
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A cyclodextrin-core star copolymer with Y-shaped ABC miktoarms and its unimolecular micelles†
Yan Wangab,
Yuyang Liu *ab,
Jianghu Liangab and
Minhao Zouab
*ab,
Jianghu Liangab and
Minhao Zouab
aKey Laboratory of Macromolecular Science and Technology of Shaanxi Province, Department of Applied Chemistry, Northwestern Polytechnical University, Xi'an 710072, P. R. China. E-mail: liu_yyang1120@nwpu.edu.cn
bThe Key Laboratory of Space Applied Physics and Chemistry, Ministry of Education, School of Science, Northwestern Polytechnical University, Xi'an 710072, P. R. China
First published on 16th February 2017
Abstract
A well-defined β-cyclodextrin-core star copolymer with Y-shaped ABC miktoarms, star poly(ethyl methacrylate(-b-poly(2-(diethylamino)ethyl methacrylate)-b-methoxypolyethylene glycols (CD-star-PEMA(-b-PDEA)-b-mPEG), was designed and synthesized via ATRP and click reactions. The copolymer could exhibit unimolecular micelles in aqueous solution. The micelles were characterized by means of DLS, 1H NMR and TEM measurements. In the micelles the PEMA blocks work as a container for storing hydrophobic guest molecules, the mPEG is used as a hydrophilic block, and the PDEA block can respond to change in environmental pH. By using Nile red, celecoxib, and ketoprofen as hydrophobic model drugs, encapsulation and release behaviors of the unimolecular micelles were investigated. It was found that the unimolecular container is a good platform for encapsulating hydrophobic guest molecules. Release of the payload from the unimolecular micelles exhibited pH-sensitivity.
1. Introduction
Amphiphilic macromolecules are able to self-assemble to form micelles in an aqueous solution. When an amphiphilic polymer self-assembles to form micelles in an aqueous solution, the formed hydrophobic cores can encapsulate hydrophobic drugs molecules.1,2 Therefore, self-assemblies based on amphiphilic macromolecules have potential application in the drug delivery field.1,2 However, owing to the existence of a critical aggregation concentration (CAC), self-assembled polymeric micelles become significantly unstable under diluted conditions and dissociate into free chains.3–10 This results in premature release of the physically encapsulated drugs.3 Also, it is difficult to accurately control the architecture of a nano-object from the self-assembly of multi-molecules. Consequently, with regard to stability and well-defined architecture, the unimolecular micelles show distinct advantages. This type of micelles demonstrates a micellar behavior as a single molecule.4–10 This implies that once this type of macromolecules is constructed the structures of their micelles are explicit. Therefore, amphiphilic unimolecular micelle is an important and interesting nano-object container.Unimolecular micelles could be generally constructed by using dendritic,9 or hyperbranched polymers10 as a core. But synthesis procedure of a perfect dendritic polymer is tedious, and it is also difficult to precisely control structure of a hyperbranched polymer. Recently, we are interesting in investigation of star copolymers,11–14 especially based on β-cyclodextrin (β-CD) core.11,12 β-CD contains 7 primary and 14 secondary hydroxyl groups,7,8,11,12,15–30 which are able to be all7,8,11,12,16–20 or selectively20–30 reacted. Therefore, well-defined β-CD-core star polymers with designed arm numbers could be synthesized. Interestingly, β-CD-core star polymers exhibit some unique properties.7,8,11,12,15–30 For example, in our recent work, we designed and synthesized well-defined β-CD-core star triblock copolymers poly(methyl methacrylate)-b-poly(2-(diethylamino)ethyl methacrylate)-b-poly(poly(ethylene glycol)methyl ether methacrylate) (CD-(PMMA-b-PDEA-b-PPEGMA)21) by the core-first ATRP method. The different morphologic self-assemblies were obtained from the star triblock copolymers. Unexpectedly, the pH-induced micelle-to-vesicle morphology transition was observed.11 In our earlier work, β-CD-core star poly(N-isopropylacrylamide) (PNIPAm) (star-PNIPAm) was synthesized and then star-PNIPAm with β-CD at arm end (star-PNIPAm-CD) were further synthesized. Interestingly, the two star polymers could self-assemble to form nanosized aggregates in aqueous solution above their lower critical solution temperatures (LCSTs), and their self-assembly behaviors showed molecular recognition capability.12 Xiao et al. synthesized a series of cationic star polymers with 21 arms (21ACSPs) by using a β-CD-based initiator. The results revealed that 21ACSPs have the ability to condense the plasmid DNA to 80–180 nm.16 Liu and co-workers synthesized Janus-type (PDEA30)7-CD-(PNIPAm25)14 heteroarm star copolymer by using β-CD as a core. The (PDEA30)7-CD-(PNIPAm25)14 copolymer could self-assemble to form two types of vesicles with “inverted” nanostructures in water by properly tuning solution pH and temperatures.21
Although star polymers based on a β-CD core have been explored widely,7,8,11,12,15–30 so far there are yet few reports on aqueous unimolecular micelles of β-CD-core star polymers,8 especially multi-functional unimolecular micelles. For multi-armed star polymers based on a β-CD core, it is possible to obtain aqueous unimolecular micelles.7,8 For example, Wang et al. synthesized β-CD-core star diblock copolymers poly(lactic acid)-b-poly(oligo(ethylene glycol) methyl ether methacrylate). The copolymers could form unimolecular micelles and were used as carriers of drug DOX.8 Herein we report a novel β-CD-core star copolymer with Y-shaped ABC miktoarms, star poly(ethyl methacrylate(-b-poly(2-(diethylamino)ethyl methacrylate)-b-methoxypolyethylene glycols (CD-star-PEMA(-b-PDEA)-b-mPEG). The copolymer exhibits unimolecular micelles in aqueous solution. The micelles can not only encapsulate hydrophobic guest molecules, but also respond to change in environmental pH. In addition, the surface charge of the micelles changes with environmental pH. To our knowledge, so far this type of β-CD-core star copolymer has not been reported yet.
