
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd–ZnO nanowire arrays as recyclable catalysts for 4-nitrophenol reduction and Suzuki coupling reactions†
Qiyan Huab,
Xiaowang Liu*a,
Lin Tangab,
Dewen Mina,
Tianchao Shia and
Wu Zhang *a
*a
aCollege of Chemistry and Materials Science, Anhui Normal University, Key Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, Wuhu 241000, P. R. China. E-mail: zhangwu@mail.ahnu.edu.cn; xwliu601@mail.ahnu.edu.cn
bSchool of Pharmacy, Wannan Medical College, Wuhu 241002, P. R. China
First published on 23rd January 2017
Abstract
In this work, we report a facile approach for the preparation of Pd nanoparticle–ZnO nanowire arrays followed by demonstrating their use as recyclable catalyst for reactions under different conditions. The facile synthesis largely relies on a spontaneous reduction of PdCl42− at the surface of oriented ZnO nanowires grown on a Zn foil. This is due to a combination of the strongly reducing ability of Zn foil and the semiconducting nature of ZnO nanowires. At room temperature and under a weak alkaline condition, the as-prepared Pd nanoparticle–ZnO nanowire arrays show a catalytic activity factor up to 76.6 s−1 g−1 in the reduction of 4-nitrophenol and no obvious decreases in the catalytic activity even after use of 10 times. Meanwhile, the as-prepared Pd nanoparticle–ZnO nanowire arrays exhibit extraordinary catalytic activity toward Suzuki and carbonylative Suzuki reactions at a higher temperature under a stronger alkaline condition. The outstanding performance of the hybrid nanowire arrays is mainly originated from the small size of the Pd nanoparticles (∼2–4 nm) with clean surfaces, as well as a strong affinity between the Pd nanoparticles and ZnO nanowires, leading to marginable catalyst loss and aggregation. Considering the multiple choices of both noble metal nanocatalyst and transition metal oxide nanowire array, we expect noble metal nanoparticle-transition metal oxide nanowire arrays to be emerged as a new class of recyclable catalysts attractive for diverse organic reactions to develop pharmaceuticals, natural products and advanced functional materials.
Introduction
Noble metal nanoparticles (NPs), such as Ag, Au and Pd, have garnered significant attention in organic synthesis as viable catalysts owing to their extraordinary catalytic nature.1 These nanocatalysts have proven effective for a diverse array of reactions, including C–C coupling, alkene hydrogenation and C–H arylation.2 However, direct utilization of noble metal NPs as catalysts usually suffers from high cost due to the lack of efficient strategies for their recollection.3 In addition, the use of pure small-sized noble metal NPs as catalysts usually leads to another issue, that is, gradual decrease in their catalytic activity as a result of nanocatalyst coalesce in the course of catalytic reactions.4To enhancing the recyclability while preserving the highly catalytic activity of nanocatalyst, considerable efforts have been devoted to growing catalytically active NPs onto the surfaces of 1- or 2-dimensional (D) nanostructure supports, including TiO2 rods, Al2O3 NPs, carbon-based nanomaterials, metal–organic frameworks and magnetic NPs.5 In fact, these studies have shown additional benefits of supported nanocatalysts besides facilitating the recollection of catalytically active noble NPs from the reaction mixture. For example, immobilization of noble metal NPs on support matrices allows the nanocatalysts to be well-separated, and effectively prevents the interparticle communication, giving rise to the persistence of the catalytic activity. Additionally, the introduction of noble metal–support interfaces in the catalysts provides new possibility for improving their catalytic activity through interface engineering.6 Despite the enormous advances, there is still room for enhancing the performance of supported nanocatalysts in terms of boosting their catalytic activity and recyclability. One promising pathway is to optimize the morphology of nanostructured support to simultaneously increase the loading of noble metal NPs and improve the interaction between the matrices and the loaded noble metal NPs.
Recent studies suggest that nanowire arrays are attractive as support for the development of highly recyclable catalysts for their outstanding features: (i) the building blocks of the nanowires in the arrays providing large surfaces for the loading of catalytically active NPs;7 (ii) the sufficient inter-nanowire space benefiting the access of reactants to the catalytic sites and the release of catalytic products from the supported catalysts;8 and (iii) the large dimensions of the array substrate up to square centimetres favouring efficient catalyst collection.9 More importantly, reliable synthesis of a variety of nanowire arrays, such as ZnO, TiO2, SnO2 and ZnMn2O4 via wet chemistry methods, promises to afford multiple choices to control of NP–nanowire interfaces for enhancing the catalytic performance of supported catalysts.10
On the basis of easy access to different types of noble metal–ZnO nanowire arrays by a interfacial reduction reaction between noble metal precursors and ZnO nanowire arrays in an aqueous solution and the excellent catalytic nature of Pd NPs,11 here we first demonstrate the preparation of Pd NP–ZnO nanowire arrays and then reveal their potential as recyclable catalysts for reactions under different conditions. Our results suggest that the as-prepared Pd NP–ZnO hybrid nanowire arrays exhibit highly recyclable and active catalysts toward 4-nitrophenol reduction at room temperature under weak alkaline conditions. More interestingly, at a higher temperature (100 °C) and under a stronger alkaline condition, the as-prepared Pd NP–ZnO nanowire arrays are also found to be suitable for both Suzuki and carbonylative Suzuki reactions. We demonstrate the crucial role of the size of the in situ formed Pd NPs in the determination of the catalytic performance of the supported nanoarray catalysts. Meanwhile, we reveal that the strong affinity between Pd NP and ZnO nanowire effectively prevents the aggregation and loss of the nanocatalysts, permitting the preservation of highly catalytic activity of the catalysts.
Results and discussion
We at first synthesized Pd NP–ZnO nanowire arrays by putting the ZnO@Zn nanowire arrays into an aqueous solution of Na2PdCl4 (5 mM) for different periods of time at room temperature (5, 15 and 25 s). We observed an immediate color change from white to black of the nanowire arrays (ESI, Fig. S1†) once they were exposed to the Pd precursor solution. After being immersed into the solution for 5 s, ZnO nanowire arrays were modified with a large number of secondary NPs (Fig. 1a). A closer scanning electron microscopy inspection (Fig. 1b) revealed that both size and density of the in situ formed NPs were decreased gradually from the tip of the ZnO nanowire. Transmission electron microscopy (TEM) suggested that the secondary NPs were mainly localized at the first 2.5 μm of the ZnO nanowire (Fig. 1c) and the average size of the secondary NP was determined to be in the range of 2–4 nm (Fig. S2a†). Noticeably, the secondary NPs at the tip of the ZnO nanowire are coalesced in some cases to form a big mesoporous one with an average diameter of 20–30 nm. The elemental mapping analysis showed that Pd is localized at the outer layer of the as-prepared ZnO hybrid nanowire (Fig. 1d–f). The outer layer NPs was verified to be Pd by energy-dispersive spectroscopy (Fig. 1g), X-ray photoelectron spectroscopy (XPS) and X-ray diffraction analysis of ZnO nanowire arrays before and after the reaction (Fig. S3†). Note that the XPS spectrum of Pd 3d5/2 and 3d3/2s were estimated to be at 335.1 and 340.5 eV, which are higher than the corresponding values for Pd(0), implying the strong affinity between the Pd NPs and the ZnO nanowires (Fig. 1h).12 The HRTEM image suggests the good crystalline nature of the in situ formed Pd NPs due to the observation of a measured interplanar distance of approximately 0.22 nm in individual nanosized grains, which is in accordance with the d-spacing in the (111) planes of cubic-phased Pd. As expected, average size of the in situ formed Pd NPs was confirmed to be highly dependent on the reaction times. For example, extension of the reaction times to 15 and 25 s offered Pd NPs with average sizes of ∼7 and ∼9 nm, respectively (Fig. S2b and c†). The in situ reduction of Na2PdCl4 can be attributed to electron transfer from the surfaces of ZnO nanowire to interfacial PdCl42− as a result of the occurrence of Zn substrate-induced electron transfer.11 | ||
| Fig. 1 (a) SEM image of the as-prepared Pd NP–ZnO hybrid nanowire arrays by immersing ZnO@Zn nanowire arrays into an aqueous solution of Na2PdCl4 (5 mM) for 5 s. (b and c) SEM and TEM images of a single hybrid Pd NP–ZnO nanowire in the array. (d–f) Corresponding elemental mapping of the single hybrid Pd NP–ZnO nanowire as indicated in (c), (d) Pd, (e) Zn, and (f) O. (g) EDS and (h) XPS profiles of the as-prepared Pd NP–ZnO nanowire arrays. Inset in (h) shows the curve fit of Pd 3d spectrum, inset in (g) showing an HRTEM image of the as-prepared Pd NP–ZnO hybrid nanowire. | ||
We next accessed the catalytic activity of the as-prepared Pd NP–ZnO nanowire arrays toward the reduction of 4-nitrophenol at room temperature. We first prepared a stock solution by addition of NaBH4 into 4-nitrophenol. Note that the addition of NaBH4 into 4-nitrophenol led to an obvious increase in the pH of the resulting solution and thus gave rise to a red shift in the maximum absorption from 317 to 400 nm due to the formation of 4-nitrophenolate ions (Fig. S4†).13 The resulting bright yellow solution exhibited superior stability at room temperature because long-term aging (one week) did not resulted in notable color changes. In contrast, a quick and complete decline (∼12 min) in the maximum absorption was observed after adding the as-prepared Pd NP–ZnO nanowire arrays into the stock solution. The absorption decline was accompanied with the generation of a new absorption peak at 295 nm due to the formation of 4-aminophenol (Fig. 2a and b). By comparison, the use of ZnO@Zn nanowire arrays alone under the same conditions led to a negligible change in the absorption spectrum (Fig. S5†). Taken together, these results suggest that the catalytic ability of the hybrid nanowire arrays is originated from the loaded Pd NPs.
