
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting human intestinal absorption with modified random forest approach: a comprehensive evaluation of molecular representation, unbalanced data, and applicability domain issues†
Ning-Ning Wang‡
a,
Chen Huang‡b,
Jie Donga,
Zhi-Jiang Yaoac,
Min-Feng Zhuac,
Zhen-Ke Denga,
Ben Lvc,
Ai-Ping Lud,
Alex F. Chenac and
Dong-Sheng Cao *acd
*acd
aXiangya School of Pharmaceutical Sciences, Central South University, Changsha, 410013, P. R. China. E-mail: oriental-cds@163.com
bSchool of Mathematics and Statistics, Central South University, Changsha 410083, P. R. China
cThe 3rd Xiangya Hospital, Central South University, Changsha, 410000, P. R. China
dInstitute for Advancing Translational Medicine in Bone & Joint Diseases, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong SAR, P. R. China
First published on 29th March 2017
Abstract
With the increase of complexity and risk in drug discovery processes, human intestinal absorption (HIA) prediction has become more and more important. Up to now, some predictive models have been constructed to estimate HIA of new drug-like compounds with acceptable accuracies, but there are still some issues to be explored including the limited and unbalanced HIA data, the performance of different types of descriptors and the application domain issues of published models. To address these problems, in this study, we collected a relatively large dataset consisting of 970 compounds, and 9 different types of descriptors were calculated for further modeling. For all the modeling processes, a parameter named samplesize in the random forest (RF) method was applied to balance the dataset. And then, classification models were established based on different training sets and different combinations of descriptors. After a series of modeling processes and various comparisons among these statistical results, we explored the aforementioned problems and evaluated the reliabilities of existing HIA classification models and subsequently obtained a robust and applicable model based on a combination of 2D, 3D, N+ and Nrule-of-five (for the training set, SE = 0.892, SP = 0.846; for the test set, SE = 0.877, SP = 0.813). Compared with other published models, our model exhibits some advantages in data size, model accuracy and model practicability to some extent. This structure–activity relationship model is necessary and useful for HIA prediction and it could be a convenient tool for virtual screening in the early stage of drug development.
Introduction
In recent years, complexity and risk have increased greatly in drug discovery and development processes which result in increasing investment in drug research.1 For an approved drug, this investment has increased from 800 million dollars in 2003 to 2.6 billion dollars in 2014. The decline in the productivity of the pharmaceutical industry is mostly due to the poor ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties.2–9 Nowadays, oral administration has become the route favored by patients because of its ease and patient compliance. For a new oral drug, bioavailability is one of the most desirable attributes, whereas the determination of oral bioavailability is very challenging due to the fact that bioavailability is a complex function of many biologic and physicochemical factors.10,11 The relationship between oral bioavailability and intestinal absorption has been proven by previous studies and we can draw the conclusion that the bioavailability of most compounds (64%) is mainly controlled by the intestinal absorption process.12 Consequently, the aforementioned phenomenon reminds us that human intestinal absorption (HIA) could be an alternative indicator for oral bioavailability to some extent and thus it also plays an important role in preclinical drug evaluation.Currently, preclinical HIA screening strategies could be classified into three categories according to different methods. In the early stage of HIA study, the evaluation of HIA was generally based on animal experiments.13,14 But considering animal protection and research status, it is necessary to develop some alternative methods to evaluate HIA. Subsequently, diverse quantitative structure–activity relationship (QSAR) models for in vitro permeability were established to estimate HIA. Among these methods, the most widely used ones are parallel artificial membrane permeability assay (PAMPA),15 human colon adenocarcinoma (Caco-2) cell lines,16 and the Madin Darby canine kidney (MDCK) cell.6,17,18 Especially, some studies have shown that there is a good sigmoidal relationship between human oral drug absorption and Caco-2 permeability but this relationship seems not so clear.19–22 Hou et al. have confirmed that Caco-2 cell lines can be used as a predictive tool to estimate oral absorption just for compounds with good intestinal absorption. While for compounds with low or medium absorption, the Caco-2 cell lines may not give a very good rank for estimating oral absorption.12,20 So even if the Caco-2 permeability predictive models have developed a lot, a high-quality and practical HIA predictive model is still in demand. To achieve this goal, major efforts have been made to build a satisfactory HIA predictive model. First of all, Lipinski proposed a “rule of five” based on several critical properties to identify compounds with possible poor absorption and permeability.23 After that, not only regression models, but also classification models have been developed a lot. For regression models, the dataset is always growing and its size ranges from 67 to 619.24,25 These models are constructed based on different types of descriptors, such as physicochemical descriptors,12,26–29 topological descriptors and so on.30–33 With regards to computational techniques, the most common mathematical methods include artificial neural networks27,28,33 and multiple linear regression.30 Furthermore, support vector machine (SVM),29,34 multivariate adaptive regression splines24 and stepwise regression25 have also been applied. Considering the fact that the distribution between a high-absorbed and poor-absorbed compound is bias, a number of classification models were established in recent years. For these classification models, the number of compounds ranges from 202 to 685. Other than these descriptors used in regression models,1,13,35,36 charge-related descriptors such as hydrogen bonding capacity and charged partial surface were proposed to improve the predictive ability for HIA models.37 In regard to statistical methods, linear discriminant analysis,1,30 SVM35,36 and Bayesian37 are the most commonly used ones in all the published classification models studies. In 2012, Taravat explained the impact of data distributions for modelling of passive intestinal absorption and attempted to address this problem of unbalanced data distribution by creating a training set through under-sampling the highly absorbed compounds. Their classification model has an accuracy of 0.798 for the training set and an accuracy of 0.958 for the test set.25 Based on the same dataset, Hou et al. and Shen et al. also built HIA classification models in 2007 and 2010 respectively (ACC = 0.97/0.98 for training set; ACC = 0.98/0.99 for test set).12,36 In addition, there have also been several QSAR models with similar statistical results for HIA classification in the last few years.13,37
Although researchers have obtained some acceptable HIA predictive models based on size-different datasets, there are still some issues to be explored. First is the bias distribution of available HIA data. For the existing models, the predictive ability for poor-absorbed compounds is usually worse than that for high-absorbed compounds due to the lack of negative molecules. Second are the descriptors used in the modeling process. Various types of descriptors have been applied to construct models as previously mentioned; we wonder which one or ones would perform better for HIA prediction. Third are the application domain and reliability of the present models. We have noticed that existing models have distinctly different predictive accuracies and those models based on Hou's dataset performed much better than models before and after them. Therefore, it is necessary to evaluate HIA classification models based on Hou's dataset through rebuilding models.
To address the above-mentioned issues, we collected a relatively large HIA dataset from the existing published literature and 9 types of descriptors were calculated for them primarily. In addition, a parameter named sample size in random forest (RF) was proposed to balance the dataset in the modeling process. Overall, there are mainly two purposes of this paper: (1) to evaluate previous HIA classification models by rebuilding HIA models and external test set and (2) to obtain a more applicable model with an optimized RF method based on a combination of different descriptors.
