
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst total synthesis of the only known 2-isopropyliden-2H-benzofuran-3-one isolated from V. luetzelburgii†
Jorgelina L. Pergomet ,
Enrique L. Larghi,
Teodoro S. Kaufman* and
Andrea B. J. Bracca*
,
Enrique L. Larghi,
Teodoro S. Kaufman* and
Andrea B. J. Bracca*
Instituto de Química Rosario (IQUIR, CONICET-UNR), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, S2002LRK Rosario, Argentina. E-mail: kaufman@iquir-conicet.gov.ar; bracca@iquir-conicet.gov.ar; Fax: +54-341-4370477; Tel: +54-341-4370477
First published on 17th January 2017
Abstract
The first total synthesis of 5-(1-hydroxy-1-ethyl)-2-isopropyliden-2H-benzofuran-3-one, is reported in its racemic and in one of its optically active [(S)-(−)] forms. This heterocycle, isolated from Verbesina luetzelburgii, is the only known 2-isopropyliden-2H-benzofuran-3-one produced by species of Verbesina. The sequence took place in eight steps and 19% overall yield (up to 33% for the chiral form), from 4′-hydroxyacetophenone. It entailed carbonyl group protection as the 1,3-dioxolane and a phenol ortho formylation, followed by a Williamson etherification with chloroacetone and an organocatalytic cross-aldolization, to afford a 2-acetyl-2,3-dihydrobenzofuran-3-ol intermediate. The latter underwent a methyl Grignard addition to the carbonyl moiety, followed by selective oxidation of the benzylic alcohol and deprotection, resulting in a β-hydroxy diketone derivative. A MsCl-assisted dehydration of the tertiary alcohol, established the isopropylidene motif, whereas the syntheses culminated by chemical or enzymatic (carrot, celeriac) selective reductions of the exocyclic carbonyl group.
Introduction
The benzofuran-3-one nucleus, a derivative of the privileged benzofuran core,1 is one of the most studied heterocyclic structural units by synthetic and medicinal chemists.2 This motif is widely found in bioactive natural and synthetic compounds, such as in the antifungal agent griseofulvin.3The aurones and the 2-isopropylidene-benzofuran-3-ones are two families of 2-methylene-benzofuran-3-one derivatives. Many of them display interesting biological activities, including anti-protozoal4 and anti-cancer.5 The aurones (1a–e),6 which are more widespread (Fig. 1), exhibit different aromatic rings and olefin configurations,7 whereas the 2-isopropylidene-benzofuran-3-ones display a tetrasubstituted alkene motif and comprise a handful of exceptional products isolated from fungi and plants.
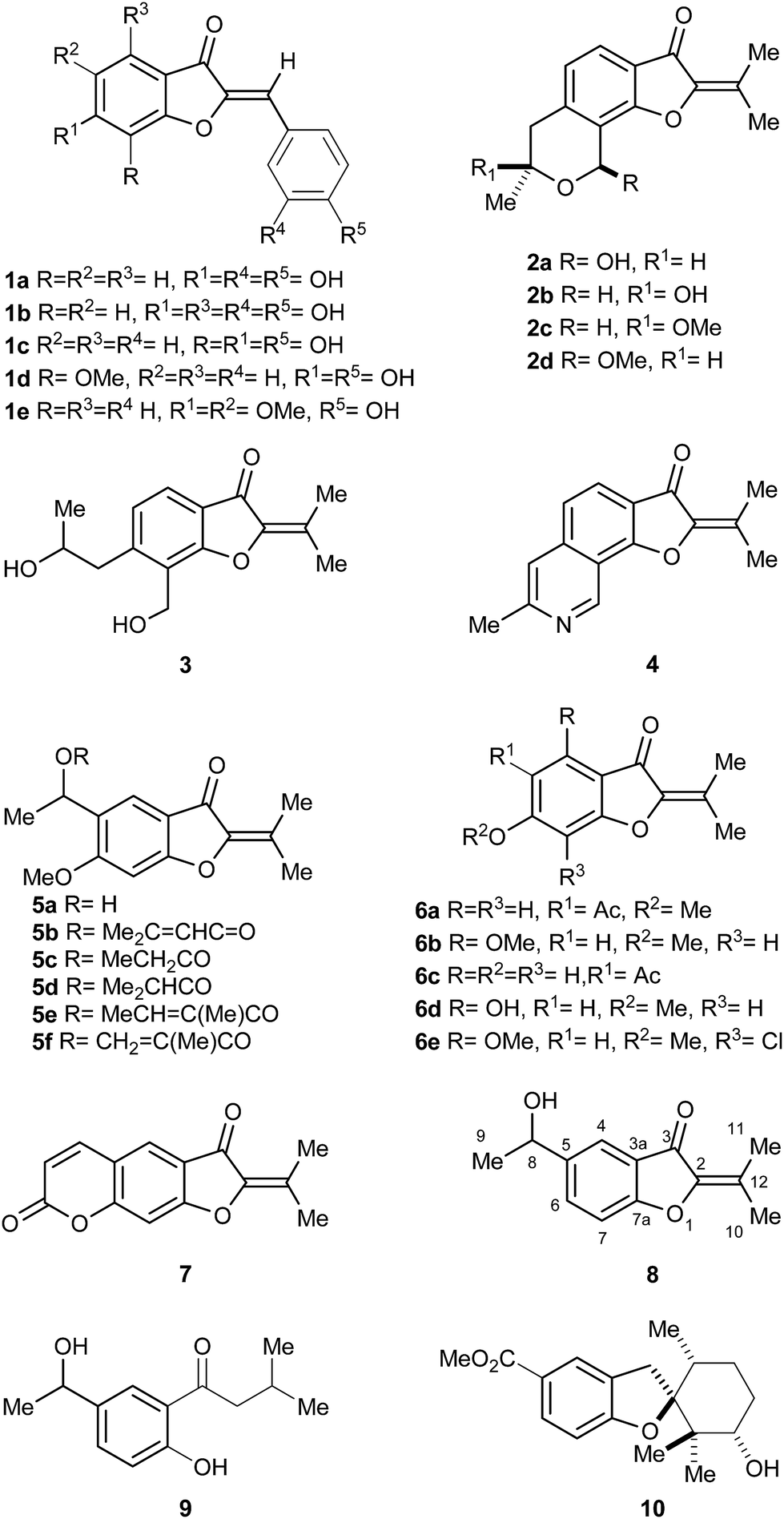 | ||
| Fig. 1 Natural products carrying the 2-isopropylidene benzofuran-3-one motif, and related structures. | ||
Fungi-derived 2-isopropylidene-benzofuran-3-ones include compound 2a,8 pergillin (2b), penicisochroman A (2c), the ustusoranes A (2d) and C (3), and TMC-120B (4). The latter is produced by terrestrial and marine-derived fungi.9 All these heterocycles have attracted marked attention for their bioactivity, and as synthetic targets.10,11
Examples of plant-produced 2-isopropylidene-benzofuran-3-ones include alcohol 5a,12a,b esters 5b–f,12b,c as well as the 5-acetyl coumaranone 6a, and the related heterocycle 6b, isolated from American specimens.13a,b It also comprises compound 6c, obtained from Chinese perennial herbs.13c,d
In addition, some 2-isopropylidene-benzofuran-3-ones have been prepared as synthetic intermediates. Compound 6d was described in connection with antitumor agents,14a,b 6e served as key intermediate toward griseofulvin analogs14c and furocoumarin 7 was used for the synthesis of poly-azamacrocycles.15
Verbesina is a large American genus within the Heliantheae, distributed from Canada to Argentina. Its members are used in folk medicine to treat digestive and respiratory conditions, and as wound healing agents;16 however, few species of Verbesina have been chemically investigated.17 V. luetzelburgii is a green bush, which grows chiefly in the State of Bahia (Brazil).18 It was chemically studied only once,19 yielding the unique 2-isopropylidene-benzofuran-3-one 8 and the ketone 9, its logical metabolic precursor, and a handful of other unrelated compounds.