2. Experimental section
2.1 Materials
1,1,4,7,10,10-Hexamethyltriethylenetetramine (HMTETA, 97.0%) was from Alfa Aesar, 1,1,4,7,7-pentamethyldiethylenetriamine (PMDETA, 98%), 2-chloropropionyl chloride (CPC, 95%), ethyl methacrylate (EMA, 99%), 3-butyn-1-ol (98%), 9-(diethylamino)benzo[a]phenoxazin-5(5H)-one (Nile red, 97.5%), 4-pentynoic acid (95%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI, 99%), and ketoprofen (98%) were from J & K Scientific Ltd. Methoxypolyethylene glycols (mPEG, average molecular weight 5000) and 2-(diethylamino)ethyl methacrylate (DEA, 98.5%) were purchased from TCI, Japan. Celecoxib (98%) was from Adamas. 4-Dimethylaminopyridine (DMAP, 99%) was from Acros. Cuprous bromide (98.5%) and anisole (chemical pure) were from Sinopharm Chemical Reagent Co., Ltd, Shanghai, China. Sodium azide (99.5%) was from Zhengzhou Paini Chemical Reagent Factory. Sodium hydride (60%) was from Chengdu Kelong Chemical Reagent Factory. Cuprous chloride (97.0%, Tianjin Hongyan Chemical Reagent Factory) and cuprous bromide were purified by being stirred in acetic acid overnight. After filtration, they were washed with anhydrous methanol and then dried in a vacuum oven at 25 °C. All of the other reagents, including pyridine, epichlorohydrin, ammonium chloride, dichloromethane (DCM), anhydrous methanol, tetrahydrofuran (THF), N,N-dimethylformamide (DMF), n-hexane, anhydrous sodium carbonate, and cupric chloride were of analytical grade and made in China. They were used as received without further purification.β-Cyclodextrin (β-CD) was recrystallized from water three times and dried under vacuum at 110 °C prior to use. 21Cl-β-CD was synthesized according to our previous studies.12 3-Butynyl-2-chloropropanoate (Alkynyl-Cl) was synthesized according to our previous work.14
2.2 Synthesis of star copolymer CD-star-PEMA-Cl
HMTETA (409 mg, 1.78 mmol), CuCl2·2H2O (15.2 mg, 0.089 mmol), EMA (11.83 g, 103.6 mmol) and 21Cl-β-CD (214.3 mg, 1.48 mmol initiating sites) were successively dissolved in 71 mL of anisole. After it was bubbled with argon gas for 80 min, CuCl (87.93 mg, 0.89 mmol) was added. It was bubbled with argon gas for an additional 20 min and the reaction system sealed was then stirred magnetically in an oil bath at 80 °C. After 1.5 h, the reaction mixture was allowed to cool to room temperature, to be exposed to air, and to be diluted with DCM (100 mL). It was passed through a neutral alumina column to remove the copper complex. The resulting solution was concentrated and was then precipitated into n-hexane. The repeated purification of the product was carried out by dissolving in DCM and precipitating in n-hexane twice. The resulting polymer was dried under vacuum at 25 °C for 48 h (yield, 3.1 g).2.3 Synthesis of star copolymer CD-star-PEMA-N3
A mixed solution containing 2.35 g of CD-star-PEMA-Cl, 9.4 mL of DMF and 0.51 g (7.8 mmol) of NaN3 was stirred in an oil bath at 60 °C for 48 h. When the stirring was completed, the resulting mixture was allowed to cool to room temperature and was then poured into 200 mL of water. The obtained precipitate was dissolved in 200 mL of DCM and it was then washed by 10% NaCl aqueous solution thrice. After dried over anhydrous sodium sulfate overnight, the organic solvent was removed by a rotary evaporator. The obtained sample was dried under vacuum at room temperature for 48 h (yield, 2.1 g).2.4 Synthesis of α-methoxy-ω-epoxypoly(ethylene oxide) (mPEG-epoxide)
20 g (4 mmol) of mPEG was dissolved in 90 mL of toluene. The solution was heated to reflux under Dean–Stark condition in an oil bath at 130 °C. When ∼40 mL of toluene was distilled, the reaction flake was set in oil bath at 30 °C and 0.48 g (12 mmol) of NaH was then added at 30 °C. After stirring of 5 h, 11.1 g (9.4 mL, 120 mmol) of epichlorohydrin was added. The reaction mixture was stirred at 40 °C for 15 h. When completion of the reaction, the mixture was allowed to cool to room temperature and small amount of water was stepwise added to consume the unreacted NaH. The resulting solution was precipitated into n-hexane (400 mL). The obtained precipitate was dissolved in 200 mL of DCM and was then washed by NaCl aqueous solution thrice. After the organic solution was dried over anhydrous sodium sulfate overnight, DCM was removed by a rotary evaporator. The obtained sample was dried under vacuum at room temperature for 48 h (yield, 16.7 g).2.5 Synthesis of mPEG(-OH)-N3
15.7 g (3.1 mmol) of mPEG-epoxide was dissolved in 63 mL of DMF. Then, 1.63 g (25.1 mmol) of NaN3 and 1.34 g (25.1 mmol) of NH4Cl were added successively. The reaction system was stirred at 50 °C for 48 h. When the mixture was cooled to room temperature, it was precipitated into a mixed ether/n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) solvent mixture. The obtained precipitate was dissolved in 200 mL of DCM and the organic solution was then washed by water thrice. After it was dried over anhydrous sodium sulfate overnight, the organic solvent was concentrated by a rotary evaporator. Afterwards, the solution was precipitated into n-hexane. The obtained sample was dried under vacuum at room temperature for 48 h (yield, 13.4 g).
:1 by volume) solvent mixture. The obtained precipitate was dissolved in 200 mL of DCM and the organic solution was then washed by water thrice. After it was dried over anhydrous sodium sulfate overnight, the organic solvent was concentrated by a rotary evaporator. Afterwards, the solution was precipitated into n-hexane. The obtained sample was dried under vacuum at room temperature for 48 h (yield, 13.4 g).
