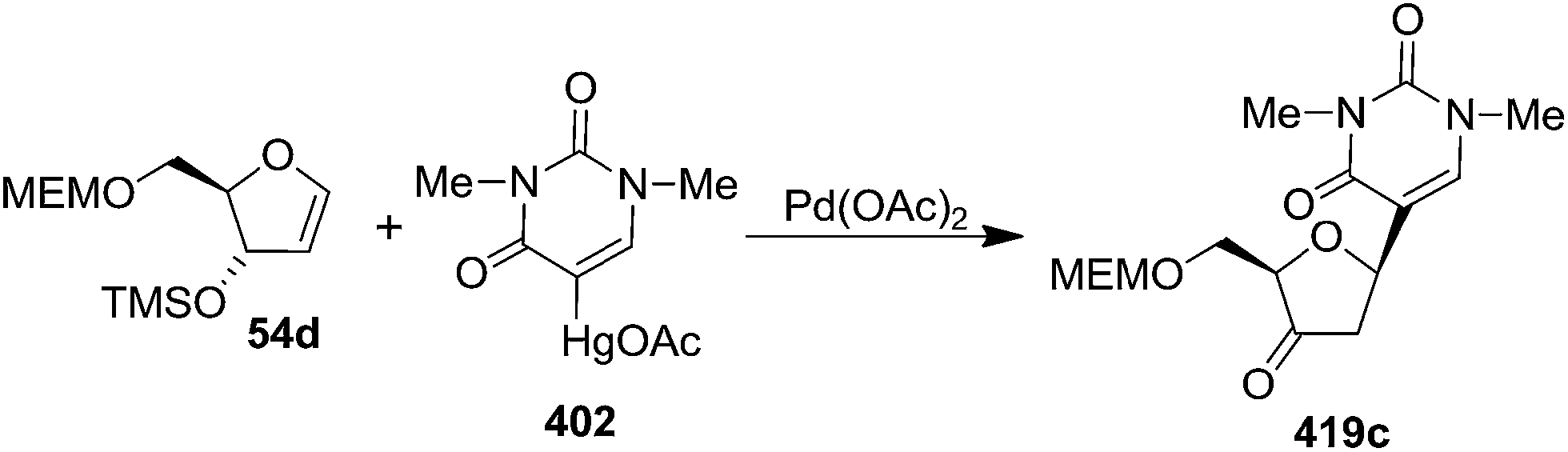
The furanoid glycal 54d on Pd-mediated coupling reaction with 402 yielded a 3′-keto-β-C-nucleoside 419c which indicated that the trimethylsilyloxy substituent effectively directed organopalladium adduct formation even though trimethylsilyl was lost during reaction (Scheme 70).
 | ||
| Scheme 70 | ||
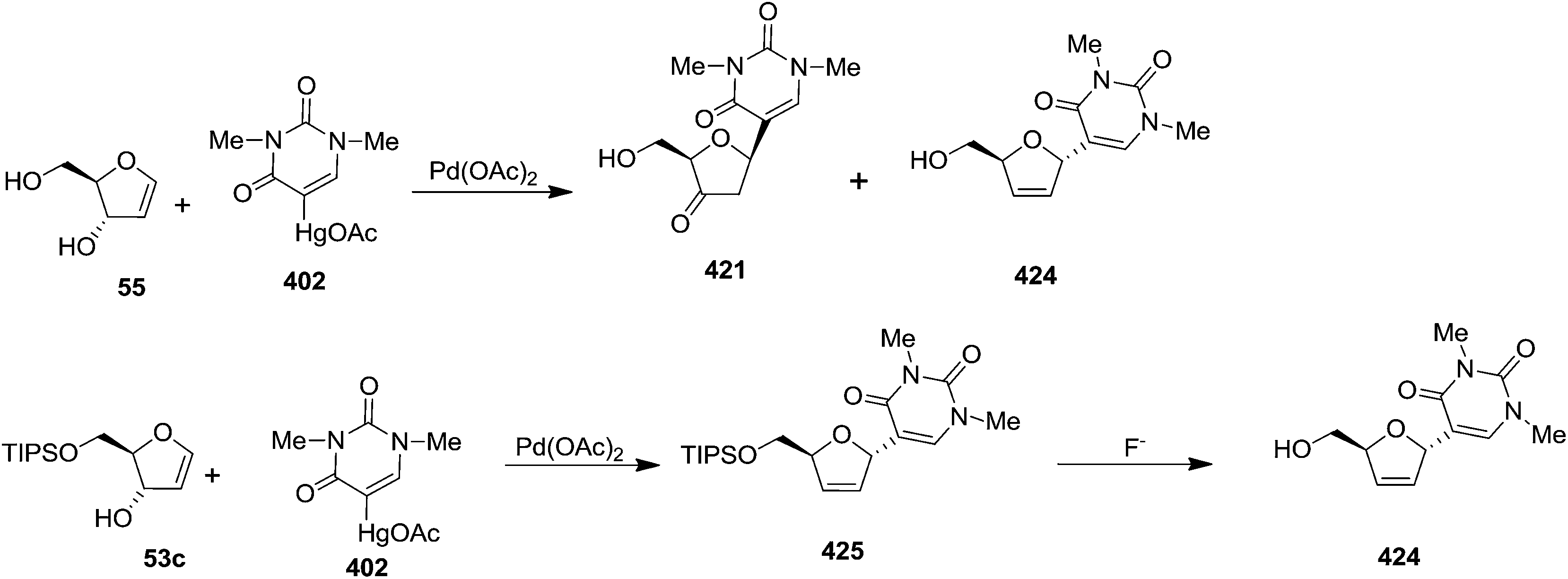
Coupling of 402 with 3,5-O-unsubstituted glycal (1,4-anhydro-2-deoxy-D-erythro-pent-1-enitol) 55 produced a mixture of the α- and β-C-nucleosides 421 and 424 in 45% and 29% yields, respectively. Coupling of 402 with 5-O-substituted glycal 53c gave α-C-nucleoside 424 as the sole product via formation of 425 followed by desilylation (Scheme 71).
 | ||
| Scheme 71 | ||
Daves Jr and Outten further utilized their developed methodology for coupling of furanoid glycals with 1-methoxy-4-(tri-n-butylstannyl)benzo[d]naphtho[1,2-b]pyran-6-one for synthesis of benzo[d]naphtho[1,2,-b]-pyran-6-one-C-glycosides related to antibiotics ravidomycin, gilvocarcins (toromycin), and chrysomycin A and B (virenomycin) for the very first time. 1-Methoxybenzo[d]-naphtho[1,2-b]pyran-6-one 426 was brominated at para position to –OMe with NBS in DMF at room temperature for 30 min to give 427 in 81% yield, which on treatment with hexa-n-butylditin (nBu3Sn)2 in the presence of 2.0 mol% of Pd(PPh3)4 in toluene (N2, 115 °C, 12 h) produced stannane 428 in 65% yield. Its coupling with furanoid glycal 54b in the presence of stoichiometric Pd(OAc)2 in CH3CN at room temperature for 24 h furnished C-glycoside 429 in 66% yield. Similar coupling of 428 with glycal 54f formed the corresponding C-glycoside 430 in a 28% yield. Silyl ether deprotection of 429 and 430 by nBu4NF in the presence of acetic acid in THF at room temperature for 2 h yielded the corresponding 3′-keto-C-glycosides 431 (94%) and 432 (92%) respectively. Reduction of the ketones 431 and 432 with NaBH4, H2O in tetrahydrofuran afforded the corresponding 2′-deoxy-C-glycosides 433 and 434 respectively (Scheme 72).62
 | ||
| Scheme 72 | ||
Daves Jr discussed coupling reaction of iodo derivatives of anthracycline aglycons with furanoid and pyranoid glycals in stoichiometric amounts in the presence of catalytic amounts of palladium(II) acetate and a tertiary amine in DMF at room temperature to get regio and stereospecific aryl C-glycosides.63 After several trial and error, they treated anthracycline iodo derivative 435 with furanoid glycal 54b in the presence of 10 mol% Pd(OAc)2, 2 equiv. of nBu3N, and 1 equiv. of NaOAc in DMF at room temperature for 48 h to furnish C-glycoside 436, which was in situ desilylated under the reaction condition to afford 3′-keto-β-C-glycoside 437 in 85% yield. This reaction was equally successful with tetra-n-butylammonium chloride and sodium bicarbonate (Scheme 73).
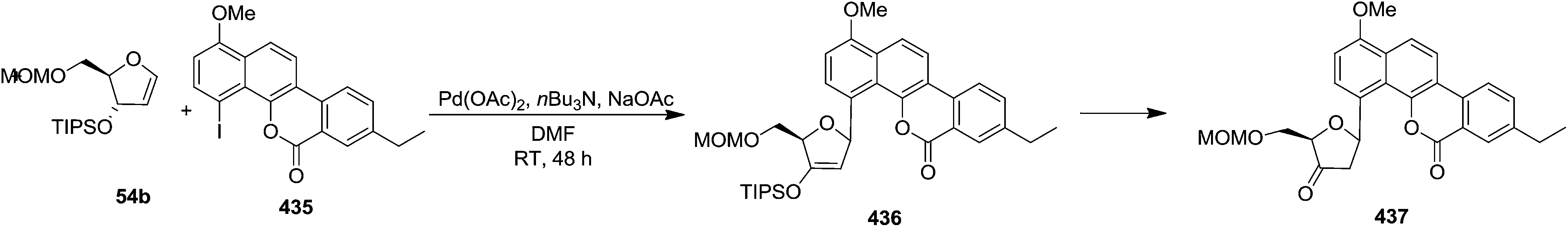 | ||
| Scheme 73 | ||
They have also synthesized C-glycoside 440 from furanoid glycal 66o in a one pot three step sequence. Under the identical palladium-catalyzed coupling reaction condition, furanoid glycal 66o was stirred with iodo anthracycline 435 for 10 h at room temperature to form 438, which was then desilylated by the nBu4NF in reaction medium to give keto derivative 439. The free –OH group was then acetylated into the same reaction medium to furnish C-glycoside 440 in 89% isolated yield by one pot three step sequence. The stereospecific reduction of a 3′-keto group of a furanosyl C-glycoside 439 with NaBH(OAc)3 gave 441 which was acetylated to C-glycoside 442 in 94% isolated yield by one-pot four-step sequence (Scheme 74).
 | ||
| Scheme 74 | ||
In 1990 Daves Jr discussed the regio- and stereospecific synthesis of C-glycosides by palladium-mediated coupling reaction of glycals (furanoid or pyranoid glycal), with suitable aglycon (heterocyclic or anthracyclic) derivatives.64 The reaction of pyrimidine mercurial derivative 402 with Pd(II) acetate led to the formation of Pd(II) organopalladium reagent 403, which underwent stereospecific coupling reaction with furanoid glycals (55, 56a, 45, 54a, 54b, 54f, 66o) to yield single product (either a α-C-glycoside or a β-C-glycoside, Table 9). They observed in the case of 56a, 45 or 66o where only one of the two glycal hydroxyls was substituted, the π-complex was formed exclusively from the face of the furanoid ring opposite to the substituted hydroxyl to give 417, 412 and 444 respectively. When both glycal hydroxyls were substituted (54a, 54b, or 54f), the organopalladium reagent attacked from the β-face of the glycal to form 413, 414, 416 respectively, indicated that the reaction was more sensitive to the steric bulk of the C3 substituent than to that at position C5 of the furanoid glycal. Only when both hydroxyls of the glycal remain unsubstituted 55 mixture of stereoisomeric C-glycosides 443a and 443b were obtained (Scheme 75, Table 9).
| Entry | % yield of α-C-nucleosides (B) | Substituents of (A) | % yield of β-C-nucleosides (C) |
|---|---|---|---|
| 1 | 29 (443a) | 55 R1 = R2 = H | 45 (443b) |
| 2 | 0 | 56a R1 = H, R2 = MOM | 65 (417) |
| 3 | 78 (412) | 45 R1 = MOM, R2 = H | 0 |
| 4 | 0 | 54a R1 = R2 = MOM | 71 (413) |
| 5 | 0 | 54b R1 = MOM, R2 = TIPS | 92 (414) |
| 6 | 0 | 54f R1 = R2 = TIPS | 51 (416) |
| 7 | 0 | 66o R1 = H, R2 = TBDPS | 84 (444) |
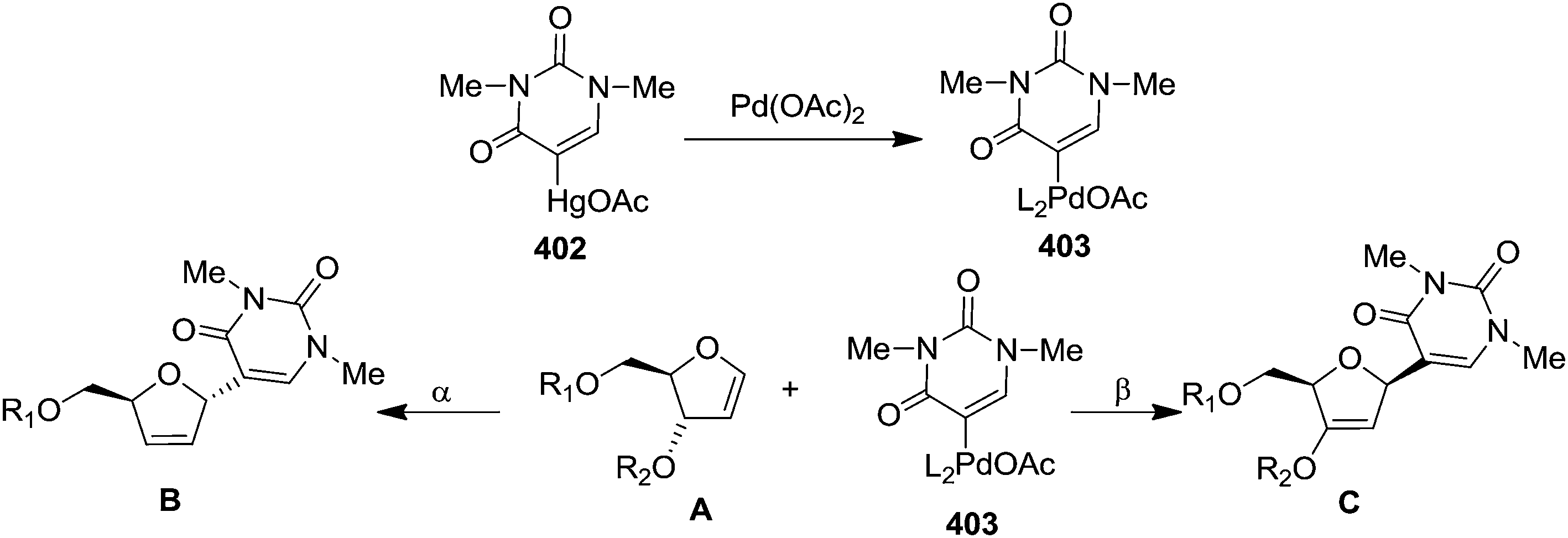 | ||
| Scheme 75 | ||
In this report, Daves Jr also showed the comparative study of coupling reaction of aglycon-mercuric acetate and tri-n-butylstannyl derivatives with a particular furanoid glycal 54b in the presence of stoichiometric Pd(II) acetate and observed there were no significant differences in the effectiveness (Scheme 76, Table 10).
 | ||
| Scheme 76 | ||
| Entry | AG-MXn | C-Glycoside product, % yield (D) |
|---|---|---|
| 1 | 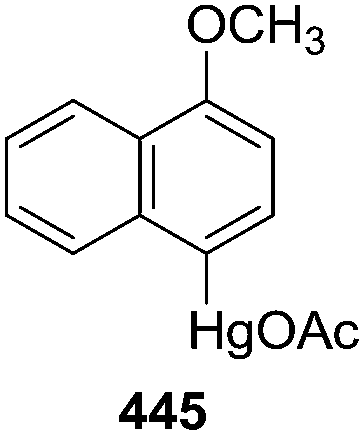 |
96 |
| 2 | 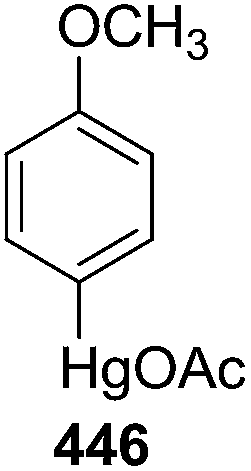 |
57 |
| 3 | 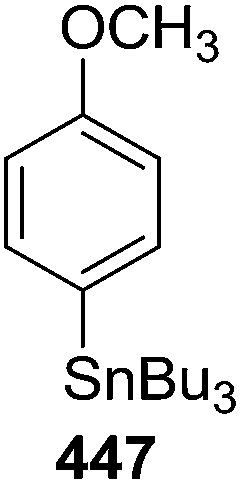 |
70 |
| 4 | 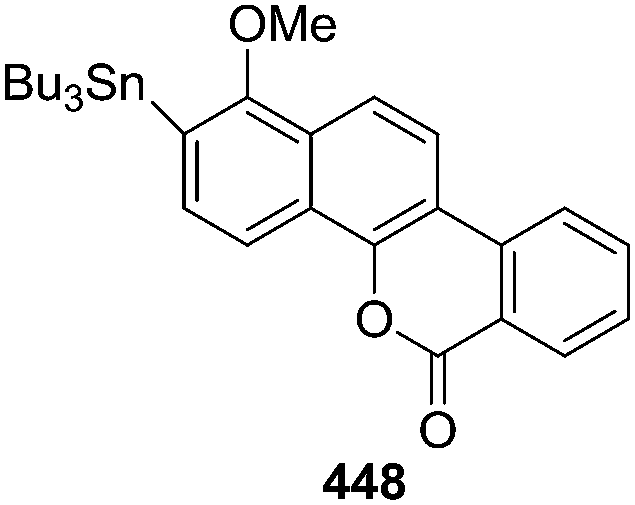 |
57 |
| 5 |  |
66 |
In 1992, this research group further reported syntheses of synthetic C-glycosides 453 and 454 structurally related to the gilvocarcin, ravidomycin, and chrysomycin antibiotics which possess the aglycon substituents (hydroxyl at C-1 and ethenyl at C-8) considered critical for the photolytic nicking of DNA. They synthesized β-C-glycosides 453 from triester 452 (Scheme 77) which was synthesized following the same one-pot four-step sequence from furanoid glycal 66o and pivaloyl protected aglycon derivative 449.65
 | ||
| Scheme 77 | ||
They further extended their study and showed the utility of regio- and stereospecific C-glycosyl bonds formation by the synthesis of C-nucleoside analogs. They synthesized 2′-deoxy-pseudouridin 458 in three steps by utilizing the palladium-mediated coupling of 5-iodouracil 455 with glycal 66o, as the key step in the presence of either triphenylphosphine or triphenylarsine ligands. The coupling reaction formed β-C-nucleoside 456 which, without isolation, was desilylated with fluoride ion to form the 2′-deoxy-3′-keto-C-nucleoside 457. Finally, stereospecific reduction of the 3′-keto group with NaBH(OAc)3 formed 2′-deoxypseudouridine 458 (Scheme 78).66
 | ||
| Scheme 78 | ||
In a similar way, they synthesized 2′-deoxyformycin B 463, 2′,3′-dideoxyformycin B 468 by palladium-mediated glycal-aglycon coupling reaction as the key step. Ribofuranoid glycal 66o and bis(tetrahydropyranyl) protected iodo aglycon derivative 459 underwent regio and stereospecific coupling reaction in the presence of Pd(dba)2 as catalyst and triphenylarsine as ligand in acetonitrile to give C-nucleoside 460 in 62% isolated yield. Following the similar reaction sequence desilylation followed by stereospecific keto group reduction, C-nucleoside 460 was converted to 462 via 461 which on treatment with pyridinium p-toluenesulfonate yielded 2′-deoxyformycin B 463 in 83% yield (Scheme 79).66
 | ||
| Scheme 79 | ||
2′-Deoxy-C-nucleoside 462 was transformed into 2′,3′-dideoxyformycin B 468 in five steps in 52% overall yield. The primary hydroxyl at C-5′ was selectively silylated to form 464 followed by protection of the C-3′ hydroxyl using O-phenyl chlorothionoformate to give intermediate 465 which on deoxygination with nBu3SnH/AIBN produced 2′,3′-dideoxy C-nucleoside 466. Its silyl ether deprotection with fluoride ion furnished 467 which on removal of tetrahydropyranyl groups with pyridinium p-toluenesulfonate afforded 2′,3′-dideoxyformycin B 468 (Scheme 80).66
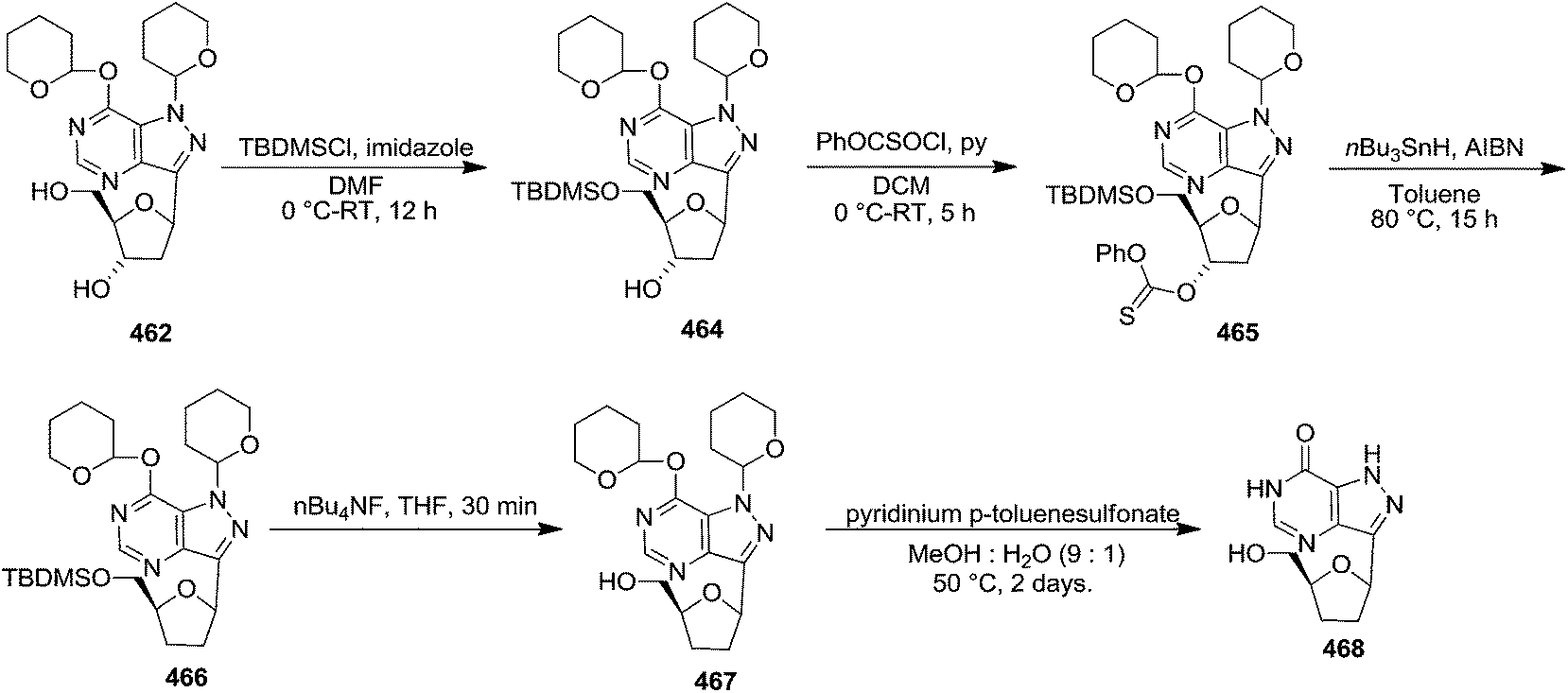 | ||
| Scheme 80 | ||
Daves Jr have reported the synthesis of 1-(tri-n-butylstannyl)furanoid glycals for the first time by lithiation of the corresponding 3-O-unsubstituted glycals (404, 469, 55, 53b) followed by reaction with nBu3SnCl. They also discussed the tri-n-butylstannaylation of 3-O-substituted hydroxy glycal 66h, which underwent elimination to yield the corresponding furan in the presence of tBuLi. Finally, they succeeded to prepare 3-O-benzyl furanoid glycal 478 from phenylthioglycoside 475, which was oxidized with m-CPBA to phenyl sulfone 476. Treatment of 476 with nBuLi furnished unsaturated sulfone 477, which was treated with nBu3SnH in the presence of AIBN formed stannylated 3-O-benzyl furanoid glycal 478. Palladium mediated coupling reaction of these stannylated furanoid glycals (470–473, 478) with iodoaglycon derivatives yielded the corresponding 1-substituted furanoid glycals in good to excellent yields (Scheme 81).67
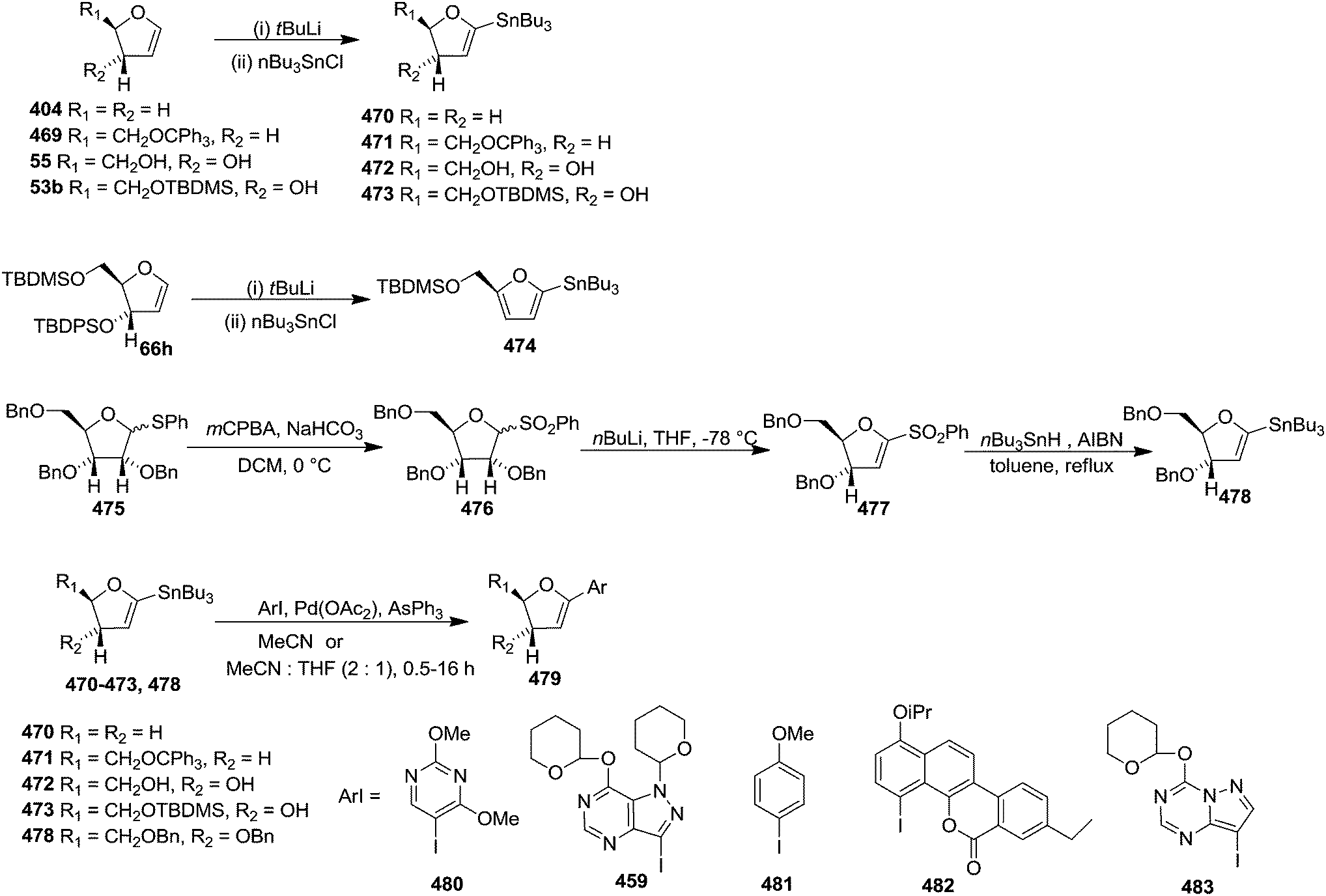 | ||
| Scheme 81 | ||
This group further extended their studies on Pd-mediated regio- and stereospecific coupling reaction of 483 with furanoid glycal 56a to obtain C-nucleosides 484.68 The coupling of iodoaglycon derivative 483 with furanoid glycal 66o followed by desilylation of 485 with TBAF and stereospecific hydroxy-activated reduction of the 3′-keto group of intermediate 486 using NaBH(OAc)3 yielded the 2′-deoxyribofuranosyl C-nucleoside 487 in 65% yield for the three steps (Scheme 82).