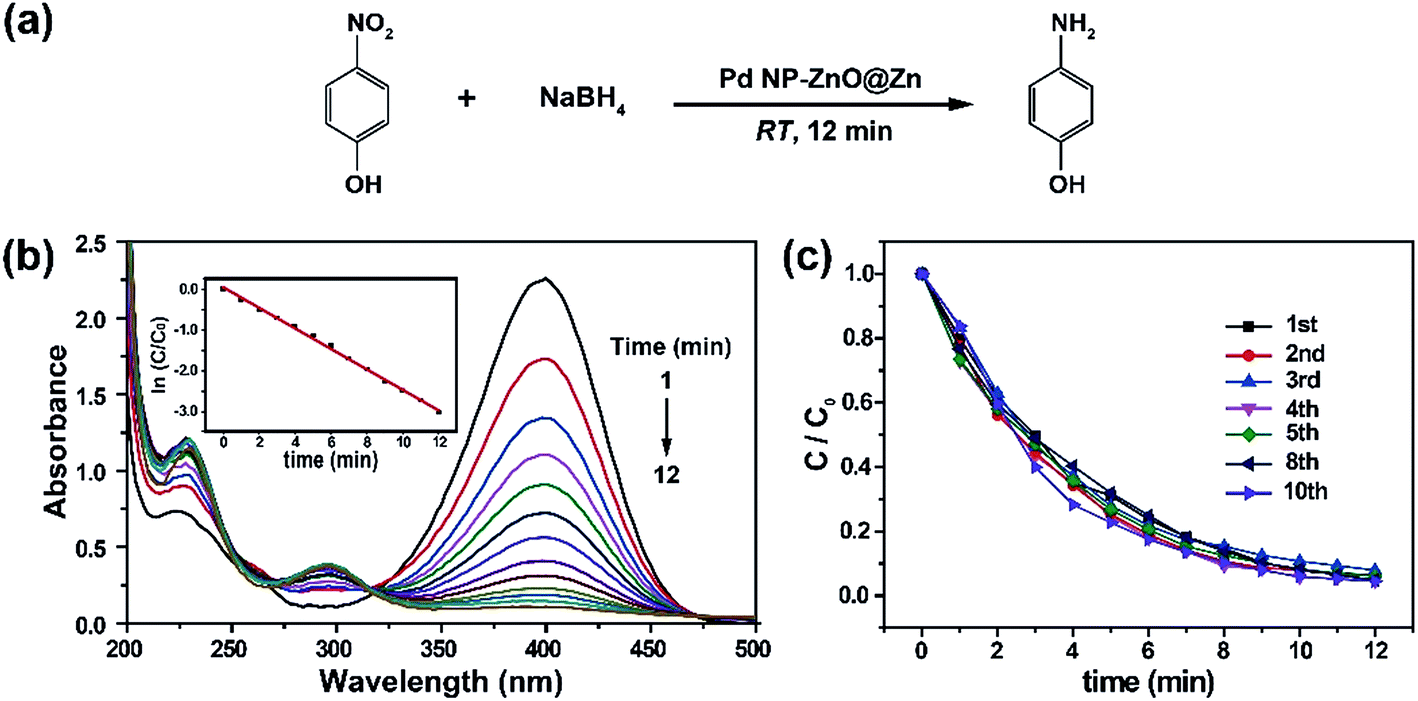 | ||
| Fig. 2 (a) Schematic representation of chemical transformation of 4-nitrophenol into 4-aminophenol in the presence of Pd NP–ZnO nanowire arrays. (b) Time-dependent UV-vis absorption spectra of a stock solution containing 4-nitrophenol and NaBH4 after the addition of Pd NP–ZnO@Zn nanowires (reaction time: 5 s). Inset: plot of ln(C/C0) at 400 nm as a function of reaction time; (c) plots of C/C0 at 400 nm as a function of reaction time obtained at different runs of the as-prepared Pd NP–ZnO@Zn in the reduction of 4-nitrophenol. | ||
As the concentration of NaBH4 in the catalytic mixture is largely higher than that of 4-nitrophenol, the reduction process can be quantitatively evaluated by pseudo-first-order kinetic model according to eqn (1):
| ln(C/C0) = −kt | (1) |
where C is the concentrations of 4-nitrophenol at different times in the course of the reduction reaction, C0 is the initial concentration of 4-nitrophenol, and k is the apparent first-order rate constant (min−1). Our reaction rate constant (k) was estimated to be ∼0.278 min−1 (inset, Fig. 2b). The inductively coupled plasma optical emission spectrometry (ICP-OES) analysis showed that the loading of Pd on the surface of the ZnO nanowire array was about 0.0126 mg cm−2, and thus the total amount of Pd NPs participated in the catalytic reduction was estimated to be 0.0605 mg. As a result, the catalytic activity factor (j) was calculated to be 76.6 s−1 g−1 by using eqn (2):
| j = k/m | (2) |
In a further set of experiments, we probed the reusability of the recycled Pd NP–ZnO nanowire arrays toward catalytic reduction of 4-nitrophenol. The experimental results (Fig. 2c) showed negligible catalytic decreases of the recollected Pd NP–ZnO nanowire arrays even after use for 10 times. The persistence of the highly catalytic performance of the collected catalysts is due to a combination of marginable catalyst loss and Pd NP coalescence in the course of catalytic reaction. Indeed, we did not detect Pd leaching from the reaction mixture after the removal of Pd NP–ZnO nanowire arrays by ICP-OES analysis. Also, we observed no obvious changes in the morphology of the used hybrid nanowire arrays and in the valence state of the Pd NPs (Fig. S6a and b†). These findings suggest that the as-prepared Pd NP–ZnO nanowire arrays rival or even exceed other types of recyclable Pd NPs toward catalytic reduction of 4-nitrophenol at room temperature.14
The successful use of the as-prepared Pd NP–ZnO nanowire arrays as recyclable catalysts under mild reaction conditions stimulates us to check their promising catalytic utility in much harsher conditions. In this case, we selected Suzuki reaction as a model system due to the need for higher reaction temperatures and stronger alkaline conditions. Furthermore, the reaction is known for its robustness and versatility in the construction of diverse molecules with exceptionally broad functional group tolerance.15 As shown in Table 1, the as-prepared Pd NP–ZnO nanowire arrays (5 s) showed excellent catalytic ability in the coupling reactions of aromatic iodides (or bromides) with aryl boronic acids. Of note, aryl iodides with electron withdrawing groups, including COOC2H5, CH3CO (Table 1, entries 10 and 11), displayed a higher reactivity compared with aryl iodide substrates bearing electron donating groups (such as CH3 and OCH3) as starting reactants (Table 1, entries 4 and 6). In contradiction to aryl iodides, aryl brominated equivalents showed a lower reactivity, and thus longer reaction times (24 h) were required for producing respectful yields (>80%) (Table 1, entries 16–18). As expected, sterically hindered or doubly functionalized aryl iodinated substrates (Table 1, entries 13–15) and heterocyclic bromide (Table 1, entry 18) could react smoothly with aryl boronic acids in the presence of the as-prepared Pd NP–ZnO nanowire arrays to afford corresponding biaryls in good yields. Notably, the nature of the functional groups on aryl boronic acids also remarkably impacts their reactivity. In general, electron donating group-substituted substrates generally showed much higher reactivity than those bearing electron withdrawing groups. On a separate note, the isolated yield of the coupling reaction of iodobenzene with phenylboronic acid by column chromatography was determined to be 96%, which is in accordance with the GC yield (Table 1, entry 1).