2.6 Synthesis of mPEG(-OH)-Cl
After a solution composed of mPEG(-OH)-N3 (5.4 g, 1.06 mmol), alkynyl-Cl (0.35 g, 2.18 mmol), PMDETA (0.2059 g, 1.19 mmol) and DMF (108 mL) was bubbled with argon gas for 90 min, CuBr (178.2 mg, 1.24 mmol) was added. The reaction mixture was bubbled with argon gas for additional 30 min. In the period of bubbling argon gas, the mixture was sonicated several times and each sonication lasted 1 min. The reaction proceeded at 40 °C. After 48 h, the mixture was cooled to room temperature and exposed to air. After stirring for 1 h, the mixture was precipitated into THF/n-hexane (1:1 by volume) solvent mixture. The obtained precipitate was dissolved in 150 mL of DCM. It was passed through a neutral alumina column to remove the copper complex. The resulting solution was concentrated and precipitated into n-hexane. The repeated purification was carried out by dissolving in DCM and precipitating in n-hexane. The resulting polymer was dried under vacuum at room temperature for 48 h (yield, 4.1 g).
2.7 Synthesis of mPEG(-alkynyl)-Cl
3.92 g (0.75 mmol) of mPEG(-OH)-Cl, 0.27 g (2.75 mmol) of 4-pentynoic acid and 0.1 g (0.82 mmol) of DMAP was successively dissolved in 4.6 mL of DCM. Under the condition of −20 °C, a solution of EDCI (0.53 g, 2.76 mmol) in 15 mL of DCM was stepwise added. After stirring for 10 min at −20 °C, the reaction mixture was stirred at room temperature for additional 24 h. Then, it was diluted with 200 mL of DCM and washed by 16.7% NaCl aqueous solution several times until water phase showing neutrality. After it was dried over anhydrous sodium sulfate overnight and the solution was concentrated by a rotary evaporator, it was precipitated into n-hexane. The obtained polymer was dried under vacuum at room temperature for 48 h (yield, 3.34 g).2.8 Synthesis of mPEG(-alkynyl)-b-PDEA
After a mixture composed of HMTETA (68.6 mg, 0.3 mmol), CuCl2·2H2O (2.54 mg, 0.015 mmol) and anisole (8.7 mL) formed a homogeneous solution under the condition of sonication at 40 °C, 3.21 g (17.3 mmol) of DEA and 1.34 g (0.25 mmol) of mPEG(-alkynyl)-Cl were successively dissolved in the solution at room temperature. After the mixture was bubbled with nitrogen gas for 40 min, CuCl (14.74 mg, 0.15 mmol) was added. After bubbling with nitrogen lasted additional 20 min, the reaction system was sealed and stirred magnetically in an oil bath at 80 °C. After 1.5 h, the reaction system was cooled to room temperature, exposed to air and diluted with DCM. The copper complex was removed by passing through a neutral alumina column. The resulting solution was concentrated by a rotary evaporator and precipitated into n-hexane at ∼−38 °C. The purification was carried out by dissolving in DCM and precipitating in n-hexane (at ∼−38 °C) twice. The obtained polymer was dried under vacuum at room temperature for 24 h (yield, 1.73 g).2.9 Synthesis of CD-star-PEMA(-b-PDEA)-b-mPEG
CD-star-PEMA-N3 (0.39 g, 0.13 mmol reaction sites), PEG(-alkynyl)-PDEA (1.49 g, 0.16 mmol) and PMDETA (26.89 mg, 0.16 mmol) were successively dissolved in 9.5 mL of DMF. Then, the mixture was bubbled with argon gas for 40 min, CuBr (22.26 mg, 0.16 mmol) was added. After completion of bubbling with argon for additional 20 min, the reaction system was sealed and stirred magnetically in an oil bath at 60 °C. After 48 h, the reaction system was cooled to room temperature, exposed to air and diluted with DCM. The mixture was precipitated into THF/n-hexane (1:1 by volume) mixture. The resulting precipitate was dissolved in DCM. The copper complex was removed by passing through a neutral alumina column. The resulting solution was concentrated by a rotary evaporator and precipitated into hexane at 35–40 °C. The further purification was carried out by dissolving in DCM and precipitating in n-hexane (at 35–40 °C) twice. The obtained polymer was dried under vacuum at room temperature for 48 h (yield, 1.1 g).
2.10 Preparation of aqueous solution of CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles
15 mg of CD-star-PEMA(-b-PDEA)-b-mPEG was dissolved in 1.7 mL of THF. After ∼8 h, 3.4 mL of pure water was added. The mixture was stirred at room temperature for 12 h. After the THF was removed by reduced pressure at 25 °C, the solution was dialyzed (molecular weight cutoff 14000) against pure water overnight. Finally, the obtained aqueous solution was diluted with water to a polymer concentration of 3 mg mL−1 for further applications.
2.11 Preparation of drug-loaded CD-star-PEMA(-b-PDEA)-b-mPEG and release studies in vitro
1 mL of CD-star-PEMA(-b-PDEA)-b-mPEG aqueous solution was added into a solution of a certain amount of guest (Nile red, celecoxib, or ketoprofen) in 0.4 mL of DCM. After the mixture was stirred at room temperature for 1 h, the DCM was removed by reduced pressure. The obtained samples loaded with Nile red, celecoxib, and ketoprofen are denoted as CDSP-NR, CDSP-CEL and CDSP-KET, respectively. Finally, CDSP-CEL and CDSP-KET were diluted with water to a polymer concentration of 1.5 mg mL−1 for further applications, respectively; CDSP-NR was diluted to a polymer concentration of 0.6 mg mL−1. The loading capacity of CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles is defined as weight ratio of guest encapsulated into micelles to the copolymer.The release behaviors of CDSP-CEL and CDSP-KET were estimated by dialysis at 37 °C. 1 mL of the loaded CD-star-PEMA(-b-PDEA)-b-mPEG solution (polymer concentration, 0.3 mg mL−1) was put into a dialysis bag (molecular weight cutoff 14000) and it was directly immersed in 50 mL of buffer solution of pH 7.4, or pH 5.0, which was shaken at ca. 100 rpm. At a specific interval, 4 mL of the solution released was withdrawn and at the same time 4 mL of fresh pH buffer solution was added. The concentrations of the guest (celecoxib or ketoprofen) released were analyzed by measuring the UV-vis absorbances at 252 nm for celecoxib and 258 nm for ketoprofen.