Materials and methods
1. Data collection
The dataset in the present study consists of 970 drug and drug-like compounds. Among them, there were 856 real drugs and the other 114 “drug-like” compounds were filtered by the “Lipinski rule of five”. We collected their HIA values from 13 HIA studies.1,3,13,20,24,26,27,32,37–41 To improve the quality and reliability of the dataset, we dealt with it as follows. When there were two or more entries for one molecule, if these values did not differ a lot (<20%), the arithmetic mean value was adopted to reduce the random error; otherwise, the corresponding HIA value will be reconfirmed again. Solvent or saline ions adhering to the molecules were removed automatically by Molecular Operating Environment software (MOE, version 2014). At the end, the dataset consists of 955 numerical and 15 categorical fraction absorption values. And then, a HIA% value of 30% was used as the criterion to divide chemical agents into the good-absorption (HIA (+)) and the poor-absorption (HIA (−)) classes.35 For the present dataset, the HIA (+) set and HIA (−) set have 818 and 152 compounds respectively.To verify the reliability and predictive ability of models, all compounds were divided into a training set (776 compounds) and a test set (194 compounds) using MOE according to their chemical structure.42 To further validate the generalization ability of our model, we applied Shen's dataset (634 oral drugs) and Hou's dataset (1013 bioavailability values) as the external validation datasets.36,43 After eliminating the duplicates, 267 oral drugs and 175 bioavailability values were obtained. Considering the fact that the value of HIA is always greater than oral bioavailability, the compounds that have bioavailability values exceeding 30% and those drugs with oral dosage formulations were considered to be included in HIA + external test set. These compounds and corresponding HIA labels can be found in the ESI.†
2. Molecular descriptor calculation
In the present study, 9 types of molecular descriptors were calculated, namely two-dimensional (2D) descriptors, three-dimensional (3D) descriptors, molecular fingerprints (ECFP2; ECFP4; ECFP6; FP2) and structural fragments (FP4; Estate; MACCS). Firstly, all the compounds were corrected using the “wash” function in MOE. And then, compounds were optimized to obtain 3D structures by MMFF94x (Merck Molecular Force Field 94X, eps = r, cutoff [8, 10]) and the gradient-threshold of potential energy was set to 0.001 kcal−1 mol−1.44 After that, 192 2D descriptors and 117 3D descriptors were computed by MOE. Moreover, ChemDes was applied to calculate these fingerprints and structural fragments.45 Thus, 1024 ECFP2/ECFP4/ECFP6/FP2 fingerprints; 307 FP4 fragments; 79 Estate fragments; and 167 MACCS fragments were prepared. Besides the above-mentioned descriptors, we recommended another two descriptors that seem to make sense in previous studies.35,36 One is the number of violations of the four rule-of-five rules (Nrule-of-five) developed by Lipinski,46 which was calculated by ChemoPy.47,48 The other is N+, which was used to represent the existence of a positively charged N atom. If N+ was found in the molecule, the label was defined to be 1; otherwise, the label was defined to be 0.3. Modified random forest and feature selection
Recent studies have suggested that RF offers several striking features which make it very attractive for QSAR/QSPR studies.49 These include relatively high accuracy of prediction, built-in descriptor selection, and a method for assessing the importance of each descriptor to the model. RF was applied to build HIA classification models in this study and it was used in the statistical computing environment R.50–53 Optimization of the training parameters was performed using R scripts which iteratively changed each parameter one-by-one and regenerated the classification model. The ranges for three main parameters to select the optimum values were as follows: ntree was set to 500; nodesize was set to 1; and mtry was in the range (2, 12) for the 2D and 3D descriptors and for the fingerprint descriptors it was in the range of (6, 58).Due to the unbalanced dataset, the obtained models may be biased if general modeling processes were applied.54,55 To obtain some more balanced classification models, we applied a parameter named sample size to achieve this goal. Sample size was used to determine the number of HIA (+) compounds and HIA (−) compounds in the process of modeling. For example, when the sample size is set to (100, 200), it means that 100 poor-absorbed compounds and 200 high-absorbed compounds were randomly selected to build a tree in each modeling process and this process repeated many times to guarantee that every compound in the training set could be used in the final RF model. The use of sample size guarantees that the number of positive samples and negative samples is relatively balanced in each bootstrap sampling process.
For 2D and 3D descriptors, two pretreatments were performed to delete some uninformative descriptors before further feature selection: (1) delete the descriptors whose variance is 0 or approaches 0; (2) if the correlation coefficient between two descriptors is higher than 0.95, only one was reserved. After the preliminary descriptor pruning, further feature selection was as follows. Firstly, all descriptors were applied to build a classification model and these involved descriptors were sorted according to their importance. Then, the last two descriptors were removed and the rest were used to rebuild the model and a new descriptor order was obtained. This process was repeated until the last two remaining descriptors were used for modeling, and finally we get a series of models based on different numbers of descriptors. Among them, we can choose a best feature combination according to the number of descriptors and the error value of the model.
4. Performance evaluation