Compound 8 is the only benzofuran derivative found among species of Verbesina. This heterocycle seems to be the prototypic and simplest 2-isopropylidene-benzofuran-3-one representative. Notably, this class of compounds has been barely studied, due to the limited amounts of isolated material and the scarcity of synthetic protocols to provide an affordable access to them.
We have previously employed filifolinol (10), a heliantaceous 2,3-dihydrobenzofuran for the synthesis of inhibitors of the human complement cascade20 and have synthesized a potential key intermediate toward TMC-120B (4).21 In pursuit of our studies on naturally-occurring benzofuran derivatives, herein we report the first total synthesis of the 2-isopropylidene-benzofuran-3-one 8 in its racemic and in one of its optically active forms, from a common intermediate and employing 4′-hydroxyacetophenone (11) as starting material.
Results and discussion
The approach toward 8 was based on a retrosynthetic analysis (Scheme 1). It was conjectured that the oxygen functionalities could be adjusted to their proper oxidation states at a late stage of the synthesis. Accordingly, compound 12 carrying a protected alcohol or ketone moiety, was postulated as a valid advanced intermediate.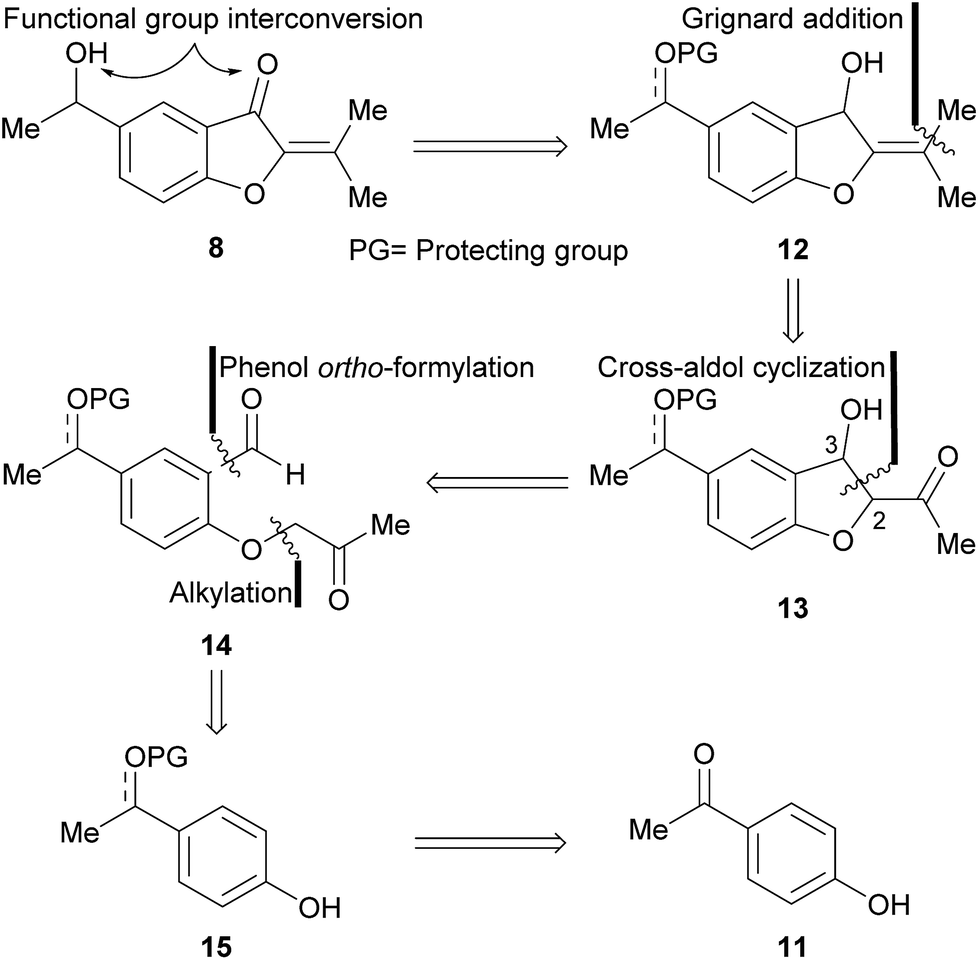 | ||
| Scheme 1 Retrosynthetic analysis of the target natural product (8). | ||
In turn, it was supposed that the isopropylidene motif could result from a Grignard addition–dehydration sequence, to a conveniently placed carbonyl moiety. This reasoning unveiled the intervention of the β-hydroxy ketone 13, which was disconnected at C-2–C-3, under the consideration that it may arise from the cross-aldol condensation of a 1,6-dicarbonyl precursor such as 14. Further analysis unearthed the protected aromatic derivative 15, as a suitable intermediate readily available from the acetophenone 11.