| Entry | Ar1–X | Ar2 | T (h) | Biaryl | GC yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: aryl halide (1.0 mmol), aryl boronic acid (1.1 mmol), K2CO3 (1.5 mmol), Pd NP–ZnO@Zn (0.01 mol% Pd) and DMF (3.0 mL), 100 °C for 5 to 24 h.b Yields were determined by GC using dodecane as an internal standard upon the completion of the reactions.c 2.2 mmol of aryl boronic acid was added into the reaction. | |||||
| 1 | 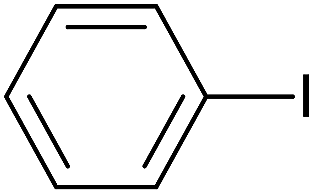 |
 |
6 |  |
99 |
| 2 | 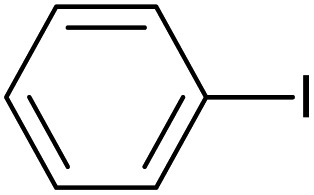 |
 |
8 |  |
99 |
| 3 |  |
 |
12 |  |
94 |
| 4 | 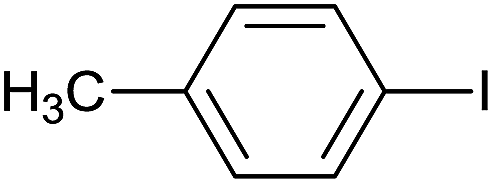 |
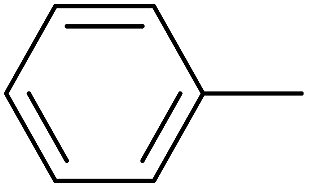 |
6 | 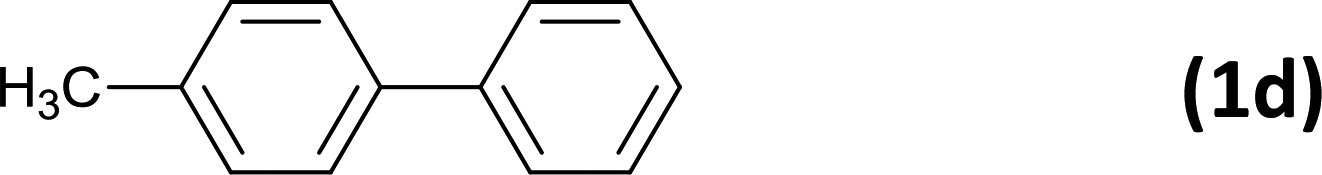 |
99 |
| 5 | 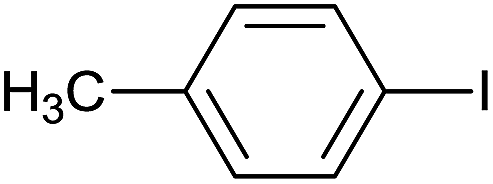 |
 |
8 |  |
99 |
| 6 |  |
 |
6 | 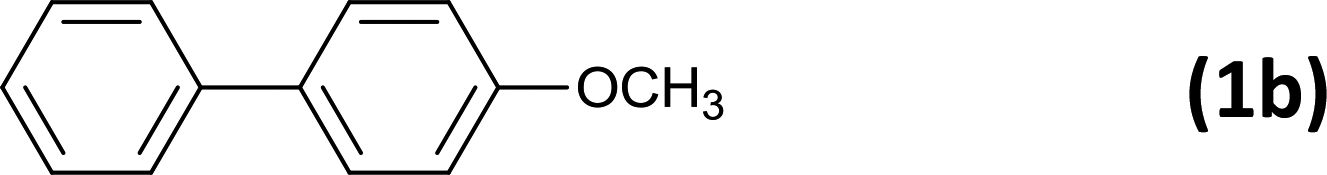 |
99 |
| 7 | 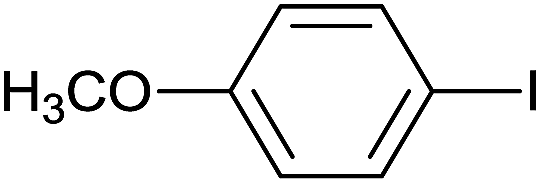 |
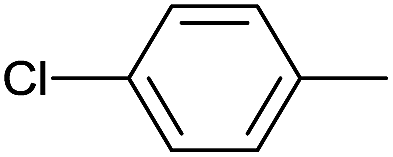 |
12 |  |
96 |
| 8 |  |
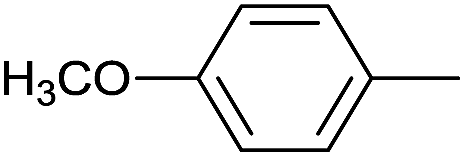 |
6 |  |
99 |
| 9 |  |
 |
12 |  |
93 |
| 10 |  |
 |
5 | 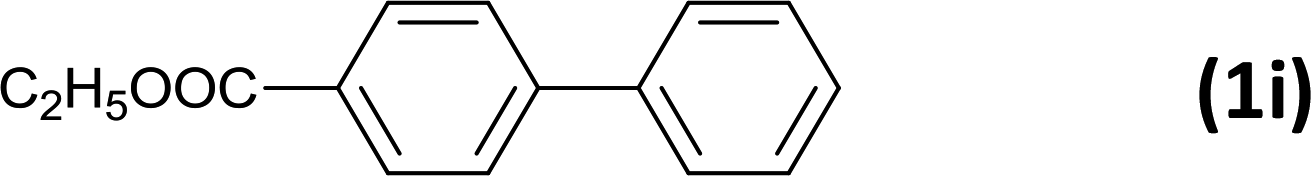 |
99 |
| 11 | 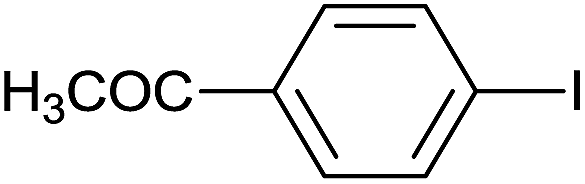 |
 |
5 | 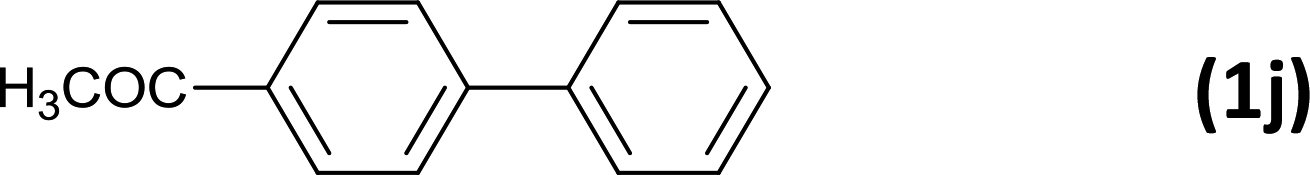 |
99 |
| 12 | 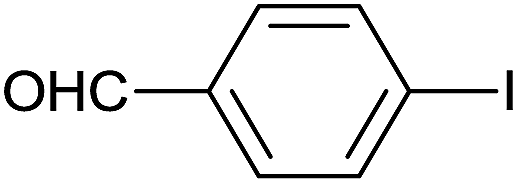 |
 |
6 |  |
99 |
| 13 |  |
 |
8 |  |
99 |
| 14 |  |
 |
12 |  |
94 |
| 15 | 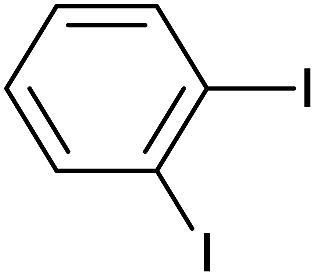 |
 |
12 |  |
92c |
| 16 | 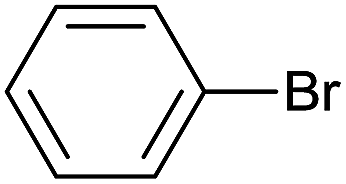 |
 |
24 |  |
91 |
| 17 | 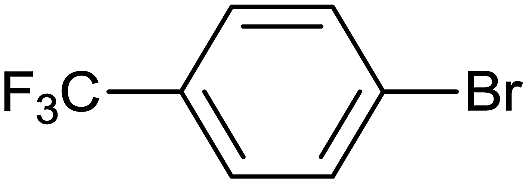 |
 |
24 | 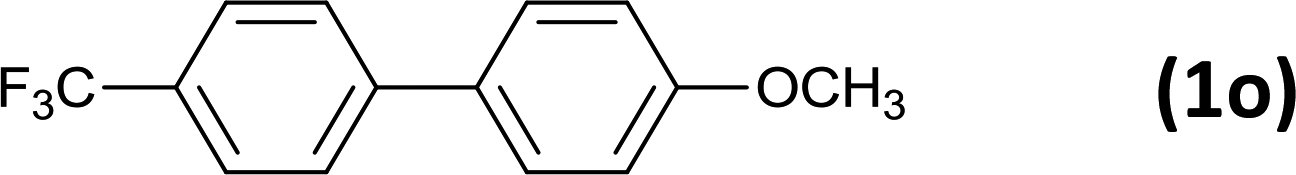 |
82 |
| 18 | 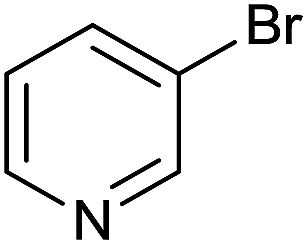 |
 |
24 | 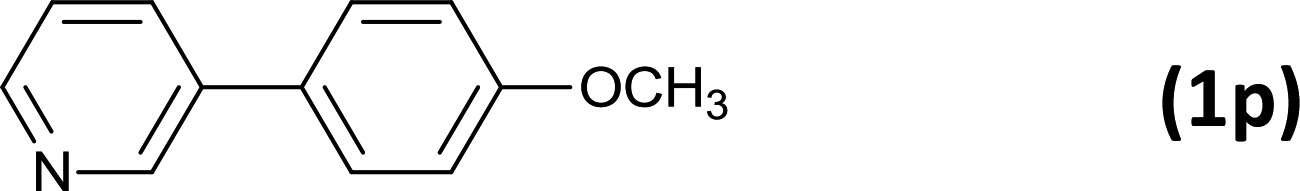 |
85 |
Based on the evaluation of Pd loading of the supported catalyst (5 s for Pd NP growth) by ICP-OES analysis (0.0126 mg cm−2), we calculated the turnover number (TON) and turnover frequency (TOF) up to be 9900 and 1980 h−1 (Table 1, entries 10 and 11), respectively. It is worth noting that scale-up reaction (10 times) of iodobenzene with phenylboronic acid afforded a yield of 99%, showing the potential utility of the as-prepared catalyst in practical chemical industry. ICP-OES analysis showed no obvious Pd leaching in the reaction system by removal of the nanoarray catalysts (<1 ppm). Taken together, these results suggest the advantage of the as-prepared Pd NP–ZnO nanowire arrays with respect to reactivity when compared with previously reported work.16