To ensure that the derived model has good generalization ability, five-fold cross validation and an individual test set were used for the validation purpose.6 The performances of the HIA classification models were evaluated using the following statistical parameters: true positive (TP); false negative (FN); true negative (TN); false positive (FP); sensitivity (SE); specificity (SP); accuracy (ACC); F value (F); area under receiver operating characteristic curve (AUC); Matthews correlation coefficient (MCC). Additionally, to deal with the possible bias from unbalanced data, we proposed SP × SE to optimize the RF model parameters and measure the whole predictive performance of the HIA classification models. These classification evaluation parameters are defined as follows: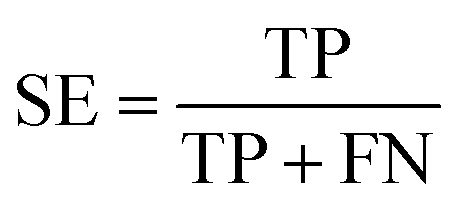
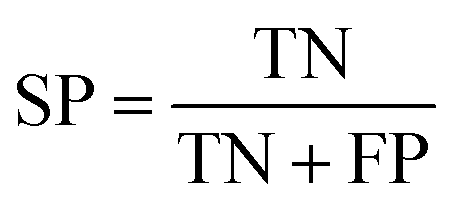
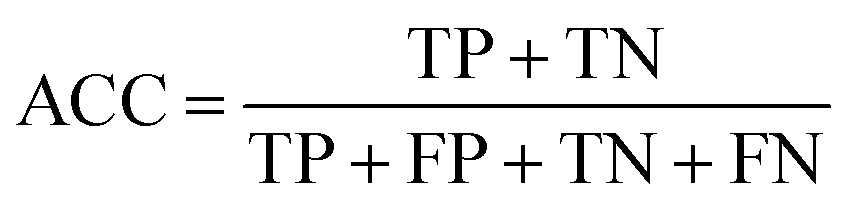

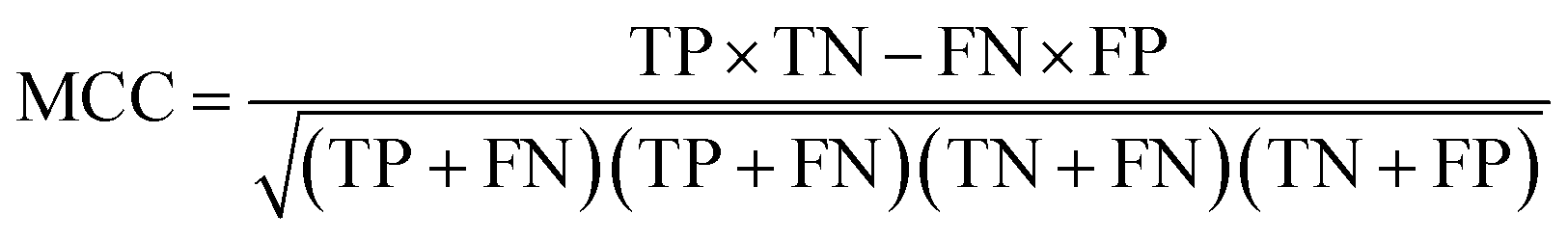
Results and discussion
1. Evaluation of models based on Hou's dataset
| Descriptor | Cross validation | Testing | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | |
| 2D(9) | 382 | 25 | 68 | 5 | 0.939 | 0.932 | 0.938 | 0.874 | 0.935 | 0.985 | 0.791 | 86 | 7 | 5 | 0 | 0.925 | 1.000 | 0.929 | 0.925 | 0.961 | 1.000 | 0.621 |
| 3D(10) | 379 | 28 | 59 | 14 | 0.931 | 0.808 | 0.913 | 0.753 | 0.865 | 0.943 | 0.689 | 88 | 5 | 5 | 0 | 0.946 | 1.000 | 0.949 | 0.946 | 0.972 | 1.000 | 0.688 |
| ECFP2 | 383 | 24 | 60 | 13 | 0.941 | 0.822 | 0.923 | 0.773 | 0.877 | 0.937 | 0.721 | 87 | 6 | 4 | 1 | 0.935 | 0.800 | 0.929 | 0.748 | 0.862 | 0.966 | 0.535 |
| ECFP4 | 386 | 21 | 54 | 19 | 0.948 | 0.740 | 0.917 | 0.702 | 0.831 | 0.926 | 0.681 | 90 | 3 | 3 | 2 | 0.968 | 0.600 | 0.949 | 0.581 | 0.741 | 0.953 | 0.521 |
| ECFP6 | 383 | 24 | 50 | 23 | 0.941 | 0.685 | 0.902 | 0.645 | 0.793 | 0.888 | 0.622 | 86 | 7 | 2 | 3 | 0.925 | 0.400 | 0.898 | 0.370 | 0.558 | 0.914 | 0.247 |
| Estate | 394 | 13 | 60 | 13 | 0.968 | 0.822 | 0.946 | 0.796 | 0.889 | 0.962 | 0.790 | 91 | 2 | 5 | 0 | 0.978 | 1.000 | 0.980 | 0.978 | 0.989 | 0.994 | 0.836 |
| FP2 | 377 | 30 | 51 | 22 | 0.926 | 0.699 | 0.892 | 0.647 | 0.797 | 0.862 | 0.599 | 83 | 10 | 5 | 0 | 0.892 | 1.000 | 0.898 | 0.892 | 0.943 | 0.969 | 0.545 |
| FP4 | 392 | 15 | 55 | 18 | 0.963 | 0.753 | 0.931 | 0.726 | 0.845 | 0.930 | 0.729 | 90 | 3 | 5 | 0 | 0.968 | 1.000 | 0.969 | 0.968 | 0.984 | 0.994 | 0.778 |
| MACCS | 392 | 15 | 62 | 11 | 0.963 | 0.849 | 0.946 | 0.818 | 0.903 | 0.960 | 0.795 | 90 | 3 | 5 | 0 | 0.968 | 1.000 | 0.969 | 0.968 | 0.984 | 0.996 | 0.778 |
 | ||
| Fig. 1 Distribution diagrams of accuracy, sensitivity and specificity for 1000 test sets (the left-hand three are based on 578 compounds; the right-hand three are based on 970 compounds). | ||
There was a strange fact that the predictive ability for the poor-absorbed compounds was better than that for good-absorbed compounds in the test set. This was not consistent with our common sense. Several studies have reported the difficulty of predicting poor-absorbed compounds due to the unbalanced issue of HIA datasets.13,25,37 In these studies, the specificity values of their classification models were near 0.60–0.70 and always smaller than their sensitivity values. After examining the original dataset, we found that there were 73 poor-absorbed compounds in the 480 training molecules but only 5 poor-absorbed compounds in the 98 test molecules. Hence, we considered that the number of poor-absorbed compounds in the test set was too little to reflect the actual predictive ability of the HIA classification model, especially for the specificity.
| Descriptor | Cross validation | Testing | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | |
| a Notes: the training set of 578 compounds is totally from Hou's dataset, and the test set of 392 compounds is collected from other literature. | ||||||||||||||||||||||
| 2D(15) | 471 | 29 | 72 | 6 | 0.942 | 0.923 | 0.939 | 0.870 | 0.932 | 0.985 | 0.778 | 253 | 61 | 47 | 31 | 0.806 | 0.603 | 0.765 | 0.486 | 0.689 | 0.796 | 0.365 |
| 3D(9) | 472 | 28 | 60 | 18 | 0.944 | 0.769 | 0.920 | 0.726 | 0.848 | 0.946 | 0.678 | 252 | 62 | 43 | 35 | 0.803 | 0.551 | 0.753 | 0.442 | 0.654 | 0.751 | 0.319 |
| ECFP2 | 471 | 29 | 61 | 17 | 0.942 | 0.782 | 0.920 | 0.737 | 0.855 | 0.946 | 0.682 | 256 | 58 | 37 | 41 | 0.815 | 0.474 | 0.747 | 0.387 | 0.600 | 0.752 | 0.270 |
| ECFP4 | 469 | 31 | 55 | 23 | 0.938 | 0.705 | 0.907 | 0.661 | 0.805 | 0.920 | 0.617 | 259 | 55 | 39 | 39 | 0.825 | 0.500 | 0.760 | 0.412 | 0.623 | 0.743 | 0.304 |
| ECFP6 | 469 | 31 | 51 | 27 | 0.938 | 0.654 | 0.900 | 0.613 | 0.771 | 0.895 | 0.580 | 253 | 61 | 43 | 35 | 0.806 | 0.551 | 0.755 | 0.444 | 0.655 | 0.756 | 0.323 |
| Estate | 485 | 15 | 67 | 11 | 0.970 | 0.859 | 0.955 | 0.833 | 0.911 | 0.959 | 0.812 | 259 | 55 | 43 | 35 | 0.825 | 0.551 | 0.770 | 0.455 | 0.661 | 0.790 | 0.347 |
| FP2 | 463 | 37 | 48 | 30 | 0.926 | 0.615 | 0.884 | 0.570 | 0.739 | 0.866 | 0.522 | 261 | 53 | 36 | 42 | 0.831 | 0.462 | 0.758 | 0.384 | 0.594 | 0.720 | 0.279 |
| FP4 | 483 | 17 | 60 | 18 | 0.966 | 0.769 | 0.939 | 0.743 | 0.856 | 0.930 | 0.739 | 274 | 40 | 36 | 42 | 0.873 | 0.462 | 0.791 | 0.403 | 0.604 | 0.729 | 0.337 |
| MACCS | 480 | 20 | 66 | 12 | 0.960 | 0.846 | 0.945 | 0.812 | 0.899 | 0.963 | 0.774 | 262 | 52 | 49 | 29 | 0.834 | 0.628 | 0.793 | 0.524 | 0.717 | 0.775 | 0.422 |
2. HIA classification models built with new training set (776 compounds)
| Descriptor | Cross validation | Testing | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | |
| 2D(18) | 582 | 77 | 96 | 21 | 0.883 | 0.821 | 0.874 | 0.725 | 0.851 | 0.899 | 0.605 | 143 | 19 | 23 | 9 | 0.883 | 0.719 | 0.856 | 0.634 | 0.792 | 0.881 | 0.542 |
| 3D(23) | 583 | 76 | 83 | 34 | 0.885 | 0.709 | 0.858 | 0.628 | 0.787 | 0.846 | 0.527 | 128 | 24 | 21 | 11 | 0.852 | 0.656 | 0.820 | 0.559 | 0.741 | 0.824 | 0.447 |
| ECFP2 | 553 | 106 | 86 | 31 | 0.839 | 0.735 | 0.823 | 0.617 | 0.784 | 0.858 | 0.476 | 136 | 26 | 23 | 9 | 0.840 | 0.719 | 0.820 | 0.603 | 0.774 | 0.870 | 0.477 |