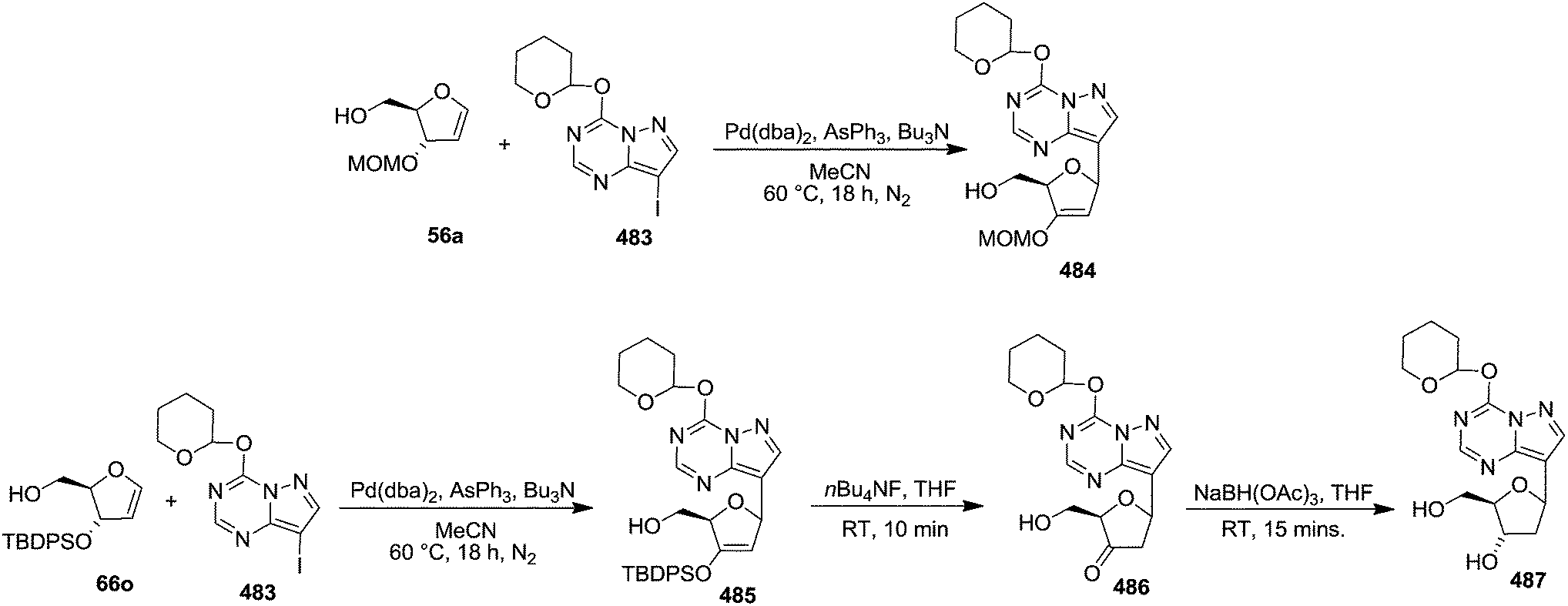 | ||
| Scheme 82 | ||
Similarly, they synthesized palladium-mediated coupling of 8-iodo-4-methoxypyrazolo[1,5-a]-1,3,5-triazine 488 and furanoid glycal 66o efficiently produced C-nucleoside intermediate 489 which was desilylated to form 3′-keto C-nucleoside 490 (Scheme 83).
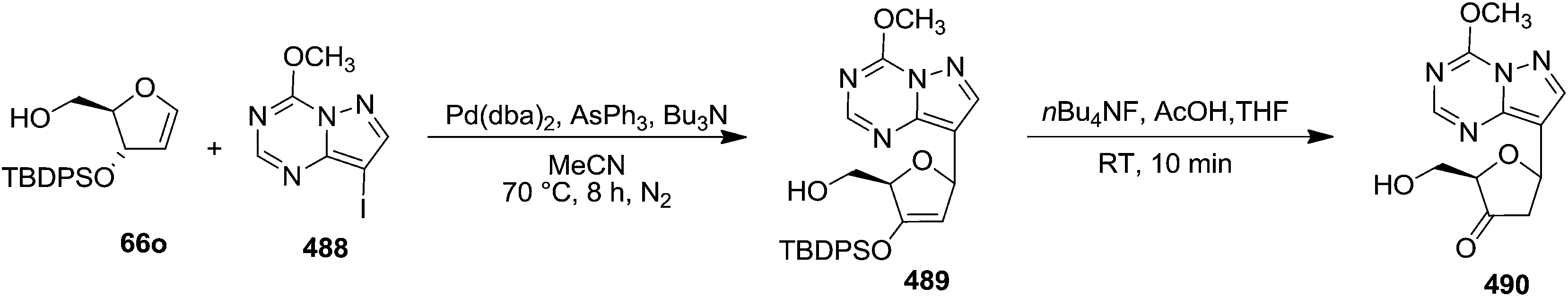 | ||
| Scheme 83 | ||
They also tried Pd-mediated coupling of iodoaglycon 491 and 492 with furanoid glycal 66o but it was futile. As a result, they prepared aglycon bis-carbamate derivative 494 by reaction of 493 with isobutyloxycarbonyl chloride in the presence of pyridine. Aglycon derivative 494, was successfully coupled with glycal 66o in the presence of catalytic Pd(dba)2 and AsPh3 to give, after desilylation of the initially formed silyl enol ether with fluoride ion, 3′-keto C-nucleoside 495 (2 steps, 50% yield) (Scheme 84).
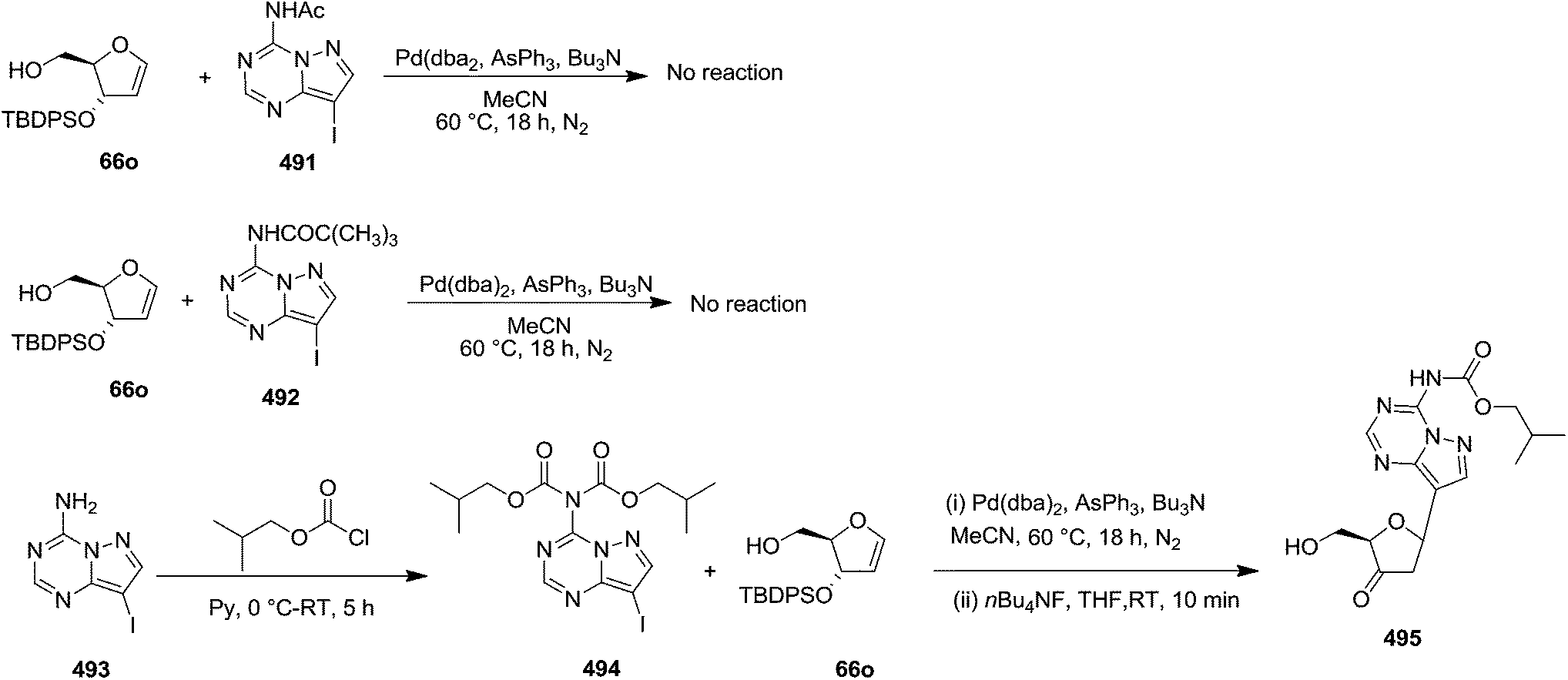 | ||
| Scheme 84 | ||
In the same year, Townsend and group reported an efficient and stereospecific synthesis of pyrazine C-nucleosides by Pd(0)-mediated cross-coupling reaction between ribofuranoid glycals 66e, 66f and 65b and iodoaglycon 496.69 The cross-coupling reaction between aglycon 496 and ribofuranoid glycal 66e resulted silyl enolether derivative 497a which was gradually converted to 498a. Pd-mediated cross-coupling reaction between iodoaglycon 496 and ribofuranoid glycal 66f furnished C-nucleoside 497b whereas under identical reaction condition furanoid glycal 65b only resulted in the isolation of the 2′-deoxy-3′-keto-C-nucleoside 498b instead of the silyl enol ether derivative 497c. They selectively deprotected the 3′-silyl group of the silyl enol ethers 497a and 497b by the fluoride ion at low temperature to give 498a and 498b, respectively. 2′-Deoxy-β-D-ribofuranoside 500 was prepared from the complete deprotection of 497a, b or 498a, b with TBAF via 499, followed by a stereospecific reduction by NaBH(OAc)3. They confirmed the β-configuration of C-nucleoside 500 by NOE analysis and also converted it to 5,5′-anhydro nucleoside 501 by diazotization reaction with iso-amyl nitrite (Scheme 85).
 | ||
| Scheme 85 | ||
Motivated by the work of Daves et al., in 1995, McLaughlin and coworkers reported the synthesis of two pyridine C-nucleosides 507 and 512, “deletion modified” analogues of dT and dC.70 After several trial and error, they prepared 2-(benzyloxy)-3-methyl-5-iodopyridine 505 from 502 in three steps. Pd-mediated coupling reaction of glycal 66o and 2-(benzyloxy)-3-methyl-5-iodopyridine 505 in the presence of ancillary ligand 1,3-bis(diphenylphosphino)propane resulted C-nucleoside 506 in 90% yield. C-Nucleoside 506 was then converted into 507 by three steps sequential reactions, silyl ether deprotection, followed by stereoselectively keto group reduction and finally benzyloxy group deprotection by catalytic hydrogenation (Scheme 86).
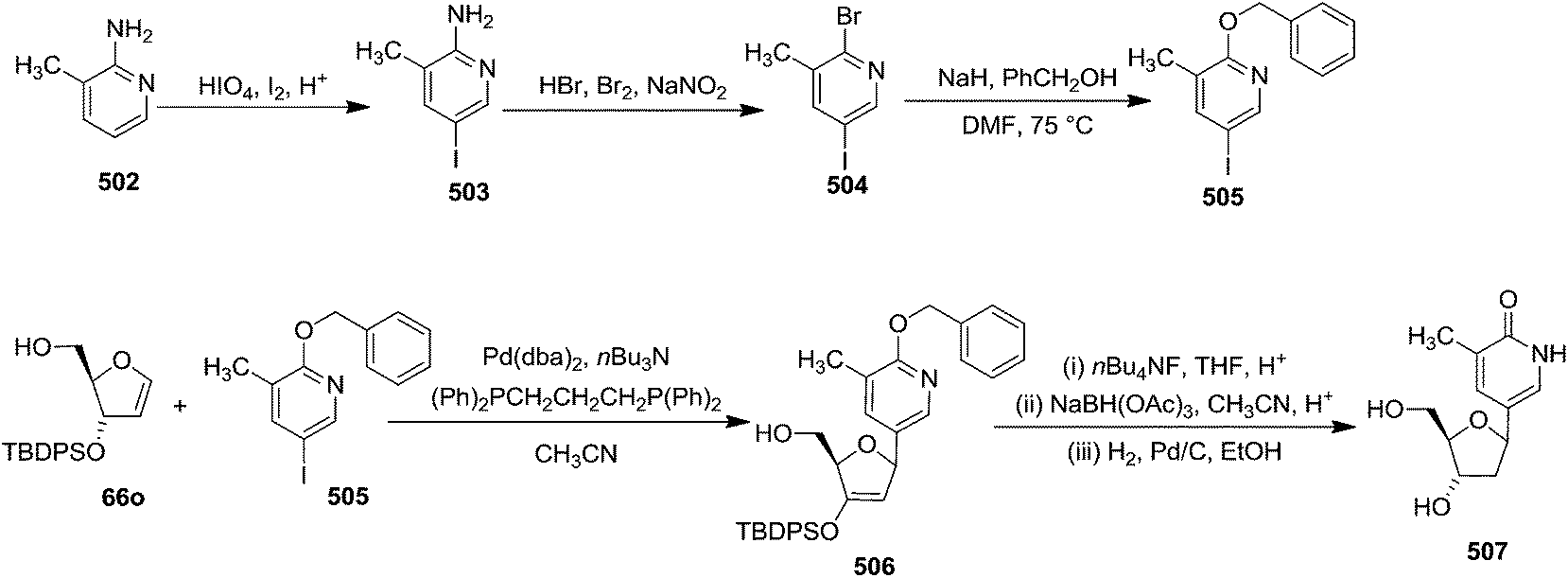 | ||
| Scheme 86 | ||
In a similar fashion, they prepared the C-nucleoside analogue of dC 512 from 2-aminopyridine 508. They prepared 510 from 508 by its iodination followed by amino group protection with BzCl of the resulting iodo derivative 509. The coupling reaction of glycal 66o with 510 in the presence of Pd and an ancillary ligand P(C6F5)3 resulted in moderate yields of product 511 (36% yield). The remaining steps to generate 512 were strictly analogous to those described in Scheme 86 for the synthesis of 507 (Scheme 87).
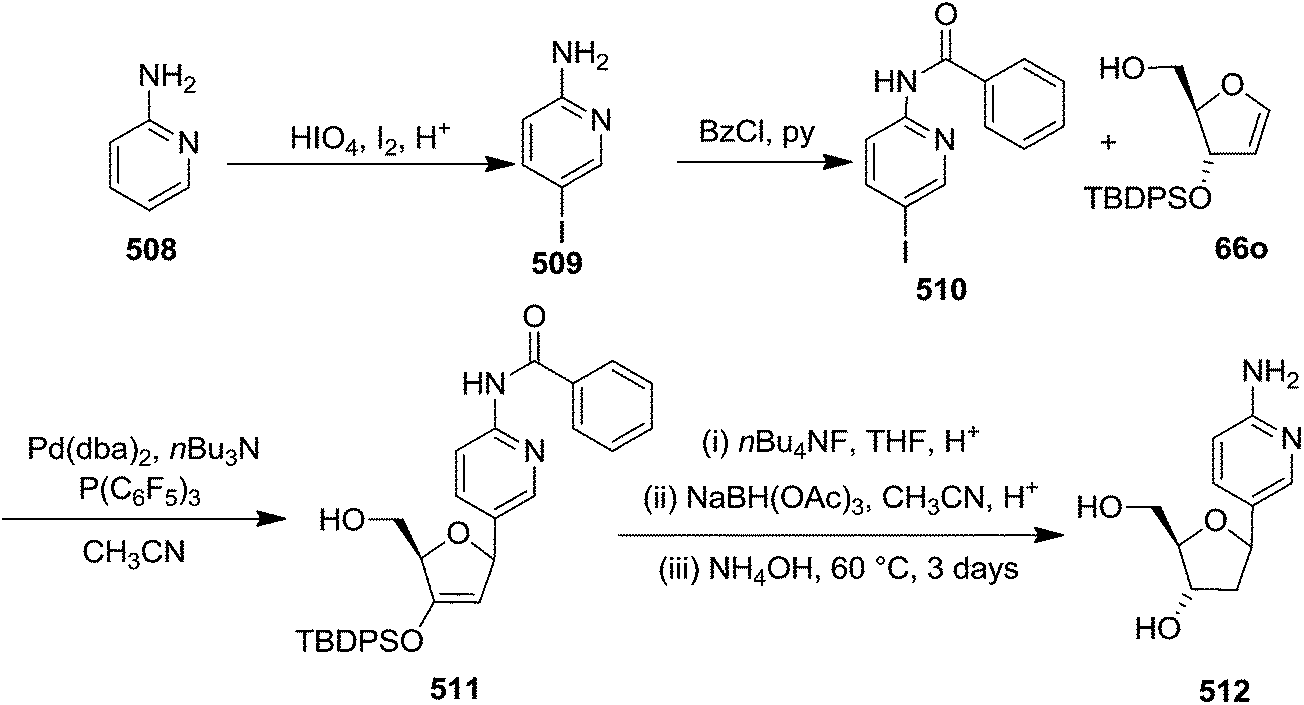 | ||
| Scheme 87 | ||
In 1998, Coleman and Madaras followed Daves Jr strategy for synthesis of coumarin β-C-riboside 522. For this, they synthesized furanoid glycal 66o,23 56a20a and 66n23 in the ususal procedure.71
Coumarin 515 (X = OH) was prepared from 8-hydroxyjulolidine 513. Reaction of 513 with bis(2,4,6-trichlorophenyl)malonate 514 in refluxing toluene effected annulation of the α-pyrone ring system to afford 515 in excellent yields (94%). Its hydroxyl functionality could be transformed to the iodide by treating it with a preformed complex of triphenylphosphine and iodine (Ph3P, I2, CH3CN, 82 °C) to form 516. Alternatively, the hydroxyl group could be acylated with trifluoromethanesulfonic anhydride (Tf2O, Et3N, DCM, 0 °C) to afford triflate 517 in 87% yield (Scheme 88). These systems were examined in the Heck coupling reaction with glycals 66o, 56a and 66n (Schemes 89 and 90).
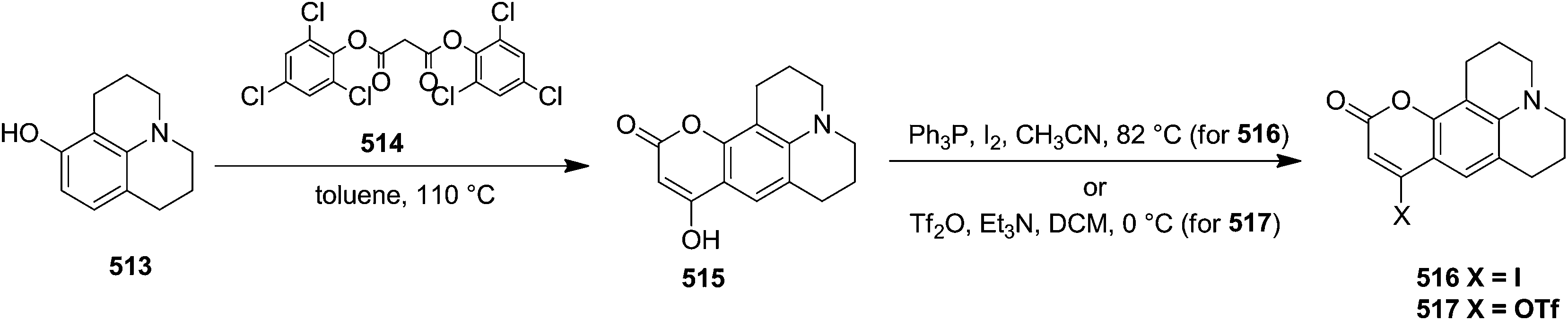 | ||
| Scheme 88 | ||
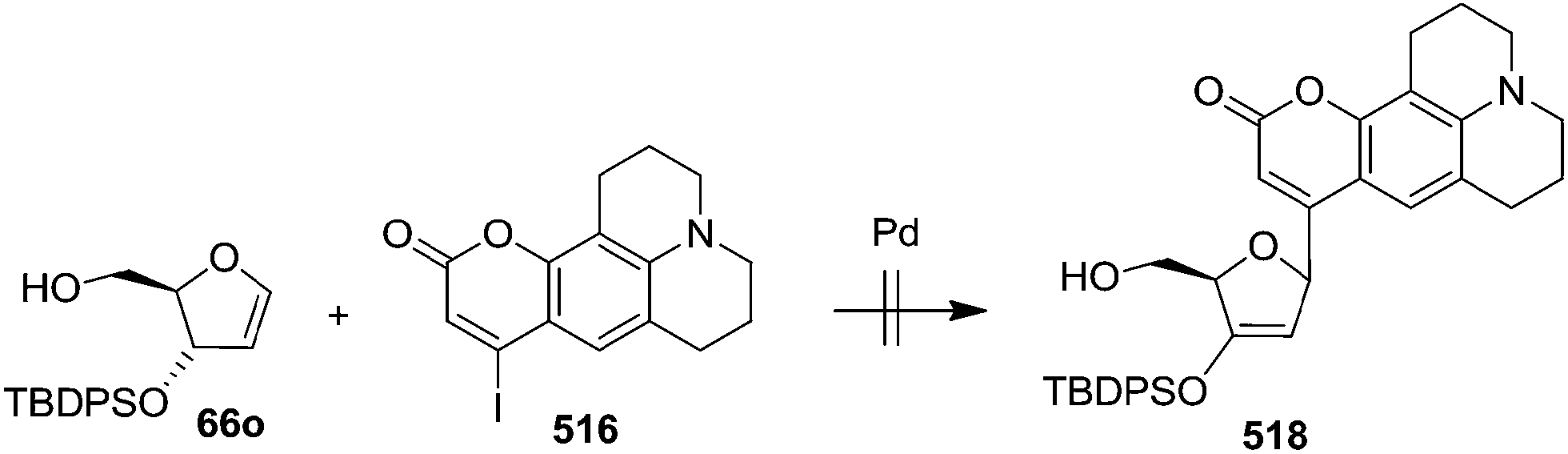 | ||
| Scheme 89 | ||
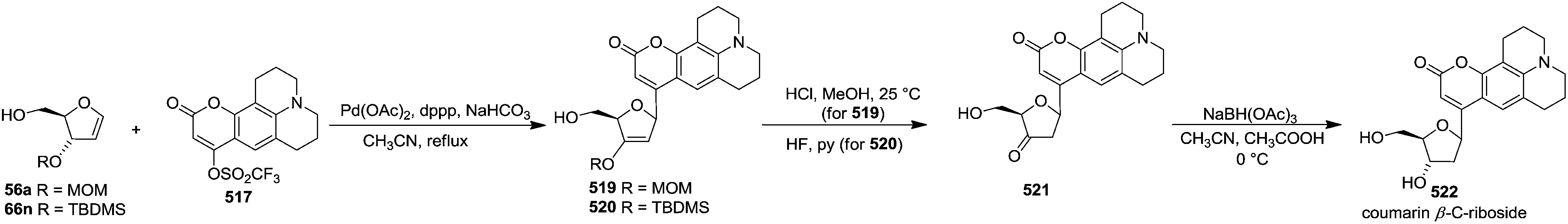 | ||
| Scheme 90 | ||
Pd-catalyzed coupling of 516 with glycal 66o was unsuccessful in providing any of the coupled product 518 (Scheme 89).
After getting unsuccessful results, they proceeded for Pd-catalyzed coupling reaction of triflate 517 with glycal 56a and 66n in the presence of 40 mol% Pd(OAc)2, 5 mol% dppp, and 3 equiv. NaHCO3 in CH3CN under refluxing condition to obtain Heck product 519 (75% yield) and 520 (79% yield) respectively. Hydrolysis of 519 under acidic conditions (HCl, CH3OH, 25 °C) afforded ketone 521. Fluoride-promoted cleavage (HF/pyridine) of the silyl ether 520 afforded ketone 521 in excellent yields. The carbonyl group of 521 was reduced stereoselectively to the ribo-glycoside 522 with NaBH(OAc)3 (Scheme 90).