We also studied the recyclability of the Pd NP–ZnO nanowire arrays in the Suzuki reactions. The results showed catalytic decrease from 99 to 80% after the use of 3 times for the coupling reaction of iodobenzene with phenylboronic acid. This may be due to the gradual dissolution of ZnO nanowires under strong alkaline and high-temperature conditions, leading to slight “fall off” of Pd NPs (Fig. S7a†). XPS analysis of the used Pd NP–ZnO nanowire arrays showed no obvious changes in the valence state of the Pd NPs (Fig. S7b†), accounting for the maintenance of the catalytic activity. Notably, the size of the in situ formed Pd NPs also exerted profound impact on their catalytic reactivity toward Suzuki reaction. For example, the use of supported Pd NP with an average size of 9 nm (25 s for Pd NP growth) led to a yield of <40% for the reaction of iodobenzene with phenylboronic acid under the same conditions.
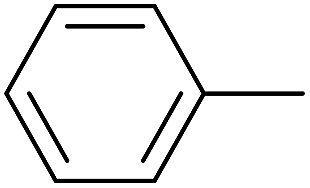
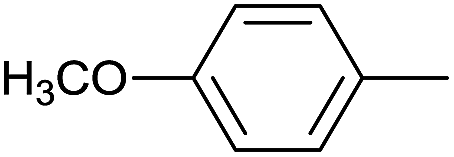
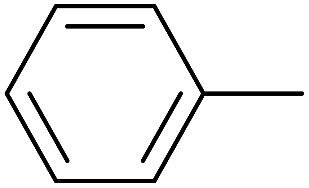
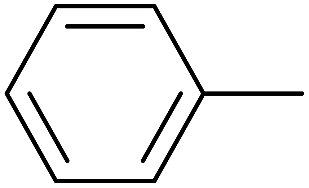
As an added benefit, the as-prepared Pd NP–ZnO nanowire arrays were found to be applicable to carbonylative Suzuki coupling reactions. Such reactions are of great importance for the development of bioactive natural products but are largely untapped.17 The limited reported methods mainly relied on the use CO as a carbonylating agent, causing practical handling difficulties.18 Instead, here we used Mo(CO)6 as an efficient solid carbonylating agent to replace CO. Reaction of aryl iodide, aryl boronic acid and Mo(CO)6 in the presence of the as-prepared Pd NP–ZnO@Zn nanowire arrays (5 s for Pd NP growth) led to a TON up to 775 and a TOF of approximately 129 (h−1) (Table 2, entry 1), respectively. This result shows the better performance of Pd NP–ZnO@Zn nanowire arrays relative to other catalysts, such as Pd/SiC and Pd–Fe3O4.19 We then examined the substrate scope and found the excellent functional group tolerance of the reaction. Similar to the findings demonstrated in Suzuki coupling reactions, aryl iodides containing electron withdrawing groups, such as CN, COOC2H5, CH3CO, and CHO (Table 2, entries 7–11) exhibited higher reactivity as well compared with reactants bearing electron donating groups including –CH3 and –OCH3 (Table 2, entries 4–6). Also, we discovered that functional groups on aryl boronic acids exerted an important influence on the reactivity of the substrates. In general, the substrates with an electron donating group exhibited much higher reactivity than those having an electron withdrawing group (Table 2, entries 1–3). In addition to yielding a variety of carbocyclic ketones, carbonylative Suzuki coupling reactions have the ability to offer heterocyclic ketone with a good yield using heterocyclic iodides such as 3-iodopyridine as the starting substrates (Table 2, entry 12).