Accordingly, the synthesis of 8 was initiated (Scheme 2) with the protection of the carbonyl moiety of 11 as the corresponding 1,3-dioxolane (16). This group was chosen on account of its improved stability over its acyclic congeners, and relative ease of hydrolytic removal.22 Conventional ketalization methods (ethyleneglycol, TsOH, 4 Å molecular sieves, PhMe)23 without previous protection of the phenol23c,d proved unsatisfactory; however, the reaction with ethyleneglycol in hexane–HC(OiPr)3 under Ce(OTf)3 catalysis afforded 92% of the protected ketone 16 [64% using HC(OEt)3], a yield that remained high even on a gram scale.24
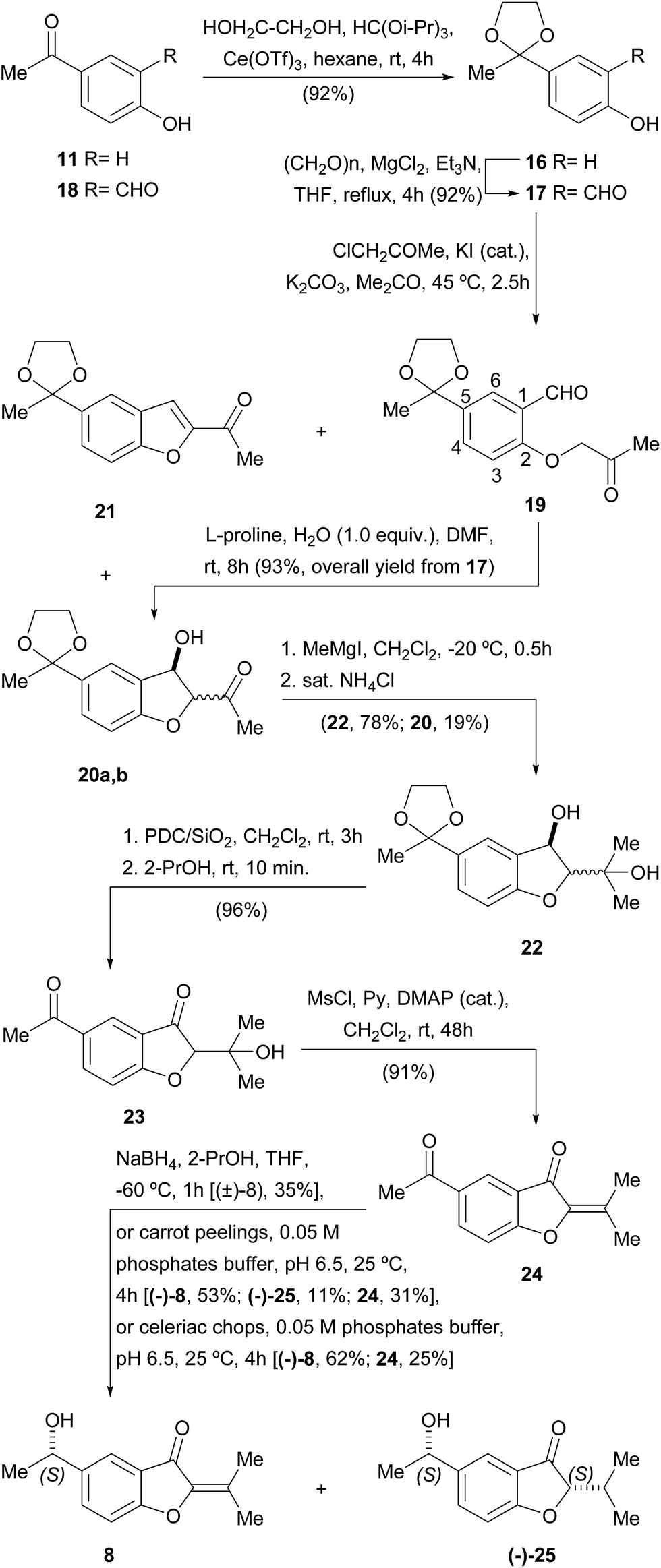 | ||
| Scheme 2 Synthesis of compounds 8 and (−)-25. | ||
Next, the ortho-formylation was undertaken, employing the procedure of Skattebøl,25 where a mixture of paraformaldehyde, Et3N, anhydrous MgCl2 and 16 was refluxed in THF, cleanly furnishing 17 in 92% yield. Contrastingly, however, it was observed that formylation of the unprotected ketone 11 gave only 20% of the related salicylaldehyde derivative 18,25a whereas literature precedents suggested that the Friedel–Crafts acylation of salicylaldehyde was not a better option toward 18.26 Hence, the Skattebøl formylation of 16 proved to be the best alternative to get salicylaldehyde 17.
The Williamson etherification of 17 with freshly distilled chloroacetone27 and K2CO3 in acetone at 45 °C smoothly gave the projected acetonyl ether 19 admixed with variable amounts (25–65%) of the diastereomeric alcohols 20a,b. The latter presumably resulted from the unwanted and uncontrolled intramolecular cross-aldol reaction of 19 under K2CO3 promotion.28
Since 19 and 20a,b formed a hardly separable mixture, where purification of 19 proved to be very difficult, attempts were made to induce completion of the cyclization, by modifying the reaction time (3–6 h) and/or temperature (50–60 °C), and by changing the promoter (NaHCO3, Li2CO3, MgCO3, Et3N). However, all those alternatives were fruitless, resulting at best in a diastereomeric mixture of 20, along with minor amounts of the benzofuran derivative 21, produced by dehydration of its benzylic alcohol moiety.
Thus, it became evident that a milder procedure was required for the cyclization stage; therefore, we turned our attention to the organocatalytic protocol of Enders.29 Luckily, treatment of the crude extract of the alkylation step, containing 19 along with some amount of 20a,b, with L-proline in DMF to which water (1 equiv.) was added, effected the desired cyclization, giving a mixture of alcohols 20a,b in 93% overall yield from 17.
In view of the structure of the target natural product (8), access to these alcohols as a diastereomeric mixture was not considered an obstacle for pursuing the synthetic sequence, which was therefore focused on the addition of the required methyl group to the ketonic carbonyl group.
Although initial experiments with MeMgI in THF at 0 °C were discouraging, even in the presence of anhydrous CeCl3,30a it was soon discovered that the outcome of the reaction was greatly improved when it was carried out in CH2Cl2 at −20 °C.30b Under these conditions, 78% yield of diol 22 was achieved, together with 19% of 20a,b that seemed to enolize, remaining refractory to the Grignard reagent and was recovered as unreacted starting material.
Next, sequential oxidation of the benzylic alcohol and deprotection of the 1,3-dioxolane were accomplished by exposing a cold solution of 22 to Jones' reagent. However, these conditions afforded only moderate yields of the diketone 23. Luckily, the secondary alcohol could be successfully oxidized with PDC/SiO2 in CH2Cl2, which also ensured the deprotection of the 1,3-dioxolane moiety, giving 96% of the diketone 23. In turn, the latter was smoothly dehydrated with MsCl and pyridine in CH2Cl2,31 which cleanly furnished 91% yield of the α,β-unsaturated ketone 24.