In 1999, Tingoli et al. showed the reaction of aromatic Grignard reagents with furanoid and pyranoid glycals in the presence of low valent Ni catalyst at low temperature. Treatment of furanoid glycal 90 with aryl magnesium bromide in the presence of Ni(0) catalyst in dry toluene at −10 °C for 5 h afforded column purified 2,3-unsaturated products (523, 524) in good yields. They further confirmed the 1,4-trans relationship between H-1 and H-4 by NOESY experiment (Scheme 91).72
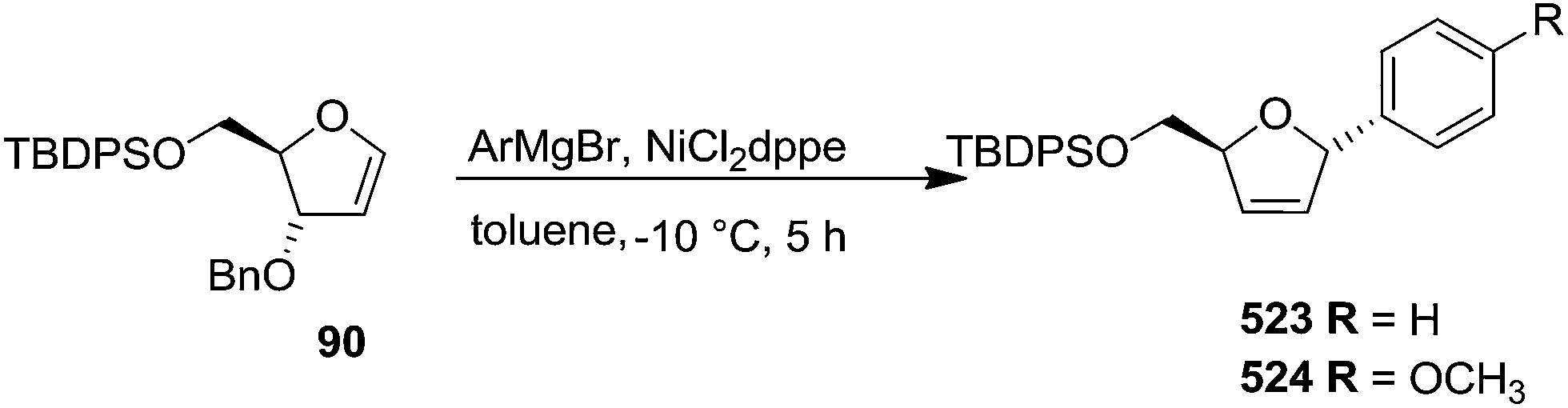 | ||
| Scheme 91 | ||
Knaus et al. synthesized furanoid glycals (146a, b) (Scheme 24), used as key intermediate for the synthesis of unnatural C-aryl 2′-deoxy-β-L-cytidine mimics (529a, b) (Scheme 92).32 The Heck coupling reaction of 2,5-difluoro-4-iodoaniline 525a or 3-fluoro-4-iodoaniline 525b with glycal 146a or 146b in the presence of Pd(OAc)2, Ph3As, and Et3N in dry CH3CN at 70 °C,32 afforded 526a (71% yield) or 526b (57% yield). After several trial and errors, 526b was subjected to Pd/C catalyzed hydrogenation in anhydrous EtOH containing several drops of Et3N at 60 °C to afford the hydroxyl ketone 527b in 61% yield. The 5-(p-chlorobenzoyl) group deprotection of 526a on treatment with NaOMe in MeOH resulted benzyl enol ether 528, which was converted to the hydroxy ketone 527a upon Pd/C catalyzed hydrogenation in anhydrous EtOH in the presence of several drops of Et3N at 60 °C. Subsequent reduction of the hydroxy ketones (527a, b) with NaBH(AcO)3 in dry CH3CN afforded the target deoxy-β-L-cytidine C-nucleoside mimics (529a, b) (Scheme 92).
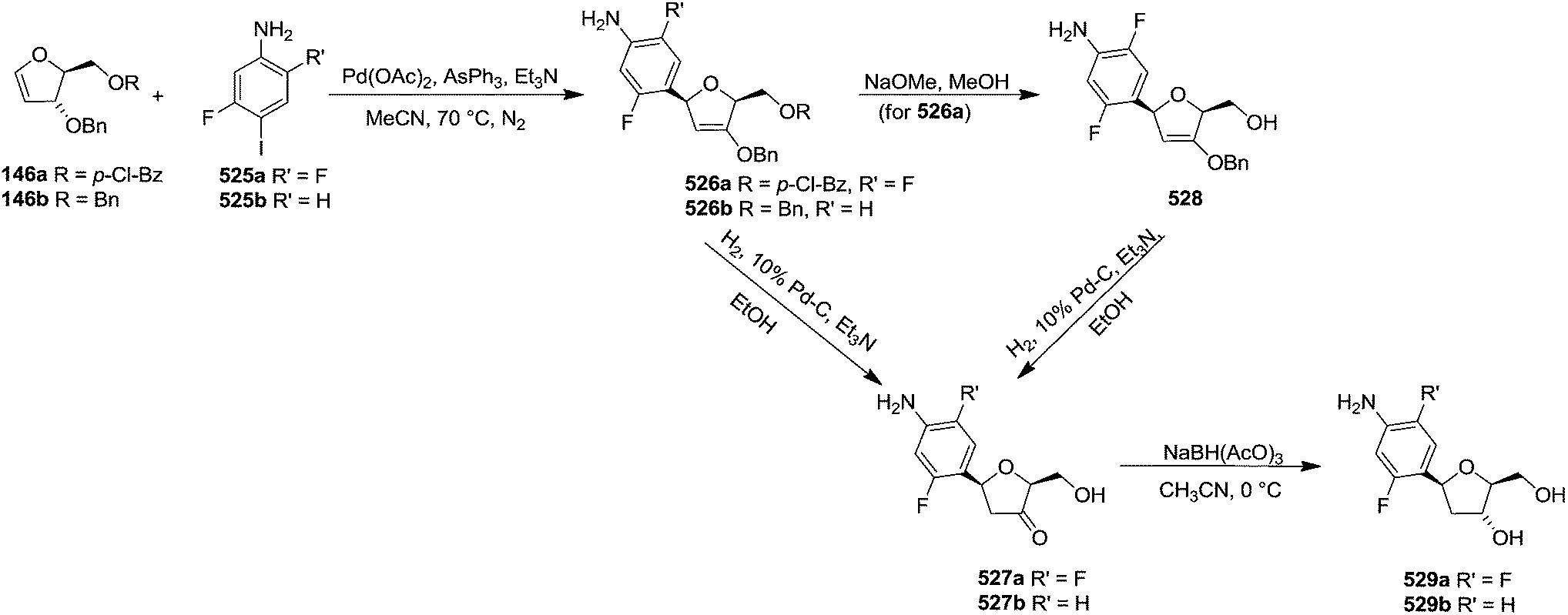 | ||
| Scheme 92 | ||
McLaughlin et al. described the synthesis of four pyrimidine C-nucleoside analogues (542–545) of natural nucleosides dC and dU.73 They synthesized desired pyridine heterocycles (530–533) necessary for the syntheses of (542–545) from the readily available differently 2,6-substituted pyridines.73 Furanoid glycal 66o and pyridine heterocycles (530–533) underwent regio and stereospecific Heck-type coupling reaction in the presence of Pd(dba)2 and (C6F5)3P in acetonitrile to give β-C-nucleoside intermediates (534–537), which, without isolation, were desilylated with nBu4NF to form 2′-deoxy-3′-keto-C-nucleosides (538–541). Stereospecific reduction of (538–541) and removal of the pNPE protecting group of 546 and 547 resulted in the target compounds (542–545) (Scheme 93).
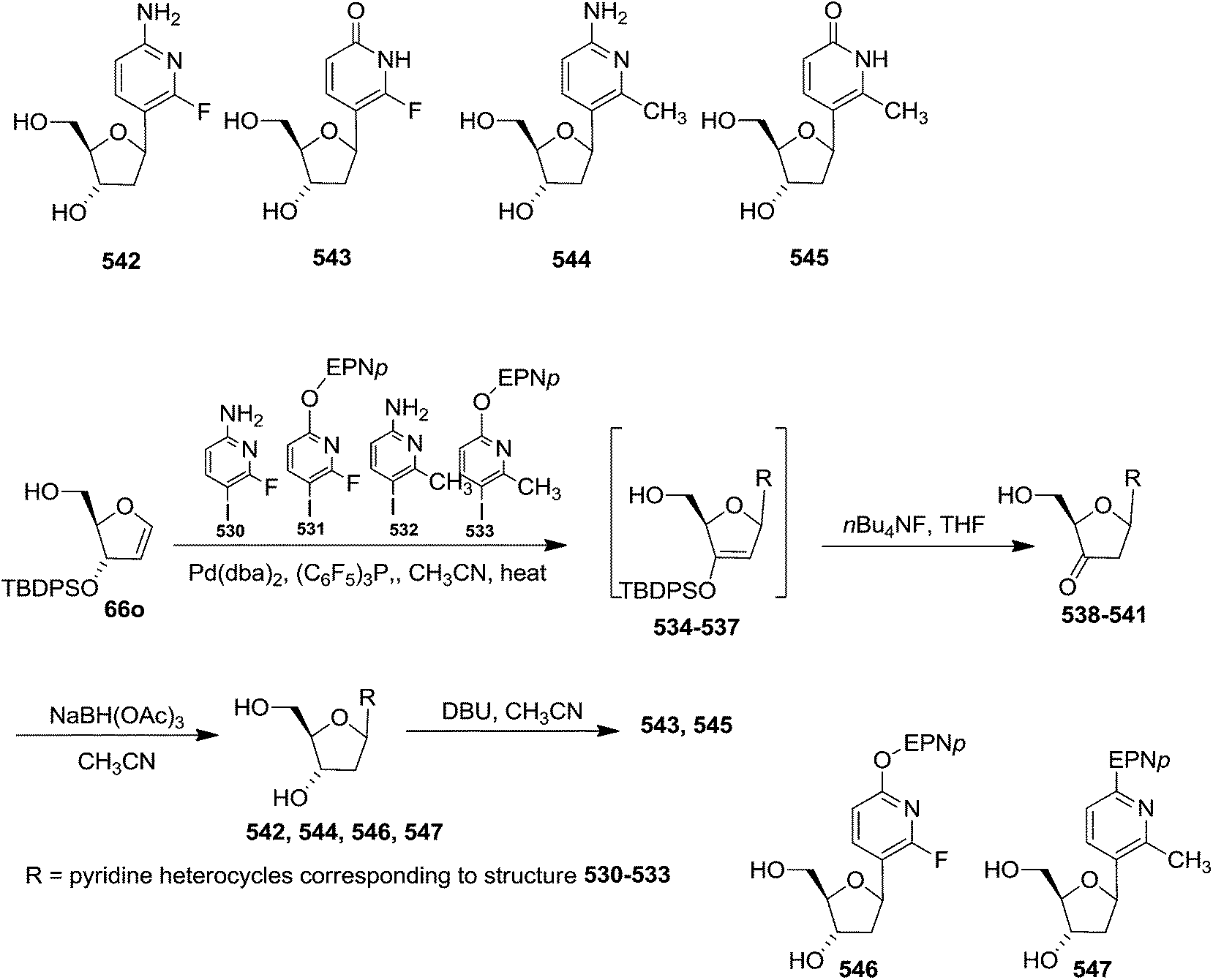 | ||
| Scheme 93 | ||
Seitz and Singh diastereoselectively synthesized β-aryl-C-2-deoxynucleosides from furanoid glycal derived glycal epoxides.74 The glycal 66f on treatment with DMDO in DCM at 0 °C was converted to epoxide 548. Its reaction with trinaphthylaluminum 549 yielded 1-naphthyl-β-C-arabinonucleoside 550 by cis opening of the epoxide ring. It was transformed into methyl xanthate 553 via phenylthionocarbonate 551 and thiocarbonylimidazole 552. They failed to reduce 553 to 554 by treatment with nBu3SnH or (TMS)3SiH in presence of AIBN due to the bulkiness of the two TBDPS groups (Scheme 94).
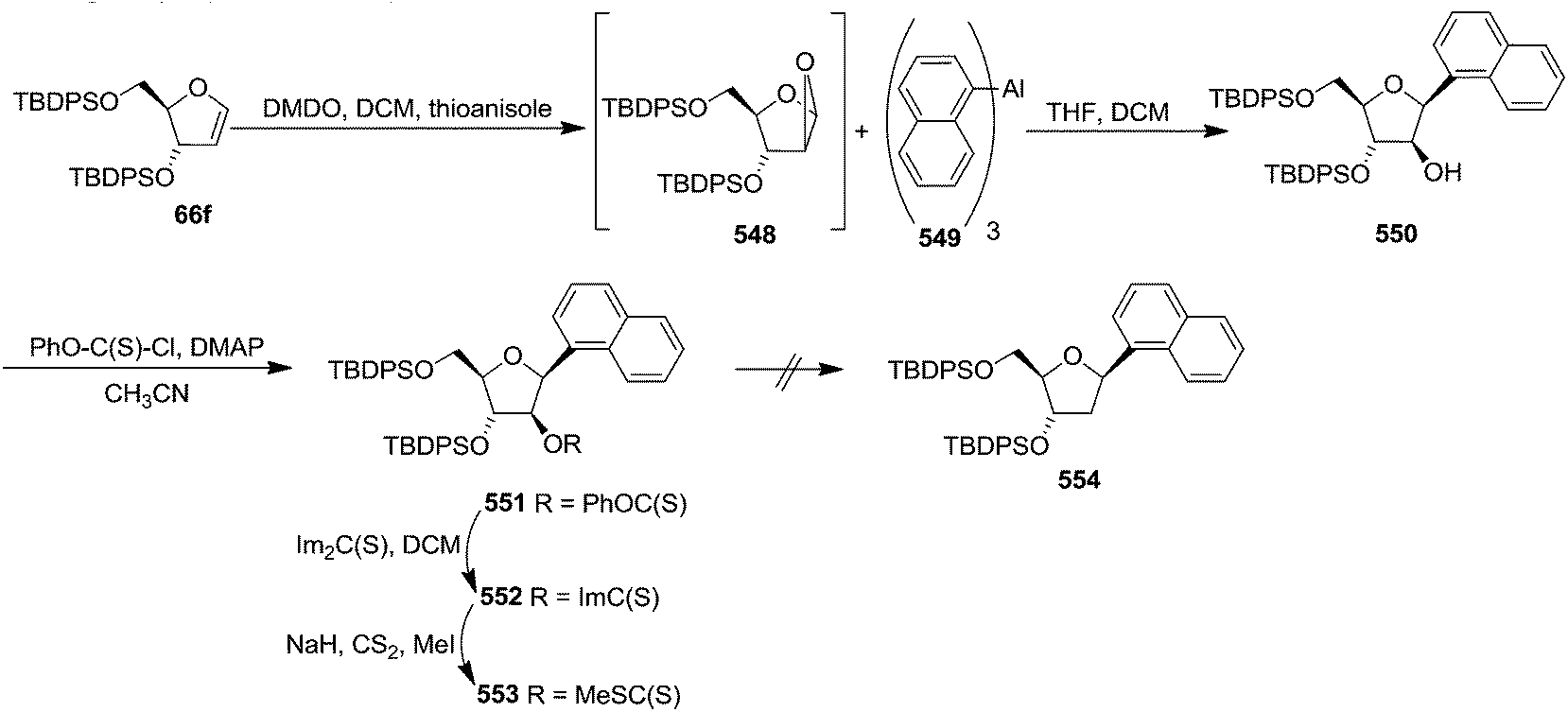 | ||
| Scheme 94 | ||
Then they selected TBDMS-protected glycal 66e.23 The required epoxide 555 was obtained from the known glycal 66e by treating it with dimethyldioxirane (DMDO). The cis opening of epoxide 555 was performed by treatment with trinaphthylaluminum to afford 1-naphthyl-β-C-arabinonucleoside 556a (method 1) in 50% yield from 66e. The nucleoside 556a was allowed to react with thiocarbonyldiimidazole to form 557a, which on reduction with (TMS)3SiH and AIBN furnished 2′-deoxynucleoside 558a in 82% yield. Finally, the silyl ether deprotection was performed by treatment with nBu4NF in THF to afford fully deprotected 2′-deoxy-1′-β-naphthyl nucleoside 559a in 98% yield. They further studied the reaction sequence by changing triarylaluminum reagent for cis opening of the glycal epoxide to dimethylarylaluminum reagent (method 2). They synthesized a series of 2′-deoxy-1′-β-naphthyl nucleoside 559a–d by employing both the two methods (Scheme 95).
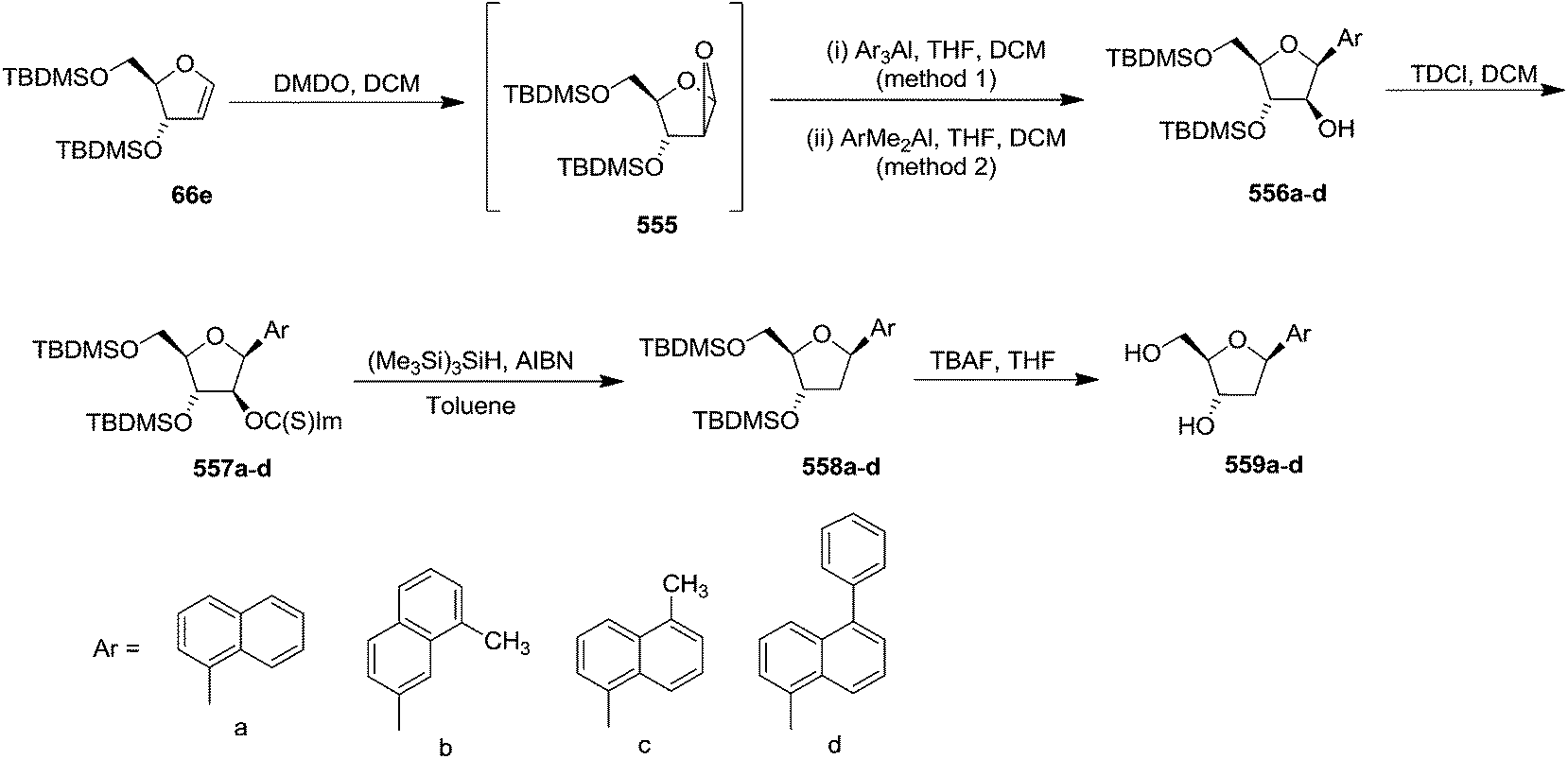 | ||
| Scheme 95 | ||
In 2007, Hocek and co-worker have developed a novel methodology for the synthesis of 6-substituted pyridin-3-yl C-nucleosides.75 After several trial experiments, they optimized the Heck reaction of 2-chloro-5-iodopyridine 560 with glycal 66n in the presence of Pd(OAc)2/AsPh3 and Ag2CO3 in chloroform to give the desired C-nucleoside precursor 561 in acceptable 65% yield. Desilylation followed by reduction of the corresponding keto 562 with NaBH(OAc)3 in a mixture of acetonitrile and acetic acid afforded 563 in a good yield of 70% for the two steps. The free hydroxyl group of nucleoside 563 was then protected with TBDMSCl with imidazole in DMF to afford the fully protected key intermediate β-C-nucleoside 564 in 87% yield (39% overall yield over four steps from glycal 66n), (Scheme 96). They prepared 6-unsubstituted pyridine nucleoside 565a by catalytic hydrogenation of 564 with H2 over Pd/C for 3 h, in a mixture of EtOH, THF, and H2O, in presence of Et3N. This key intermediate 564 was then subjected to a series of palladium catalyzed cross-coupling reactions to form new pyridine C-nucleosides 565b–g bearing diverse C (6-alkyl, 6-aryl, or 6-hetaryl) groups. They performed Hartwig-Buchwald aminations, and alkoxylations on 564 to give a series of protected 1β-(6-amino-, and 6-tert-butoxypyridin-3-yl)-2′-deoxyribonucleosides 565h–j in good yields. All the silylated nucleosides 565a–i were deprotected using Et3N·3HF in THF to give the free 6-substituted pyridine C-nucleosides 566a–i in good yields (Scheme 96).
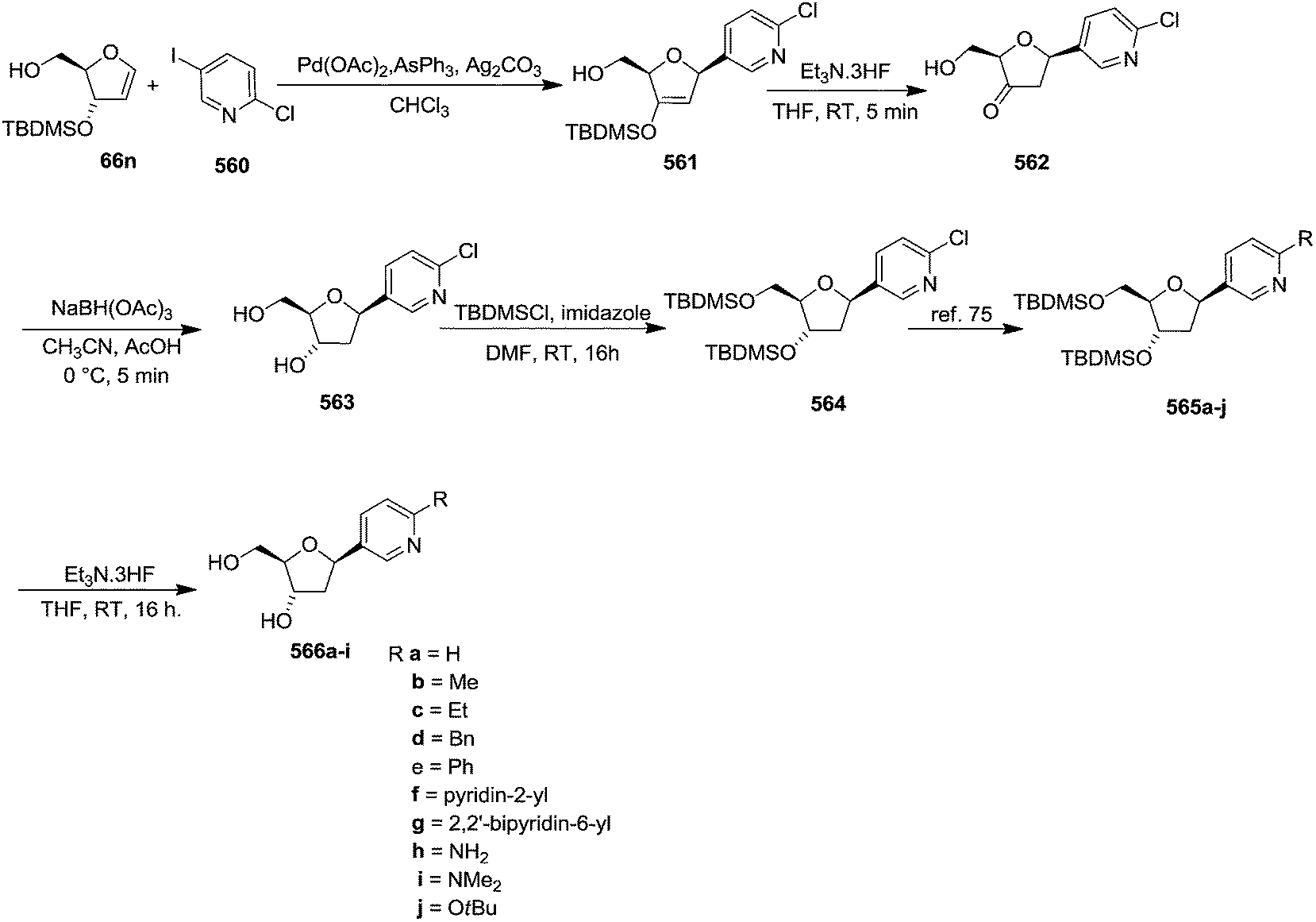 | ||
| Scheme 96 | ||
6-Oxopyridine C-nucleoside 567 was synthesized in 84% yield by this research group from 6-(tert-butoxy)pyridine derivative 566j on treatment with TFA for 20 min. They also synthesized C-nucleoside 567 from aminopyridine C-nucleoside 566h by reacting it with isopentyl nitrite in 80% aqueous AcOH at 70 °C for 100 min. However, here 567 was isolated along with some inseparable impurities (Scheme 97).75
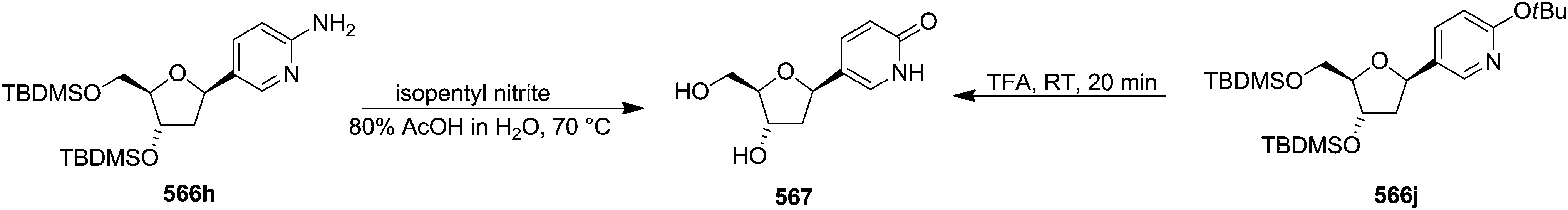 | ||
| Scheme 97 | ||
6. Synthesis of N-nucleosides
In 1990 Danishefsky and Chow proposed the epoxidation of furanoid glycals and showed their application towards the synthesis of nucleosides.8 The epoxidation of furanoid glycal 65b with DMDO (dimethyldioxirane) was highly face selective to give 568. The treatment of the epoxide 568 with (TMS)2thymine provided a mixture of 569 (52% yield) and 570 (30% yield). The mixture was deprotected with TBAF in THF to afford 571. Its acetylation with Ac2O-DMAP afforded C1-epi-arabinonucleoside triacetate 572 in 94% yield. On the other hand, reaction of furanoid glycal 66b (having free hydroxyl group at C-3) with DMDO in acetone with a minimal amount of DCM afforded 1:1 mixture of anhydro sugars 573 and 574. They further investigated reaction of 66b with a mixture of acetone/DCM in 6:1 ratio to furnish mixture of epoxides 573 and 574 in 9:1 ratio. Treatment of the mixture with (TMS)2thymine in acetonitrile followed by desilylation with TBAF in THF and acetylation with acetic anhydride/DMAP resulted in a mixture of 575 and 572 in 4:1 ratio with 36% yield (Scheme 98).