| Entry | Ar1–X | Ar2 | T (h) | Product | GC yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: aryl halide (0.5 mmol), aryl boronic acid (0.6 mmol), K2CO3 (1.0 mmol), Mo(CO)6 (0.3 mmol), DMF (3.0 mL), and Pd NP–ZnO@Zn (0.12 mol%), 100 °C for 6–24 h.b Yields were determined by GC using dodecane as an internal standard upon the completion of the reactions. | |||||
| 1 |  |
 |
6 | 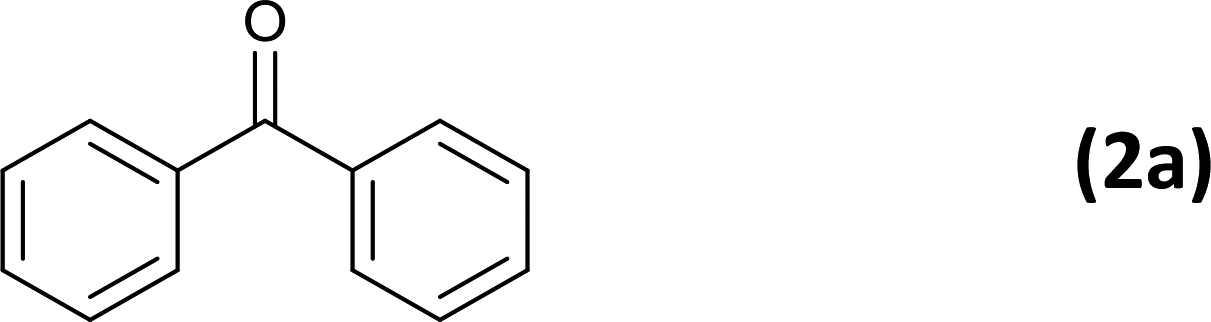 |
93 |
| 2 |  |
 |
12 | 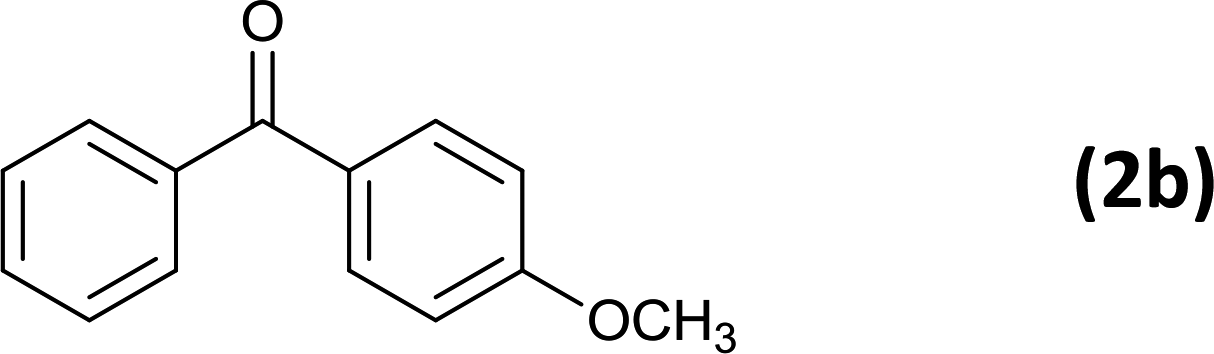 |
85 |
| 3 | 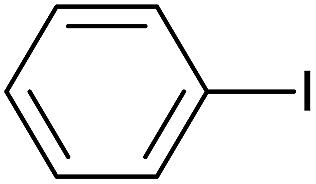 |
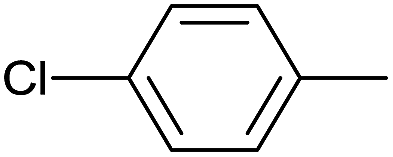 |
24 | 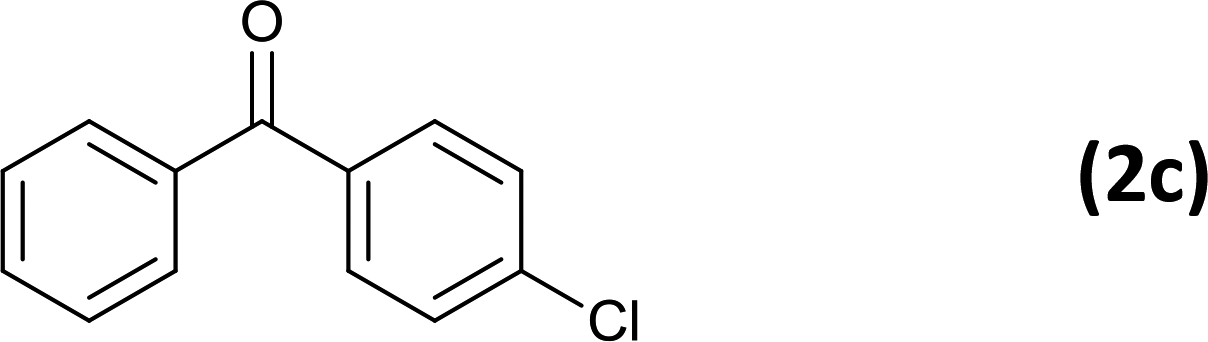 |
81 |
| 4 |  |
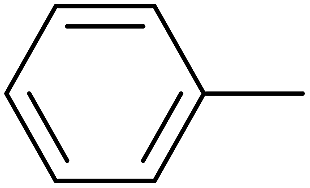 |
6 | 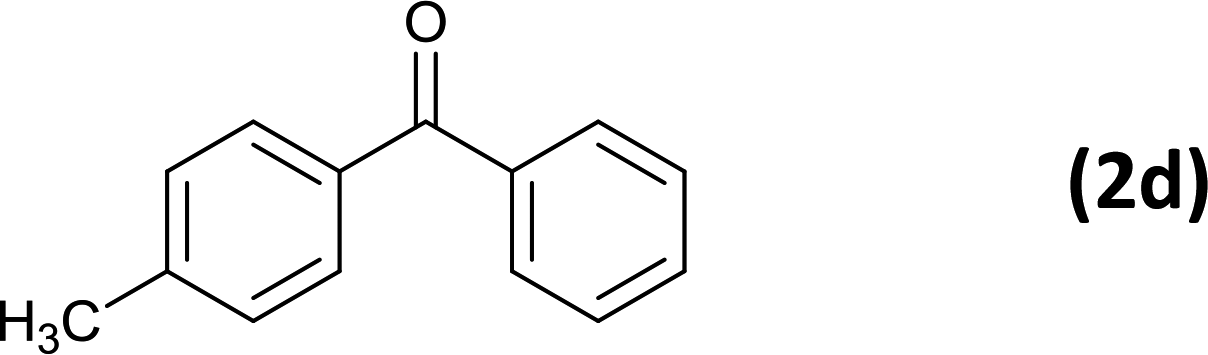 |
86 |
| 5 | 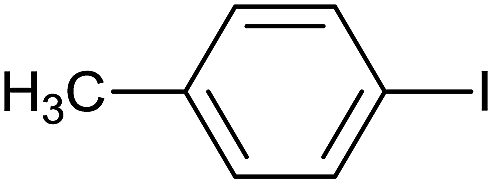 |
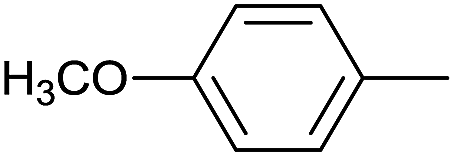 |
12 | 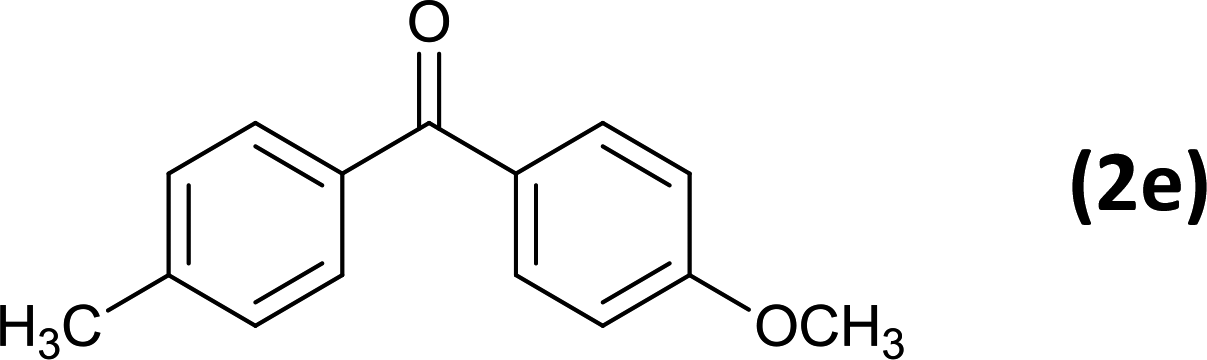 |
82 |
| 6 | 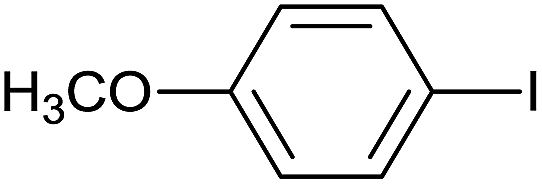 |
 |
6 |  |
87 |
| 7 | 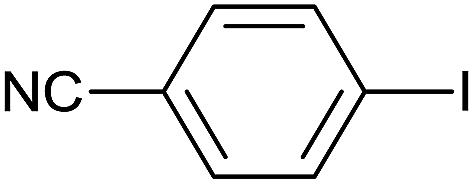 |
 |
6 | 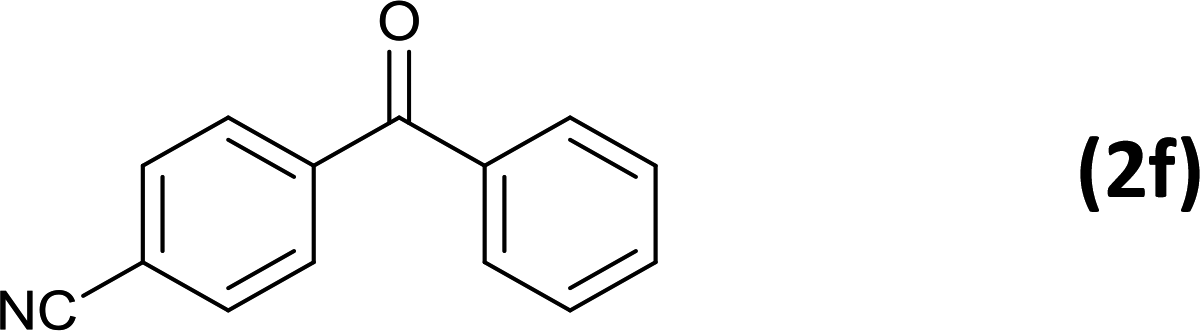 |
92 |
| 8 | 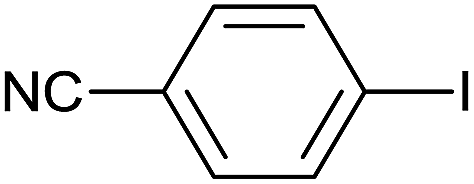 |
 |
12 | 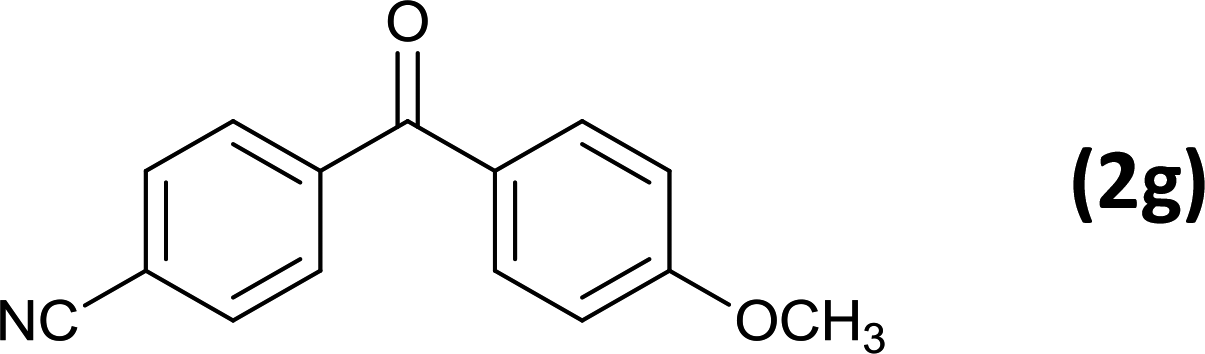 |
90 |
| 9 |  |
 |
6 |  |
89 |
| 10 |  |
 |
6 | 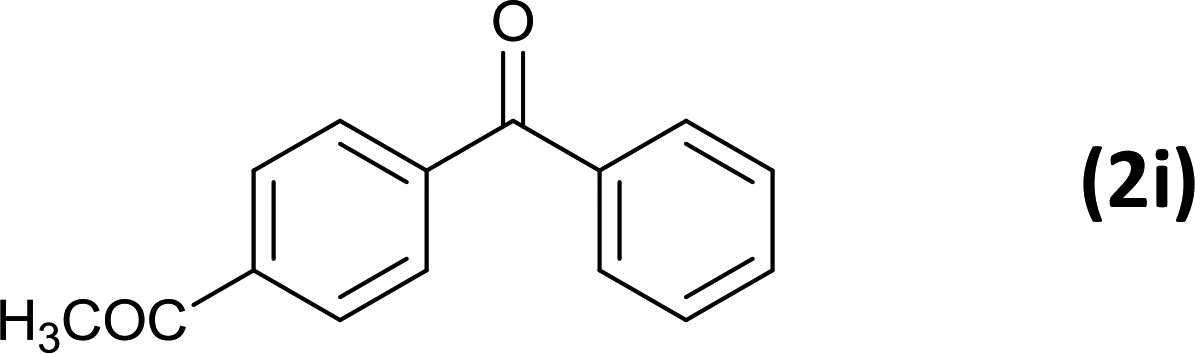 |
88 |
| 11 |  |
 |
6 |  |
86 |
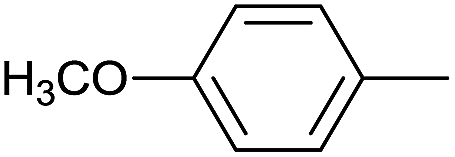
| 12 | 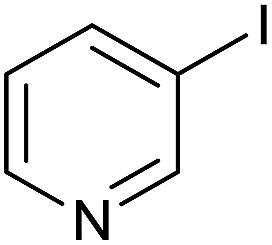 |
 |
12 | 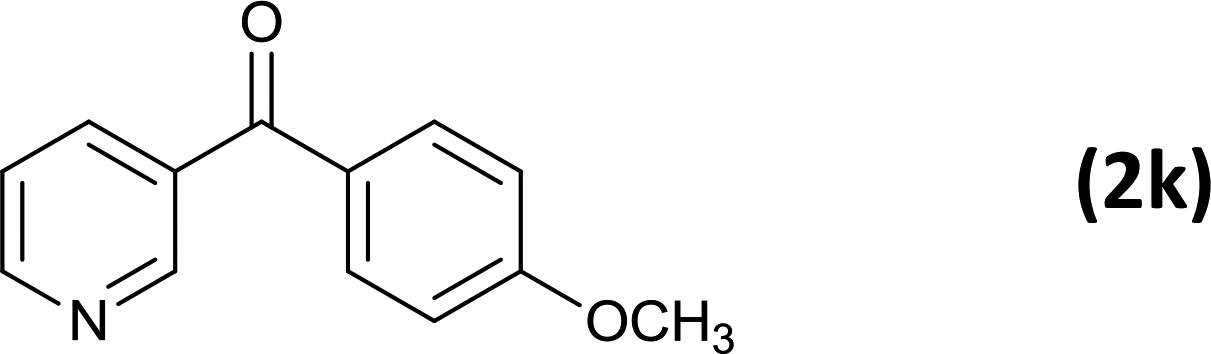 |
82 |
Conclusions
In summary, we have examined the catalytic utility of Pd NP–ZnO nanowire arrays under different catalytic condition on the basis of their easy synthesis by immersing ZnO@Zn nanowire arrays into an aqueous solution of Na2PdCl4 for varied periods of time. The as-prepared Pd NP–ZnO nanowire arrays with an immersion time of 5 s have been found to be applicable as recyclable catalysts for both 4-nitrophenol reduction and (carbonylative) Suzuki reaction, which were performed under mild and relatively harsh conditions, respectively. These findings imply the feasibility of the development of highly recyclable catalysts through a combination of transition metal oxide nanowire array supports and viable catalytically active noble metal NPs.Experimental section
General
Zn foil (≥99.99%), ammonium hydroxide solution (28.0–30.0%), Na2PdCl4 (≥99.99%), dimethylformamide (DMF, analytic grade), 4-nitrophenol (≥98%) and NaBH4 (98%) were purchased from Shanghai Chemical Reagents Company. Aromatic iodides and bromides (≥98%), aryl boronic acid (≥98%) and Mo(CO)6 (98%) were purchased from Alfa Aesar. Unless otherwise stated, all chemicals were used as received without further purification.Physical measurements
Transmission electron microscopy (TEM) characterization was performed on a FEI Tecnai G2 20 operated at 200 kV. The energy-dispersive X-ray (EDX) spectroscopic analysis was recorded with an Oxford INCA energy system operated at 200 kV. Scanning electron microscopy (SEM) measurements were carried out by using a field-emission scanning electron microscope (Hitachi S-4800). X-ray photoelectron spectroscopy (XPS) analysis was conducted on a Thermo ESCALAB 250 electron spectrometer with the use of a monochromated Al Ka X-ray source. ICP-OES measurement was performed on a Dual-view Optima 5300 DV ICP-OES system (PerkinElmer). UV-visible absorption profiles were obtained on a Shimadzu UV-2450 spectrometer. The organic products were analyzed by GC7890F equipped with a flame ionization detector and a GC (Thermo Trace 1300)-MS (Thermo ISQ QD) system. Note that during the analysis, temperatures at the column, injector and flame ionization detector were respectively set at 300, 250 and 300 °C. 1H and 13C NMR spectra were recorded at 25 °C on a Bruker Avance-300 at 300 and 75 MHz by using tetramethylsilane as an internal standard, respectively.Synthesis of ZnO@Zn nanowire arrays