Finally, the dicarbonylic compound 24 was submitted to a challenging and scarcely precedented selective reduction of its exocyclic ketone in the presence of the reactive α,β-unsaturated ketone system containing an endocyclic carbonyl moiety. Following the literature, this transformation was performed with NaBH4 at −60 °C, in anhydrous THF to which a small amount of 2-propanol was added.32 Fortunately, 35% of the target α-phenethyl alcohol (±)-8 was obtained under these conditions.
Taking into account that the tiny amounts of the natural product that were isolated originally prevented from acquiring suitable optical rotation data, a biotransformation-based synthesis of 8 was also put in place. This choice was based on the facts that the bioreduction of acetophenone derivatives is known to be a highly enantioselective and eco-friendly process.33
Thus, the dicarbonylic intermediate 24 was exposed to a suspension of carrot (Daucus carota L., Apiaceae) peelings in 0.05 M phosphate buffer, pH 6.5. In this way, the natural product (−)-8 was obtained in 53% yield, along with 11% of the 1,4-reduced compound (−)-25, unveiling an ene-reductase activity, and unreacted starting material. Unfortunately, longer reaction times produced increased amounts of the unwanted compound (−)-25, forcing termination of the biotransformation before all the starting material was consumed.
Better yields were obtained when the biotransformation was carried out with chopped pieces of celeriac (Apium graveolens L., var. rapaceum, Apiaceae) in place of the carrot peelings; in this case, the benzofuranone (−)-8 {[α]23D = −8.1 (c = 4.00, CHCl3)} was accessed in 62% yield, along with only 25% of unreacted starting material. Unfortunately, longer reaction times also lead to an increased production of (−)-25.
Noteworthy, despite the wide distribution of the ene reductase activity among microorganisms and plants, its presence in carrot and celeriac has not been specifically reported to date.34 This observation may have impact on the synthesis of the related 2-isopropyl furanone-type natural products.10a–c,12c,35
According to literature precedents, the selective bioreduction of aromatic methyl ketones by carrots and celeriac follows “Prelog behaviour”,33e,f to afford the corresponding S-enantiomers.33a Hence, this configuration was attributed to the α-phenethyl alcohol centers of (−)-8 and (−)-25. In addition, the chiral HPLC analysis of (−)-8 determined its enantiomeric excess (ee) as 87%. The IR and 1H NMR spectral data of the thus synthesized compound 8 in both, racemic and optically active forms, were in full agreement with those informed for the natural product.19 and its 13C NMR spectral data agreed with the proposed structure.
On the other hand, the absolute configuration of the C-2 center of (−)-25 was established as S employing electronic circular dichroism (ECD) measurements and comparing the results with theoretical ECD calculations.36 These results were also in agreement with the modified octant rule, defining the relationship between the chirality of aryl ketone derivatives and the sign of their high wavelength Cotton effect.37
Conclusions
We have developed efficient first total syntheses of the naturally occurring 5-(1-hydroxy-1-ethyl)-2-isopropyliden-2H-benzofuran-3-one (8), isolated from the Brazilian plant Verbesina luetzelburgii in its racemic form and as one of its enantiomers. The syntheses were achieved in 8 steps and 19% overall yield [up to 33% for (−)-8] from commercially available 4′-hydroxyacetophenone.Protecting the carbonyl moiety of the starting ketone, as the 1,3-dioxolane derivative, enabled the development of an efficient ortho-formylation of the phenol and a low-temperature Grignard 1,2-addition to a carbonyl group in CH2Cl2, as pivotal synthetic steps.
The first critical transformation was complemented with an etherification and a proline-mediated aldolization, which afforded the heterocyclic ring, whilst a secondary alcohol oxidation and a MsCl-assisted dehydration supplemented the second key reaction, and installed the conjugated isopropylidene side chain.
The stability of the cyclic ketal motif combined with its facile deprotection reduced the burden associated to the use of protecting groups during the synthetic sequence, whereas the challenging differentiation between the endocyclic and exocyclic carbonyl groups was performed by chemical (NaBH4/iPrOH) and enzymatic (carrot peels, celeriac chops) means. The strategy developed toward 8 is a useful starting point to gain access to other, still non-synthesized congeners and to unveil their potentially interesting bioactivities. Work in this direction is under way and the results will be disclosed in due time.
Experimental section
General information
The reactions were executed under anhydrous argon atmospheres, employing oven-dried glassware and freshly distilled anhydrous solvents. Anhydrous THF and toluene were obtained by reflux of AR solvents over sodium metal (benzophenone as indicator), followed by distillation. Anhydrous acetone was prepared by refluxing 4 h the AR grade product over dry K2CO3 followed by distillation. Anhydrous CH2Cl2 was obtained from an M. Braun solvent purification and dispenser system. Anhydrous Et3N was prepared by refluxing the solvent over CaH2 for 4 h, followed by distillation. 2-Propanol was dried by reaction with sodium and distillation from the so formed sodium isopropoxide. Anhydrous DMF was prepared by heating and reduced pressure distillation from dry BaO. All the anhydrous solvents were transferred via cannula and stored in dry Young ampoules containing molecular sieves. Fresh carrot and celeriac were purchased in a local market. HPLC separations were performed with HPLC grade solvents. All other reagents were used as received.The reactions were monitored by TLC developed in different hexane–EtOAc solvent mixtures. The chromatographic spots were detected by exposure to 254 nm UV light, and by spraying with ethanolic p-anisaldehyde/sulfuric acid reagent, followed by careful heating to improve selectivity. Flash column chromatography was performed with silica gel 60 H (particle size 63–200 μm), eluting with hexane–EtOAc mixtures, under positive pressure and employing solvent gradients.
Equipment
The melting points were measured on an Ernst Leitz Wetzlar model 350 hot-stage microscope. The FT-IR spectra were recorded on a Shimadzu Prestige 21 spectrophotometer, as solid dispersions in KBr disks or as thin films held between NaCl cells.The nuclear magnetic resonance spectra were acquired in CDCl3 unless otherwise noted, on a FT-NMR Bruker Avance 300 spectrometer, at 300.13 (1H) and 75.48 (13C) MHz. The chemical shifts are consigned in parts per million in the δ scale. TMS was used as the internal standard (resonances of CHCl3 in CDCl3: δ 7.26 and 77.0 for 1H and 13C NMR, respectively). An asterisk (*) designates signals which attribution can be exchanged. The coupling constants (J) and half-width (w1/2) values are given in hertz. Some 2D-NMR experiments were also performed.
The high resolution mass spectra were obtained from UMYMFOR (Buenos Aires, Argentina) with a Bruker MicroTOF-Q II instrument. Detection of the ions was performed in electrospray ionization, positive ion mode.