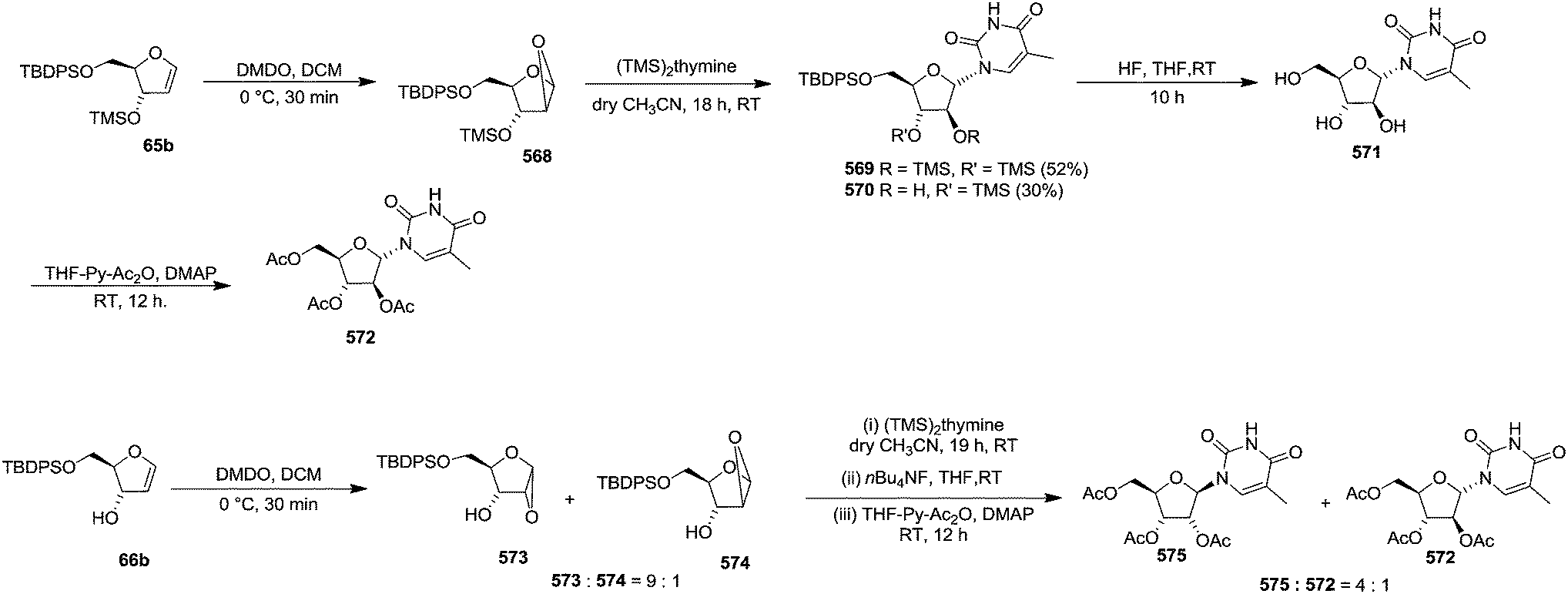 | ||
| Scheme 98 | ||
In 1991 Kim et al. prepared phosphonate isosteres of 577 (d4T), 578 (d4A), and 579 (ddA) (Fig. 5) monophosphates using regiospecific and highly stereoselective electrophilic addition to furanoid glycals as the key step.76 The starting material for this study was the glycal 582, which was readily prepared from thymidine 64a via thymidine-5′-carboxylic acid 581 in two steps by adopting the reported procedure of Horwitz and coworkers.77 Glycal 582 was treated with PhSeCl at −70 °C, to give a 12:1 mixture of 583 and 584 in high yield. This mixture was allowed to react with silver perchlorate in the presence of dimethyl(hydroxymethyl)phosphonate to afford the phosphonate 585 in 41% overall yield. It was then transformed into the d4T phosphonate analogue 587 by the sequential oxidation with sodium periodate in methanol followed by phosphonate ester removal of the resulting 586 with TMSBr in DMF and finally by neutralization with NaHCO3 in overall 52% yield (Scheme 99). They also showed phosphonates 586 exhibited a potent antiviral activity comparable to that of 577 (d4T).
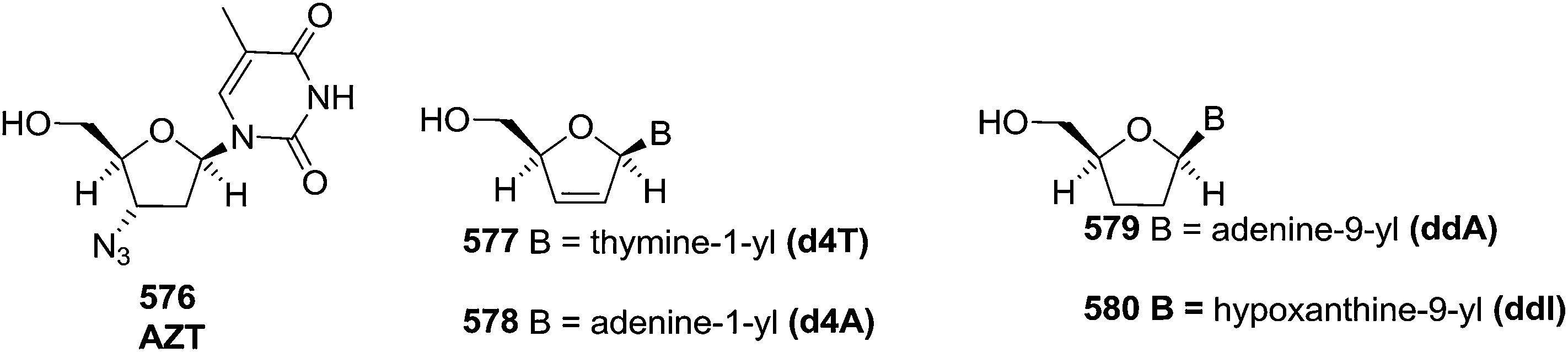 | ||
| Fig. 5 Structures of AZT 576, d4T 577, d4A 578, ddA 579 and ddI 580. | ||
 | ||
| Scheme 99 | ||
They also described the synthesis of glycal 591 from 2′-deoxyadenosine 588 by following the same reaction sequence described for glycal 582. Glycal 591 on treatment with dimethyl(hydroxymethyl)phosphonate in the presence of N-(phenylseleno)phthalimide or IBr afforded 592 (65% yield) or 593 (95% yield) in a regiospecific and a highly stereoselective manner. Oxidative elimination of the phenylselenyl group in 592 or base (DBU) promoted elimination of hydrogen iodide in 593 gave olefin 594 in high yield, which on deprotection was converted to 595, phosphonate isostere of d4A (578) monophosphate, by following the same reaction sequence for the conversion of 586 to 587. The tetrahydrofuranyl derivative 596, a phosphonate isostere of ddA (579) monophosphate was also prepared by catalytic hydrogenation of the olefin 595. Dihydroxylation of the double bond in 594 with catalytic OsO4 and NMO as the oxidant gave diol 597 as a single isomer in high yield. The deblocking of the protecting groups in 597 led to 598, which is a phosphonate isostere of adenosine monophosphate. These d4T and d4A phosphonate analogues 587 and 595 exhibited potent anti HIV activity (Scheme 100).
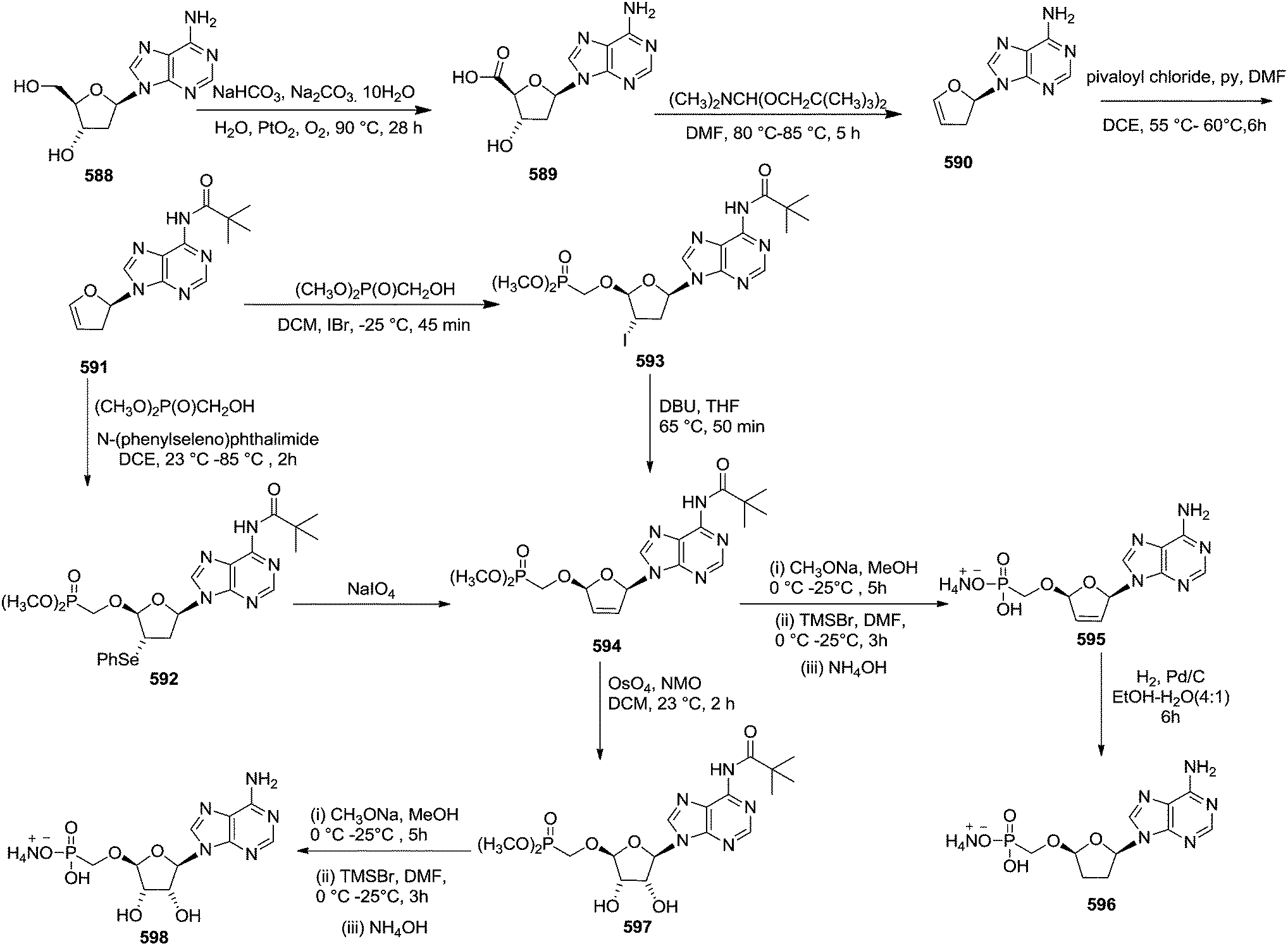 | ||
| Scheme 100 | ||
In 1992, Kim and Misco, demonstrated highly stereoselective synthesis of d4T 577 and ddA 579, antiviral nucleosides, from L-glutamic acid 269 via furanoid glycal intermediate.9 The synthesis of the requisite furanoid glycal 106 was derived from the known lactone 599 which was in turn, readily available by the diazotation-lactonization of L-glutamic acid 269.78 Pivaloyl group protection of the free hydroxyl group of 599 with pivaloyl chloride gave 600, which on DIBALH reduction afforded 601. Its chlorination with SOCl2 followed by elimination of chloride 602 with KOtBu gave the glycal 106 (overall 52% yield). Addition of acetic acid to glycal 106 in the presence of NIS produced a mixture of 603 and 604 in a ratio of 14:1. This mixture without further purification was coupled with silylated N6-benzoyladenine in the presence of SnCl4 to give the adenosine analogue 605 (45% yield over 2 steps) after chromatographic purification. Hydrogenolysis of iodide 605 followed by removal of the pivaloyl group by saponification gave ddA 579 in 75% yield (Scheme 101).
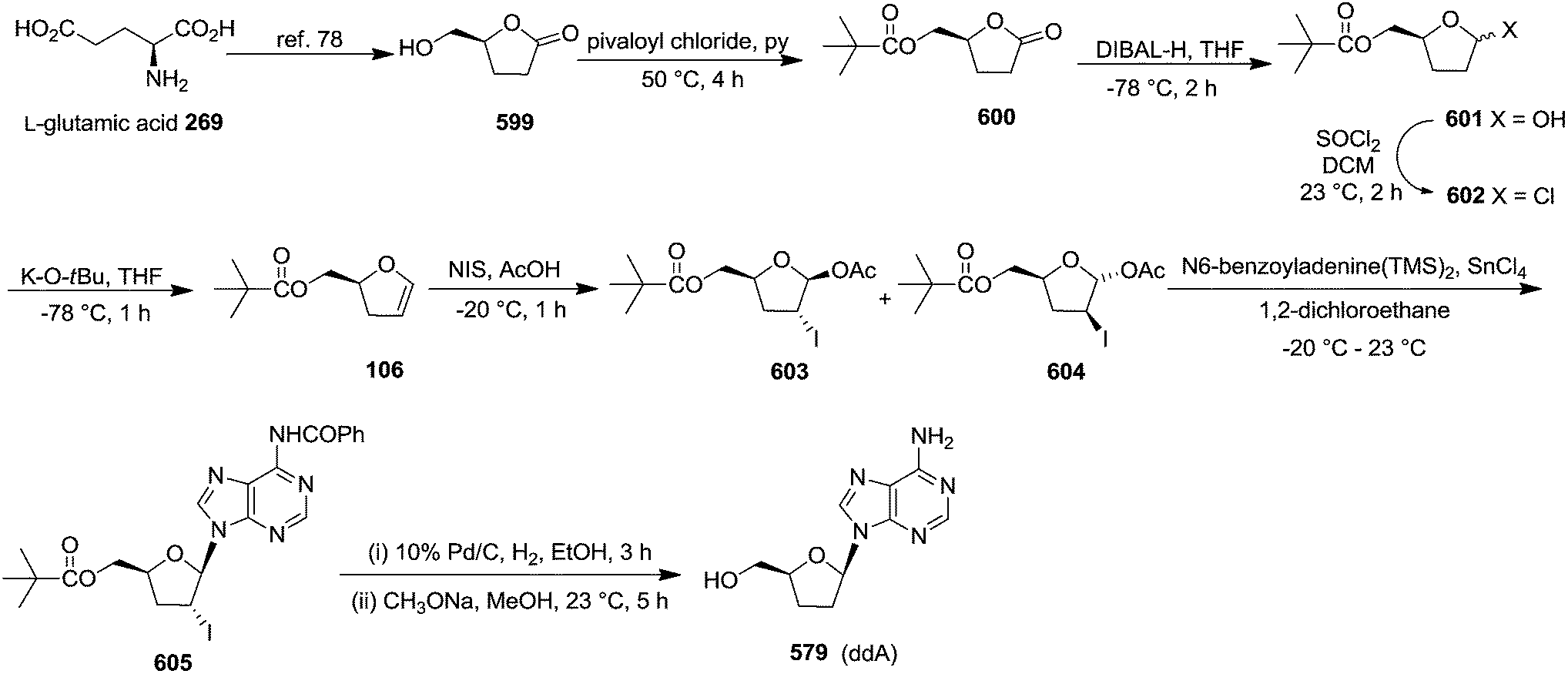 | ||
| Scheme 101 | ||
Then they repeated this reaction sequences with pyrimidine series. But in this case, they directly coupled the pyrimidine base and the furanoid glycal in the presence of NIS. When NIS was added to a mixture of glycal 106 and silylated thymine in DCM, the desired thymidine analogue 606 was formed as a major product (Scheme 102), which without purification, on treatment with DBU furnished the anhydro intermediate 607 in 52% overall yield. Its treatment with KOtBu in THF produced olefin 608 (82% yield) which on acyl deprotection with NaOMe yielded 577 (d4T) in 95% yield.
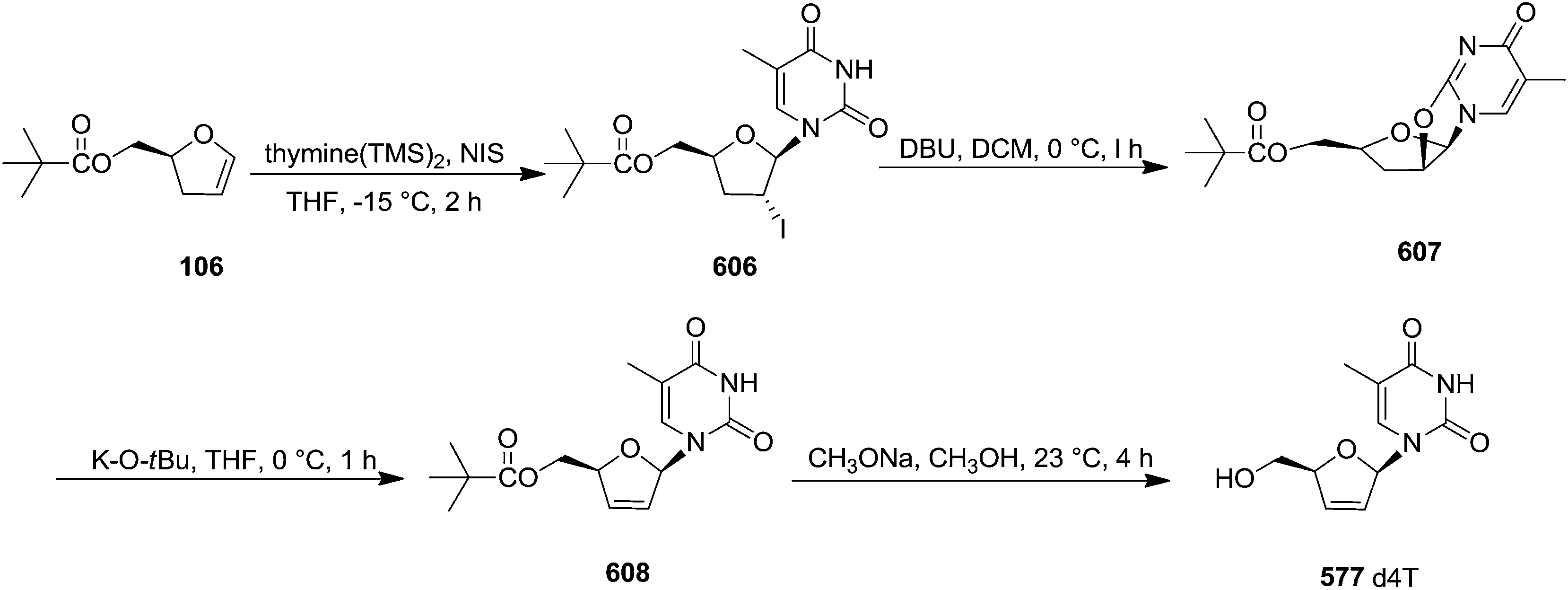 | ||
| Scheme 102 | ||
Liotta et al. described the synthesis of nucleosides via region- and stereoselective electrophilic addition to furanoid glycals 610 which was prepared from lactone 609 in a straightforward fashion.79 The DIBAL-H reduction of 609 followed by addition of SOCl2 and Et3N to the corresponding lactol afforded furanoid glycal 610. Its exposure to an appropriate source of electrophilic sulfur in the presence of a silylated nucleoside base and Lewis acids produced the pyrimidine and purine derivatives 611 in a stereo- and regioselective fashion by judicious choice of the Lewis acid, solvent and temperature (Scheme 103, Table 11).
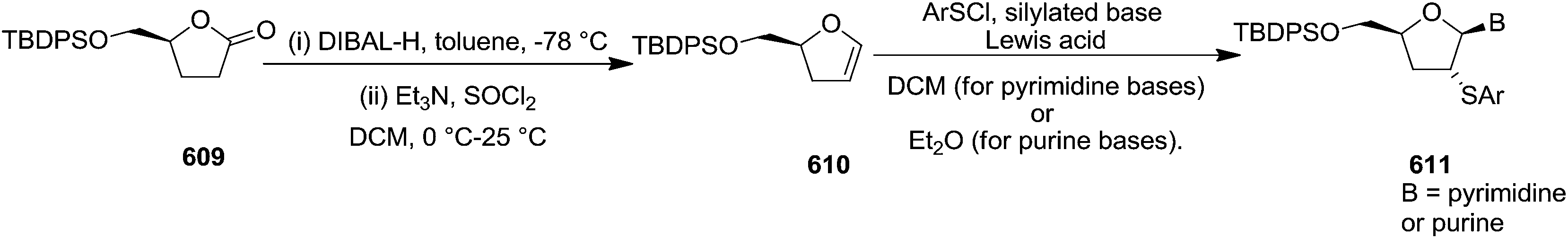 | ||
| Scheme 103 | ||
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | ArSCl | Lewis acida | Silylated base | Conditionsb (°C) | Ratio (β:α) |
Time (h) | Yield (%) |
| a The reaction utilized 1 .15 eq. of Lewis acid.b (i)−78 to 0 to 25 °C: after addition of the sulfenyl chloride, the reaction was kept at −78 °C for 30 min and then warmed to 0 °C. The silylated base and Lewis acid were then introduced, and the reaction was allowed to warm to room temperature (25 °C). (ii) −78 to 25 °C: after addition of the sulfenyl chloride, the reaction was kept at −78 °C for 30 min. The silylated base and Lewis acid were then introduced and the reaction was allowed to warm to room temperature. (iii) The solvent of choice for pyrimidine bases was DCM and diethyl ether for purine bases. TIPP = 2,4,6-triisopropylphenyl. | |||||||
| 1 | PhSCl | SnCl4 | N-Ac-Cytosine | −78 to 25 | 18:1 |
2 | 65 |
| 2 | PhSCl | TMSOTf | N-Ac-Cytosine | −78 to 0 to 25 | 5:1 |
1.5 | 60 |
| 3 | TIPPSCl | SnCl4 | N-Ac-Cytosine | −78 to 25 | 23:1 |
2 | 70 |
| 4 | TIPPSCl | TMSOTf | N-Ac-Cytosine | −78 to 0 to 25 | 6:1 |
2 | 73 |
| 5 | PhSCl | SnCl4 | Thymine | −78 to 25 | 42:1 |
2 | 68 |
| 6 | PhSCl | TMSOTf | Thymine | −78 to 0 to 25 | 4:1 |
2 | 50 |
| 7 | TIPPSCl | SnCl4 | Thymine | −78 to 25 | 44:1 |
2 | 60 |
| 8 | TIPPSCl | SnCl4 | Uracil | −78 to 25 | >99:1 |
2 | 52 |
| 9 | TIPPSCl | TMSOTf | 6-Cl-Purine | −78 to 25 | 5:1 |
5 | 80 |
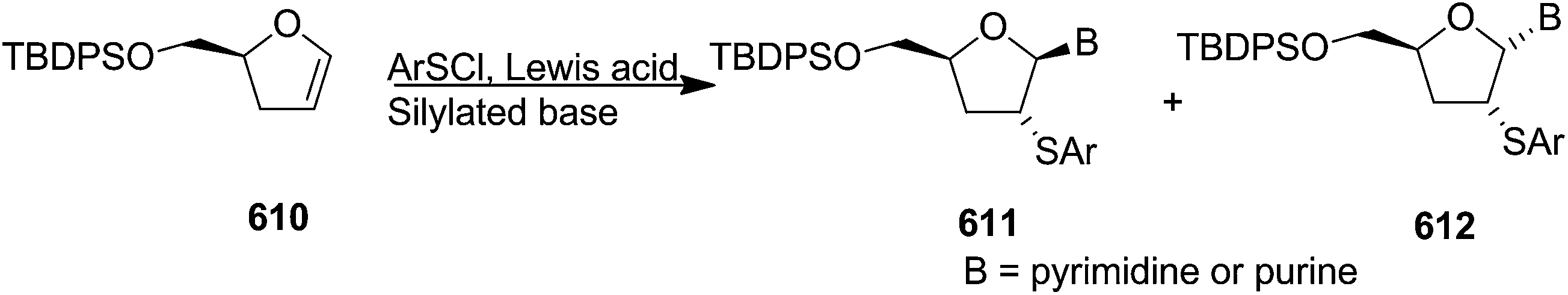
In 1993, Castillón and co workers showed selenium controlled stereoselective synthesis of a series of 2′-deoxy nucleosides and a formal synthesis of 3′-azido-3′-deoxythymidine (AZT, 576) and 3′-fluoro-3′-deoxythymidine (FDT, 621), a powerful anti HIV agent, starting from furanoid glycals. Furanoid glycals 139 and 132c were derived from 2,3:5,6-di-O-isopropyliden-manno furanose31 and 2,3-O-isopropylidene-lyxo-furanose30 respectively.10 These glycals (139 and 132c) on treatment with PhSeCl in presence of AgOTf followed by glycosylation with pyrimidine bases in non polar solvents accomplished 2′-deoxy-2′-phenylselenenyl nucleosides 613–615. In all cases, removal of the phenylselenenyl group was carried out by reaction with nBu3SnH in refluxing toluene to afford 2′-deoxynucleosides 616–618 in 80–90% overall yields from the glycal. Hydrogenolysis of compounds 617 and 618 using Pd/C as the catalyst gave the unprotected nucleosides 619 and 620 in quantitative yield (Scheme 104).