1 mL of Nile red loaded unimolecular solution (polymer concentration, 0.6 mg mL−1) was mixed with 70 mL of buffer solution of pH 7.4, or pH 5.0. Then, the mixture was incubated at 37 °C, which was shaken at ca. 100 rpm. At predetermined time intervals, 2 mL of the solution was drawn to measure the fluorescence spectroscopy, and the release profiles were obtained. Nile red was excited at 485 nm and its emission was recorded at 580 nm.
2.12 Analyses
1H NMR measurements were conducted on Bruker Ascend 400M spectrometer. The molecular structure parameters of the star copolymers were determined on a DAWN EOS size exclusion chromatography/multiangle laser light scattering (SEC/MALLS). HPLC-grade THF was used as the eluent at a flow rate of 0.5 mL min−1. The refractive index increment (dn/dc) value of a sample in THF solution was determined by an Optilab rEX detector at 25 °C through a batch model. The Z-average diameter (Dz) and zeta-potential of micelle were characterized by Zetasizer Nano-ZS dynamic light scattering (DLS) (Malvern, Britain). The morphologies of the micelles were observed by transmission electron microscopy (TEM) (JEM-3010, Japan). All samples were stained with 3% phosphotungstic acid before TEM observation. Fluorescence spectra were recorded using a Hitachi F-4600 spectrometer.3. Results and discussion
3.1 Synthesis and characterization of CD-star-PEMA(-b-PDEA)-b-mPEG
Synthesis route to CD-star-PEMA(-b-PDEA)-b-mPEG is shown in Scheme 1. | ||
| Scheme 1 Synthesis route to CD-star-PEMA(-b-PDEA)-b-mPEG. | ||
| Samples | Mn,theo (kg mol−1) | Mn,SEC/MALLS (kg mol−1) | Mw/Mn | dn/dc (mL g−1) |
|---|---|---|---|---|
| a Determined by monomer conversion; the monomer conversion was measured by gravimetric method.b Calculated by Mn,theo of CD-star-PEMA-Cl and change of end group.c Calculated by molecular weight of mPEG and additive end groups.d Determined by Mn,theo of mPEG(-alkynyl)-Cl and degree of polymerization of DEA units.e Determined by 1H NMR spectrum of CD-star-PEMA(-b-PDEA)-b-mPEG. | ||||
| CD-star-PEMA-Cl | 63.34a | 61.03 | 1.07 | 0.0779 |
| CD-star-PEMA-N3 | 63.48b | 54.07 | 1.09 | 0.0754 |
| mPEG-epoxide | 5.06c | 5.18 | 1.04 | 0.0644 |
| mPEG(-OH)-N3 | 5.10c | 5.59 | 1.03 | 0.0628 |
| PEG(-OH)-Cl | 5.26c | 5.68 | 1.05 | 0.0633 |
| mPEG(-alkynyl)-Cl | 5.36c | 5.24 | 1.09 | 0.0658 |
| PEG(-alkynyl)-PDEA | 9.58d | 11.7 | 1.21 | 0.0749 |
| CD-star-PEMA(-b-PDEA)-b-PEG | 263.02e | 200.03 | 1.09 | 0.0744 |
Subsequently, CD-star-PEMA-N3 was synthesized by azide groups replacing the chlorine groups at the arm ends of CD-star-PEMA-Cl (Scheme 1b).32,33 The perfect occurrence of the later azide–alkyne click reaction (this is discussed later) seems to imply that the chlorine groups should be replaced successfully by azide groups and the substitution degree may be close to 100%. In the 1H NMR spectrum of CD-star-PEMA-N3 (Fig. 1a), the resonance intensity ratio of different protons still conforms to the chemical structure of PEMA. In addition, the molecular weight distribution of CD-star-PEMA-N3 remained narrow (Fig. 2a, Table 1). These mean that the substitution reaction did not generate evident side result. The incorporation of azide groups should lead to an slight increase in molecular weight of the resulting product, but the Mn,SEC/MALLS value of CD-star-PEMA-N3 is 54.07 kg mol−1, which is lower than that of CD-star-PEMA-Cl (Table 1). This may be due to different end groups exerting influence on the measurement result.
 | ||
| Fig. 1 1H NMR spectrum of CD-star-PEMA(-b-PDEA)-b-mPEG and its precursors. | ||
 | ||
| Fig. 2 DRI signals of SEC/MALLS chromatograms of CD-star-PEMA(-b-PDEA)-b-mPEG and its precursors. | ||
:1, indicating the fact that the synthesis of epoxy-ended mPEG was carried out successfully. Secondly, the reaction of the epoxy group of mPEG-epoxide with NaN3 afforded the mPEG to carry a hydroxyl group and an azide group,36–38 i.e. mPEG(-OH)-N3 (see Scheme 1d). Compared with the 1H NMR spectrum of precursor mPEG-epoxide, the new resonance peaks at δ = 3.36 and 3.96 are from the protons of –CH2N3 and –CH(OH)CH2N3 groups of mPEG(-OH)-N3, respectively (ESI, Fig. S4†). The resonance intensity ratio of the protons of different moieties of mPEG(-OH)-N3 confirms its molecular structure (ESI, Fig. S4†). Thirdly, the azide–alkyne click reaction39,40 of mPEG(-OH)-N3 with alkynyl-Cl produced mPEG(-OH)-Cl that contains an ATRP initiating group (see Scheme 1e). The 1H NMR spectrum of mPEG(-OH)-Cl verifies its molecular structure (ESI, Fig. S5†). Fourthly, the hydroxyl-carboxyl condensation reaction of mPEG(-OH)-Cl with 4-pentynoic acid in the presence of EDCI/DMAP41 created mPEG(-alkynyl)-Cl with an alkynyl group (see Scheme 1f). Compared with the1H NMR spectrum of precursor mPEG(-OH)-Cl (ESI, Fig. S5†), new resonance peaks at δ = 2.02, 2.49 and 2.58 ppm from the protons of CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–, CCH2– and CHCCH2CH2– groups, respectively, could be observed clearly (ESI, Fig. S6†). The resonance intensity ratio of the protons of different moieties of mPEG(-alkynyl)-Cl is consistent with the molecular structure. Also, molecular weights and distributions of mPEG-epoxide, mPEG(-OH)-N3, mPEG(-OH)-Cl, and mPEG(-alkynyl)-Cl were estimated by SEC/MALLS measurements (ESI, Fig. S7†). The Mn,SEC/MALLS values of the mPEG-based derivative are all around 5000 and are in well agreement with their theoretic ones (Fig. S7 in ESI† and Table 1). The narrow molecular weight distributions also evidence that the modifications were successful.