ZnO@Zn nanowire arrays were prepared by a hydrothermal method.20 In a typical synthesis, ammonia (6.0 mL) and distilled water (34.0 mL) were first added into a 60 mL Teflon-lined autoclave to form a homogeneous solution. After the addition of four pieces of clean Zn foil (1.5 cm × 1 cm) into the resulting solution, the autoclave was heated at 110 °C for 14 h.Synthesis of Pd NP–ZnO nanowire arrays
The as-prepared ZnO@Zn nanowire arrays were first washed with distilled water, and then dried under a nitrogen atmosphere. The dried ZnO@Zn NW arrays were then immersed into an aqueous solution of Na2PdCl4 (5.0 mM) for different periods of time (5, 15 and 25 s) to control the size distribution of Pd NPs. The resulting ZnO@Zn NW arrays were finally washed with distilled water and dried under a nitrogen atmosphere before further use.Determination of Pd loading on the surface of ZnO nanowire arrays
Two pieces of as-prepared Pd–ZnO nanowire arrays (1.5 cm × 1 cm) were dissolved with aqua regia. The content of Pd in the sample was determined by ICP-OES. On the basis of ICP-OES analysis and the total size of the hybrid nanowire arrays, Pd loading per square centimetre was obtained.Pd NP–ZnO nanowire arrays for 4-nitrophenol reduction
A stock solution was first prepared by mixing NaBH4 (2.0 mL, 0.2 M) and 4-nitrophenol (2.0 mL, 2 × 10−4 M) in a vial (5.0 mL). Two pieces (∼4.8 cm2 in total) of Pd NP–ZnO were then added into the resulting solution to initialize the reduction of 4-nitrophenol. The kinetics of the catalytic reduction of 4-nitrophenol was in situ monitored by UV-visible absorption spectroscopy. At the end of the reaction, Pd NP–ZnO nanowire arrays were separated from the mixed solution by tweezers, washed three times with ethanol, and finally dried at room temperature for recyclably catalytic studies.Pd NP–ZnO nanowire arrays for Suzuki reaction
Five small pieces (∼1.0 cm2 in total) of Pd NP–ZnO were added into a 25 mL tube reactor containing aryl halide (1.0 mmol), aryl boronic acid (1.1 mmol), K2CO3 (1.5 mmol) and DMF (3.0 mL), and then the mixture was sealed and heated at 100 °C for 5–24 h under gentle stirring. At the end of the reactions, Pd NP–ZnO nanowire arrays were recollected from the catalytic mixture by centrifugation. The organic phase was subsequently obtained by extraction through the use of ethyl acetate as solvent. The obtained solution was further washed with brine (3 × 10.0 mL), dried over anhydrous magnesium sulphate, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/petroleum ether = 1/20) to afford a pure product.Recycling test for Suzuki reaction
The supported Pd catalyst was recollected by centrifugation at 4000 rpm for 3 min. After being washed with deionized water and ethanol for several times, the supported catalyst was reused in the second run of the reaction. Other procedure is essentially identical to that mentioned in the first run of catalytic study.Pd NP–ZnO nanowire arrays for carbonylative Suzuki reaction
The procedure used for performing carbonylative Suzuki reaction was similar to that used for realizing Suzuki reaction. Typically, several small pieces (∼5.0 cm2 in total) of freshly prepared Pd NP–ZnO NW arrays were added into a mixture of aryl halide (0.5 mmol), aryl boronic acid (0.6 mmol), K2CO3 (1.0 mmol), Mo(CO)6 (0.3 mmol) and DMF (3.0 mL), and the resulting mixture was then sealed and heated at 100 °C for 6–12 h under gentle stirring. The process for catalyst collection and product purification was identical to that described in catalytic study of the hybrid nanowire arrays in Suzuki reactions.Determine the Pd leaching by ICP-OES analysis