| ECFP4 | 580 | 79 | 80 | 37 | 0.880 | 0.684 | 0.851 | 0.602 | 0.770 | 0.861 | 0.500 | 142 | 20 | 20 | 12 | 0.877 | 0.625 | 0.835 | 0.548 | 0.730 | 0.854 | 0.460 |
| ECFP6 | 583 | 76 | 74 | 43 | 0.885 | 0.632 | 0.847 | 0.560 | 0.738 | 0.843 | 0.469 | 135 | 27 | 20 | 12 | 0.833 | 0.625 | 0.799 | 0.521 | 0.714 | 0.843 | 0.397 |
| Estate | 597 | 62 | 89 | 28 | 0.906 | 0.761 | 0.884 | 0.689 | 0.827 | 0.603 | 0.901 | 137 | 25 | 25 | 7 | 0.846 | 0.781 | 0.835 | 0.661 | 0.812 | 0.858 | 0.532 |
| FP2 | 571 | 88 | 81 | 36 | 0.866 | 0.692 | 0.840 | 0.600 | 0.770 | 0.834 | 0.484 | 131 | 31 | 19 | 13 | 0.809 | 0.594 | 0.773 | 0.480 | 0.685 | 0.750 | 0.341 |
| FP4 | 584 | 75 | 81 | 36 | 0.886 | 0.692 | 0.857 | 0.614 | 0.777 | 0.852 | 0.517 | 144 | 18 | 21 | 11 | 0.889 | 0.656 | 0.851 | 0.583 | 0.755 | 0.866 | 0.505 |
| MACCS | 583 | 76 | 93 | 24 | 0.885 | 0.795 | 0.871 | 0.703 | 0.837 | 0.876 | 0.589 | 137 | 25 | 25 | 7 | 0.846 | 0.781 | 0.835 | 0.661 | 0.812 | 0.893 | 0.532 |
To further ensure the reasonability of our model, 1000 randomized predictive models have also been obtained based on 970 compounds and the distribution diagrams of accuracy, sensitivity and specificity for the validation set are also shown in Fig. 1. In this process, sample size was set to (90, 100) uniformly. From the figure, we can see that the accuracy values of these randomly shuffled models were mainly located in the range of 0.883 ± 0.02. Subsequently, their sensitivity values were mainly in the range of 0.902 ± 0.02 and the specificity values were gathered in the range of 0.778 ± 0.072. Compared with the statistical results from our predictive model and those derived from Hou's dataset, it is apparent that our primary models were more rational than other models from the perspective of subdivision for the training set and test set. These results also indicated the importance of the creation of training set and test set for a modeling process.
| Descriptor | Cross validation | Testing | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | TP | FN | TN | FP | SE | SP | ACC | SP × SE | F | AUC | MCC | |
| 2D + 3D | 589 | 70 | 98 | 19 | 0.894 | 0.838 | 0.885 | 0.749 | 0.865 | 0.915 | 0.635 | 143 | 19 | 24 | 8 | 0.883 | 0.750 | 0.861 | 0.662 | 0.811 | 0.862 | 0.565 |
| 2; 3 + N | 587 | 72 | 98 | 19 | 0.891 | 0.838 | 0883 | 0.746 | 0.863 | 0.919 | 0.630 | 140 | 22 | 26 | 6 | 0.864 | 0.813 | 0.856 | 0.702 | 0.838 | 0.858 | 0.582 |
| 2; 3 + NR | 588 | 71 | 99 | 18 | 0.892 | 0.846 | 0.885 | 0.755 | 0.869 | 0.920 | 0.639 | 142 | 20 | 26 | 6 | 0.877 | 0.813 | 0.866 | 0.702 | 0.843 | 0.861 | 0.601 |
| 2; 3 + E | 593 | 66 | 95 | 22 | 0.900 | 0.812 | 0.887 | 0.731 | 0.854 | 0.917 | 0.628 | 140 | 22 | 25 | 7 | 0.864 | 0.781 | 0.851 | 0.675 | 0.821 | 0.887 | 0.559 |
| 2; 3 + E + NR | 590 | 69 | 96 | 21 | 0.895 | 0.821 | 0.884 | 0.735 | 0.856 | 0.924 | 0.626 | 141 | 21 | 25 | 7 | 0.870 | 0.781 | 0.856 | 0.680 | 0.823 | 0.887 | 0.569 |
| 2; 3 + M | 595 | 64 | 93 | 24 | 0.903 | 0.795 | 0.887 | 0.718 | 0.845 | 0.909 | 0.622 | 142 | 20 | 23 | 9 | 0.877 | 0.719 | 0.851 | 0.630 | 0.790 | 0.890 | 0.532 |
| 2; 3 + M + NR | 598 | 61 | 94 | 23 | 0.907 | 0.803 | 0.892 | 0.729 | 0.852 | 0.910 | 0.636 | 143 | 19 | 22 | 10 | 0.883 | 0.688 | 0.851 | 0.607 | 0.773 | 0.891 | 0.518 |
| 2; 3 + E + M | 593 | 66 | 94 | 23 | 0.900 | 0.803 | 0.885 | 0.723 | 0.849 | 0.908 | 0.622 | 139 | 23 | 25 | 7 | 0.858 | 0.781 | 0.845 | 0.670 | 0.818 | 0.899 | 0.550 |
| 2; 3 + EMNR | 592 | 67 | 95 | 22 | 0.898 | 0.812 | 0.885 | 0.729 | 0.853 | 0.917 | 0.625 | 140 | 22 | 25 | 7 | 0.864 | 0.781 | 0.851 | 0.675 | 0.821 | 0.880 | 0.559 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||||||||||||||||
| No sample | ||||||||||||||||||||||
| 2; 3 + NR | 639 | 20 | 70 | 47 | 0.970 | 0.598 | 0.914 | 0.580 | 0.740 | 0.925 | 0.635 | c | 5 | 15 | 17 | 0.969 | 0.469 | 0.887 | 0.454 | 0.632 | 0.851 | 0.534 |
| 2; 3 + EMNR | 642 | 17 | 73 | 44 | 0.974 | 0.624 | 0.921 | 0.608 | 0.761 | 0.928 | 0.668 | 158 | 4 | 19 | 13 | 0.975 | 0.594 | 0.912 | 0.579 | 0.738 | 0.890 | 0.653 |
From the table, some interesting phenomena were drawn out. Firstly, compared with the model based on only 2D descriptors, the model derived from a combination of 2D and 3D descriptors has better performance for both training and test set, especially for specificity (SP = 0.838/0.750). This may imply that the 3D descriptors could give some ESI† for HIA prediction. Secondly, after adding N+, the predictive performance for the test set improved to some extent (SP: 0.750 to 0.813). We speculated that this is in accordance with previous studies which have demonstrated that a compound with N+ has poor intestinal absorption generally. Similarly, the new model with Nrule-of-five also has a better predictive ability. Thirdly, for the other classification models based on combinations of 2D/3D and structural fragments, the specificity values were smaller for both training set and test set after adding structural fragments. This result was probably because the predictive model from the individual type of structural fragment generally has a relatively poor specificity value. Fourthly, the statistical results for models based on 2/3D + E and 2/3D + E + M + N + R were the same, which may imply that the Estate fragments cover the useful information contained in MACCS fragments, N+ and Nrule-of-five. In short, the final HIA classification model (highlighted in bold) derived from seven 2D descriptors, two 3D descriptors, N+ and Nrule-of-five was the best one after a series of attempts for descriptor combination. In addition, we also rebuilt models based on the same descriptors with 2; 3 + NR and 2; 3 + EMNR models without an optimized sample size in RF; their results are also shown in Table 4. The two models have excellent abilities to predict positive compounds but they are very short of predictive abilities for poor-absorbed compounds. Compared with these models using sample size, the importance of a balanced classification model was self-evident.