C–, CCH2– and CHCCH2CH2– groups, respectively, could be observed clearly (ESI, Fig. S6†). The resonance intensity ratio of the protons of different moieties of mPEG(-alkynyl)-Cl is consistent with the molecular structure. Also, molecular weights and distributions of mPEG-epoxide, mPEG(-OH)-N3, mPEG(-OH)-Cl, and mPEG(-alkynyl)-Cl were estimated by SEC/MALLS measurements (ESI, Fig. S7†). The Mn,SEC/MALLS values of the mPEG-based derivative are all around 5000 and are in well agreement with their theoretic ones (Fig. S7 in ESI† and Table 1). The narrow molecular weight distributions also evidence that the modifications were successful.Finally, using mPEG(-alkynyl)-Cl as the ATRP macroinitiator and DEA as the monomer, block copolymer mPEG(-alkynyl)-PDEA was synthesized (see Scheme 1g). Fig. 1b shows 1H NMR spectrum of mPEG(-alkynyl)-PDEA. In Fig. 1b, the peaks at δ = 2.63 and 2.75 are assigned to proton resonances of methylene of –CH2N-(CH2-)2 group from PDEA block.13 According to the resonance intensity ratio of –OCH3 group from mPEG block to –CH2N-(CH2-)2 group from PDEA block, the DP of the PDEA block of mPEG(-alkynyl)-PDEA was calculated to be 22.8. According to the DP value, Mn,thero of mPEG(-alkynyl)-PDEA was calculated to be 9.58 kg mol−1, whereas Mn,SEC/MALLS is 11.7 kg mol−1. The Mw/Mn value of the copolymer is 1.21.
3.2 CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles
CD-star-PEMA(-b-PDEA)-b-mPEG could not directly dissolve in aqueous solution even at pH 2.0, probably due to the presence of the hydrophobic intermolecular interaction. Therefore, to prepare unimolecular micelles, CD-star-PEMA(-b-PDEA)-b-mPEG was first dissolved in THF. THF is a common solvent for all building blocks of the star copolymer, guaranteeing its molecular dissolution. And then 2-fold amount of water (relative to the used amount of THF) was added. After stirring several hours, the THF was slowly removed by reduced pressure. After dialyzing against pure water overnight, an aqueous micellar solution was obtained. Z-Average diameter (Dz) of the micelles (3 mg mL−1) was determined to be 26.7 nm, whereas that of CD-star-PEMA(-b-PDEA)-b-mPEG in THF (3 mg mL−1) is 28.9 nm. These almost identical sizes in water and THF demonstrate that the micelles should be a unimolecular state in water.8 In addition, Dzs of the micelles were determined under dilution. It was found that Dz values at polymer concentrations of 2, 1, 0.5, 0.1, and 0.05 mg mL−1 were determined to 28.1, 28.3, 28.1, 27.9 and 28.2 nm, respectively. The less change of size with decrease in polymer concentration supports that CD-star-PEMA(-b-PDEA)-b-mPEG was unimolecular micelles in water. Fig. 3a shows TEM image from the aqueous CD-star-PEMA(-b-PDEA)-b-mPEG solution. As seen from Fig. 3a, the micelles are approximately spherical and their sizes are ∼20 nm in diameter, which are smaller than that from DLS measurement (Fig. 3a′). This means that CD-star-PEMA(-b-PDEA)-b-mPEG could indeed exhibit unimolecular micelles aqueous solution. Therefore, the aid method of THF is good for preparing aqueous CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles. | ||
| Fig. 3 TEM images of (a) blank unimolecular micelles, (b) CDSP-NR, (c) CDSP-CEL and (d) CDSP-KET; (a′)–(d′) for their corresponding DLS measurements. | ||
To investigate architecture of CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles in aqueous solution, we utilized deuterated tetrahydrofuran (THF-d8) and deuterium oxide (D2O) to replace THF and water, respectively, and prepared micelles through the similar method for 1H NMR analysis. That is, CD-star-PEMA(-b-PDEA)-b-mPEG was first dissolved in THF-d8. D2O was then added and finally THF-d8 was slowly removed by reduced pressure. CD-star-PEMA(-b-PDEA)-b-mPEG in THF-d8, THF-d8/D2O and D2O were analyzed by 1H NMR, respectively, and the results are shown in Fig. S8 (ESI†). As seen from Fig. S8a (ESI†), the characteristic resonance signals of PEMA, PDEA and mPEG blocks of CD-star-PEMA(-b-PDEA)-b-mPEG in THF-d8 could be observed clearly (the resonance signals of the characteristic protons were labeled in Fig. S8a of ESI†). This means that THF is good solvent for all blocks of the copolymer and the blocks could be solvated fully. After addition of D2O, although the baseline of the spectrum is not good possible due to selecting D2O as the solvent mode (in fact at this moment the solvent was composed of D2O and THF-d8), the characteristic resonance proton signals of the mPEG and PDEA blocks were clearly observed. In contrast, the characteristic signal of PEMA block at δ = 1.28 ppm was weaken evidently (ESI, Fig. S8b†). This suggests that in the THF-d8/D2O mixture the PEMA moieties were desolvated partly, whereas mPEG and PDEA moieties still remained solvated. After THF-d8 solvent was removed, only resonance signals of mPEG block could clearly be observed, whereas the characteristic resonance proton peaks of PEMA and PDEA blocks disappeared (ESI, Fig. S8c†). This indicates that in D2O, only mPEG block was solvated, whereas PEMA and PDEA blocks had been desolvated.6,31 In the D2O solution Dz of CD-star-PEMA(-b-PDEA)-b-mPEG was measured by DLS to be 29.6 nm, indicating the copolymer being a unimolecular state. The results demonstrate the formation of the unimolecular micelles. According to the results, a formation process of CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles is depicted as follows. Owing to the fact that THF is common solvent and water is selective one, addition of water into the THF solution of CD-star-PEMA(-b-PDEA)-b-mPEG resulted in the PEMA block being firstly desolvated partly, and in contrast, the mPEG and PDEA blocks still remained solvated in the mixed solvent. This led to the PEMA blocks collapsing into β-CD core partly. At this moment, the mPEG and PDEA blocks were still in a stretched state. When THF was removed step by step, the PEMA blocks would further collapsed toward β-CD core. And the same time the PDEA blocks also began to collapse toward the inside. Finally, the PEMA and PDEA blocks desolvated fully and exhibited a collapsed state, whereas the mPEG block remained solvated and exhibited a stretched state. As a result, a core–shell CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelle formed. The PEMA blocks constituted the hydrophobic inner core of the micelle and the hydrophilic mPEG blocks formed the shell. The PDEA blocks should hydrophobically collapse at the interfaces of mPEG shell and PEMA core and formed outer core of the micelle. Therefore, the objective of the utilization of common solvent THF is to cause the hydrophobic PEMA blocks to collapse toward the β-CD core, avoiding the occurrence of intermolecular hydrophobic interaction.