The specific optical rotations were measured with a Jasco DIP-1000 polarimeter using a 1 cm microcell. The circular dichroism measurements were carried out in a Jasco J-710 circular dichroism spectrometer, with a 1 cm cell and the sample dissolved in acetonitrile, at a concentration of 0.25 mg mL−1.
The chiral HPLC determinations were carried out on a Varian Prostar 210 instrument consisting of two pumps, a manual injector fitted with a 20 μL loop and a Prostar 325 variable dual-wavelength UV-vis detector. The determinations were performed on a Chiralcel OD analytical column (250 mm × 4.6 mm I.D., 5 μm particle size) thermostated at 30 °C. The mobile phase was a hexanes![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-propanol mixture (91/9, v/v), pumped at 1.0 mL min−1. The detection wavelengths were 254 and 360 nm. All samples were filtered through 0.45 μm nylon filters before injection.
:2-propanol mixture (91/9, v/v), pumped at 1.0 mL min−1. The detection wavelengths were 254 and 360 nm. All samples were filtered through 0.45 μm nylon filters before injection.
A portion of the dried extract containing the keto-aldehyde 19 along with 20a,b (500 mg, 1.88 mmol) was dissolved in DMF (7.5 mL), and successively treated with distilled water (0.035 mL, 1.88 mmol) and L-proline (88 mg, 0.75 mmol). The reaction was stirred for 8 h at 25 °C, when brine (10 mL) was added and the organic products were extracted with EtOAc (4 × 20 mL). The combined organic extracts were washed with brine (10 mL), dried over MgSO4, and concentrated under reduced pressure. Filtration of the residue through a short pad of silica afforded a barely separable mixture of 20a and 20b (475 mg, 95%), as a pale yellow oil (20a:20b = 1:1.6). Compound 20a: oil; IR (film, ν): 3418, 2986, 2891, 1715, 1614, 1487 and 1038 cm−1. 1H NMR: δ 7.52 (d, 1H, J = 2.0, H-4), 7.43 (dd, 1H, J = 2.0 and 8.2, H-6), 6.91 (d, 1H, J = 8.2, H-7), 5.51 (d, 1H, J = 6.7, H-3), 4.94 (d, 1H, J = 6.7, H-2), 3.98–4.02 (m, 2H, OCH2CH2O), 3.73–3.77 (m, 2H, OCH2CH2O), 2.35 (s, 3H, H-10) and 1.61 (s, 3H, H-9). 13C NMR: δ 206.4 (C-12), 159.2 (C-7a), 137.4 (C-5), 128.5 (C-6), 126.8 (C-3a), 123.0 (C-4), 110.4 (C-7), 108.7 (C-8), 90.6 (C-2), 72.9 (C-3), 64.4 (2C, OCH2CH2O), 28.4 (C-10) and 27.7 (C-9). HRMS: calcd for C14H17O5 [M + H]+ 265.1071; found 265.1073. Compound 20b: oil; IR (film, ν): 3418, 2986, 2891, 1715, 1614, 1487 and 1038 cm−1. 1H NMR: δ 7.50 (d, 1H, J = 2.0, H-4), 7.40 (dd, 1H, J = 2.0 and 8.2, H-6), 6.91 (d, 1H, J = 8.2, H-7), 5.38 (d, 1H, J = 3.4, H-3), 4.88 (d, 1H, J = 3.4, H-2), 3.98–4.02 (m, 2H, OCH2CH2O), 3.73–3.77 (m, 2H, OCH2CH2O), 2.24 (s, 3H, H-10) and 1.60 (s, 3H, H-9). 13C NMR: δ 207.2 (C-12), 159.0 (C-7a), 137.4 (C-5), 128.3 (C-6), 126.7 (C-3a), 122.8 (C-4), 110.2 (C-7), 108.7 (C-8), 94.3 (C-2), 75.1 (C-3), 64.4 (2C, OCH2CH2O), 27.7 (C-9) and 26.6 (C-10). HRMS: calcd for C14H17O5 [M + H]+ 265.1071; found 265.1073.
:3 mixture of 22a and 22b (323 mg, 78%), as a thick yellow oil. Compound 22a: IR (film, ν): 3418, 2984, 2930, 1715, 1690, 1614, 1487, 1373, 1258, 1038 and 824 cm−1. 1H NMR: δ 7.49 (d, 1H, J = 1.9, H-4), 7.37 (dd, 1H, J = 1.9 and 8.5, H-6), 6.80 (d, 1H, J = 8.5, H-7), 5.37 (d, 1H, J = 4.9, H-3), 4.29 (d, 1H, J = 4.9, H-2), 3.99–4.04 (m, 2H, OCH2CH2O), 3.77–3.81 (m, 3H, OCH2CH2O, OH), 2.88 (br s, 1H, OH), 1.64 (s, 3H, H-9), 1.36 (s, 3H, H-10)* and 1.29 (s, 3H, H-11).* 13C NMR: δ 159.5 (C-7a), 136.6 (C-5), 128.5 (C-3a), 127.8 (C-6), 122.3 (C-4), 109.7 (C-7), 108.8 (C-8), 97.1 (C-2), 73.6 (C-3), 71.5 (C-12), 64.5 (2C, OCH2CH2O), 27.7 (C-9), 25.9 (C-10)* and 24.4 (C11).* HRMS: calcd for C15H20NaO5 [M + Na]+ 303.1203; found 303.1171. Compound 22b: IR (film, ν): 3418, 2984, 2930, 1715, 1690, 1614, 1487, 1373, 1258, 1038 and 824 cm−1. 1H NMR: δ 7.52 (d, 1H, J = 1.9, H-4), 7.39 (dd, 1H, J = 1.9 and 8.5, H-6), 6.85 (d, 1H, J = 8.5, H-7), 5.29 (d, 1H, J = 6.0, H-3), 4.21 (d, 1H, J = 6.0, H-2), 3.99–4.04 (m, 2H, OCH2CH2O), 3.77–3.81 (m, 3H, OCH2CH2O, OH), 2.88 (br s, 1H, OH), 1.64 (s, 3H, H-9), 1.57 (s, 3H, H-11)* and 1.51 (s, 3H, H-10).* 13C NMR: δ 159.1 (C-7a), 136.8 (C-5), 129.7 (C-3a), 127.8 (C-6), 122.5 (C-4), 110.0 (C-7), 108.8 (C-8), 89.4 (C-2), 72.9 (C-3),* 72.7 (C-12),* 64.5 (2C, OCH2CH2O), 28.4 (C-9), 27.7 (C-10)* and 25.5 (C-11).* HRMS: calcd for C15H20NaO5 [M + Na]+ 303.1203; found 303.1170.Acknowledgements
The authors thank Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and financial support (PICTs No. 2011-0399 and 2014-0445 and PIP No. 2012-0471). J. L. P. also acknowledges ANPCYT and CONICET for her fellowships.Notes and references
- (a) H. Sundén and R. Olsson, Org. Biomol. Chem., 2010, 7, 4831–4833 RSC; (b) N. Leflemme, A. R. Stoit and A. Borghese, Tetrahedron Lett., 2012, 53, 2432–2435 CrossRef CAS; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed; (d) L. Costantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65–85 CrossRef CAS PubMed; (e) R. W. DeSimone, K. S. Currie, S. A. Mitchell, J. W. Darrow and D. A. Pippin, Comb. Chem. High Throughput Screening, 2004, 7, 473–493 CrossRef CAS; (f) H. Kwiecie, A. Goszczyska and P. Rokosz, Curr. Pharm. Des., 2016, 22, 879–894 CrossRef CAS.