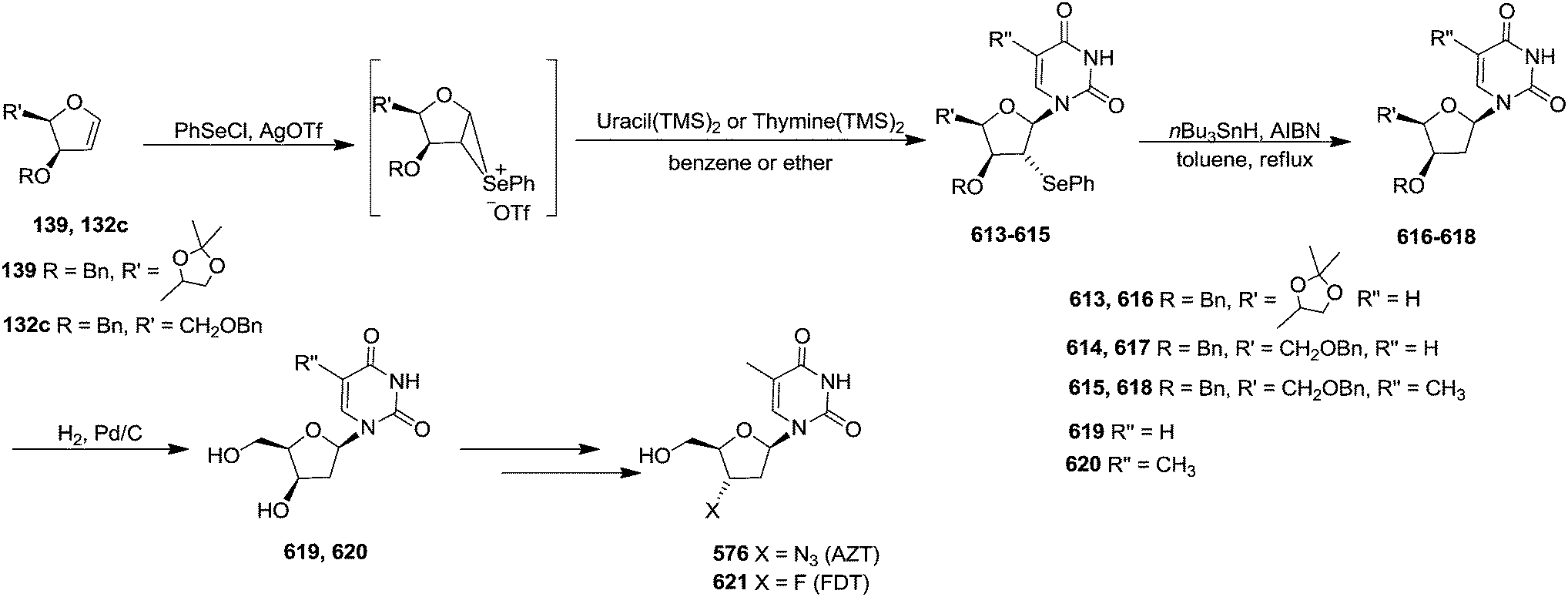 | ||
| Scheme 104 | ||
In 1995, Florent and coworkers also showed the utilization of furanoid glycal 97a (synthesized from α-D-isosaccharino-1,4-lactone 93, Scheme 16) for the synthesis of 2′,3′-dideoxy-2′-C-methylidene-5-methyl uridine 623, 3′-deoxy analog of DMDC 626 and 627.27 The glycosylation of bis-(trimethylsilyl)thymine with crude furanoid glycal in the presence of Pd2(dba)3 and PPh3 gave nucleoside 622 in 25% overall yield. Deprotection by ammonolysis led to the 2′,3′-dideoxy-2′-C-methylidene-5-methyl uridine 623 (80% yield). Glycosylation of silylated cytosine with 97a under the identical reaction conditions afforded β-nucleoside 624 along with the corresponding α-anomer 625 (40% overall yield and ratio β:α = 8:2) after column purification of the crude product mixture. Removal of the acetyl group produced 3′-deoxy analog of DMDC 626 and 627 (Scheme 105).
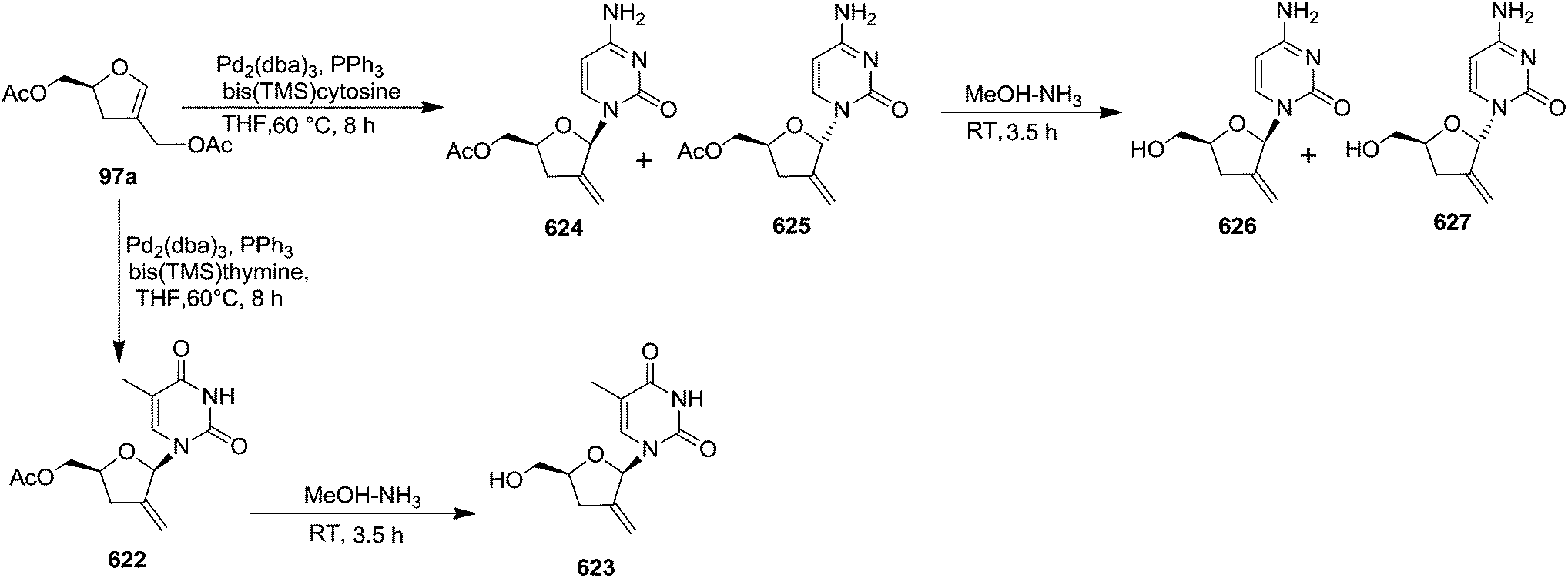 | ||
| Scheme 105 | ||
McDonald and coworkers synthesized Stavudine (d4T, 577), an anti-AIDS compound,80 from allyl alcohol 103 derived furanoid glycal key intermediate 106, whose synthesis already described in Scheme 18.29,80 Iodine-mediated addition of (TMS)2thymine to 106 gave iodonucleoside 606 which on without further purification treated with freshly prepared NaOMe to accomplish stavudine 577 (d4T) (Scheme 106).
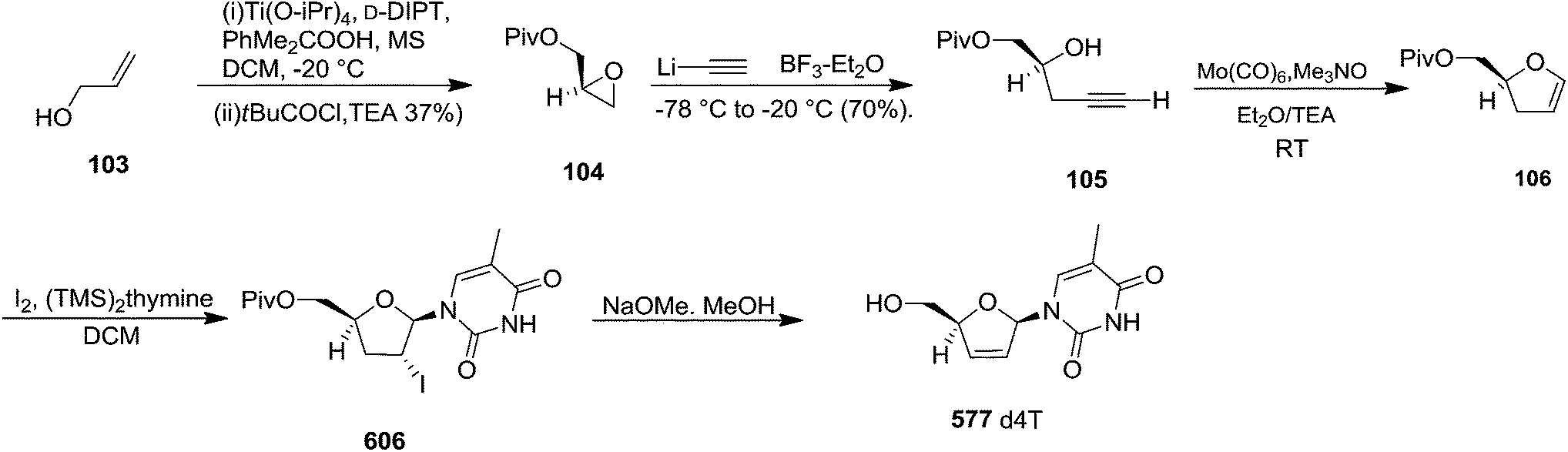 | ||
| Scheme 106 | ||
Dihydroxylation of 106 with OsO4 in presence of NMO followed by acylation of the crude diol provided a 13:4:3:1 mixture of diacetylated products favouring 628 (Scheme 107). This mixture was subjected to Lewis acid catalyzed adenine glycosylation to give a 9:1 mixture of stereoisomers favouring 629. Methanolysis of acyl groups followed by column purification produced the synthetic cordycepin 630 (Scheme 107).
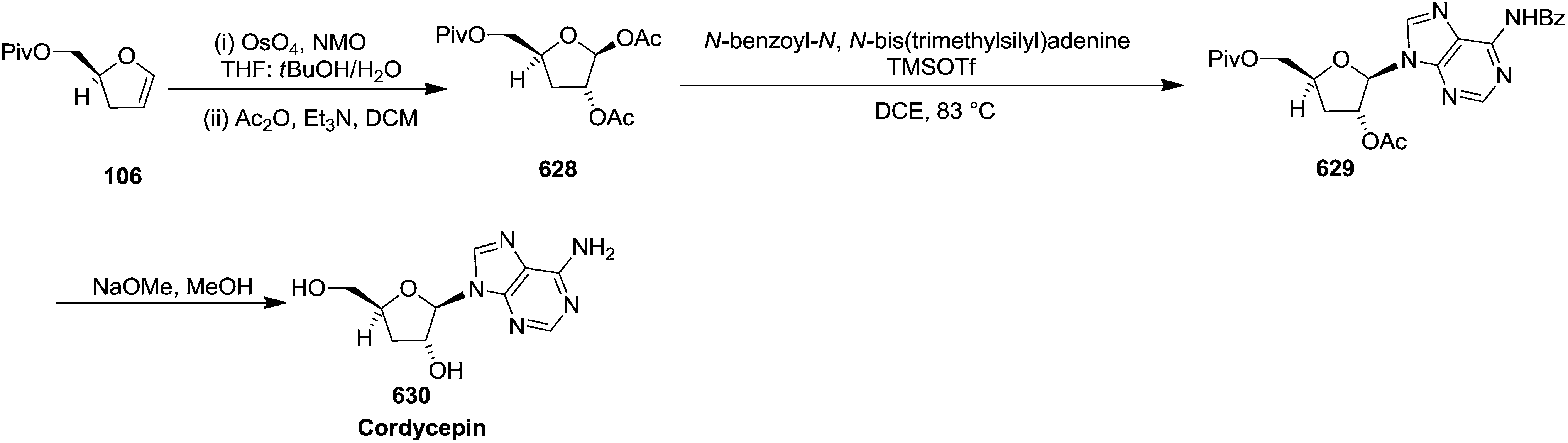 | ||
| Scheme 107 | ||
After reporting the synthesis of d4T 577 and cordycepin 630 from 106, McDonald and Gleason then optimized the glycosylation of 3-amidofuranose glycal 121 or 122 (whose synthesis already discussed in Scheme 19, Table 4) with pyrimidine and purine bases and found that reaction of 121 with CF3SO3H and silylated thymine at room temperature with acetonitrile as solvent afforded predominantly the β-nucleoside 631T (Scheme 108, Table 12, entry 1).29 Treatment of 121 with acetic acid gave a more highly reactive glycosyl donor 632, which underwent high-yielding TMSOTf-induced glycosylation with silylated pyrimidine bases in the presence of acetonitrile to afford the desired β-nucleoside 631T, U, C (Table 12, entries 2–4). They obtained similar results by using CF3SO3H as the activating agent (Table 12, entries 5 and 6); possibly CF3SO3H was also generated by in situ hydrolysis of trimethylsilyl trifluoromethanesulfonate.
 | ||
| Scheme 108 | ||
| Entry | Silylated base | Glycosyl donar | Conditions | Nucleoside, isolated yield (β:α ratio) |
|---|---|---|---|---|
| a (Method a) CF3SO3H, silylated base, 3 Å MS, MeCN, 20 °C (entry 1); (Method b) TMSOTf, silylated base, 3 Å MS, MeCN, 0 °C to 20 °C (entries 2–4); (Method c) CF3SO3H, silylated base, 3 Å MS, MeCN, 20 °C (entries 5, 6); (Method d) NIS, TfOH, silylated base, 3 Å MS, MeCN, −40 °C (entry 7). | ||||
| 1 | (TMS)2-thymine | 121 | a | 631T, 50% (>20:1) |
| 2 | (TMS)2-thymine | 632 | b | 631T, 85% (4.7:1) |
| 3 | (TMS)2-uracil | 632 | b | 631U, 85% (21:1) |
| 4 | N–Ac(TMS)2-cytosine | 632 | b | 631C, 77% (8.7:1) |
| 5 | (TMS)2-thymine | 632 | c | 631T, 87% (8.4:1) |
| 6 | N–Ac(TMS)2-cytosine | 632 | c | 631C, 84% (3.3:1) |
| 7 | N-Bz(TMS)2-adenine | 633 | d | 631A, 42% (>10:1) |
For the addition of purine bases, they treated the more reactive thioglycoside donor 633 obtained from 121 with NIS and CF3SO3H at significantly lower temperature giving the purine β-nucleoside 631A with high stereoselectivity (Table 12, entry 7). The deblocking of ester and amide protective groups of 631T with NaOMe in MeOH gave the 3′-amino-2′,3′-dideoxythymidine 634T.
In the case of guanine glycosylations, the reaction of 633 under kinetic conditions gave primarily the N-7 regioisomer 631G* as the major nucleoside product (Scheme 109, Table 13, entry 1). They observed that when glycosylation of guanine with 632 was carried out at room temperature, the proportion of N-9 regioisomer 631G increased along with α-nucleoside isomers (entry 2) and were the major product when the glycosylation was conducted in refluxing acetonitrile (entry 3).
 | ||
| Scheme 109 | ||
| Entry | Glycosyl donar | Condition | Nucleosides, combined yield | Relative ratio of products 631G:631G*:α-nucleosides:121 |
|---|---|---|---|---|
| a (Method a) N–Ac(TMS)3-guanine, NIS, TfOH, EtCN, 3 Å MS, −78 °C to 0 °C, 2 h (entry 1); (Method b) N–Ac(TMS)3-guanine, TMSOTf, MeCN, 3 Å MS, 20 °C, 3 h (entry 2); (Method c) N–Ac(TMS)3-guanine, TMSOTf, MeCN, 3 Å MS, 81 °C, 1.5 h (entry 3). | ||||
| 1 | 633 | a | 35% | 1.0:7.3:0:0 |
| 2 | 632 | b | 38% | 3.4:1.4:1.0:0 |
| 3 | 632 | c | 50% | 2.2:1.0:12:3.2 |
Epoxidation of 121 with peroxyacids gave 635 (Scheme 110). Acylation of free hydroxyl group yielded 636 which permitted trans-glycosylation under Lewis acid conditions to give purine β-nucleosides including 637. Basic methanolysis of 637 yielded the deprotected puromycin aminonucleoside 638.
 | ||
| Scheme 110 | ||
Peroxyacetic acid epoxidation followed by acetylation of 3-trifluoroacetamide glycal 122 furnished diacetate 640 via 639 (Scheme 111). Under the thermodynamic conditions of these glycosylations, the naturally occurring N-9 regioisomers 641A, 641G, and 641A′ were the major products (Table 14, entries 1–3).
 | ||
| Scheme 111 | ||
| Entry | Silylated base | Glycosyl donar | Conditions | Nucleoside, isolated yield |
|---|---|---|---|---|
| a (a) silylated base, TMSOTf, 3 Å MS, DCE, 83 °C, (entries 1–3); (b) silylated base, MeCN, 20 °C, aq. AcOH/THF work up (entries 4–7). | ||||
| 1 | N-Bz(TMS)2-adenine | 640 | a | 641A, 90% |
| 2 | N-Ac(TMS)3-guanine | 640 | a | 641G, 77% (10:1) |
| 3 | N,N–Me2(TMS)-adenine | 640 | a | 641A′, 71% |
| 4 | (TMS)2-uracil | 642 | b | 641U, 80% |
| 5 | (TMS)2-thymine | 642 | b | 641T, 86% |
| 6 | N-Ac(TMS)2-cytosine | 642 | b | 641C, 71% |
| 7 | N-Bz(TMS)2-adenine | 642 | b | 641A, 16% |
DMDO epoxidation of 122 was also directed by the amide when epoxidation was conducted in solvent DCM. The crude glycal epoxide 642 reacted stereospecifically with silylated pyrimidine bases to give 641T, U, C in good yields (Scheme 111, Table 14, entries 4–6), whereas in the case of pyrimidine bases, benzoyladenine gave 641A in a very low isolated yield (Table 14, entry 7).
In 1997, Castillón et al. also reported the formation of 2′,3′-dideoxy nucleosides by electrophilic addition of selenium to furanoid glycal 610, which was synthesized from 2-deoxyribose 67.81 They further discussed the synthesis of d4T 577 via precursor 656 through selenium-mediated glycosylation and selenoxide elimination.
Thus, 2-deoxyribose 67 was converted into the phenyl-1-seleno-glycoside 645 (mixture α/β = 1.9:1) in four steps involving methyl glycoside synthesis, selective 5-OH protection, Barton deoxygenation, and treatment with PhSeH in the presence of BF3·OEt2. Oxidation of 645 gave glycal 610 with a yield of 52%. The reaction of glycal 610 with (TMS)2uracil, PhSeCl, and AgOTf at room temperature in ether led to a mixture of 2′-phenylselenenyl nucleosides 648 and 649 in a 99:1 ratio in good yield. Similarly, they treated glycal 610 with (TMS)2thymine to obtain 2′-phenylselenenyl nucleosides 650 and 651 with a ratio of 90:10 in excellent yields.
Under identical reaction condition, the reaction of glycal 610 with silylated 6-chloropurine showed a lower stereoselectivity (ratio 652/653, 3:1). In order to increase the β-stereoselectivity of the glycosylation step, they synthesized glycal 647 in a similar way the glycal 610 was synthesised. In this case a mixture of 654/655 (β/α = 89:11) was obtained in a yield of 78% (Scheme 112).
 | ||
| Scheme 112 | ||
Compound 650 on oxidative elimination afforded the corresponding didehydro derivative 656 in 85% yield by using tBuOOH/Ti(OiPr)4 as an oxidative system; the deprotection of the TBDPS group gave d4T 577 (Scheme 113).
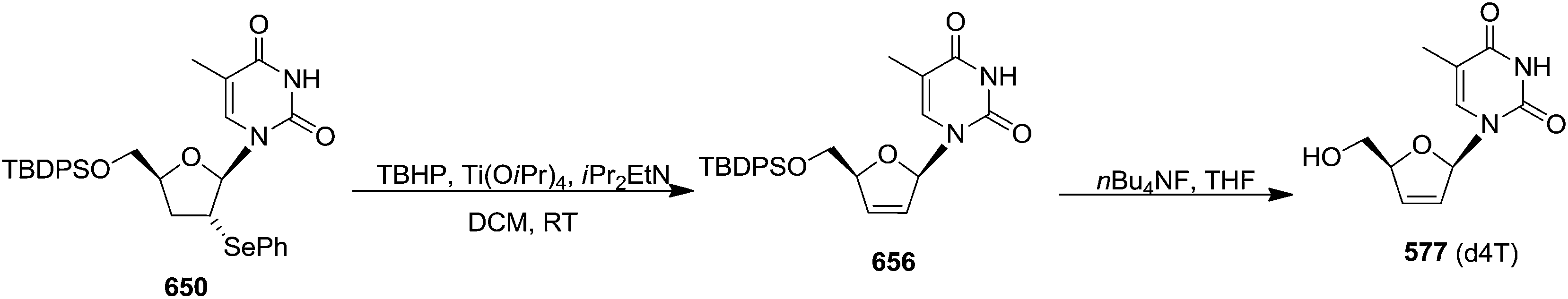 | ||
| Scheme 113 | ||
In the same year, this group also reported stereoselective synthesis of 2′-deoxy-2′-phenylselenenyl nucleosides from furanoid glycals in a “one pot” reaction and efficiently converted them into 2′-deoxy nucleosides.83 They also showed the stereoselectivity of the reaction was affected by some of the factors such as stereochemistry at position 3, the nature of the protecting groups, the phenylselenenyl reagent and the solvent. For this purpose they synthesized a series of furanoid glycals. Glycal 139 and 214 (ref. 5) from D-mannose derived furanoid glycal 26. The glycal 132c was prepared from D-mannose by degradation of the side chain, in a similar way to 26,21 or from 2-deoxyribose 67.24
Treatment of 139 with PhSeCl and (TMS)2uracil in the presence of AgOTf in ether at room temperature yielded β-gluco nucleoside 613 and α-gluco nucleoside 657 in 81% yield (ratio 613/657 = 90:10) (Scheme 114, Table 15, entry 1).
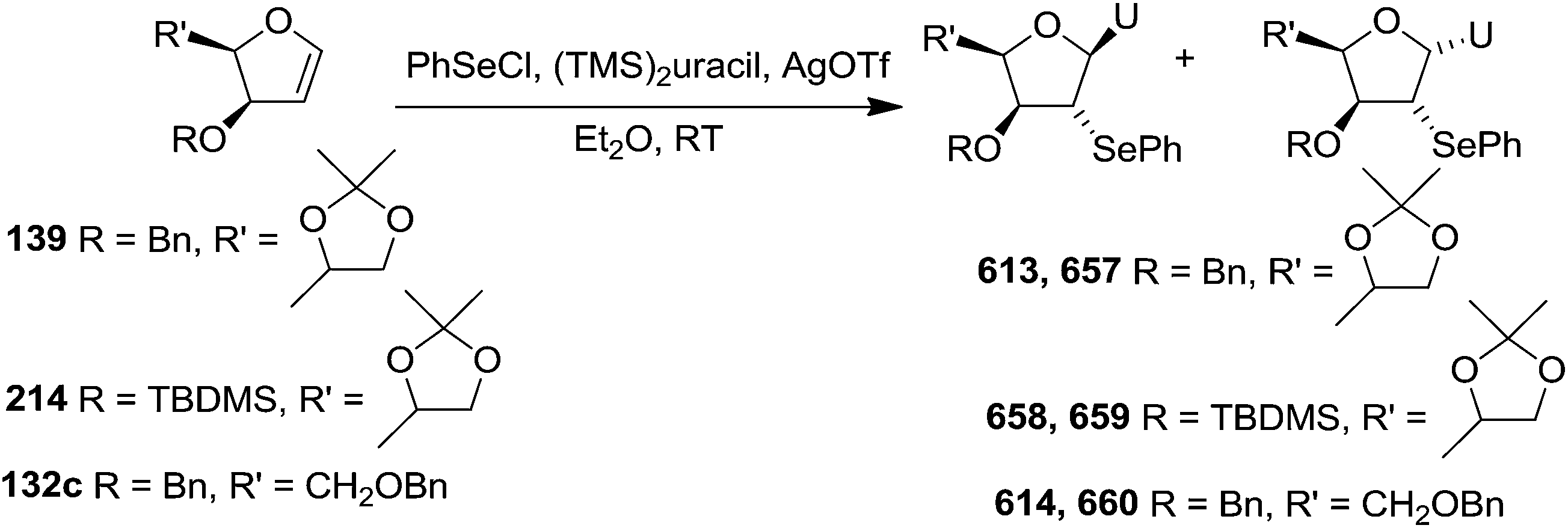 | ||
| Scheme 114 | ||
| Starting glycals | Time (h) | Yieldb (%) | 2′-Selenenyl nucleosidesc (diastereomeric ratio) | |
|---|---|---|---|---|
| a Reactions were carried out using the molar ratio glycal/PhSeCl/AgOTf/Uracil(TMS)2 = 1/1.5/1.7/2.b Expressed as a percentage of recovered mixture of products after chromatography.c Determined by integration of the H-1′ protons in the 1H NMR spectrum of the reaction mixture. | ||||
| 139 | 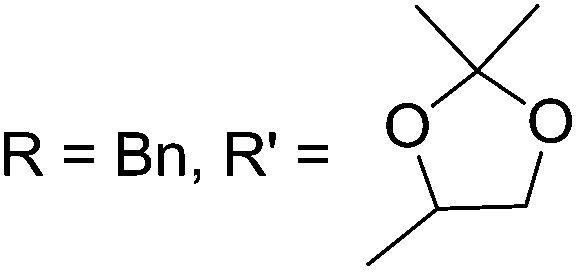 |
1 | 81 | β-Gluco 613:α-Gluco 657 (90:10) |
| 214 | 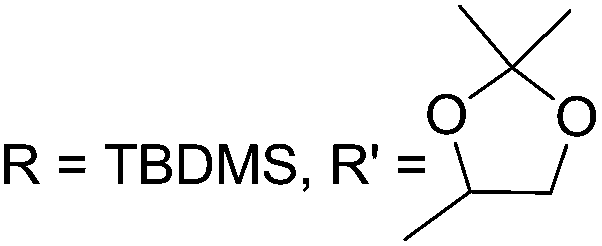 |
1 | 95 | β-Gluco 658:α-Gluco 659 (86:14) |
| 132c | R = Bn, R′ = BnOCH2 | 0.5 | 90 | β-xylo 614:α-xylo 660 (91:9) |
Under the identical reaction condition treatment of glycal 214 and 132c afforded 2′-deoxy-2′-phenylselenenyl nucleosides β-gluco 658/α-gluco 659 (86:14) in 95% yield and β-xylo 614/α-xylo 660 (91:9) in 91% yield respectively (Table 15, entries 2 and 3).