After a run of Suzuki reaction, the reaction mixture was washed with H2O after the removal of Pd NP–ZnO nanowire arrays. The resulting aqueous phase was separated and further diluted for ICP-OES measurement.Acknowledgements
This study was supported by the National Natural Science Foundation of PR China (No. 21272006, 21471007), and the research grant for innovative experiments of graduate students (No. 2016yks053).References
- (a) P. Hervés, M. Pérez-Lorenzo, L. M. Liz-Marzán, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC; (b) S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen and J. Zhao, J. Am. Chem. Soc., 2013, 135, 5793–5801 CrossRef CAS PubMed; (c) S. Cai, H. Rong, X. Yu, X. Liu, D. Wang, W. He and Y. Li, ACS Catal., 2013, 3, 478–486 CrossRef CAS; (d) H. Guo, X. Yan, Y. Zhi, Z. Li, C. Wu, C. Zhao, J. Wang, Z. Yu, Y. Ding, W. He and Y. Li, Nano Res., 2015, 8, 1365–1372 CrossRef CAS; (e) Q. Zhang, S. Cai, L. Li, Y. Chen, H. Rong, Z. Niu, J. Liu, W. He and Y. Li, ACS Catal., 2013, 3, 1681–1684 CrossRef CAS; (f) Y. M. A. Yamada, T. Arakawa, H. Hocke and Y. Uozumi, Chem.–Asian J., 2009, 4, 1092–1098 CrossRef CAS PubMed.
- (a) L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed; (b) M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed; (c) Z. Niu, Q. Peng, Z. Zhuang, W. He and Y. Li, Chem.–Eur. J., 2012, 18, 9813–9817 CrossRef CAS PubMed; (d) C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed; (e) A. J. Reay and I. J. S. Fairlamb, Chem. Commun., 2015, 51, 16289–16307 RSC; (f) H. A. Elazab, A. R. Siamaki, S. Moussa, B. F. Gupton and M. S. El-Shall, Appl. Catal., A, 2015, 491, 58–69 CrossRef CAS; (g) S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall, ACS Catal., 2012, 2, 145–154 CrossRef CAS; (h) A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS.
- (a) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC; (b) J. Mondal, Q. T. Trinh, A. Jana, W. K. H. Ng, P. Borah, H. Hirao and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 15307–15319 CrossRef CAS PubMed.
- (a) V. Chechik and R. M. Crooks, J. Am. Chem. Soc., 2000, 122, 1243–1244 CrossRef CAS; (b) J. Jin, R. Li, H. Wang, H. Chen, K. Liang and J. Ma, Chem. Commun., 2007, 386–388 RSC; (c) V. K. Sharma, J. Filip, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 8410–8423 RSC; (d) N. B. Golovinaa and L. M. Kustov, Mendeleev Commun., 2013, 23, 59–65 CrossRef.
- (a) M. B. Gawande, P. S. Brancoa and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC; (b) C. Wang, L. Yin, L. Zhang, N. Liu, N. Lun and Y. Qi, ACS Appl. Mater. Interfaces, 2010, 2, 3373–3377 CrossRef CAS PubMed; (c) Z. Li, J. Liu, Z. Huang, Y. Yang, C. Xia and F. Li, ACS Catal., 2013, 3, 839–845 CrossRef CAS; (d) Y. Xie, K. Ding, Z. Liu, R. Tao, Z. Sun, H. Zhang and G. An, J. Am. Chem. Soc., 2009, 131, 6648–6649 CrossRef CAS PubMed; (e) Q. M. Kainz, R. Linhardt, R. N. Grass, G. Vilé, J. Pérez-Ramírez, W. J. Stark and O. Reiser, Adv. Funct. Mater., 2014, 24, 2020–2027 CrossRef CAS; (f) M. Shekhar, J. Wang, W.-S. Lee, W. D. Williams, S. M. Kim, E. A. Stach, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Am. Chem. Soc., 2012, 134, 4700–4708 CrossRef CAS PubMed; (g) M. Shokouhimehr, Catalysts, 2015, 5, 534–560 CrossRef CAS; (h) A. Kim, S. M. Rafiaei, S. Abolhosseini and M. Shokouhimehr, Energy and Environment Focus, 2015, 4, 18–23 CrossRef; (i) K.-H. Choi, M. Shokouhimehr and Y.-E. Sung, Bull. Korean Chem. Soc., 2013, 34, 1477–1480 CrossRef CAS.