- (a) R. Kuppusamy, P. Gandeepan and C. H. Cheng, Org. Lett., 2015, 17, 3846–3849 CrossRef CAS PubMed; (b) A. S. Van Dyk, J. P. Petzer, A. Petzer and L. J. Legoabe, Drug Des., Dev. Ther., 2015, 9, 5479–5489 Search PubMed; (c) M. Alipour, M. Khoobi, H. Nadri, A. Sakhteman, A. Moradi, M. Ghandi, A. Foroumadi and A. Shafiee, Arch. Pharm., 2013, 346, 577–587 CrossRef CAS PubMed; (d) J. Alvarado, J. Fournier and A. Zakarian, Angew. Chem., Int. Ed., 2016, 55, 11625–11628 CrossRef CAS PubMed; (e) A. Hiremathad, M. R. Patil, K. R. Chethana, K. Chand, M. A. Santos and R. S. Keri, RSC Adv., 2015, 5, 96809–96828 RSC; (f) H. Khanam and Shamsuzzaman, Eur. J. Med. Chem., 2014, 97, 483–504 CrossRef PubMed.
- A. B. Petersen, M. H. Rønnest, T. O. Larsen and M. H. Clausen, Chem. Rev., 2014, 114, 12088–12107 CrossRef CAS PubMed.
- (a) O. Kayser, W. R. Waters, K. M. Woods, S. J. Upton, J. S. Keithly and A. F. Kiderlen, Planta Med., 2001, 67, 722–725 CrossRef CAS PubMed; (b) B. S. Sepuvelda and B. K. Cassels, Planta Med., 1996, 62, 98–105 CrossRef PubMed; (c) O. Kayser, A. F. Kiderlen, U. Folkens and H. Kolodziej, Planta Med., 1999, 65, 316–319 CrossRef CAS PubMed; (d) O. Kayser, A. F. Kiderlen and R. Brun, Planta Med., 2001, 67, 718–721 CrossRef CAS PubMed.
- (a) B. M. Lee, S. K. Lee and H. S. Kim, Cancer Lett., 1998, 132, 219–227 CrossRef CAS PubMed; (b) A. D. Kinghorn, H. H. S. Fong, N. R. Farnsworth, R. G. Mehta, R. C. Moon, R. M. Moriarty and J. M. Pezzuto, Curr. Org. Chem., 1998, 2, 597–612 CrossRef CAS.
- (a) C. Shu, R. Liu, S. Liu, J.-Q. Li, Y.-F. Yu, Q. He, X. Lu and L.-W. Ye, Chem.–Asian J., 2015, 10, 91–95 CrossRef CAS PubMed; (b) R. Haudecoeur, A. Gouron, C. Dubois, H. Jamet, M. Lightbody, R. Hardre, A. Milet, E. Bergantino, L. Bubacco, C. Belle, M. Reglier and A. Boumendjel, ChemBioChem, 2014, 15, 1325–1333 CrossRef CAS PubMed; (c) Y.-K. Li, J.-Q. Sun, X.-M. Gao and C. Lei, Helv. Chim. Acta, 2014, 97, 414–419 CrossRef CAS; (d) Y. Okada, M. Okita, Y. Murai, Y. Okano and M. Nomura, Nat. Prod. Res., 2014, 28, 201–204 CrossRef CAS PubMed; (e) X. Gao, L. Yang, L. Shu, Y. Shen, Y. Zhang and Q. Hu, Heterocycles, 2012, 85, 1925–1931 CrossRef CAS.
- (a) C. Zwergel, F. Gaascht, S. Valente, M. Diederich, D. Bagrel and G. Kirsch, Nat. Prod. Commun., 2012, 7, 389–394 CAS; (b) R. Haudecoeur and A. Boumendjel, Curr. Med. Chem., 2012, 19, 2861–2875 CrossRef CAS PubMed; (c) A. Boumendjel, Curr. Med. Chem., 2003, 10, 2621–2630 CrossRef CAS PubMed.
- (a) J. Kohno, M. Sakurai, N. Kameda, M. Nishio, K. Kawano, N. Kishi, T. Okudaand and S. Komatsubara, J. Antibiot., 1999, 52, 913–916 CrossRef CAS PubMed; (b) T. Kumemura, T. Choshi, A. Hirata, M. Sera, Y. Takahashi, J. Nobuhiro and S. Hibino, Heterocycles, 2003, 61, 13–17 CrossRef CAS; (c) T. Kumemura, T. Choshi, A. Hirata, M. Sera, Y. Takahashi, J. Nobuhiro and S. Hibino, Chem. Pharm. Bull., 2005, 53, 393–397 CrossRef CAS PubMed.
- (a) S. O. Simonetti, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, Org. Biomol. Chem., 2012, 10, 4124–4134 RSC; (b) S. O. Simonetti, E. L. Larghi, A. B. J. Bracca and T. S. Kaufman, Nat. Prod. Rep., 2013, 30, 941–969 RSC.
- (a) N. Bunbamrung, C. Intaraudom, N. Boonyuen, P. Rachtawee, P. Laksanacharoen and P. Pittayakhajonwut, Phytochem. Lett., 2014, 10, 13–18 CrossRef CAS; (b) K. Trisuwan, V. Rukachaisirikul, Y. Sukpondma, S. Phongpaichit, S. Preedanon and J. Sakayaroj, Tetrahedron, 2010, 66, 4484–4489 CrossRef CAS; (c) Z. Lu, Y. Wang, C. Miao, P. Liu, K. Hong and W. Zhu, J. Nat. Prod., 2009, 72, 1761–1767 CrossRef CAS PubMed; (d) A. Ogawa, C. Murakami, S. Kamisuki, I. Kuriyama, H. Yoshida, F. Sugawara and Y. Mizushina, Bioorg. Med. Chem. Lett., 2004, 14, 3539–3543 CrossRef CAS PubMed; (e) J. Kohno, H. Hiramatsu, M. Nishio, M. Sakurai, T. Okuda and S. Komatsubara, Tetrahedron, 1999, 55, 11247–11252 CrossRef CAS.