To show the stereoselectivity in the formation of 2′-deoxy-2′-phenylselenenyl nucleosides derived from erythro configured furanoid glycals, they synthesized glycals 661, 662, 66e and 665 (Table 16) from D-ribonic-γ-lactone18 and 71a, 90, 663, 664 and 66j from 2-deoxyribose.24,28 For the glycals (661, 662, 66e, 71a, 90, 663, 664, 665 and 66j) with an erythro configuration, stereoselectivity was seen to depend on the protecting groups at positions 3 and 5 (Scheme 115, Table 16).
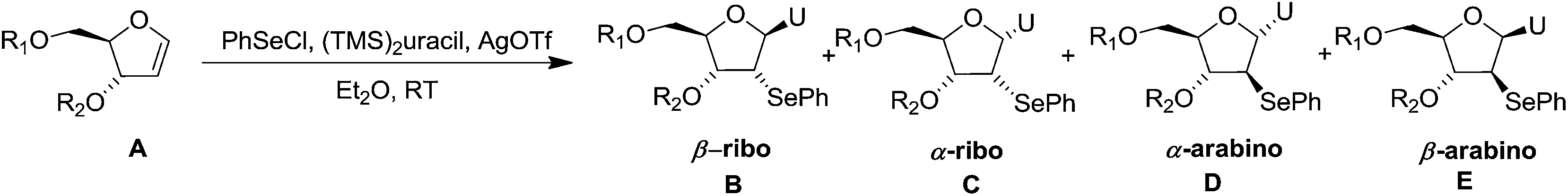 | ||
| Scheme 115 | ||
| Starting glycals | Time (h) | Yield (%) | 2′-Selenenyl nucleosides | (Diastereomeric ratio) (β-ribo:α-ribo:α-arabino:β-arabino) B:C:D:E |
|||
|---|---|---|---|---|---|---|---|
| a Reactions were carried out at room temperature using the molar ratio glycal/PhSeCl/AgOTf/Uracil(TMS)2 = 1/1.5/1.7/2.b R = SePh stands for selenenylation of nucleosides at position 5. | |||||||
| 661 R1 = MEM, R2 = Bn | 2 | 82 | 666 R1 = MEM, R2 = Bn | 14 | — | 49 | — |
| 666a R1 = OH, R2 = Bn | — | — | 37 | — | |||
| 662 R1 = TBDMS, R2 = Bn | 1.5 | 87 | 667 R1 = TBDMS, R2 = Bn | 32 | 16 | 16 | — |
| 667a R1 = OH, R2 = Bn | 18 | 5 | 13 | — | |||
| 66e R1 = TBDMS, R2 = TBDMS | 1.5 | 88 | 668 R1 = TBDMS, R2 = TBDMS | 28 | 21 | 15 | — |
| 668a R1 = OH, R2 = TBDMS | 22 | 8 | 6 | — | |||
| 71a R1 = Bn, R2 = Bn | 1 | 89 | 669 R1 = Bn, R2 = Bn | 30 | — | 70 | — |
| 90 R1 = TBDPS, R2 = Bn | 2 | 58 | 670 R1 = TBDPS, R2 = Bn | 44 | 9 | — | — |
| 670a R1 = TBDPS, R2 = Bn, R = SePhb | 19 | 8 | 6 | 14 | |||
| 663 R1 = Bn, R2 = TBDPS | 2 | 85 | 671 R1 = Bn, R2 = TBDPS | 43 | 11 | 32 | 14 |
| 664 R1 = TBDPS, R2 = MEM | 2 | 83 | 672 R1 = TBDPS, R2 = MEM | 54 | 18 | 15 | 4 |
| 672a R1 = TBDPS, R2 = MEM, R = SePhb | 9 | — | — | — | |||
| 665 R1 = Ac, R2 = TBDPS | 2 | 84 | 673 R1 = Ac, R2 = TBDPS | 23 | 14 | 16 | 6 |
| 673a R1 = Ac, R2 = TBDPS, R = SePhb | 26 | 5 | 5 | 5 | |||
| 66j R1 = TBDPS, R2 = TBDMS | 2 | 87 | 674 R1 = TBDPS, R2 = TBDMS | 66 | 20 | 14 | — |
They further synthesized 2′-deoxy nucleosides from 2′-deoxy-2′-phenylselenenyl nucleosides viz. β-gluco nucleoside 613, β-xylo 614, and α-arabino 669 and β-ribo 672 by their treatment with nBu3SnH and AIBN in refluxing benzene to give 2′-deoxy nucleosides 616, 617 and 675, 676 respectively (Scheme 116).
 | ||
| Scheme 116 | ||
In 1997, Robles and coworkers have shown synthesis of a series of 2′-deoxy-2′-iodo nucleosides (678–684), from furanoid glycals (133c, 76b, 132c and 136c), which were synthesized from differently O-protected D-xylo 133a, 677, 132a and D-gluco 136a configured furanoid 1,2-diols respectively on treatment with I2/PPh3/imidazole. NIS-mediated glycosylation of furanoid glycals (133c, 76b, 132c and 136c) with pyrimidine bases afforded 2′-deoxy-2′-iodo nucleosides (678–684) (Scheme 117).84
 | ||
| Scheme 117 | ||
In 1999, Kim and coworker described stereoselective synthesis of 1′-β-2′,3′-dideoxy-2′-bis(ethoxycarbonyl)methyluridine nucleosides (687a–e) and (688a–e) in good yields from furanoid glycals 404 and 610. Cyclopropanation of furanoid glycals 404, 610 with diethyl diazomalonate and dirhodium tetraacetate (N2C(CO2Et)2:Rh2(OAc)4:glycal = 2:0.01:1) afforded stereoselectively cyclopropanated sugars 685 and 686 respectively. Lewis acid mediated glycosylations of 685 and 686 with 5-substituted uracils afforded 1′β-2′,3′-dideoxy-2′-bis(ethoxycarbonyl)methyluridine nucleosides (687a–e) and (688a–e) respectively in good yields (Scheme 118).85
 | ||
| Scheme 118 | ||
In 1999, Paquette and group showed furanoid glycals which are amenable to C-5 metalation in the presence of tBuLi, were readily coupled to N-protected 2,3-azetidinediones.86 L-Glutamic acid derived79 (S)-(+)-dihydro-5-(hydroxymethyl)-2-(3,4)-furanone 599 was converted to 689 and 690 by TrCl and TBDMSCl respectively. Their DIBALH reduction followed by acetylation of the resulting lactols yielded 691 and 692 respectively whose vacuum pyrolysis in a Kugelrohr apparatus afforded 693 and 270 respectively (Scheme 119). Also, exposure of the lithium derivative of 693 to excess nBu3SnCl afforded 694 in 60% yield. They also treated 270 with KOtBu in the presence of PhSCl and PhSeCl to afford 695 and 696 respectively.
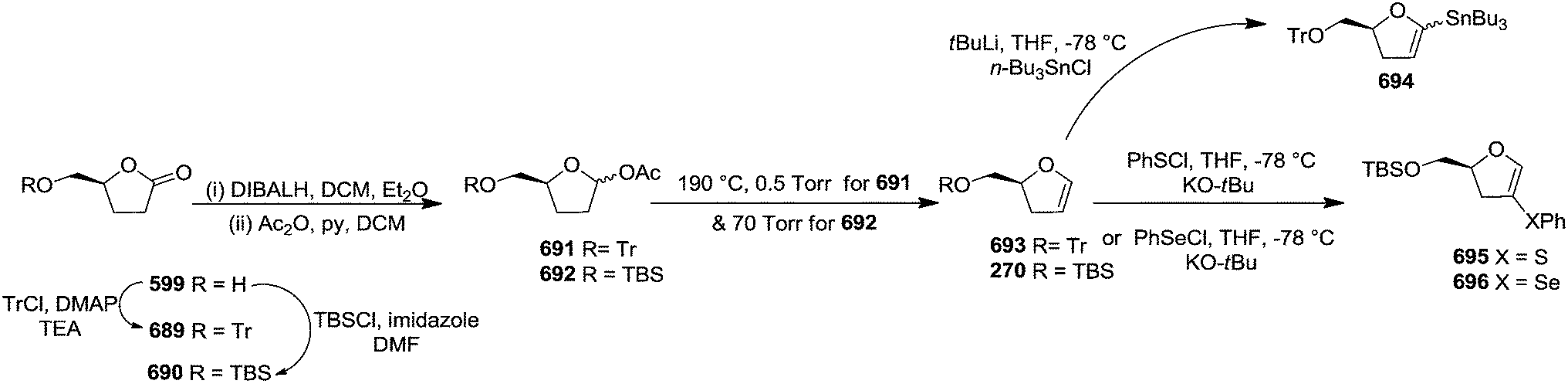 | ||
| Scheme 119 | ||
These furanoid glycals (693, 270, 404, 695, 696 and 610) in the presence of tBuLi were readily coupled to N-protected 2,3-azetidinediones (697 and 698) at low temperature in THF containing BF3·Et2O to give the desired epimeric mixture of carbinols, i.e. (699–704) respectively. Treatment of (699–703) with pyridinium p-toluenesulfonate (PPTS) in benzene afforded spirocyclic keto amides (705, 706, 707, 708, 709, 710, 711) respectively (Scheme 120).
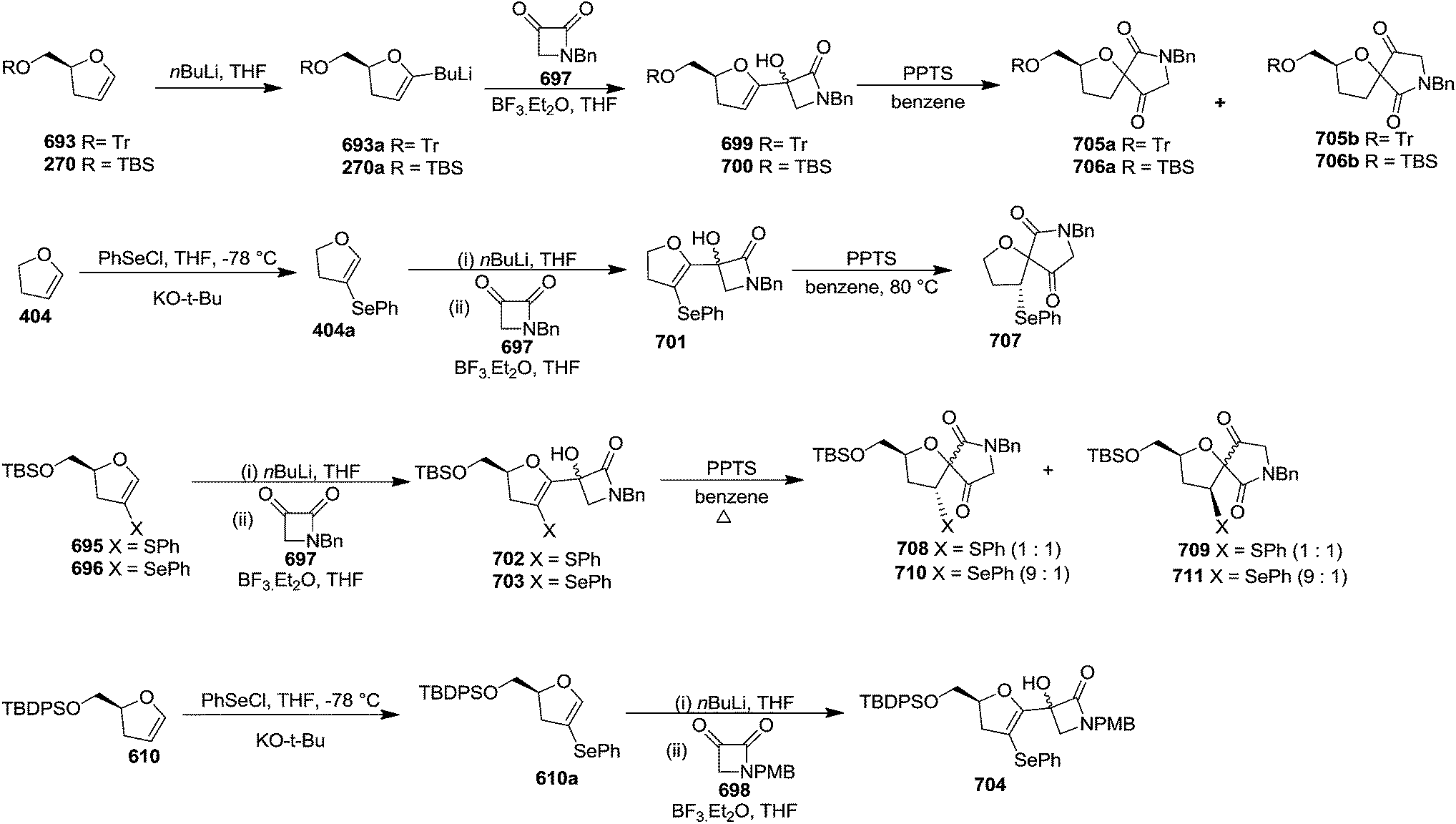 | ||
| Scheme 120 | ||
In 2001 Quirion and group reported the synthesis of 2′-deoxy-2′-difluoromethyluridine 716.87
They described two methods A and B for its synthesis. One of them was started from thymidine 64a which was converted into benzylated furanoid glycal 71a (56% overall yield) in two steps involving the treatment of 64a with an excess of HMDS in the presence of (NH4)2SO4 followed by benzylation with BnBr of the resulting 65a. Then they applied Miethchen method88 on 71a to obtain 712 which was acetylated to 713 (47% yield from 71a). It was then converted to 714 via a radical reductive process (nBu3SnH, AIBN). Addition of (TMS)2uracil to 714 in the presence of TMSOTf furnished a 4:1 mixture of isomeric nucleosides in 76% yield. The major one was 715α (NMR) whose hydrogenolysis afforded 2′-deoxy-2′-difluoromethyluridine 716α (Scheme 121, method A).
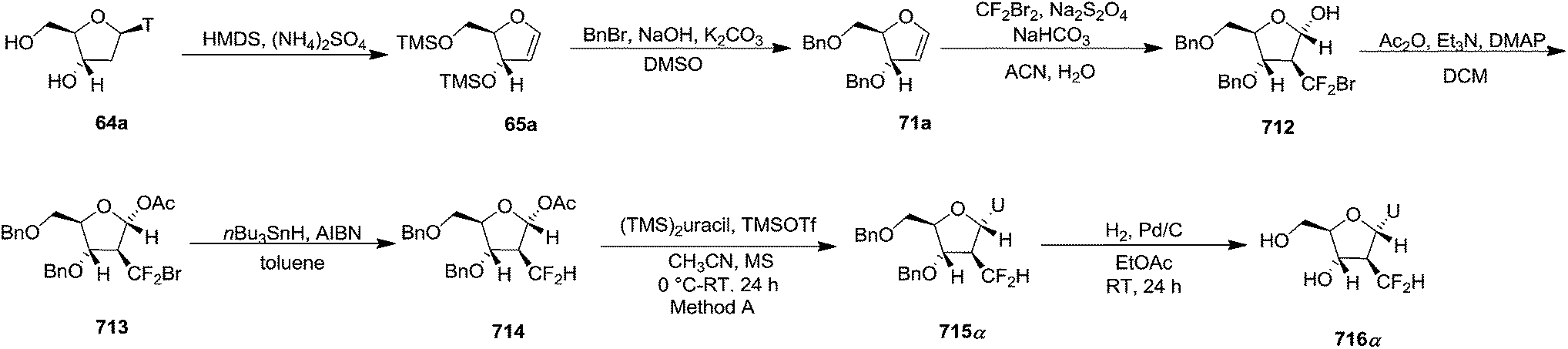 | ||
| Scheme 121 | ||
In order to change the α/β ratio in favour of β, they synthesized α-halodeoxyarabinose 717 from 714 on treatment with HCl. The condensation of 717 in DCM with (TMS)2uracil gave a 57:43 mixture of 715β and 715α isomers (NMR), respectively, via a SN2 type reaction. Finally, the deprotection of the benzyl groups was easily achieved by hydrogenolysis of 715β and 715α to give the two desired nucleosides 716β and 716α with good yields (Scheme 122, method B).
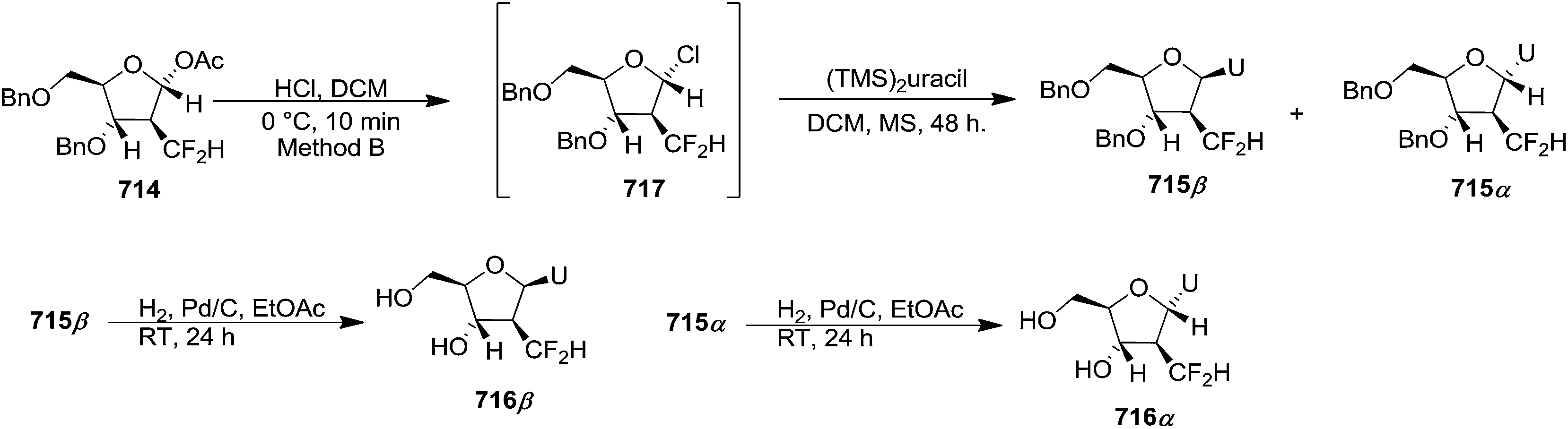 | ||
| Scheme 122 | ||
In 2005, Choudhury and Pierce et al. synthesized D-D4FC 724 from an aromatization prone xylo-furanoid glycal 721, by development of a palladium mediated Ferrier rearrangement-type glycosidation.89 For the synthesis of xylo-furanoid glycal 721, they chose commercially available 1,2-isopropylidine D-(+)-xylofuranose 718, as the starting material. The free hydroxyl groups of 718 were then protected with p-anisoyl chloride in pyridine to afford 719. The acetonide deprotection followed by treatment of the diols 720 with I2/resin bound Ph3P/imidazole afforded xylo-furanoid glycal 721 in more than 90% yield. After several trial and error for the choice of solvent, base and different mol% of Pd(Ph3P)4, the glycosylation reaction of glycal 721 with an unprotected nucleoside base fluoro cytosine 722 was optimized with DBU as the base, NMP as the solvent with 3 mol% of Pd(Ph3P)4 as the catalyst at 33 °C for 2 days to afford 5′-anisoyl-D-D4FC 723 in good isolated yield. Deprotection of 723 provided the D-D4FC 724 in 82% yield (Scheme 123).
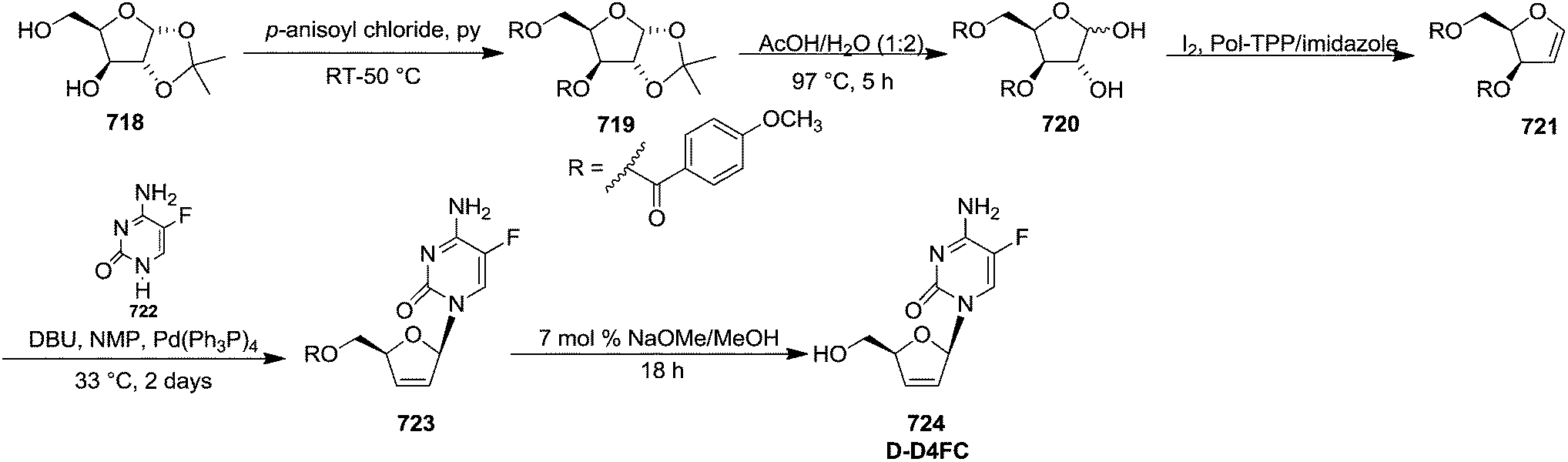 | ||
| Scheme 123 | ||
In 2006, Lequeux and group reported an alternative strategy based on group transfer reaction of S-alkyl dithiocarbonates (xanthates) followed by substitution reactions to prepare 2,3-trans disubstituted tetrahydrofuran derivatives.90 They described the additions of alkyl radicals to 2,3-dihydrofuran derivatives using S-alkyl dithiocarbonates and the nucleophilic displacements of the resulting anomeric S-alkyl dithiocarbonate function by various nucleophiles in the presence of Lewis acid to form a new carbon–carbon or carbon–heteroatom bond which presented a new route for the preparation of modified 2′-β-C-branched nucleoside analogues.
To achieve the goal, first they synthesized xanthate 726 following known procedure from commercially available ethyl bromofluoroacetate 725 (Scheme 124) by a nucleophilic substitution of its bromine atom with O-ethyl potassium dithiocarbonate.91
 | ||
| Scheme 124 | ||
Then they studied the ethyl fluoroacetate group transfer reaction with the slow addition of lauroyl peroxide (30%) over 1 h in the mixture of different terminal alkenes (1.1 equiv.) and xanthate 726 in refluxing DCM (Scheme 124) to form fluoroesters (727a–d) in fair to good yields as a mixture of diastereomers (1:1 ratio).
They further repeated the same reaction (described for Scheme 124) with cyclopentene 728 and 2,3-dihydrofuran 729 in the presence of lauroyl peroxide. The best results were obtained from these alkenes (728, 729) and the fluoroxanthate 726 (Scheme 125, Table 17, entries 1 and 2, method A) when lauroyl peroxide was added slowly to a mixture of alkene and xanthate (method A). In case of cyclopentene (X = CH2) 728, only two diastereomers were detected in a 1:1 ratio, and purification of the crude product afforded 2,3-trans disubstituted cyclopentane derivatives 730 in 79% yield. Similar results were observed from the 2,3-dihydrofuran (X = O) 729 and xanthate 726, and a mixture of two trans diastereomers of 731 (2,3-trans disubstituted tetrahydrofuran derivatives, 1H{19F} HOESY) was obtained in 81% yield. These two isomers of 731 differed only by the relative configuration of the third stereogenic center bearing the fluorine atom. The cis products were not reported in both cases (cyclopentene and 2,3-dihydrofuran). 2,3-trans Isomers 732 were obtained from 729 in the case of xanthate 734 in 30% isolated yield (Scheme 125, Table 17, entry 3, method A).
 | ||
| Scheme 125 | ||
| Entry | Alkene | Xanthate R1OC(S)SR2 | Method | Yield (%) | Trans/cis dr | Product |
|---|---|---|---|---|---|---|
| a Method (A): slow addition of 0.3 equiv. of lauroyl peroxide, refluxed DCE, 1–2 h. Method (B): addition of 3 × 0.1 equiv. of Et3B, DCM, room temperature, 1–2 h.b Stirring was maintained overnight at room temperature. | ||||||
| 1 | X = CH2 | 726: R1 = Et, R2 = CHFCO2Et | A | 79 | >98:2 |
730 |
| B | 67 | |||||
| 2 | X = O | 726 | A | 81 | >98:2 |
731 |
| B | 73 | |||||
| 3 | X = O | 734: R1 = Ph(CH2)2, R2 = CF3 | A | 30 | >98:2 |
732 |
| Bb | 52 | |||||
| 4 | X = O | 735: R1 = Et, R2 = CH2CN | B | 55 | 9:1 |
733 |
They also repeated the reaction with Et3B, as an alternative free radical initiator of lauroyl peroxide, in deoxygenated DCM under N2 atmosphere (method B) to afford 2,3-trans products 730 in 67% isolated yield from 728 and xanthate 726 (Table 17, entry 1, method B). While 2,3-trans disubstituted furans 731 were isolated in 73% yield from 2,3-dihydrofuran 729 and xanthate 726 (Table 17, entry 2, method B), the reaction between 729 and the trifluoromethylxanthate 734 afforded the trans adduct 732 in 52% yield (Table 17, entry 3, method B). The reaction proceeded smoothly with xanthate 735 and afforded the 2,3-trans isomers 733 as major products in 55% yield (Table 17, entry 4, method B).