To define the structural domain the model does not cover, we rebuilt classification models based on the total of 970 compounds and five-fold cross validation was used to evaluate the quality of a model. This process was repeated 1000 times. From these predictive results, we found some anomalous compounds that always are wrongly categorized by all predictive models. Among them, there were eight representative molecules and their structures are shown in Fig. 2. For the eight compounds, cyclosporine has the largest molecular weight of 1202 and is composed of 11 amino acids. Cyclosporine is a kind of immune inhibitor widely used to prevent organ transplant rejection.56 We speculated that the incorrect prediction may due to its particularly large molecular weight and its macrocycle structure. Both azithromycin and viomycin are macrolide antibiotics and they can inhibit protein synthesis through blocking peptide and displacement of mRNA.57 This class of antibiotics has a big lactonic ring commonly and it may make HIA prediction much harder. Fosinopril, a new angiotensin converting enzyme inhibitor, is widely used to relieve light, medium and severe hypertension and various types of heart failure.58 From its structure, we found that there are an L-proline and a phosphoryl which are unusual in the whole dataset. As to the other four compounds, although there is nothing abnormal from their 2D structures, their partial charges and potential energies or other properties may not be in the application domain of the predictive model. After removing these compounds from the initial dataset, we obtained a new classification model: TP = 583, FN = 76, TN = 100, FP = 17, SP = 0.855, SE = 0.885, ACC = 0.880, AUC = 0.921.
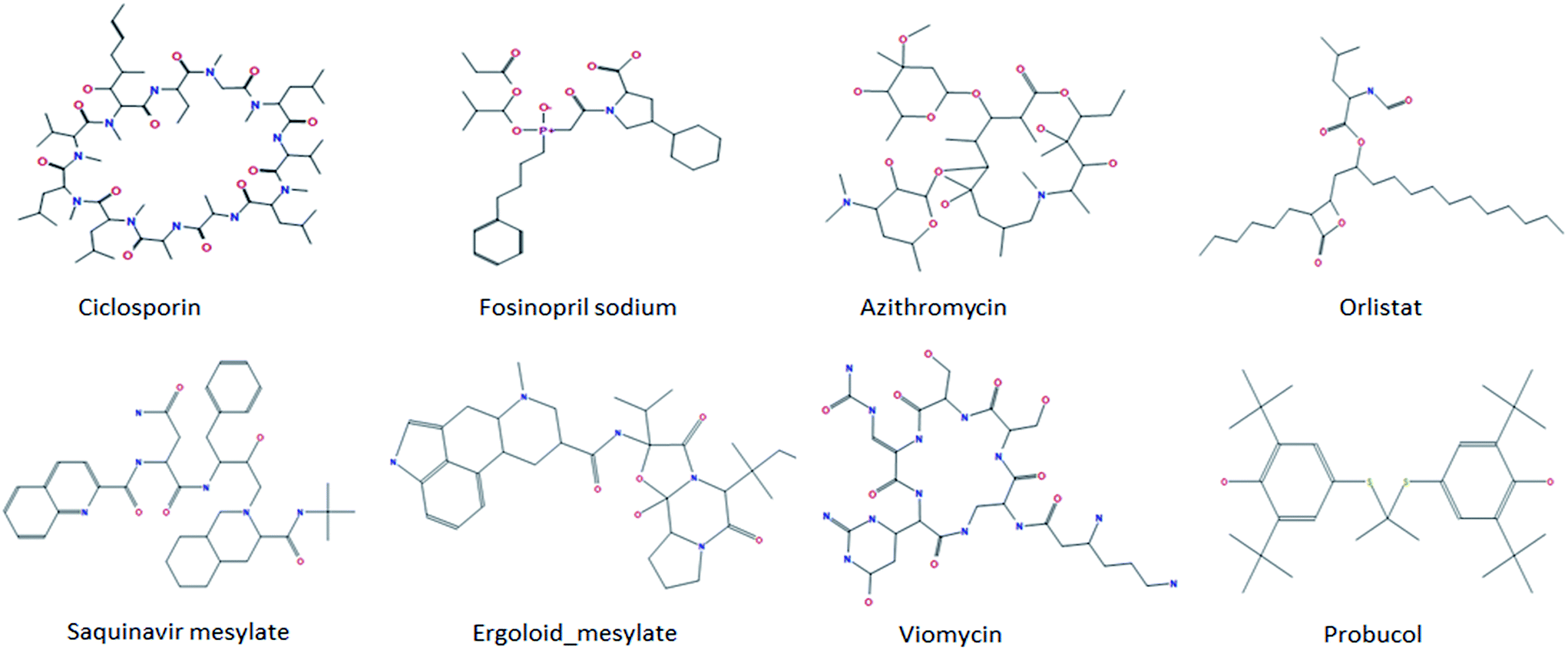 | ||
| Fig. 2 Structures of 8 compounds that always are wrongly categorized. | ||
| Data | TP | FN | SE | ACC |
|---|---|---|---|---|
| Oral drug | 222 | 45 | 0.831 | 0.831 |
| F | 146 | 29 | 0.834 | 0.834 |
| Index | Code | Description |
|---|---|---|
| 1 | a_donacc | Number of hydrogen bond donor and acceptor atoms |
| 2 | a_base | Number of basic atoms |
| 3 | KierFlex | Molecular flexibility index |
| 4 | PEOE_VSA_PPOS | Total positive polar van der Waals surface area |
| 5 | PEOE_VSA_POL | Total polar van der Waals surface area |
| 6 | PEOE_VSA_FHYD | Fractional hydrophobic van der Waals surface area |
| 7 | PEOE_VSA_FPPOS | Fractional positive polar van der Waals surface area |
| 8 | DCASA | Absolute value of the difference between CASA+ and CASA− |
| 9 | E_Sol | Solvation energy |
| 10 | N+ | Existence of positively charged N atom |
| 11 | Nrule-of-five | Number of violations of rule-of-five rules |
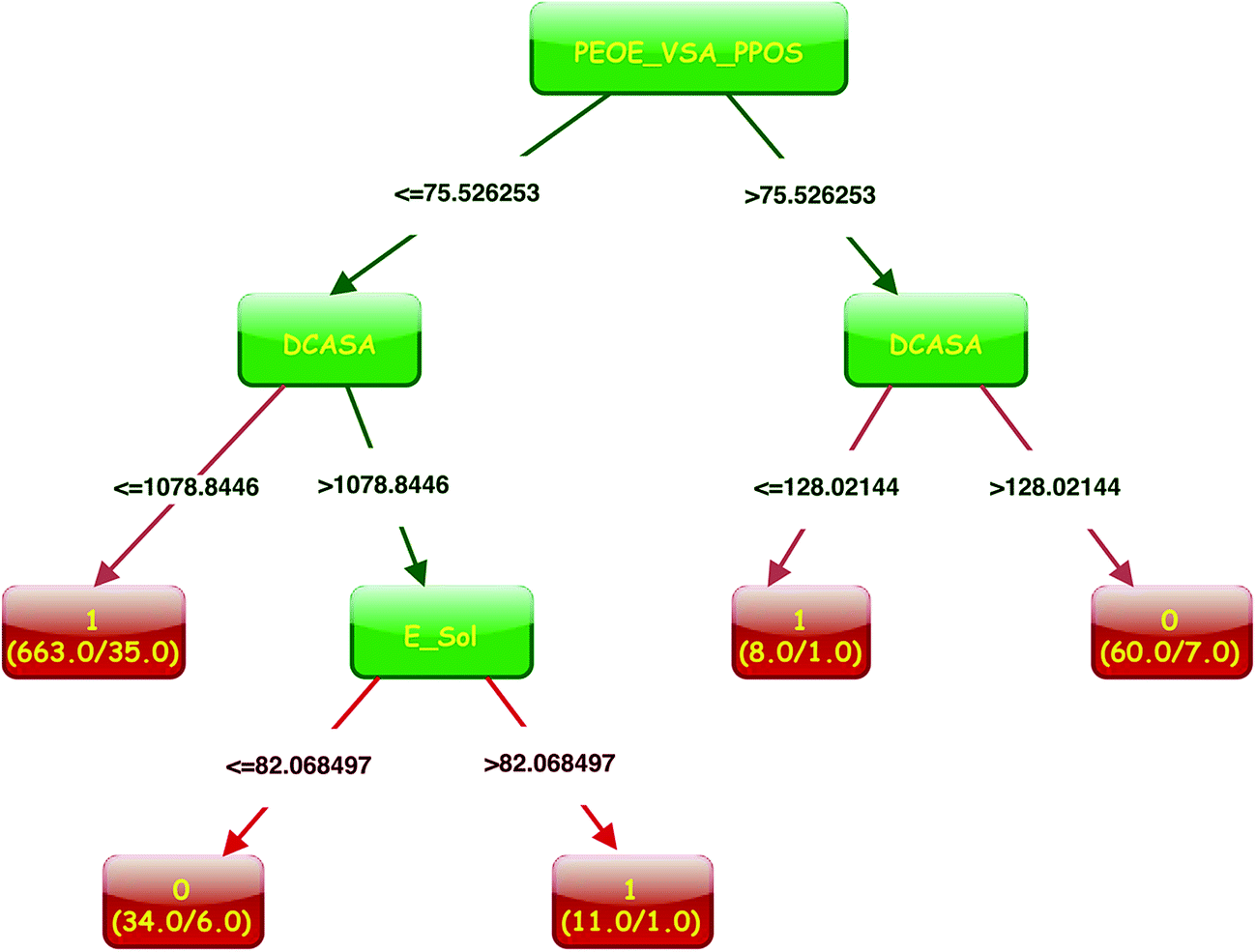 | ||
| Fig. 3 HIA classification model based on same training set using CART. | ||
Table 7 lists the first five MACCS and Estate fragments and their corresponding description information. From the table, the first three fragments of MACCS and Estate, which represent the existence of ammonium and oxygen, are the same, indicating that these structural fragments are of high importance for HIA classification. For MACCS 31 and Estate 33, they represent the existence of a positively charged N atom, only 19/119 poor-absorbed compounds have this structural fragment in the training set, all high-absorbed compounds do not have this fragment. For the test set, only 4 compounds have this fragment. Accompanying the above-mentioned descriptors, we speculated that these fragments play a very important role in predicting poor-absorbed drug compounds. In addition to the two fragments, the distributions of the rest were biased to highly absorbed compounds. This is in accordance with the fact that the models derived from MACCS and Estate structural fragments have slightly worse predictive ability for highly absorbed compounds than those derived from other descriptors or fingerprints.