Owing to PDEA exhibiting pH-sensitivity (pKa of homoPDEA, ∼7.3),11,13,21 influence of change in environmental pH on the micelles was investigated. Dz and zeta-potential of the micelles (0.1 mg mL−1) were measured at pHs 3.0, 5.0, 6.0, 7.4, and 9.0. It was found that Dz of CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles were 33.9, 33.2, 32.4, 29.9, and 29.6 nm at the pHs, respectively. The zeta-potentials at five pHs corresponded to 9.3, 7.3, 2.5, −1.64, and −3.1 mV. This means that at acidic condition the surface of unimolecular micelle carried some positive charge. This should be attributed to the protonation of the PDEA blocks at acidic aqueous solution,11,13,21,42,43 which lead to the PDEA blocks exhibiting hydrophilicity and producing a transformation from collapsed state to extended one due to electrostatic repulsion of the protoned groups.13,44 Therefore, at acidic condition, the PDEA and mPEG built shell of the micelles. At pHs 7.4 and 9.0 at the PDEA blocks collapsed on the PEMA core. Therefore, the location of the PDEA blocks relies on the surrounding pH. This kind of phenomenon has been observed in the self-assemblies from environmental-sensitive miktoarm star copolymer.38,44 It is noted that the pH-dependent surface charge of the micelles may be beneficial to their application in biomaterial field like drug delivery.43
3.3 Drug loading and release
Whether are the core–shell CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles able to encapsulate hydrophobic guest compounds? To test this, herein Nile red,45–49 celecoxib13,14,50,51 and ketoprofen52 were selected as model hydrophobic molecules. It was found that the loading capacities of CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles for Nile red, celecoxib and ketoprofen were 6–7%, 20–25% and 30–35%, respectively. In the loading experiments, the guest molecules were first dissolved in DCM, and CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelle solution was added into the DCM solution. After stirring, the DCM was then removed by reduced pressure. The obtained loaded micelles were denoted as CDSP-NR, CDSP-CEL and CDSP-KET, respectively. In following studies, the encapsulation amounts are 6% Nile red for CDSP-NR, 20% celecoxib for CDSP-CEL and 30% ketoprofen for CDSP-KET. If aqueous solution without the copolymer was added into DCM solution with Nile red, celecoxib, or ketoprofen, the guest molecules could easily be precipitated out when the DCM component was removed. This means that the above-mentioned guest molecules were encapsulated into the unimolecular micelles. It was found that Dzs of samples CDSP-NR, CDSP-CEL and CDSP-KET were determined to be 30.5, 29 and 31.2 nm, respectively (Fig. 3b′–d′). The fact that sizes of the loaded samples are only slightly higher than that of blank sample means that they were still a unimolecular state in aqueous solution. The results from TEM measurements shown in Fig. 3b–d further confirm this conclusion. As seen clearly from Fig. 3b–d, incorporation of the guest molecules into the unimolecular micelles did not evidently change morphologies and size of the micelles. This suggests that CD-star-PEMA(-b-PDEA)-b-mPEG is indeed a good unimolecular container for hydrophobic guest molecules. Among three guest molecules, the amount of ketoprofen encapsulated into CD-star-PEMA(-b-PDEA)-b-mPEG molecules is largest, probably due to it being acidic compound (pKa = 4.4).52 The acid–base reaction of ketoprofen with the PDEA moieties53 may enhance its loading. Therefore, for Nile red and celecoxib, the encapsulations of the star copolymer should mainly depend on hydrophobic interaction with the guest molecules. For ketoprofen, there existed hydrophobic and electrostatic interactions in the encapsulation.Nile red is fluorescent when loaded into a hydrophobic environment but its fluorescence emission is very small in water.45–49 Therefore, when Nile red was released from the hydrophobic moieties into aqueous solution, its fluorescent intensity reduce greatly. Consequently, release behaviors of Nile red are directly monitored by fluorescent spectra. Fig. 4 shows normalized fluorescence intensity of Nile red-loaded CD-star-PEMA(-b-PDEA)-b-mPEG under incubation at 37 °C and pH 5.0 or 7.4. As seen from Fig. 4, decrease rate of the fluorescence intensity of Nile red with time at pH 5.0 was faster than at pH 7.4. This means that Nile red was faster released from CDSP-NR at pH 5.0. Owing to pKa of Nile red being ∼1,46 the protonation of the amine group of Nile red should be very low at pH 5.0. This suggests that variation of pH from 7.4 to 5.0 may produce small influence on water-solubility of Nile red. Therefore, the pH-tunable release of Nile red should ascribe to pH-responsiveness of the PDEA blocks, i.e. pH-dependent protonation/deprotonation of the PDEA moieties. At pH 5.0, the protonation of the amine groups of the PDEA blocks cause them to exhibit hydrophilicity, whereas the PDEA exhibits hydrophobicity at pH 7.4. Consequently, on the one hand, at pH 5.0 the Nile red encapsulated in the PDEA moieties could be released out quickly due to the hydrophilicity of the PDEA moieties; on the other hand, at pH 5.0 the decrease in the content of hydrophobic component of micelles also led to an increase in diffusivity of the loaded molecules.