Afterward, they explored the formation of a new carbon–carbon or carbon–heteroatom bond by displacement of dithiocarbonate function of 731 by a variety of nucleophiles in the presence of Lewis acid (Scheme 126, Tables 18 and 19).
 | ||
| Scheme 126 | ||
| Entry | Nu-X | Conditions | Lewis acids | Products | Yields (%) | Trans/cis dr |
|---|---|---|---|---|---|---|
| 1 | EtOH | Toluene, 20 °C, 15 min | AgOTf |  |
72 | 7:3 |
| 2 | tBuCH2OH | Toluene, −17 °C, 1 h | AgOTf |  |
73 | 3:2 |
| 3 | Me3SiCN | Toluene, −78 °C, 2.5 h | SnCl4 | 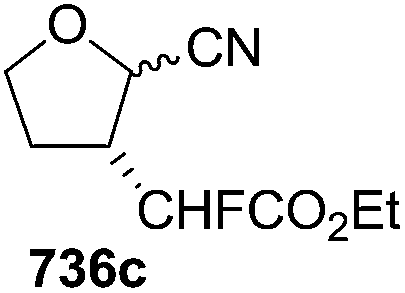 |
83 | 9:1 |
| 4 | 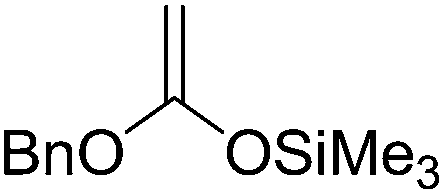 |
Toluene, −17 °C, 2 h | AgOTf | 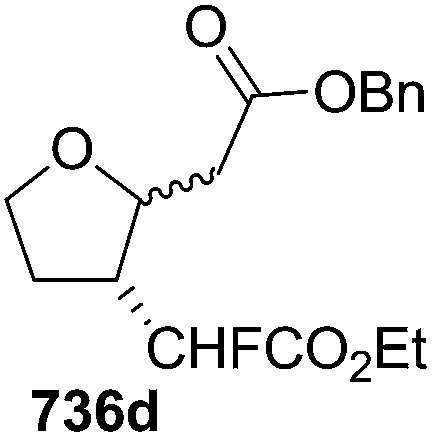 |
36 | 4:1 |
| 5 | 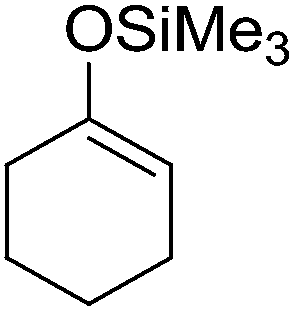 |
Toluene, −17 °C, 1 h | AgOTf | 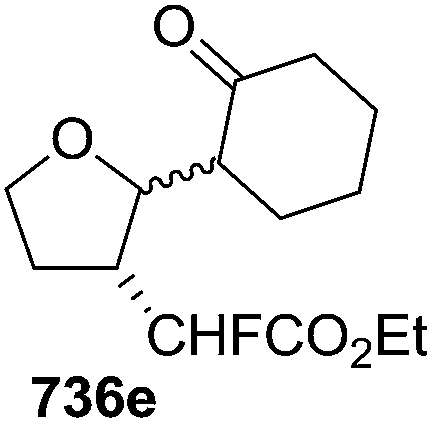 |
32 | >98:2 |
| Entry | Nu-X | Conditions | Lewis acids | Products | Yields (%) | Trans/cis dr |
|---|---|---|---|---|---|---|
| 1 | Me3SiN3 | Toluene, −17 °C, 1 h | AgOTf | 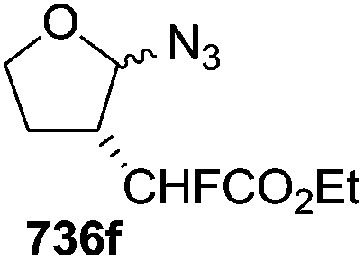 |
68 | 3:2 |
| 2 | 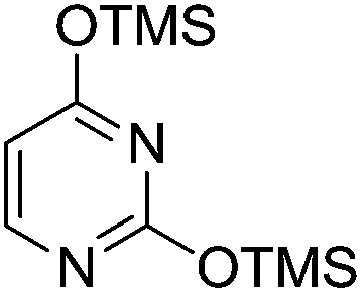 |
Toluene, −17 °C, 2 h | AgOTf | 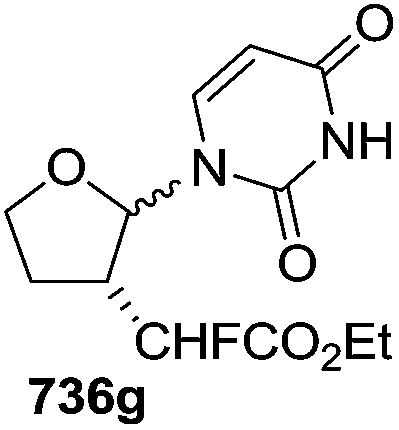 |
63 | 9:1 |
| 3 | 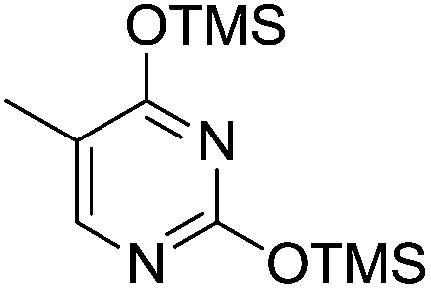 |
Toluene, −17 °C, 1.5 h | AgOTf |  |
83 | 9:1 |
| 4 | 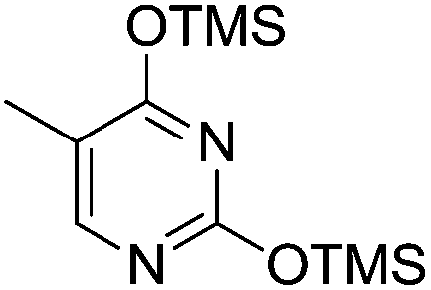 |
Toluene, −17 °C, 4 h | Cu(OTf)2 |  |
76 | 9:1 |
| 5 | 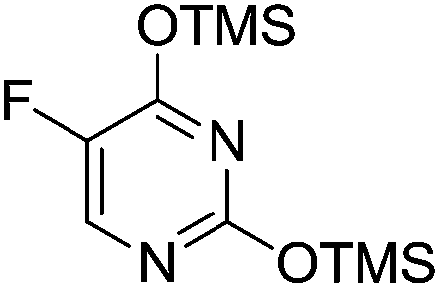 |
Toluene, −17 °C, 1.5 h | AgOTf | 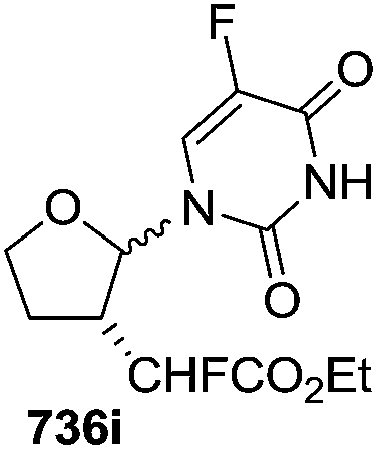 |
45 | 4:1 |
| 6 | 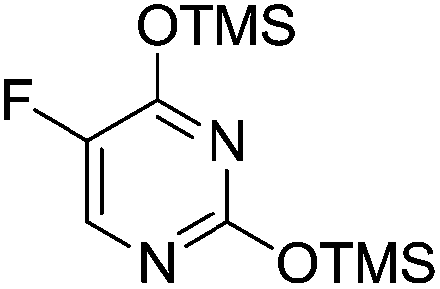 |
Toluene, −17 °C, 3.5 h | Cu(OTf)2 | 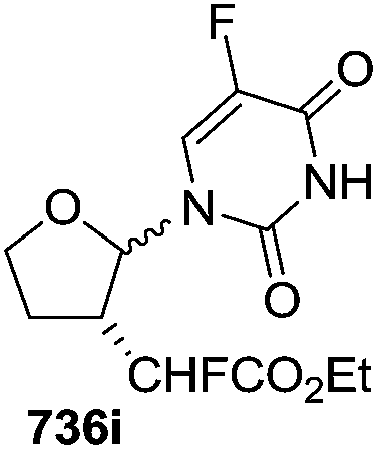 |
25 | 4:1 |
By following this strategy, they have synthesized 2′-deoxy-2′-C-β-alkyl nucleoside analogues 739 from furanoid glycal 71a and xanthate 726 (Scheme 127). Lauroyl peroxide was added slowly in the reaction mixture of furanoid glycal 71a and xanthate 726 in DCE and the reaction mixture was refluxed for 5 h to obtain a diastereomeric mixture of 2,3-trans addition products 737 (1:1) in 57% isolated yield. Treatment of (TMS)2thymine with 737 in the presence of AgOTf at 0 °C for 3 h afforded a mixture of protected 2′-deoxy-2′-C-β-alkyl nucleoside analogues 738 in 61% yield (only trans products). These were subjected to hydrogenation to give the corresponding diol 739.
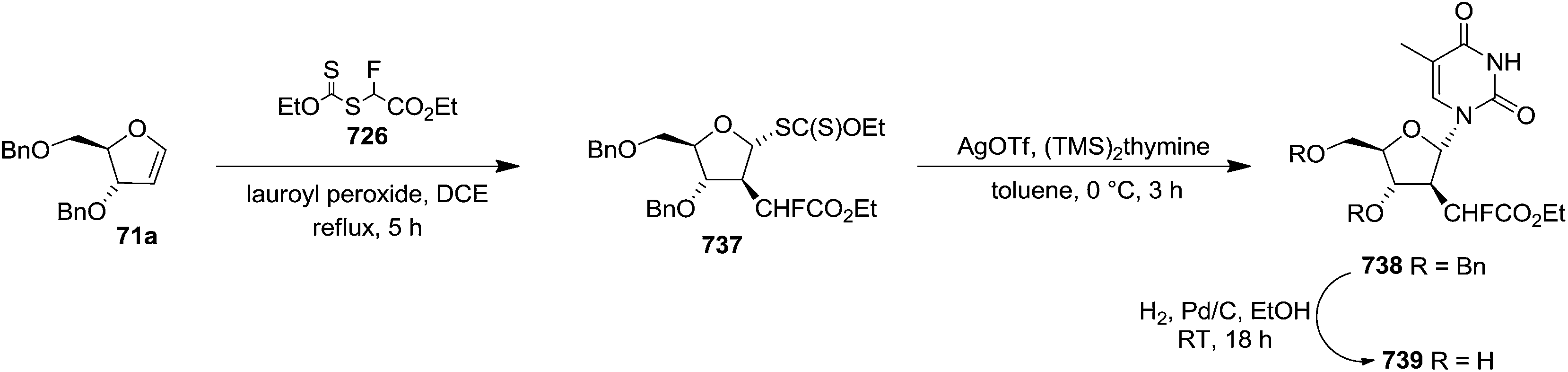 | ||
| Scheme 127 | ||
The synthesis of furanoid glycals (171, 173) proposed by Haraguchi and group has been already discussed in Scheme 30.38 Now, the utilization of their synthesized furanoid glycals (66e, 171 and 173) is being discussed here for the synthesis of 2′-deoxynucleosides and its 1′-branched analogues. They performed NIS-mediated electrophilic glycosidation between protected erythro-furanoid glycals (66e, 171 and 173) and silylated thymine in CH3CN/DCM at room temperature and observed that only the glycal 173 selectively furnished β-anomer 742 exclusively in 76% yield. Whereas, under identical reaction condition formation of the α-anomer (740α, 62% yield) dominated over that of the β-anomer (740β, 15% yield) in the case of glycal 66e. On the other hand, glycal 171 gave equal amounts of the β-(741β, 35% yield) and α-(741α, 35% yield) anomers (Scheme 128, Table 20).
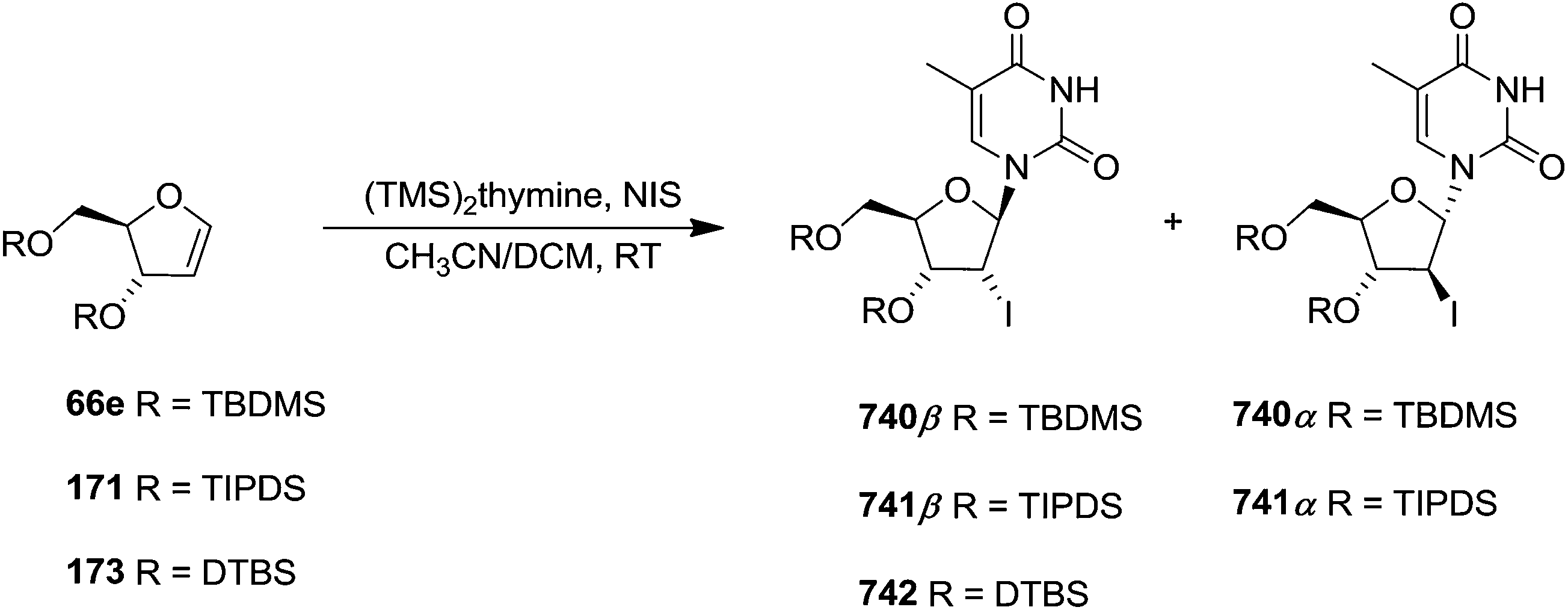 | ||
| Scheme 128 | ||
| Entry | Glycal | Products (isolated yield) | Ratio of β-anomer/α-anomer |
|---|---|---|---|
| a All reactions were carried out in CH3CN/DCM at rt for 12 h by using (TMS)2thymine (3.0 equiv.) and NIS (1.5 equiv.). | |||
| 1 | 66e | 740β and 740α (77%) | 1:4 |
| 2 | 171 | 741β (35%) and 741α (35%) | 1:1 |
| 3 | 173 | 742 (76%) | — |
They further showed the electrophilic glycosidation of silylated uracil, N4-(acetyl)cytosine with glycal 173 also gave β-anomer respectively in exclusive amount: 743 (76% yield); 744 (55% yield) (Fig. 6). But in the case of silylated N6-(benzoyl)adenine desired N9-glycoside 745 was formed only 26% yield along with N7-(746, 17% yield) and N1-(747, 13% yield) isomers.
 | ||
| Fig. 6 Compounds (743–747). | ||
The glycosidation products (742–745) were converted to the corresponding 2′-deoxynucleosides (748–751) in good yields by reacting each with nBu3SnH, Et3B/O2 in toluene at room temperature (Scheme 129).
 | ||
| Scheme 129 | ||
They further showed the scope of this glycosidation method by the formation of several 1-alkyl and 1-(ω-hydroxy)alkyl erythro-furanoid glycals which also gave the respective β-anomer exclusively. Furanoid glycal 173 was lithiated with tBuLi (3 equiv.) in THF, which on treatment with several carbon electrophiles (Scheme 130, Table 21), afforded 1-alkyl erythro-furanoid glycals (752, 753) and 1-(ω-hydroxy)alkyl erythro-furanoid glycals (754–759). To obtain derivatives suitable for glycosidation, 1-(ω-hydroxy)alkyl erythro-furanoid glycals (754–759) were converted to their O-triethylsilyl derivatives (760–765) respectively on silylation (Scheme 130).
 | ||
| Scheme 130 | ||
| Entry | Electrophile (equiv.) | R | Product (isolated yield) |
|---|---|---|---|
| a After addition of the respective electrophile, the reaction mixture was stirred below −70 °C for 0.5 h, except entry 2.b After addition of the electrophile,the reaction mixture was stirred at −40 °C for 11 h.c The product was obtained as a mixture of diastereomers.d The starting material (173) was recovered in 78% yield. | |||
| 1 | MeI (10)/HMPA (5) | Me | 752 (56%) |
| 2 | PhCH2Br (5)/HMPA (10)b | CH2Ph | 753 (56%) |
| 3 | DMF (5) then NaBH4 (1.5) | CH2OH | 754 (82%) |
| 4 | PhCHO (3) | CH(OH)Ph | 755 (93%)c |
| 5 | MeCHO (5) | CH(OH)Me | 756 (90%)c |
| 6 | CH3COCH3 (5) | C(OH)Me2 | 757 (19%)d |
| 7 | Ethylene oxide (5) & BF3·OEt2 (5) | CH2CH2OH | 758 (62%) |
| 8 | Trimethylene oxide (5) & BF3·OEt2 (3) | CH2CH2CH2OH | 759 (65%) |
NIS-initiated electrophilic glycosidation of silylated thymine with these glycals (752, 753, and 760–765) formed exclusively the β-anomers of 1′-branched 2′-iodothymidine derivatives (766–773) respectively. Compounds (766–773) were transformed to 1′-branched thymidines (774–781) in good yields (Scheme 131, yields are given in parentheses) by reacting them with nBu3SnH, Et3B/O2, in toluene at room temperature.
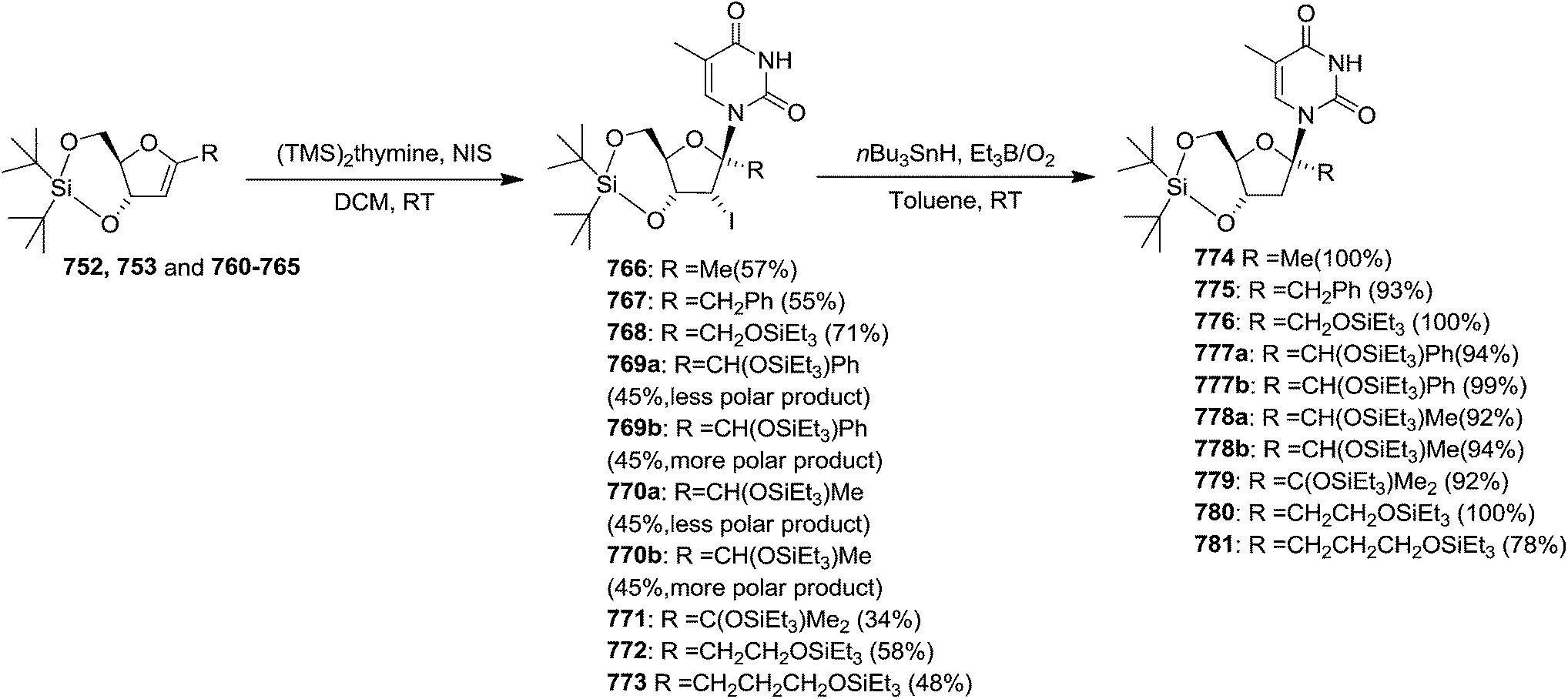 | ||
| Scheme 131 | ||
Pal and Shaw have already reported the synthesis of four stereochemically different enantiomerically pure, furanoid glycal building blocks (162a–c, 139) (Scheme 28) (Fig. 3)35 and also shown their synthetic utility to obtain some natural products such as the aggregation pheromones brevicomins (285a–d) (Schemes 47–50) and styryllactones (+)-cardiobutanolide (290a, Scheme 51), (−)-cardiobutanolide (290b, Scheme 52) and (+)-goniofufurone (295a, Scheme 53).2h Thus, inspired by literature reports on the interesting and important biological activities of 2′-deoxynucleosides we decided to undertake the synthesis of six 2′-deoxynucleoside analogue building blocks (Fig. 7) from the stereochemically different furanoid glycals (162a–c, 139) by adopting a methodology reported earlier by Kim and Misco.9 We have synthesized three pairs of enantiomeric 2′-deoxynucleoside analogues (783a, 783b), (783c, 783e) and (783d, 783f) respectively as building blocks from furanoid glycals (162a–c, 139) by involving the similar synthetic strategy.2i
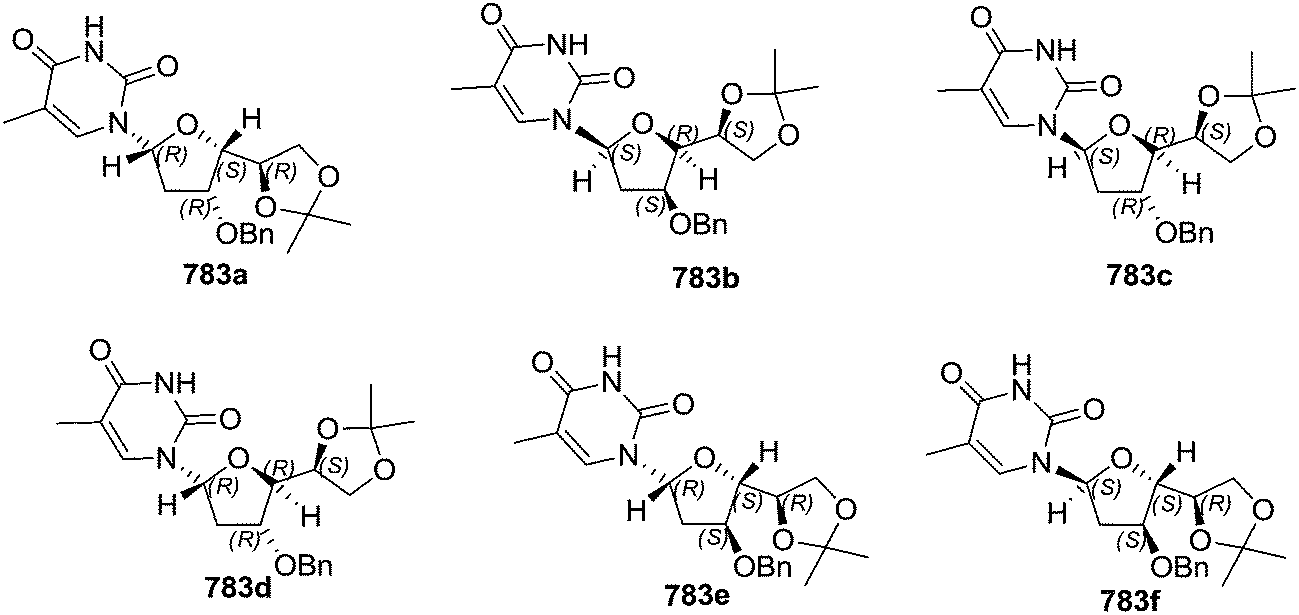 | ||
| Fig. 7 Structures of 2′-deoxynucleoside analogues (783a–f). | ||
Furanoid glycal 139 on treatment with (TMS)2thymine (2.0 equiv.) in the presence of NIS (2.5 equiv.) in dry DCM at −30 °C → rt for overnight afforded glycosylated product 782a, which, without purification, was directly converted into 2′-deoxynucleoside analogue 783a in 26% yield (over two steps) in the presence of nBu3SnH and a radical initiator ABCN (1,1′-Azobis-(cyclohexanecarbonitrile)) (Scheme 132). The stereochemistry of compound 783a was determined on the basis of its NOE spectrum.
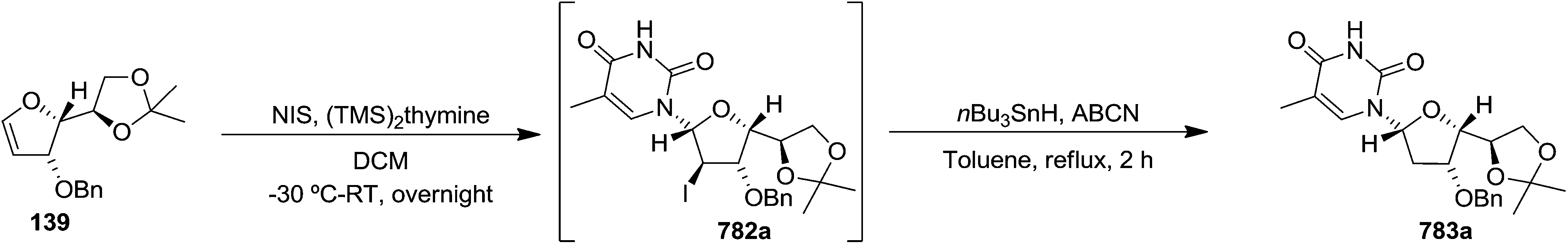 | ||
| Scheme 132 | ||
Having this result in hand, we further extended our study on furanoid glycal 162a which was an optical antipode of 139. Thus 2′-deoxynucleoside analogue 783b (Scheme 133) was obtained from 162a following the identical reaction pathway as it was shown in the case of 783a (Scheme 132). The [α]D of 2′-deoxynucleoside analogue 783b [[α]25D + 17.7 (c 0.27, MeOH)] was just opposite to that of 783a [[α]25D − 23.4 (c 1.67, MeOH)].