| Index | Structure | Description |
|---|---|---|
| MACCS 140 | –O– or O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) |
Number of oxygens > 3, key = 1; otherwise, key = 0 |
| MACCS 31 | 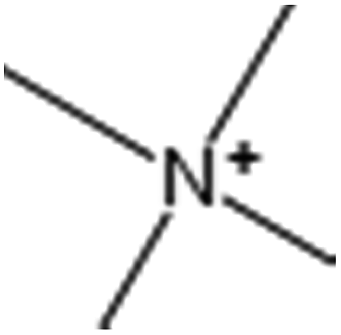 |
Non-c Q4 bonded to ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) 3C 3C |
| MACCS 53 | 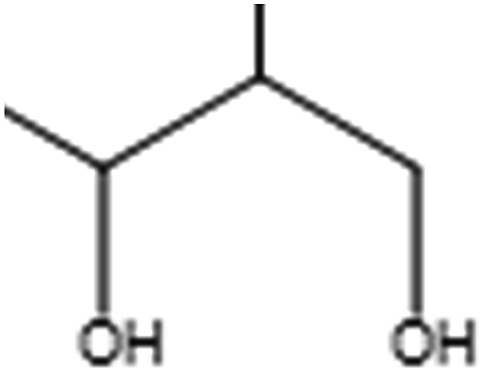 |
QH, 4 bonds away from another QH |
| MACCS 139 | –OH | OH groups |
| MACCS 91 | 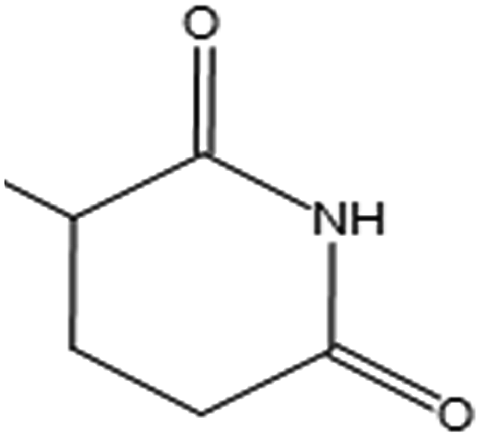 |
OH or NH, 4 bonds away from CH2 |
| Estate 33(SssssNp) | 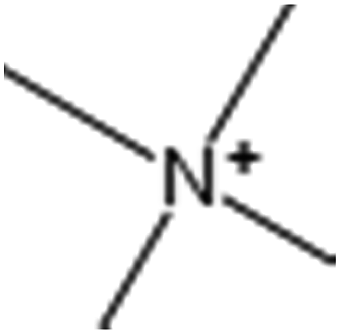 |
Sum of (N+![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ) E-states ) E-states |
| Estate 34(SsOH) | –OH | Sum of (–OH) E-states |
| Estate 35(SdO) | O |
Sum of (O) E-states |
| Estate 13(SsssCH) | CH– |
Sum of (CH–) E-states |
| Estate 16(SdssC) | C |
Sum of (C) E-states |
| Index | Author | Dataset | Descriptor | Method | Result |
|---|---|---|---|---|---|
| a LDA: linear discriminant analysis.b SVM: support vector machine.c GBT: gradient boosted tree. | |||||
| 1 | Miguel Angel Cabrera Pérez | Tr: 82, Te: 127, Ex: 109(F) | Sub-structural descriptors | LDAa | Tr:ACC = 0.89/0.89, Te:ACC = 0.93/0.80, Ex:ACC = 0.94/0.92 |
| 2 | Tingjun Hou | Tr: 481, Te: 98 | Physicochemical descriptors | Recursive partitioning | Tr:ACC = 0.96/0.95, Te:ACC = 0.97/1.00 |
| 3 | Tingjun Hou | Tr: 480, Te: 98 | Physicochemical descriptors | SVMb | Tr:SE/P = 0.98/0.95, Te:SE/P = 0.98/1.00 |
| 4 | Claudia Suenderhauf | Tr: 458 | Charge; constitutional; topological | Bayesian | SP = 0.685, SE = 0.941, MCC = 0.643 |
| 5 | Claudia Suenderhauf | Tr: 458 | Charge; constitutional; topological | Multilayer perceptron | SP = 0.619, SE = 0.840, MCC = 0.461 |
| 6 | Jie Shen et al. | Tr: 480, Te: 98, Ex: 634(+) | FP4, MACCS | SVM | Tr:ACC = 0.985/0.99, Te:ACC = 1.00/0.98, Ex:ACC = 0.938/0.94 |
| 7 | A. Guerra | Tr: 37, Te: 165 | CODES, 2D descriptors | Neural network | Tr:ACC = 0.79, Te:ACC = 0.75 |
| 8 | Nikita Basant | Tr: 403, Te: 87, Ex: 87 | Constitutional, topological descriptors | GBTc | Tr:ACC = 0.9975, Te:ACC = 0.9885, Ex:ACC = 0.977 |
| 9 | Taravat Ghafourian | Tr: 94, Te: 502, Ex: 89 | 215 descriptors | Discriminant analysis | Tr:ACC = 0.872, Te:ACC = 0.876, Ex:ACC = 0.798 |
Conclusion
In the present study, we collected a relatively large HIA dataset consisting of 970 compounds to evaluate the reliability of HIA classification models based on Hou's dataset and then to obtain a practical classification model. In this process, 9 different types of descriptors and the modified RF model with a parameter named sample size were applied. The obtained results indicated that some previous studies may lead to the over-satisfactory performance for HIA prediction, especially for the poorly absorbed compounds. From these models using 9 types of descriptors, we found that the predictive models based on 2D, 3D, Estate and MACCS descriptors perform better than the others. In addition, sample size is a useful parameter for getting a balanced model based on a biased dataset. Finally, we obtained a classification model derived from seven 2D descriptors, two 3D descriptors, N+ and Nrule-of-five. Furthermore, the test set and external dataset validated the robustness and reliability of our proposed model. Compared with other published models, our model has some advantages in dataset size, model accuracy and model practicability to some extent. All in all, this study has evaluated the existing HIA models and generated a robust and practical model for HIA classification prediction. It could be a convenient tool for virtual screening in the early stage of drug development.Declarations