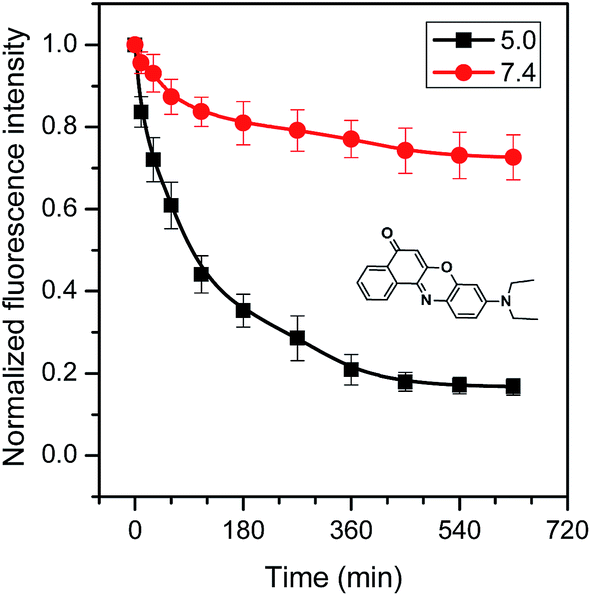 | ||
| Fig. 4 Normalized fluorescence intensity of Nile red-loaded CD-star-PEMA(-b-PDEA)-b-mPEG under incubation at 37 °C and pHs 5.0 and 7.4. | ||
Fig. 5 shows release profiles of celecoxib and ketoprofen from their loaded samples at pHs 7.4 and 5.0, respectively. As can be seen from (a), in the first 25 min, the cumulative amounts of the celecoxib released were 52.9% at pH 5.0 and 31.1% at pH 7.4, respectively. Over 170 min, the cumulative amount of the celecoxib released were 85.1% at pH 5.0 and 62.4% at pH 7.4, respectively. Obviously, the release rate of celecoxib at pH 5.0 was still faster than at pH 7.4. Similar with release mechanism of Nile red, it was attributed to the pH-responsiveness of the PDEA blocks.
 | ||
| Fig. 5 Release profiles of (a) celecoxib and (b) ketoprofen from their loaded samples at pH 7.4 and 5.0 and 37 °C. | ||
With regard to ketoprofen, influence of change in pH on its release involves the following several points. (1) Owing to ketoprofen with pKa ∼ 4.4,52 at pH 7.4 the carboxyl group of ketoprofen becomes carboxylate, and this should be able to evidently enhance water-solubility of ketoprofen to promote its release from the copolymer. But at this pH the PDEA block exhibit hydrophobicity, which could delay the drug release by hydrophobic interaction with ketoprofen. (2) According to the pKa values of ketoprofen and PDEA, their electrostatic interaction should be well constructed in the pH range from 4.4 to 7.3, and the interaction also delay diffusion and release of ketoprofen, but the presence of salt ionic in buffer solution could weaken the interaction.53 (3) At pH 5.0, the PDEA blocks show hydrophilicity, and this could enhance release of ketoprofen due to decrease in content of hydrophobic component of the unimolecular micelles. Therefore, release kinetics of ketoprofen is a balance result of the above-mentioned factors. Fig. 5b clearly shows that the release rate of ketoprofen at pH 5.0 is faster than at pH 7.4. This means that the pH-dependent hydrophilicity/hydrophobicity of the PDEA moieties should be a dominating factor for ketoprofen release in our conditions.
4. Conclusion
A well-defined cyclodextrin-core star copolymer with Y-shaped ABC miktoarms, star poly(ethyl methacrylate(-b-poly(2-(diethylamino)ethyl methacrylate)-b-methoxypolyethylene glycols, was synthesized via ATRP and click reactions. Aqueous CD-star-PEMA(-b-PDEA)-b-mPEG unimolecular micelles was obtained under the aid of THF. Z-Average diameter of the micelles in water was ∼27 nm. The unimolecular micelles could encapsulate hydrophobic drug molecules like Nile red, celecoxib and ketoprofen. Release of the payloads from the unimolecular micelles exhibited pH-tunability due to pH-sensitive PDEA blocks. And the same time, pH-dependent the surface charge of the micelles were observed.References
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- Y. Li, K. Xiao, W. Zhu, W. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58–73 CrossRef CAS PubMed.
- H. Gao, Macromol. Rapid Commun., 2012, 33, 722–734 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS PubMed.
- Y. Cai and Y.-Y. Liu, Macromol. Chem. Phys., 2013, 214, 882–891 CrossRef CAS.
- X. Pang, L. Zhao, M. Akinc, J. K. Kim and Z. Lin, Macromolecules, 2011, 44, 3746–3752 CrossRef CAS.
- Z. Xu, S. Liu, H. Liu, C. Yang, Y. Kang and M. Wang, Chem. Commun., 2015, 51, 15768–15771 RSC.
- M. Liu, K. Kono and J. M. J. Fréchet, J. Controlled Release, 2000, 65, 121–131 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Bioconjugate Chem., 2010, 21, 496–504 CrossRef CAS PubMed.
- L. Niu, Y. Liu, Y. Hou, W. Song and Y. Wang, Polym. Chem., 2016, 7, 3406–3415 RSC.
- Y.-Y. Liu, Y.-B. Zhong, J.-K. Nan and W. Tian, Macromolecules, 2010, 43, 10221–10230 CrossRef CAS.