- (a) K. Kuramochi, F. Saito, A. Nakazaki, T. Takeuchi, K. Tsubaki, F. Sugawara and S. Kobayashi, Biosci., Biotechnol., Biochem., 2010, 74, 1635–1640 CrossRef CAS PubMed; (b) T. Maegawa, K. Otake, K. Hirosawa, A. Goto and H. Fujioka, Org. Lett., 2012, 14, 4798–4801 CrossRef CAS PubMed; (c) M. Tobe, T. Tashiro, M. Sasaki and H. Takikawa, Tetrahedron, 2007, 63, 9333–9337 CrossRef CAS; (d) Y. Sato, K. Kuramochi, T. Suzuki, A. Nakazaki and S. Kobayashi, Tetrahedron Lett., 2011, 52, 626–629 CrossRef CAS.
- (a) F. Bohlmann and C. Zdero, Phytochemistry, 1979, 18, 492–493 CrossRef CAS; (b) A. Mitsakos, M. Breuer, H. Budzikiewicz and P. Proksch, Phytochemistry, 1986, 25, 2243–2244 CrossRef CAS; (c) H. Budzikiewicz, G. Laufenberg, C. Clark and P. Proksch, Phytochemistry, 1984, 23, 2625–2627 CrossRef CAS.
- (a) J. Jakupovic, A. Schuster, T. V. Chau-Thi, F. Bohlmann and X. A. Domínguez, Phytochemistry, 1988, 27, 2235–2240 CrossRef CAS; (b) V. Castro, G. Tamayo-Castillo and J. Jakupovic, Phytochemistry, 1989, 28, 2415–2418 CrossRef CAS; (c) H. Nagano, R. Hanai, H. Yamada, M. Matsushima, Y. Miura, T. Hoya, M. Ozawa, M. Fujiwara, H. Kodama, A. Torihata, H. Onuki, Y. Nezu, S. Kawai, M. Yamazaki, H. Hirota, Y. Saito, M. Tori, A. Ohsaki, X. Gong and C. Kuroda, Chem. Biodiversity, 2012, 9, 789–805 CrossRef CAS PubMed; (d) Y. Zhao, Z. Jia and L. Yang, Phytochemistry, 1994, 37, 1149–1152 CrossRef CAS.
- (a) S. Bauer, R. Endele, G. Fertig, W.-G. Friebe, M. Koerner and H.-W. Krell, Patent WO 2004/76444, 2004; (b) G. De Cillis, R. Di Domenico, B. Könic and A. Oliva, US Pat. 6200989, 2001; (c) H. Tomozane, Y. Takeuchi, T. Choshi, S. Kishida and M. Yamato, Chem. Pharm. Bull., 1990, 38, 925–929 CrossRef CAS PubMed.
- (a) A. V. Lipeeva, E. E. Shul'ts, M. M. Shakirov and G. A. Tolstikov, Russ. J. Org. Chem., 2013, 49, 99–107 CrossRef CAS; (b) A. V. Lipeeva, E. E. Shul'ts, M. M. Shakirov and G. A. Tolstikov, Russ. J. Org. Chem., 2010, 46, 1858–1868 CrossRef CAS; (c) S. A. Osadchii, E. E. Shul'ts, M. M. Shakirov and G. A. Tolstikov, Russ. Chem. Bull., 2006, 55, 375–379 CrossRef CAS; (d) A. V. Lipeeva, E. E. Shults, M. M. Shakirov, M. A. Pokrovsky and A. G. Pokrovsky, Molecules, 2014, 19, 7881–7900 CrossRef PubMed; (e) A. V. Lipeeva, E. E. Shul'ts, M. M. Shakirov and G. A. Tolstikov, Chem. Nat. Compd., 2009, 45, 338–345 CrossRef CAS.
- (a) L. Dalla Via, M. Mejia, A. N. García-Argáez, A. Braga, A. Toninello and M. Martínez-Vázquez, Bioorg. Med. Chem., 2015, 23, 5816–5828 CrossRef CAS PubMed; (b) P. Giovannini and M. Heinrich, J. Ethnopharmacol., 2009, 121, 383–399 CrossRef PubMed; (c) A. J. Alonso-Castro, M. L. Villarreal, L. A. Salazar-Olivo, M. Gómez-Sánchez, F. Domínguez and A. García-Carranca, J. Ethnopharmacol., 2011, 133, 945–972 CrossRef CAS PubMed; (d) M. Gonzáles de la Cruz, S. B. Malpartida, H. B. Santiago, V. Jullian and G. Bourdy, J. Ethnopharmacol., 2014, 155, 1093–1117 CrossRef PubMed.
- (a) M. R. J. R. Albuquerque, K. M. Canuto, O. D. L. Pessoa, E. P. Nunes, R. F. Nascimento and E. R. Silveira, Flavour Fragrance J., 2006, 21, 634–636 CrossRef CAS; (b) P. Carrillo-Reyes, J. Rzedowski and G. Calderón de Rzedowski, Acta Botanica Mexicana, 2010, 93, 127–143 CrossRef.
- http://reflora.jbrj.gov.br/reflora/herbarioVirtual/ConsultaPublicoHVUC/ConsultaPublicoHVUC.do?idTestemunho=2693667, access 4 January 2017.
- F. Bohlmann, M. Grenz, R. K. Gupta, A. K. Dhar, M. Ahmed, R. B. King and H. Robinson, Phytochemistry, 1980, 19, 2391–2397 CrossRef CAS.
- (a) E. L. Larghi, M. A. Operto, R. Torres and T. S. Kaufman, Eur. J. Med. Chem., 2012, 55, 74–84 CrossRef CAS PubMed; (b) E. L. Larghi, M. A. Operto, R. Torres and T. S. Kaufman, Bioorg. Med. Chem. Lett., 2009, 19, 6172–6175 CrossRef CAS PubMed; (c) M. Useglio, P. M. Castellano, M. A. Operto, R. Torres and T. S. Kaufman, Bioorg. Med. Chem. Lett., 2006, 16, 5097–5101 CrossRef CAS PubMed.
- J. L. Pergomet, T. S. Kaufman and A. B. J. Bracca, Helv. Chim. Acta, 2016, 99, 398–404 CrossRef CAS.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, Wiley, NY, USA, 4th edn, 2007 Search PubMed.