Availability of data and materials
All the compounds and their basic structural information and their HIA classification are listed in the ESI.† It includes three training sets and three test sets: 480 + 98; 578 + 392; 776 + 194. In addition, two external validation datasets and the detailed information of scaffolds and carbon skeletons can also be found in ESI† as the external validation datasets.Conflict of interest
The authors declare that they have no competing interests.Acknowledgements
This work is financially supported by the National Key Basic Research Program (2015CB910700), the National Natural Science Foundation of China (grant no. 81402853), the Central South University Innovation Foundation for Postgraduate (2016zzts498), the Hunan Provincial Innovation Foundation for Postgraduate (CX2016B058), the Project of Innovation-driven Plan in Central South University, and the Postdoctoral Science Foundation of Central South University, the Chinese Postdoctoral Science Foundation (2014T70794, 2014M562142). The studies meet with the approval of the university's review board.References
- M. A. C. Pérez, M. B. Sanz, L. R. Torres, R. G. Ávalos, M. P. González and H. G. Díaz, Eur. J. Med. Chem., 2004, 39, 905–916 CrossRef PubMed.
- B. Booth and R. Zemmel, Nat. Rev. Drug Discovery, 2004, 3, 451–456 CrossRef CAS PubMed.
- S. B. Gunturi and R. Narayanan, QSAR Comb. Sci., 2007, 26, 653–668 CAS.
- T. Hou, J. Wang, W. Zhang, W. Wang and X. Xu, Curr. Med. Chem., 2006, 13, 2653–2667 CrossRef CAS PubMed.
- J.-B. Wang, D.-S. Cao, M.-F. Zhu, Y.-H. Yun, N. Xiao and Y.-Z. Liang, J. Chemom., 2015, 29, 389–398 CrossRef CAS.
- N.-N. Wang, J. Dong, Y.-H. Deng, M.-F. Zhu, M. Wen, Z.-J. Yao, A.-P. Lu, J.-B. Wang and D.-S. Cao, J. Chem. Inf. Model., 2016, 56, 763–773 CrossRef CAS PubMed.
- J. Dong, Z.-J. Yao, M. Wen, M.-F. Zhu, N.-N. Wang, H.-Y. Miao, A.-P. Lu, W.-B. Zeng and D.-S. Cao, J. Cheminf., 2016, 8, 34 Search PubMed.
- Z.-J. Yao, J. Dong, Y.-J. Che, M.-F. Zhu, M. Wen, N.-N. Wang, S. Wang, A.-P. Lu and D.-S. Cao, J. Comput.-Aided Mol. Des., 2016, 30, 413–424 CrossRef CAS PubMed.
- D.-S. Cao, J. Dong, N.-N. Wang, M. Wen, B.-C. Deng, W.-B. Zeng, Q.-S. Xu, Y.-Z. Liang, A.-P. Lu and A. F. Chen, Chemom. Intell. Lab. Syst., 2015, 146, 494–502 CrossRef CAS.
- T. Hou and X. Xu, Curr. Pharm. Des., 2004, 10, 1011–1033 CrossRef CAS PubMed.
- T. Kennedy, Drug discovery today, 1997, 2, 436–444 CrossRef.
- T. Hou, J. Wang, W. Zhang and X. Xu, J. Chem. Inf. Model., 2007, 47, 208–218 CrossRef CAS PubMed.
- A. Guerra, N. E. Campillo and J. Páez, Eur. J. Med. Chem, 2010, 45, 930–940 CrossRef CAS PubMed.
- C. Clemedson, A. Kolman and A. Forsby, ATLA, Altern. Lab. Anim., 2007, 35, 33–38 CAS.
- A. Avdeef, S. Bendels, L. Di, B. Faller, M. Kansy, K. Sugano and Y. Yamauchi, J. Pharm. Sci., 2007, 96, 2893–2909 CrossRef CAS PubMed.
- P. Artursson, K. Palm and K. Luthman, Adv. Drug Delivery Rev., 2001, 46, 27–43 CrossRef CAS PubMed.
- J. D. Irvine, L. Takahashi, K. Lockhart, J. Cheong, J. W. Tolan, H. Selick and J. R. Grove, J. Pharm. Sci., 1999, 88, 28–33 CrossRef CAS PubMed.
- H. Bohets, P. Annaert, G. Mannens, K. Anciaux, P. Verboven, W. Meuldermans and K. Lavrijsen, Curr. Top. Med. Chem., 2001, 1, 367–383 CrossRef CAS PubMed.