- (a) K. An, S. Alayoglu, N. Musselwhite, K. Na and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 6830–6833 CrossRef CAS PubMed; (b) S. Chen, R. Si, E. Taylor, J. Janzen and J. Chen, J. Phys. Chem. C, 2012, 116, 12969–12976 CrossRef CAS; (c) L. Wang, H. Wang, A. E. Rice, W. Zhang, X. Li, M. Chen, X. Meng, J. P. Lewis and F.-S. Xiao, J. Phys. Chem. Lett., 2015, 6, 2345–2349 CrossRef CAS PubMed; (d) M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed; (e) K. Qadir, B. T. P. Quynh, H. Lee, S. Y. Moon, S. H. Kim and J. Y. Park, Chem. Commun., 2015, 51, 9620–9623 RSC; (f) S. O. Moussa, L. S. Panchakarla, M. Q. Ho and M. S. El-Shall, ACS Catal., 2014, 4, 535–545 CrossRef CAS; (g) Y. Lin, Z. Wu, J. Wen, K. Ding, X. Yang, K. R. Poeppelmeier and L. D. Marks, Nano Lett., 2015, 15, 5375–5381 CrossRef CAS PubMed.
- (a) Y. M. A. Yamada, Y. Yuyama, T. Sato, S. Fujikawa and Y. Uozumi, Angew. Chem., Int. Ed., 2014, 53, 127–131 CrossRef CAS PubMed; (b) Q. Simon, D. Barreca, A. Gasparotto, C. Maccato, T. Montini, V. Gombac, P. Fornasiero, O. I. Lebedev, S. Turnere and G. V. Tendeloo, J. Mater. Chem., 2012, 22, 11739–11747 RSC; (c) Y. G. Lin, Y. K. Hsu, S. Y. Chen, Y. K. Lin, L. C. Chen and K. H. Chen, Angew. Chem., Int. Ed., 2009, 48, 7586–7590 CrossRef CAS PubMed; (d) Y. G. Lin, Y. K. Hsu, Y. C. Chen, S. B. Wang, J. T. Miller, L. C. Chen and K. H. Chen, Energy Environ. Sci., 2012, 5, 8917–8922 RSC.
- (a) S. H. Lee, E. J. Lim, Y.-R. Jo, B.-J. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 20634–20642 CrossRef CAS PubMed; (b) C. Cao, Y. Zhang, D. Liu and M. Meng, Small, 2015, 11, 3659–3664 CrossRef CAS PubMed.
- (a) H. Zeng, X. Xu, Y. Bando, U. K. Gautam, T. Zhai, X. Fang, B. Liu and D. Golberg, Adv. Funct. Mater., 2009, 19, 3165–3172 CrossRef CAS; (b) L. E. Greene, M. Law, J. Goldberger, F. Kim, J. C. Johnson, Y. Zhang, R. J. Saykally and P. Yang, Angew. Chem., Int. Ed., 2003, 42, 3031–3034 CrossRef CAS PubMed; (c) T. J. Athauda, P. Hari and R. R. Ozer, ACS Appl. Mater. Interfaces, 2013, 5, 6237–6246 CrossRef CAS PubMed.
- (a) K. Deng, H. Lu, Z. Shi, Q. Liu and L. Li, ACS Appl. Mater. Interfaces, 2013, 5, 7845–7851 CrossRef CAS PubMed; (b) C. P. Tsangarides, H. Ma and A. Nathan, Nanoscale, 2016, 8, 11760–11765 RSC; (c) W. Q. Wu, Y. F. Xu, C. Y. Su and D. B. Kuang, Energy Environ. Sci., 2014, 7, 644–649 RSC; (d) L. Zhang, K. Zhao, W. Xu, Y. Dong, R. Xia, F. Liu, L. He, Q. Wei, M. Yan and L. Mai, Phys. Chem. Chem. Phys., 2015, 17, 7619–7623 RSC; (e) S. S. Mali, C. S. Shim, H. K. Park, J. Heo, P. S. Patil and C. K. Hong, Chem. Mater., 2015, 27, 1541–1551 CrossRef CAS; (f) S. H. Lee, W. Park, B. H. Lee and W. B. Kim, J. Mater. Chem. A, 2015, 3, 13492–13499 RSC; (g) J. G. Kim, S. H. Lee, Y. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 11321–11328 CrossRef CAS PubMed.
- Q. Hu, X. Liu, C. Wu, Q. You, T. Shi and W. Zhang, RSC Adv., 2016, 6, 1542–1548 RSC.
- (a) Y. Xu, T. Wang, Z. He, A. Zhong and K. Huang, RSC Adv., 2016, 6, 39933–39939 RSC; (b) L. Yu, Y. Huang, Z. Wei, Y. Ding, C. Su and Q. Xu, J. Org. Chem., 2015, 80, 8677–8683 CrossRef CAS PubMed.
- (a) E.-M. Felix, M. Antoni, I. Pause, S. Schaefer, U. Kunz, N. Weidler, F. Muench and W. Ensinger, Green Chem., 2016, 18, 558–564 RSC; (b) B. K. Barman and K. K. Nanda, Dalton Trans., 2015, 44, 4215–4222 RSC; (c) G. Zheng, L. Polavarapu, L. M. Liz-Marzán, I. Pastoriza-Santos and J. Pérez-Juste, Chem. Commun., 2015, 51, 4572–4575 RSC.
- (a) Z. Wang, C. Xu, G. Gao and X. Li, RSC Adv., 2014, 4, 13644–13651 RSC; (b) L. Huang, L. Ao, X. Xie, G. Gao, M. F. Foda and W. Su, Nanoscale, 2015, 7, 806–813 RSC; (c) M. Atarod, M. Nasrollahzadeh and S. M. Sajadi, J. Colloid Interface Sci., 2016, 465, 249–258 CrossRef CAS PubMed.
- (a) C. Peter, A. Derible, J.-M. Becht, J. Kiener, C. L. Drian, J. Parmentier, V. Fierro, M. Girleanu and O. Ersen, J. Mater. Chem. A, 2015, 3, 12297–12306 RSC; (b) Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Mater., 2015, 27, 1921–1924 CrossRef CAS; (c) A. K. Rathi, M. B. Gawande, J. Pechousek, J. Tucek, C. Aparicio, M. Petr, O. Tomanec, R. Krikavova, Z. Travnicek, R. S. Varmac and R. Zboril, Green Chem., 2016, 18, 2363–2373 RSC; (d) L. Zhong, A. Chokkalingam, W. S. Cha, K. S. Lakhi, X. Su, G. Lawrence and A. Vinu, Catal. Today, 2015, 243, 195–198 CrossRef CAS; (e) F. Yang, C. Chi, S. Dong, C. Wang, X. Jia, L. Ren, Y. Zhang, L. Zhang and Y. Li, Catal. Today, 2015, 256, 186–192 CrossRef CAS; (f) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6723–6737 Search PubMed.
- (a) Y. Masuyama, Y. Sugioka, S. Chonan, N. Suzuki, M. Fujita, K. Ara and A. Fukuoka, J. Mol. Catal. A: Chem., 2012, 352, 81–85 CrossRef CAS; (b) R. Cano, D. J. Ramón and M. Yus, Tetrahedron, 2011, 67, 5432–5436 CrossRef CAS.
- (a) K. M. Bjerglund, T. Skrydstrup and G. A. Molander, Org. Lett., 2014, 16, 1888–1891 CrossRef CAS PubMed; (b) F. Jin and W. Han, Chem. Commun., 2015, 51, 9133–9136 RSC; (c) X. F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 282–285 CrossRef CAS PubMed; (d) X. Qi, L. B. Jiang, H. P. Li and X. F. Wu, Chem.–Eur. J., 2015, 21, 17650–17656 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC.
- (a) A. Ahlburg, A. T. Lindhardt, R. H. Taaning, A. E. Modvig and T. Skrydstrup, J. Org. Chem., 2013, 78, 10310–10318 CrossRef CAS PubMed; (b) Y. Long, K. Liang, J. Niu, X. Tong, B. Yuan and J. Ma, New J. Chem., 2015, 39, 2988–2996 RSC; (c) Y. Cui, X. Guo, Y. Wang and X. Guo, Chin. J. Catal., 2015, 36, 322–327 CrossRef CAS.
- H. Yang, Y. Song, L. Li, J. Ma, D. Chen, S. Mai and H. Zhao, Cryst. Growth Des., 2008, 8, 1039–1043 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28467a |
| This journal is © The Royal Society of Chemistry 2017 |