- (a) B. Altava, M. I. Burguete, B. Escuder, S. V. Luis and R. V. Salvador, J. Org. Chem., 1997, 62, 3126–3134 CrossRef CAS PubMed; (b) P. Kahnberg, C. W. Lee, R. H. Grubbs and O. Sterner, Tetrahedron, 2002, 58, 5203–5208 CrossRef CAS; (c) A. Kuboki, T. Yamamoto and S. Ohira, Chem. Lett., 2003, 32, 420–421 CrossRef CAS; (d) N. Shohji, T. Kawaji and S. Okamoto, Org. Lett., 2011, 13, 2626–2629 CrossRef CAS PubMed.
- (a) F. Ono, H. Takenaka, T. Fujikawa, M. Mori and T. Sato, Synthesis, 2009, 1318–1322 CAS; (b) F. Ono, H. Takenaka, Y. Eguchi, M. Endo and T. Sato, Synlett, 2009, 487–489 CAS; (c) S. Ma and L. M. Venanzi, Synlett, 1993, 751–752 CrossRef CAS.
- (a) N. U. Hofslokken and L. Skattebøl, Acta Chem. Scand., 1999, 53, 258–262 CrossRef CAS; (b) T. V. Hansen and L. Skattebøl, Org. Synth., 2005, 82, 64–68 CrossRef CAS.
- Z. Zhang, C. Pan and Z. Wang, Chem. Commun., 2007, 4686–4688 RSC.
- (a) S. Clamens and R. Teíssíer, FR Patent 2633614, 1988; (b) G. A. Hiegel and K. B. Peyton, Synth. Commun., 1985, 15, 385–392 CrossRef CAS; (c) K. Terao, Bull. Inst. Chem. Res., Kyoto Univ., 1992, 70, 338–377 CAS.
- (a) Y. Lin, G. B. Jones, G.-S. Hwang, L. Kappen and I. H. Goldberg, Org. Lett., 2005, 7, 71–74 CrossRef CAS PubMed; (b) B. M. Williams and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 2191–2194 CrossRef CAS PubMed; (c) J. R. Falck, S. C. Manna, L. Alcaraz and C. Mioskowski, Tetrahedron Lett., 1994, 35, 2013–2016 CrossRef CAS.
- (a) D. Enders, J. Fronert, T. Bisschops and F. Boeck, Beilstein J. Org. Chem., 2012, 8, 1112–1117 CrossRef CAS PubMed; (b) D. Enders, O. Niemeier and L. Straver, Synlett, 2006, 3399–3402 CrossRef CAS; (c) M. K. Georgieva, F. J. S. Duarte, M. V. B. Queirós and A. Gil Santos, J. Org. Chem., 2012, 77, 5569–5576 CrossRef CAS PubMed; (d) V. Gauchot and A. R. Schmitzer, J. Org. Chem., 2014, 79, 2694–2701 CrossRef CAS PubMed.
- (a) T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392–4398 CrossRef CAS; (b) D. A. Bianchi, G. Schmeda Hirschmann, C. Theoduloz, A. B. J. Bracca and T. S. Kaufman, Bioorg. Med. Chem. Lett., 2005, 15, 2711–2715 CrossRef CAS PubMed.
- (a) K. Tatsuta, K. Akimoto and M. Kinoshita, Tetrahedron, 2014, 37, 4365–4369 CrossRef; (b) T. Patonay, J. Jeko and E. Juhasz-Toth, Eur. J. Org. Chem., 2008, 1441–1448 CrossRef CAS; (c) P. Belanger and P. Prasit, Tetrahedron Lett., 1988, 29, 5521–5524 CrossRef CAS.
- (a) M. Kawada, M. Watanabe, K. Ukamoto, H. Sugihara, T. Hirata, Y. Maki, I. Imada and Y. Sanno, Chem. Pharm. Bull., 1984, 32, 3532–3550 CrossRef CAS PubMed; (b) J. R. Pfister, I. T. Harrison and J. H. Fried, US Pat. 3963752, 1976.
- (a) W. K. Mazczka and A. Mironowicz, Tetrahedron: Asymmetry, 2004, 15, 1965–1967 CrossRef; (b) Z.-H. Yang, R. Zeng, G. Yang, Y. Wang, L.-Z. Li, Z.-S. Lv, M. Yao and B. Lai, J. Ind. Microbiol. Biotechnol., 2008, 35, 1047–1051 CrossRef CAS PubMed; (c) J. S. Yadav, S. Nanda, P. Thirupathi Reddy and A. Bhaskar Rao, J. Org. Chem., 2002, 67, 3900–3903 CrossRef CAS PubMed; (d) X. Liu, Z. G. Pan, J. H. Xu and H. X. Li, Chin. Chem. Lett., 2010, 21, 305–308 CrossRef CAS; (e) V. Aldabalde, P. Arcia, A. González and D. González, Green Chem. Lett. Rev., 2007, 1, 25–30 CrossRef CAS; (f) V. Prelog, Pure Appl. Chem., 1964, 9, 119–130 CrossRef CAS.
- (a) C. K. Winkler, G. Tasnádia, D. Clay, M. Hall and K. Faber, J. Biotechnol., 2012, 162, 381–389 CrossRef CAS PubMed; (b) H. S. Toogood, D. Mansell, J. M. Gardiner and N. S. Scrutton, 7.11 Reduction: Enantioselective Bioreduction of C–C Double Bonds, in Comprehensive Chirality, Elsevier, Amsterdam, The Netherlands, 2012, vol. 7, pp. 216–255, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering Search PubMed.
- F. Bohlmann, U. Fritz, R. M. King and H. Robinson, Phytochemistry, 1980, 19, 2500–2501 CrossRef CAS.
- (a) G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466–474 CrossRef CAS PubMed; (b) G. Pescitelli, T. Kurtán, U. Flörke and K. Krohn, Chirality, 2009, 21, E181–E201 CrossRef CAS PubMed; (c) K. Ling-Yi and W. Peng, Chin. J. Nat. Med., 2013, 11, 193–198 Search PubMed.
- (a) G. Snatzke, Tetrahedron, 1965, 21, 439–448 CrossRef CAS; (b) S. Lin, Y. Zhang, M. Liu, S. Yang, M. Gan, J. Zi, W. Song, X. Fan, S. Wang, Y. Liu, Y. Yang, X. Chen, Y. Guo, W. Wang and J. Shi, J. Nat. Prod., 2010, 73, 1914–1921 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected spectra of intermediates and final product. See DOI: 10.1039/c6ra28587b |
| This journal is © The Royal Society of Chemistry 2017 |