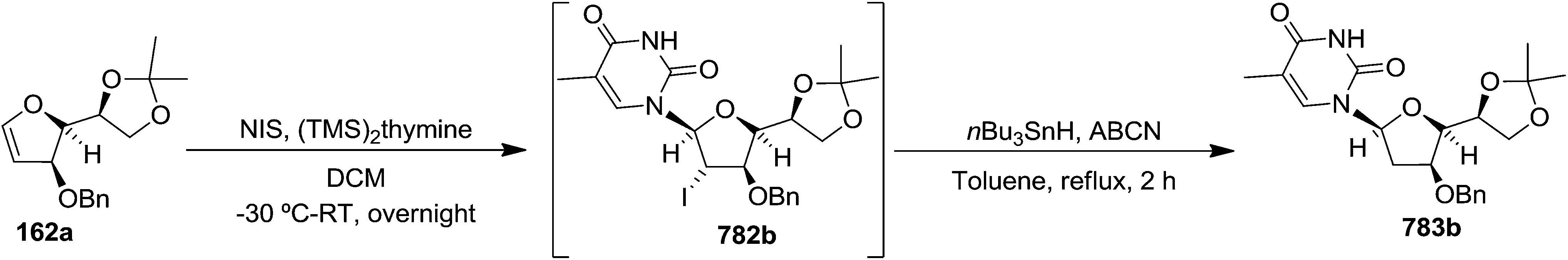 | ||
| Scheme 133 | ||
Our further study on erythro-furanoid glycal 162b, under the identical reaction conditions furnished a mixture of two compounds. Column chromatographic purification of the mixture led to the isolation of 783c and 783d in 8% and 32% yields respectively (over two steps, Scheme 134). The NOE experiment was also carried out for the complete characterization of 2′-deoxy-β-nucleoside analogue 783c [[α]25D − 1.91 (c 0.10, MeOH)] and 2′-deoxy-α-nucleoside analogue 783d [[α]26D − 10.2 (c 0.40, MeOH)] in a 1:4 ratio.
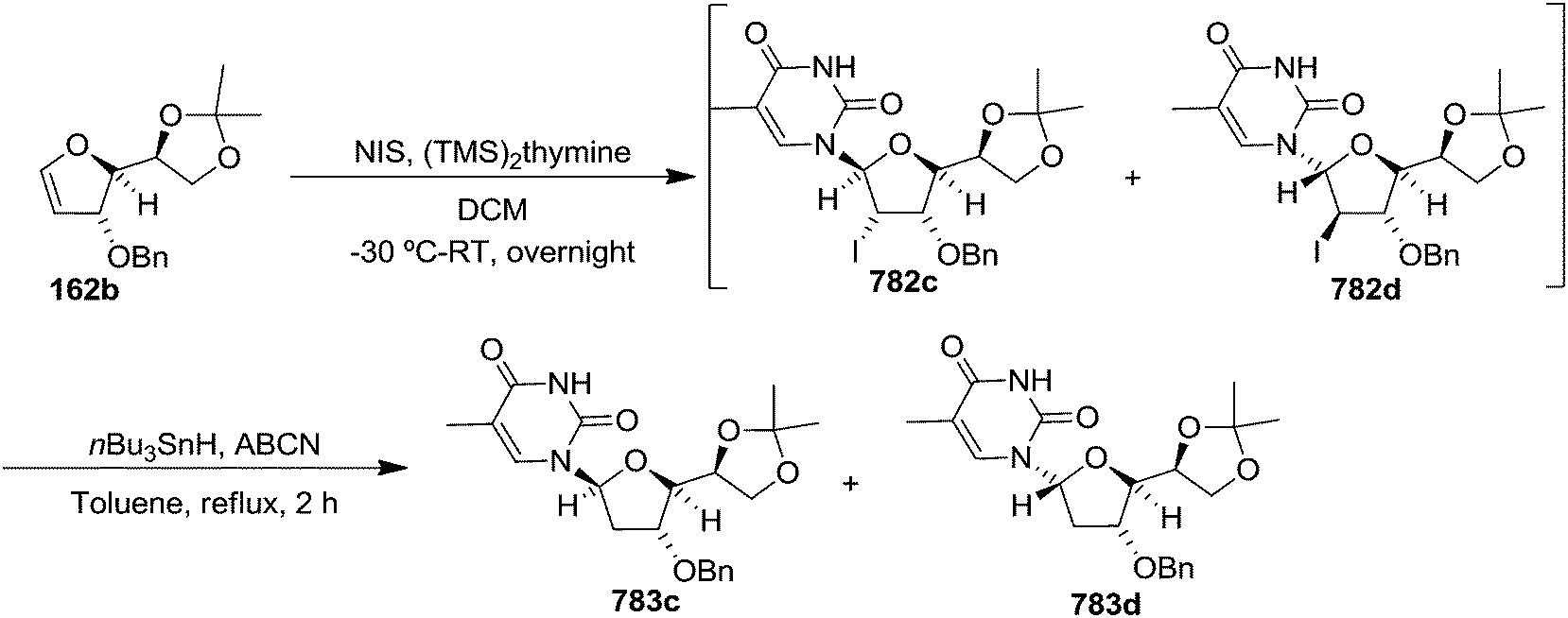 | ||
| Scheme 134 | ||
Under the identical reaction conditions formation of mixture of nucleosides was quite obvious from erythro-furanoid glycal 162c having two anti bulky groups at C-3′ and C-4′ which was enantiomeric to 162b. The chromatographic purification of the mixture containing the isomeric nucleoside analogues led to the isolation of 2′-deoxynucleoside analogues 783f [[α]26D + 12.1 (c 0.57, MeOH)] as the major product in 27% yield (over two steps) and 783e [[α]25D + 1.42 (c 0.37, MeOH)] as minor product in 9% yield (over two steps) (Scheme 135) which were the enantiomers of 783d and 783c respectively.
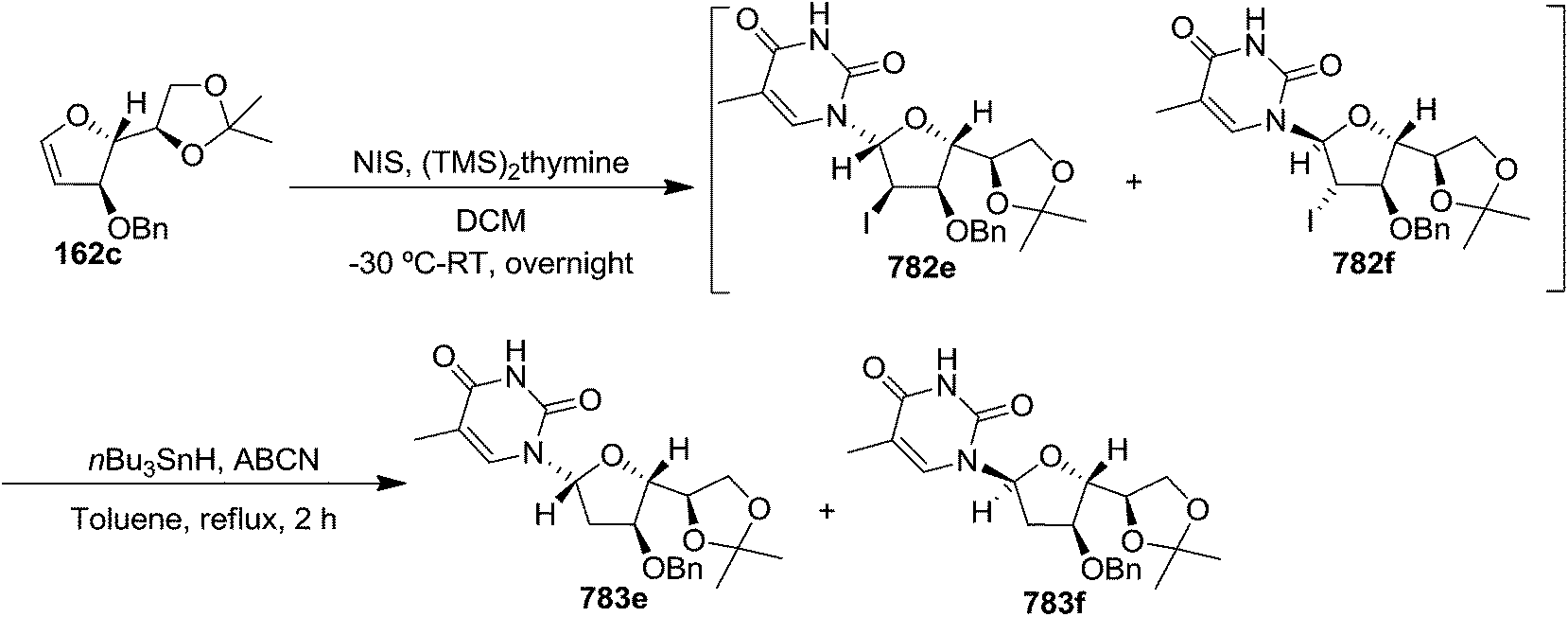 | ||
| Scheme 135 | ||
7. Conclusion
In summary, this review is an attempt to describe the various synthetic approaches to obtain both erythro and threo furanoid glycals since their discovery. Emphasis has also been given to their synthetic applications towards the syntheses of natural products, natural product like molecules, important “building blocks” and C-and N-nucleosides. One of the purposes, of this review is to attract the attention of the synthetic community to develop new approaches for furanoid glycals syntheses and to exploit this inexpensive and widely available chiral building blocks for broader applications both in synthetic as well as medicinal chemistry. We hope that this review will be useful to those who have an interest in furanoid glycals.Acknowledgements
Dr Pinki Pal is highly grateful to CSIR, New Delhi, for fellowships in the form of junior research fellow and senior research fellow.Notes and references
- Z. J. Witczak, Curr. Med. Chem., 1999, 6, 165 CAS.
- (a) M. Saquib, M. K. Gupta, R. Sagar, Y. S. Prabhakar, A. K. Shaw, R. Kumar, P. R. Maulik, A. N. Gaikwad, S. Sinha, A. K. Srivastava, V. Chaturvedi, R. Srivastava and B. S. Srivastava, J. Med. Chem., 2007, 50, 2942 CrossRef CAS PubMed; (b) L. V. R. Reddy, P. V. Reddy and A. K. Shaw, Tetrahedron: Asymmetry, 2007, 18, 542 CrossRef CAS; (c) P. V. Reddy, L. V. R. Reddy, B. Kumar, R. Kumar, P. R. Maulik and A. K. Shaw, Tetrahedron, 2008, 64, 2153 CrossRef CAS; (d) L. V. R. Reddy, G. N. Swamy and A. K. Shaw, Tetrahedron: Asymmetry, 2008, 19, 1372 CrossRef CAS; (e) M. Saquib, I. Hussain, B. Kumar and A. K. Shaw, Chem.–Eur. J., 2009, 15, 6041 CrossRef CAS PubMed; (f) L. V. R. Reddy, P. V. Reddy, N. N. Mishra, P. K. Shukla, G. Yadav, R. Srivastava and A. K. Shaw, Carbohydr. Res., 2010, 345, 1515 CrossRef PubMed; (g) P. V. Reddy, B. Bajpai, B. Kumar and A. K. Shaw, Eur. J. Org. Chem., 2011, 1575 CrossRef CAS; (h) P. Pal and A. K. Shaw, Tetrahedron, 2011, 67, 4036 CrossRef CAS; (i) P. Pal, P. Singh, B. Kumar, H. M. Gauniyal and A. K. Shaw, Tetrahedron: Asymmetry, 2011, 22, 992 CrossRef CAS.
- (a) L. V. R. Reddy, V. Kumar, R. Sagar and A. K. Shaw, Chem. Rev., 2013, 113, 3605 CrossRef CAS PubMed; (b) S. J. Danishefsky and M. T. Bilodeau, Angew. Chem., Int. Ed., 1996, 35, 1380 CrossRef CAS; (c) R. J. Ferrier and O. A. Zubkov, Org. React., 2003, 62, 569 CAS.
- R. E. Ireland, S. Thaisrivongs, N. Vanier and C. S. Wilcox, J. Org. Chem., 1980, 45, 48 CrossRef CAS and references cited therein.
- E. J. Corey and G. Goto, Tetrahedron Lett., 1980, 21, 3463 CrossRef CAS.
- U. Hacksell and G. D. Daves Jr, J. Org. Chem., 1983, 48, 2870 CrossRef CAS.
- U. Hacksell and G. D. Daves Jr, Prog. Med. Chem., 1985, 22, 1 CrossRef CAS PubMed.
- K. Chow and S. Danishefsky, J. Org. Chem., 1990, 55, 4211 CrossRef CAS.
- C. U. Kim and P. F. Misco, Tetrahedron Lett., 1992, 33, 5733 CrossRef CAS.
- A. El-Laghdach, Y. Diáz and S. Castillón, Tetrahedron Lett., 1993, 34, 2821 CrossRef CAS.
- E. Fischer and K. Zach, Sitzber Kgl Preuss Akad Wiss, 1913, 16, 311 Search PubMed.
- R. K. Ness and H. G. Fletcher Jr, J. Am. Chem. Soc., 1958, 80, 2007 CrossRef CAS.
- R. K. Ness and H. G. Fletcher Jr, J. Org. Chem., 1963, 28, 435 CrossRef.
- M. Haga and R. K. Ness, J. Org. Chem., 1965, 30, 158 CrossRef CAS PubMed.
- (a) K. Bischofberger and R. H. Hall, Carbohydr. Res., 1975, 42, 175 CrossRef CAS; (b) A. K. M. Anisuzzaman and R. L. Whistler, J. Org. Chem., 1972, 37, 3187 CrossRef CAS PubMed; (c) B. Iselin and T. Reichstein, Helv. Chim. Acta, 1944, 27, 1146 CrossRef CAS; (d) K. Bischofberger and R. H. Hall, Carbohydr. Res., 1976, 52, 223 CrossRef CAS.
- (a) S. J. Eitelman and A. Jordaan, J. Chem. Soc., Chem. Commun., 1977, 552 RSC; (b) S. Hanessian, M. M. Ponpipom and P. Lavallee, Carbohydr. Res., 1972, 24, 45 CrossRef CAS.
- (a) S. J. Eitelman, R. H. Hall and A. Jordaan, J. Chem. Soc., Perkin Trans. 1, 1978, 595 RSC; (b) J. B. Lee and T. J. Nolan, Tetrahedron, 1967, 23, 2789 CrossRef CAS PubMed; (c) B. D. Kohn, P. Kohn and A. Dubin, Carbohydr. Res., 1971, 18, 349 CrossRef CAS PubMed.
- R. E. Ireland, C. S. Wilcox and S. Thaisrivongs, J. Org. Chem., 1978, 43, 786 CrossRef CAS.
- R. E. Ireland, D. W. Norbeck, G. S. Mandel and N. S. Mandel, J. Am. Chem. Soc., 1985, 107, 3285 CrossRef CAS.
- (a) J. C.-Y. Cheng, U. Hacksell and G. D. Daves Jr, J. Org. Chem., 1985, 50, 2778 CrossRef CAS; (b) R. F. Cunico and L. Bedell, J. Org. Chem., 1980, 45, 4797 CrossRef CAS.
- M. Obayashi and M. Schlosser, Chem. Lett., 1985, 1715 CrossRef CAS.
- E. Larsen, P. T. Jørgensen, M. A. Sofan and E. B. Pederson, Synthesis, 1994, 1037 CrossRef CAS.
- M. A. Cameron, S. B. Cush and R. P. Hammer, J. Org. Chem., 1997, 62, 9065 CrossRef CAS.
- M. Kassou and S. Castillón, Tetrahedron Lett., 1994, 35, 5513 CrossRef CAS.
- F. Bravo, M. Kassou and S. Castillón, Tetrahedron Lett., 1999, 40, 1187 CrossRef CAS.
- F. Bravo, M. Kassou, Y. Díaz and S. Castillón, Carbohydr. Res., 2001, 336, 83 CrossRef CAS PubMed.
- R. Pontikis, J. Wolf, C. Monneret and J.-C. Florent, Tetrahedron Lett., 1995, 36, 3523 CrossRef CAS.
- J. A. Walker II, J. J. Chen, D. S. Wise and L. B. Townsend, J. Org. Chem., 1996, 61, 2219 CrossRef.
- F. E. McDonald and M. M. Gleason, J. Am. Chem. Soc., 1996, 118, 6648 CrossRef CAS.
- R. R. Diaz, C. R. Melgarejo, I. I. Cubero and M. T. P. López-Espinosa, Carbohydr. Res., 1997, 300, 375 CrossRef CAS.
- C. Kim, R. Hoang and E. A. Theodorakis, Org. Lett., 1999, 1, 1295 CrossRef CAS.
- Z.-X. Wang, L. I. Wiebe, J. Balzarini, E. D. Clercq and E. E. Knaus, J. Org. Chem., 2000, 65, 9214 CrossRef CAS PubMed.
- B. Schmidt and H. Wildemann, Eur. J. Org. Chem., 2000, 3145 CrossRef CAS.
- A. M. Gómez, M. Casillas, A. Barrio, A. Gawel and J. C. López, Eur. J. Org. Chem., 2008, 3933 CrossRef.
- (a) P. Pal, B. Kumar and A. K. Shaw, Eur. J. Org. Chem., 2009, 2179 CrossRef CAS; (b) F. Gonzalez, S. Lesage and A. S. Perlin, Carbohydr. Res., 1975, 42, 267 CrossRef CAS; (c) R. Sagar, R. Pathak and A. K. Shaw, Carbohydr. Res., 2004, 339, 2031 CrossRef CAS PubMed; (d) M. Saquib, R. Sagar and A. K. Shaw, Carbohydr. Res., 2006, 341, 1052 CrossRef CAS PubMed; (e) R. Sagar, L. V. R. Reddy and A. K. Shaw, Tetrahedron: Asymmetry, 2006, 17, 1189 CrossRef CAS.
- G. V. M. Sharma and K. Krishnudu, Carbohydr. Res., 1995, 268, 287 CrossRef CAS and references cited therein.
- T. Oishi, K. Ando and N. Chida, Chem. Commun., 2001, 1932 RSC and references cited therein.
- K. Haraguchi, K. Konno, K. Yamada, Y. Kitagawa, K. T. Nakamura and H. Tanaka, Tetrahedron, 2010, 66, 4587 CrossRef CAS.
- S. Hanessian, Total Synthesis of Natural Products; The ‘Chiron’ Approach, Oxford, Pergamon, 1983 Search PubMed.
- R. E. Ireland, S. Thaisrivongs and C. S. Wilcox, J. Am. Chem. Soc., 1980, 102, 1155 CrossRef CAS and references cited therein.
- R. E. Ireland, R. C. Anderson, R. Badoud, B. J. Fitzsimmons, G. J. Mcgarvey, S. Thaisrivongs and C. S. Wilcox, J. Am. Chem. Soc., 1983, 105, 1988 CrossRef CAS and references cited therein.
- E. J. Corey, D. A. Clark, G. Goto, A. Marfat, C. Mioskowski, B. Samuelsson and S. Hammarström, J. Am. Chem. Soc., 1980, 102, 1436 CrossRef CAS.
- S. D. Haveli, P. R. Sridhar, P. Suguna and S. Chandrasekaran, Org. Lett., 2007, 9, 1331 CrossRef CAS PubMed.
- (a) U. Ravid, R. M. Silverstein and L. R. Smith, Tetrahedron, 1978, 34, 1449 CrossRef CAS; (b) A. Takle and P. Kocienski, Tetrahedron, 1990, 46, 4503 CrossRef CAS.
- P. R. R. Meira, A. V. Moro and C. R. D. Correia, Synthesis, 2007, 2279 CAS.
- K. Bischofberger, S. J. Eitelman and A. Jordaan, Carbohydr. Res., 1979, 74, 145 CrossRef CAS.
- K. Dax, B. I. Glänzer, G. Schulz and H. Vyplel, Carbohydr. Res., 1987, 162, 13 CrossRef CAS.
- B. J. Fitzsimmons, Y. Leblanc and J. Rokach, J. Am. Chem. Soc., 1987, 109, 285 CrossRef CAS.
- Y. Leblanc, B. J. Fitzsimmons, J. P. Springer and J. Rokach, J. Am. Chem. Soc., 1989, 111, 2995 CrossRef CAS.
- W. Abramski, K. Badowska-Rosbnek and M. Chmielewski, Bioorg. Med. Chem. Lett., 1993, 3, 2403 CrossRef CAS.
- P. Bertrand, J.-P. Gesson, B. Renoux and I. Tranoy, Tetrahedron Lett., 1995, 36, 4073 CrossRef CAS.
- K. A. Parker and D.-S. Su, J. Org. Chem., 1996, 61, 2191 CrossRef CAS.
- J. D. Bois, C. S. Tomooka, J. Hong and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 3179 CrossRef.
- V. Kirsch, C. Wolff, C. Näther and W. Tochtermann, Eur. J. Org. Chem., 2000, 1741 CrossRef CAS.
- V. Y. Mizuno, The Organic Chemistry of Nucleic Acids, Elsevier, Amsterdam, 1986 Search PubMed.
- (a) H. Mitsuya, R. Yarchoan and S. Broder, Science, 1990, 249, 1533 CAS; (b) E. DeClercq, Design of anti-AIDS Drugs, Elsevier, New York, 1990 Search PubMed; (c) S. Broder, AIDS: Modern Concepts and Therapeutic Challenges, Marcel Dekker, New York, 1987 Search PubMed.
- P. Herdewijn, J. Balzarini, E. De Clercq, R. Pauwels, M. Baba, S. Broder and H. Vanderhaeghe, J. Med. Chem., 1987, 30, 1270 CrossRef CAS PubMed.
- J. A. Montgomery, T. P. Johnston and Y. F. Shealy, in Burger's Medicinal Chemistry, ed. M. E. Wolff, Wiley, New York, 4th edn, 1980, Part 2 Search PubMed.
- M. I. Johnston and D. F. Hoth, Science, 1993, 260, 1286 CAS.
- Q. Wu and C. Simons, Synthesis, 2004, 1533 CrossRef CAS.
- J. C.-Y. Cheng, U. Hacksell and G. D. Daves Jr, J. Org. Chem., 1986, 51, 3093 CrossRef CAS.
- R. A. Outten and G. D. Daves Jr, J. Org. Chem., 1987, 52, 5064 CrossRef CAS.
- R. N. Farr, R. A. Outten, J. C.-Y. Cheng and G. D. Daves Jr, Organometallics, 1990, 9, 3151 CrossRef CAS.
- G. D. Daves Jr, Acc. Chem. Res., 1990, 23, 201 CrossRef.
- R. N. Farr, D.-I. Kwok and G. D. Daves Jr, J. Org. Chem., 1992, 57, 2093 CrossRef CAS.
- H.-C. Zhang and G. D. Daves Jr, J. Org. Chem., 1992, 57, 4690 CrossRef CAS.
- H.-C. Zhang, M. Brakta and G. D. Daves Jr, Tetrahedron Lett., 1993, 34, 1571 CrossRef CAS.
- H.-C. Zhang, M. Brakta and G. D. Daves Jr, Nucleosides Nucleotides, 1995, 14, 105 CAS.
- J. J. Chen, J. A. Walker II, W. Liu, D. S. Wise and L. B. Townsend, Tetrahedron Lett., 1995, 36, 8363 CrossRef CAS.
- H.-P. Hsieh and L. W. McLaughlin, J. Org. Chem., 1995, 60, 5356 CrossRef CAS.
- R. S. Coleman and M. L. Madaras, J. Org. Chem., 1998, 63, 5700 CrossRef CAS.
- M. Tingoli, B. Panunzi and F. Santacroce, Tetrahedron Lett., 1999, 40, 9329 CrossRef CAS.
- Z. Sun, S. Ahmed and L. W. McLaughlin, J. Org. Chem., 2006, 71, 2922 CrossRef CAS PubMed and references cited therein.
- I. Singh and O. Seitz, Org. Lett., 2006, 8, 4319 CrossRef CAS PubMed.
- N. Joubert, R. Pohl, B. Klepetářová and M. Hocek, J. Org. Chem., 2007, 72, 6797 CrossRef CAS PubMed.
- C. U. Kim, B. Y. Luh and J. C. Martin, J. Org. Chem., 1991, 56, 2642 CrossRef CAS.
- J. Žemlička, R. Gasser, J. V. Freisler and J. P. Horwitz, J. Am. Chem. Soc., 1972, 94, 3213 CrossRef.
- M. Taniguchi, K. Koga and S. Yamada, Tetrahedron, 1974, 30, 3547 CrossRef CAS.
- J. Wang, J. A. Wurster, L. J. Wilson and D. Liotta, Tetrahedron Lett., 1993, 34, 4881 CrossRef CAS.
- F. E. McDonald and M. M. Gleason, Angew. Chem., Int. Ed. Engl., 1995, 34, 350 CrossRef CAS.
- Y. Díaz, A. El-Laghdach, M. I. Matheu and S. Castillón, J. Org. Chem., 1997, 62, 1501 CrossRef.
- P. Hansen and E. B. Pedersen, Acta Chem. Scand., 1990, 44, 522 CrossRef CAS.
- Y. Díaz, A. El-Laghdach and S. Castillón, Tetrahedron, 1997, 53, 10921 CrossRef.
- R. Robles, C. Rodríguez, I. Izquierdo, M. T. Plaza and A. Mota, Tetrahedron: Asymmetry, 1997, 8, 2959 CrossRef CAS.
- J. Lim and Y. H. Kim, J. Chem. Soc., Perkin Trans. 1, 1999, 3239 RSC.
- L. A. Paquette, S. Brand and C. Behrens, J. Org. Chem., 1999, 64, 2010 CrossRef CAS PubMed.
- S. Marcotte, B. Gérard, X. Pannecoucke, C. Feasson and J.-C. Quirion, Synthesis, 2001, 929 CrossRef CAS.
- (a) R. Miethchen, M. Hein and H. Reinke, Eur. J. Org. Chem., 1998, 919 CrossRef CAS; (b) F. H. Wu, B. N. Huang, L. Lu and W. Y. Huang, J. Fluorine Chem., 1996, 80, 91 CrossRef CAS.
- A. Choudhury, M. E. Pierce, D. Nguyen, L. Storace and P. N. Confalone, Tetrahedron Lett., 2005, 46, 8099 CrossRef CAS.
- L. Jean-Baptiste, S. Yemets, R. Legay and T. Lequeux, J. Org. Chem., 2006, 71, 2352 CrossRef CAS PubMed.
- (a) S. Z. Zard, in Radical Reactions in Organic Synthesis, Oxford Chemistry Masters, Oxford, 2003 Search PubMed; (b) S. Z. Zard, Angew. Chem., Int. Ed. Engl., 1997, 36, 672 CrossRef.
Footnotes |
| † CDRI Communication Number is 9482. |
| ‡ Present address: Department of Chemistry, Mount Carmel College Autonomous, 58, Palace Road, Bangalore-560052, India. E-mail: pinkipal@gmail.com; Tel: +91-7760512715 |
| This journal is © The Royal Society of Chemistry 2017 |