- P. Artursson and J. Karlsson, Biochem. Biophys. Res. Commun., 1991, 175, 880–885 CrossRef CAS PubMed.
- D. Newby, A. A. Freitas and T. Ghafourian, Eur. J. Med. Chem., 2015, 90, 751–765 CrossRef CAS PubMed.
- M.-C. Grès, B. Julian, M. Bourrié, V. Meunier, C. Roques, M. Berger, X. Boulenc, Y. Berger and G. Fabre, Pharm. Res., 1998, 15, 726–733 CrossRef.
- V. Pade and S. Stavchansky, J. Pharm. Sci., 1998, 87, 1604–1607 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- E. Deconinck, H. Ates, N. Callebaut, E. Van Gyseghem and Y. Vander Heyden, J. Chromatogr. A, 2007, 1138, 190–202 CrossRef CAS PubMed.
- T. Ghafourian, A. A. Freitas and D. Newby, Int. J. Pharm., 2012, 436, 711–720 CrossRef CAS PubMed.
- Y. H. Zhao, J. Le, M. H. Abraham, A. Hersey, P. J. Eddershaw, C. N. Luscombe, D. Boutina, G. Beck, B. Sherborne and I. Cooper, J. Pharm. Sci., 2001, 90, 749–784 CrossRef CAS PubMed.
- S. Agatonovic-Kustrin, R. Beresford and A. P. M. Yusof, J. Pharm. Biomed. Anal., 2001, 25, 227–237 CrossRef CAS PubMed.
- M. J. Polley, F. R. Burden and D. A. Winkler, Aust. J. Chem., 2006, 58, 859–863 CrossRef.
- A. Yan, Z. Wang and Z. Cai, Int. J. Mol. Sci., 2008, 9, 1961–1976 CrossRef CAS PubMed.
- N. Basant, S. Gupta and K. P. Singh, Comput. Biol. Chem., 2016, 61, 178–196 CrossRef CAS PubMed.
- R. Jones, P. C. Connolly, A. Klamt and M. Diedenhofen, J. Chem. Inf. Model., 2005, 45, 1337–1342 CrossRef CAS PubMed.
- G. Klopman, L. R. Stefan and R. D. Saiakhov, Eur. J. Pharm. Sci., 2002, 17, 253–263 CrossRef CAS PubMed.
- A. Talevi, M. Goodarzi, E. V. Ortiz, P. R. Duchowicz, C. L. Bellera, G. Pesce, E. A. Castro and L. E. Bruno-Blanch, Eur. J. Med. Chem., 2011, 46, 218–228 CrossRef CAS PubMed.
- M. Wen, B.-C. Deng, D.-S. Cao, Y.-H. Yun, R.-H. Yang, H.-M. Lu and Y.-Z. Liang, Analyst, 2016, 141, 5586–5597 RSC.
- T. Hou, J. Wang and Y. Li, J. Chem. Inf. Model., 2007, 47, 2408–2415 CrossRef CAS PubMed.
- J. Shen, F. Cheng, Y. Xu, W. Li and Y. Tang, J. Chem. Inf. Model., 2010, 50, 1034–1041 CrossRef CAS PubMed.
- C. Suenderhauf, F. Hammann, A. Maunz, C. Helma and J. R. Huwyler, Mol. Pharmaceutics, 2010, 8, 213–224 CrossRef PubMed.
- E. Deretey, M. Feher and J. M. Schmidt, Quant. Struct.-Act. Relat., 2002, 21, 493–506 CrossRef CAS.
- M. V. Varma, K. Sateesh and R. Panchagnula, Mol. Pharmaceutics, 2005, 2, 12–21 CrossRef CAS PubMed.
- M. De Vrieze, P. Janssens, R. Szucs, J. Van der Eycken and F. Lynen, Anal. Bioanal. Chem., 2015, 407, 7453–7466 CrossRef CAS PubMed.
- E. L. Cunha, C. F. Santos, F. S. Braga, J. S. Costa, R. C. Silva, H. A. Favacho, L. I. Hage-Melim, J. C. Carvalho, C. H. da Silva and C. B. Santos, J. Comput. Theor. Nanosci., 2015, 12, 3682–3691 CrossRef CAS.
- OECD-QSAR-07-ENV-JM-MONO, 2007, 2.
- S. Tian, Y. Li, J. Wang, J. Zhang and T. Hou, Mol. Pharmaceutics, 2011, 8, 841–851 CrossRef CAS PubMed.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- J. Dong, D.-S. Cao, H.-Y. Miao, S. Liu, B.-C. Deng, Y.-H. Yun, N.-N. Wang, A.-P. Lu, W.-B. Zeng and A. F. Chen, J. Cheminf., 2015, 7, 1 CrossRef.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- D.-S. Cao, Q.-S. Xu, Q.-N. Hu and Y.-Z. Liang, Bioinformatics, 2013, btt105 Search PubMed.
- D. S. Cao, Y. Z. Liang, J. Yan, G. S. Tan, Q. S. Xu and S. Liu, J. Chem. Inf. Model., 2013, 53, 3086–3096 Search PubMed.
- D. S. Cao, Y. N. Yang, J. C. Zhao, J. Yan, S. Liu, Q. N. Hu, Q. S. Xu and Y. Z. Liang, J. Chemom., 2012, 26, 7–15 CrossRef CAS.
- L. Breiman, Machine Learning, 2001, 45, 5–32 CrossRef.
- V. Svetnik, A. Liaw, C. Tong, J. C. Culberson, R. P. Sheridan and B. P. Feuston, J. Chem. Inf. Model., 2003, 43, 1947–1958 CrossRef CAS PubMed.
- L. Breiman, J. Friedman, C. J. Stone and R. A. Olshen, Classification and regression trees, CRC press, 1984 Search PubMed.
- A. Liaw and M. Wiener, Chem. Eng. News, 2002, 2, 18–22 Search PubMed.
- M. D. Wessel, P. C. Jurs, J. W. Tolan and S. M. Muskal, J. Chem. Inf. Model., 1998, 38, 726–735 CrossRef CAS PubMed.
- Y. H. Zhao, M. H. Abraham, J. Le, A. Hersey, C. N. Luscombe, G. Beck, B. Sherborne and I. Cooper, Pharm. Res., 2002, 19, 1446–1457 CrossRef CAS.
- J. Borel, in Ciclosporin, Karger Publishers, 1986, pp. 9–18 Search PubMed.
- S. Omura, Macrolide antibiotics: chemistry, biology, and practice, Academic press, 2002 Search PubMed.
- F. W. Asselbergs, G. F. Diercks, H. L. Hillege, A. J. van Boven, W. M. Janssen, A. A. Voors, D. de Zeeuw, P. E. de Jong, D. J. van Veldhuisen and W. H. van Gilst, Circulation, 2004, 110, 2809–2816 CrossRef CAS PubMed.
- G. W. B. And and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef PubMed.
- Y. J. Xu and M. Johnson, J. Chem. Inf. Model., 2002, 42, 912–926 CrossRef CAS PubMed.
- J. P. Bai, A. Utis, G. Crippen, H.-D. He, V. Fischer, R. Tullman, H.-Q. Yin, C.-P. Hsu, L. Jiang and K.-K. Hwang, J. Chem. Inf. Model., 2004, 44, 2061–2069 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28442f |
| ‡ The first two authors equally contribute to this paper. |
| This journal is © The Royal Society of Chemistry 2017 |