- P. Zhou, Y.-Y. Liu, L.-Y. Niu and J. Zhu, Polym. Chem., 2015, 6, 2934–2944 RSC.
- J. Zhu, Y. Liu, L. Xiao and P. Zhou, Macromol. Chem. Phys., 2016, 217, 773–782 CrossRef CAS.
- W. Wu, W. Wang and J. Li, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS.
- J. Li, Z. Guo, J. Xin, G. Zhao and H. Xiao, Carbohydr. Polym., 2010, 79, 277–283 CrossRef CAS.
- K. Ohno, B. Wong and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2206–2214 CrossRef CAS.
- J. Li, H. Xiao, Y. S. Kim and T. L. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6345–6354 CrossRef CAS.
- X. Chen, W. Wu, Z. Guo, J. Xin and J. Li, Biomaterials, 2011, 32, 1759–1766 CrossRef CAS PubMed.
- P. Chmielarz, S. Park, A. Sobkowiak and K. Matyjaszewski, Polymer, 2016, 88, 36–42 CrossRef CAS.
- Z. Ge, J. Xu, J. Hu, Y. Zhang and S. Liu, Soft Matter, 2009, 5, 3932–3939 RSC.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, Biomacromolecules, 2010, 11, 934–943 CrossRef CAS PubMed.
- Y. Miura, A. Narumi, S. Matsuya, T. Satoh, Q. Duan, H. Kaga and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4271–4279 CrossRef CAS.
- M. Adeli, Z. Zarnegar and R. Kabiri, Eur. Polym. J., 2008, 44, 1921–1930 CrossRef CAS.
- P.-F. Gou, W.-P. Zhu and Z.-Q. Shen, Polym. Chem., 2010, 1, 1205–1214 RSC.
- K. M. Xiu, J. J. Yang, N. N. Zhao, J. S. Li and F. J. Xu, Acta Biomater., 2003, 9, 4726–4733 CrossRef PubMed.
- Y. Li, H. Guo, J. Zheng, J. Gan, K. Wu and M. Lu, Colloids Surf., A, 2015, 471, 178–189 CrossRef CAS.
- T. Kakuchi, A. Narumi, T. Matsuda, Y. Miura, N. Sugimoto, T. Satoh and H. Kaga, Macromolecules, 2003, 36, 3914–3920 CrossRef CAS.
- Z.-X. Zhang, X. Liu, F. J. Xu, X. J. Loh, E.-T. Kang, K.-G. Neoh and J. Li, Macromolecules, 2008, 41, 5967–5970 CrossRef CAS.
- S. Shao, J. Si, J. Tang, M. Sui and Y. Shen, Macromolecules, 2014, 47, 916–921 CrossRef CAS.
- M.-C. Jones, M. Ranger and J.-C. Leroux, Bioconjugate Chem., 2003, 14, 774–781 CrossRef CAS PubMed.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2011, 133, 13421–13429 CrossRef CAS PubMed.
- K. Van Butsele, F. Stoffelbach, R. Jérôme and C. Jérôme, Macromolecules, 2006, 39, 5652–5656 CrossRef CAS.
- R. Alizadeh and M. Ghaemy, Polymer, 2015, 66, 179–191 CrossRef CAS.
- W. Sun, X. He, C. Gao, X. Liao, M. Xie, S. Lin and D. Yan, Polym. Chem., 2013, 4, 1939–1949 RSC.
- Y.-Y. Yuan, Y.-C. Wang, J.-Z. Du and J. Wang, Macromolecules, 2008, 41, 8620–8625 CrossRef CAS.
- H. Liu, C. Li, H. Liu and S. Liu, Langmuir, 2009, 25, 4724–4734 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 114, 2708–2711 CrossRef.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- A. Gopin, S. Ebner, B. Attali and D. Shabat, Bioconjugate Chem., 2006, 17, 1432–1440 CrossRef CAS PubMed.
- Y. Q. Yang, B. Zhao, Z. D. Li, W. J. Lin, C. Y. Zhang, X. D. Guo, J. F. Wang and L. J. Zhang, Acta Biomater., 2013, 9, 7679–7690 CrossRef CAS PubMed.
- Y. Shen, Y. Zhan, J. Tang, P. Xu, P. A. Johnson, M. Radosz, E. A. V. Kirk and W. J. Murdoch, AIChE J., 2008, 54, 2979–2989 CrossRef CAS.
- K. V. Butsele, C. A. Fustin, J. F. Gohy, R. Jérôme and C. Jérôme, Langmuir, 2009, 25, 107–111 CrossRef PubMed.
- M. K. Gupta, J. R. Martin, T. A. Werfel, T. Shen, J. M. Page and C. L. Duvall, J. Am. Chem. Soc., 2014, 136, 14896–14902 CrossRef CAS PubMed.
- E. M. Moreno and D. Levy, Chem. Mater., 2000, 12, 2334–2340 CrossRef CAS.
- Q. Zhang, N. Vanparijs, B. Louage, B. G. D. Geest and R. Hoogenboom, Polym. Chem., 2014, 5, 1140–1144 RSC.
- C. Xiao, J. Ding, L. Ma, C. Yang, X. Zhuang and X. Chen, Polym. Chem., 2015, 6, 738–747 RSC.
- B. Mo, H. Liu, X. Zhou and Y. Zhao, Polym. Chem., 2015, 6, 3489–3501 RSC.
- N. V. Seedher and S. Bhatia, AAPS PharmSciTech, 2003, 4(3), 33 CrossRef PubMed.
- M. Bachar, A. Mandelbaum, I. Portnaya, H. Perlstein, S. Even-Chen, Y. Barenholz and D. Danino, J. Controlled Release, 2012, 160, 164–171 CrossRef CAS PubMed.
- N. H. Hashim and S. J. Khan, J. Chromatogr. A, 2011, 1218, 4746–4754 CrossRef CAS PubMed.
- W. Song, Y. Liu, Y. Hou and X. Fan, Soft Mater., 2016, 14, 228–237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra and SEC/MALLS measurements were provided. See DOI: 10.1039/c6ra28456f |
| This journal is © The Royal Society of Chemistry 2017 |