
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pharmacokinetics of rosmarinic acid in rats by LC-MS/MS: absolute bioavailability and dose proportionality
Jingxian Wang†
ab,
Guoyuan Li†ab,
Tianqi Ruiab,
An Kangab,
Guochun Lic,
Tingming Fuab,
Junsong Li *ab,
Liuqing Diab and
Baochang Caia
*ab,
Liuqing Diab and
Baochang Caia
aCollege of Pharmacy, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Nanjing 210023, China. E-mail: lijunsong1964@163.com; Fax: +86-25-85811517; Tel: +86-25-85811517
bJiangsu Provincial TCM Engineering Technology Research Center of High Efficient Drug Delivery System (DDS), Nanjing 210023, China
cThe Center of Statistical Research and Consultation of TCM, Nanjing University of Chinese Medicine, Nanjing 210023, China
First published on 30th January 2017
Abstract
Rosmarinic acid (RA), one of the main bioactive compounds of Rosmarinus officinalis L., exhibits diverse pharmacological effects. However, its oral absolute bioavailability and dose proportionality in vivo have not been comprehensively studied. In the present study, a validated LC-MS/MS method was developed for the determination of RA in rat plasma. Pharmacokinetic parameters were obtained following oral and intravenous dosing using DAS 3.0 software. Absolute bioavailability in rats was determined by comparing pharmacokinetic data after administration of single oral (12.5, 25 and 50 mg kg−1) and intravenous (0.625 mg kg−1) doses of RA. The dose proportionality of AUC and Cmax were analyzed using a power model. After single-dose oral administration of RA, Cmax values for groups with 12.5, 25 and 50 mg kg−1 doses ranged from 215.29 to 790.96 ng mL−1, with AUC0–t values from 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 789.84 to 96070.00 min ng mL−1, while Tmax and t1/2 values ranged from 8.33 to 18.33 and from 332.34 to 295.32 min, respectively. The power model showed RA lacked dose proportionality over the dose range from 12.5–50 mg kg−1. Oral absolute bioavailability was calculated to range from 0.91% to 1.69%. All the results demonstrated that the pharmacokinetic properties of RA in rats after oral administration were characterized as rapid absorption, middle-speed elimination and poor absolute bioavailability. Systemic exposure exhibited lack of dose proportionality over the dose range from 12.5 to 50 mg kg−1. These presented data could provide useful information for the rational clinical application and optimal dosage form design of RA.
789.84 to 96070.00 min ng mL−1, while Tmax and t1/2 values ranged from 8.33 to 18.33 and from 332.34 to 295.32 min, respectively. The power model showed RA lacked dose proportionality over the dose range from 12.5–50 mg kg−1. Oral absolute bioavailability was calculated to range from 0.91% to 1.69%. All the results demonstrated that the pharmacokinetic properties of RA in rats after oral administration were characterized as rapid absorption, middle-speed elimination and poor absolute bioavailability. Systemic exposure exhibited lack of dose proportionality over the dose range from 12.5 to 50 mg kg−1. These presented data could provide useful information for the rational clinical application and optimal dosage form design of RA.
Introduction
Rosemary, Rosmarinus officinalis L. (Labiatae), is used in folk medicine worldwide for initial health care among a variety of plants. It is an evergreen perennial shrub native to Asia Minor and southern Europe. Today it has been cultivated in many parts of the world.1,2 The powerful antioxidant activity of its constituents supports protection against damage induced by free radicals.3,4 Furthermore, it has been demonstrated that Rosmarinus officinalis produces an antinociceptive effect in experimental models of pain like acetic acid and formalin, but also in the pain-induced functional impairment model in rats.5 According to phytochemical reports, rosmarinic acid (RA) (Fig. 1) is the main effective, water-soluble polyphenolic acid from Rosemary, which was found to have various curative activities, such as anti-inflammatory,6,7 antioxidant,8 anticancer,9,10 antimicrobial11 and anti-diabetes12 properties. Recent studies also showed that it could improve cognitive performance.13 For safety and efficacy reasons, it is important to understand the real-time effective levels of phytochemicals in the systemic circulation and the time course concentrations following oral administration. Besides, research on bioavailability plays a very important role in the development of a drug and its clinical use. | ||
| Fig. 1 Typical chromatograms of a blank rat plasma sample (A), a blank rat plasma sample spiked with RA (448.8 ng mL−1) and I.S. (B), a rat plasma sample 10 min after intravenous administration of RA (C), a rat plasma sample 10 min after receiving oral doses of 50 mg kg−1 (D) and the chemical structures of RA and I.S. | ||
To date, several pharmacokinetic studies for RA have been reported in human or animals. Most of these studies on pharmacokinetics of RA were accomplished by oral administration of a single herb,14,15 compound prescription extract16,17 or mixture containing RA.18,19 On the other hand, the pharmacokinetics of RA after pure RA treatment only has been roughly or partially carried out.20,21 These pharmacokinetics studies have enabled us to understand the absorption characteristics of RA in vivo. But a systemic pharmacokinetic study of RA, especially with gradient dosages, after single RA treatment still remains to be done.
Phenolic acids were usually reported with low oral bioavailability.22,23 In Caco-2 cells' model, RA transport was mainly via paracellular diffusion with low intestinal absorption efficiency.24,25 Intestinal flora and intestinal epithelial cells usually transform polyphenols into low molecular weight metabolites, which decrease the intestinal absorption of polyphenols, such as catechins found in tea.26 In order to achieve rational clinical application and optimal dosage form design of drug, the study of absolute bioavailability is indispensable. Furthermore, dose proportionality of a drug is important for its safety and efficiency, because it could indicate whether dose–response relation is linear or not, which may affect effectiveness and toxicity. To the best of our knowledge, there is no report concerning bioavailability or dose proportionality of RA in vivo.
In the present paper, a validated LC-MS/MS method was developed for the determination of RA in rat plasma. Pharmacokinetic parameters were obtained following oral and intravenous dosing using DAS 3.0 software. Absolute bioavailability in rats was determined by comparing pharmacokinetic data after administration of single oral (12.5, 25 and 50 mg kg−1) and intravenous (0.625 mg kg−1) doses of RA. The dose proportionality of AUC and Cmax were analyzed by power model.
Materials and methods
Materials
Rosmarinic acid (raw material) was purchased from Nanjing Zelang Medical Technology Co., Ltd. (Nanjing, China) and had a purity of mass fraction of more than 98.0%. Rosmarinic acid and silibinin (purity > 99%), used as a reference standard and an internal standard (I.S.), were obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Acetonitrile and formic acid (HPLC grade) were purchased from Merck and Tedia Company Inc. (Merk, Darmstadt, Germany). Purified water was prepared using a Milli-Q purification system (Millipore, Billerica, MA, USA). Other chemical reagents of analytical grade or better were obtained from Sinopharm Chemical Reagent Co. (Nanjing, China).Animal treatment
Twenty-four Sprague Dawley male rats weighing 220–250 g were supplied by the Animal Center of Nanjing Medical University. Animals were housed in environmentally controlled conditions at 25 ± 2 °C and 50 ± 10% relative humidity under a 12 h dark–light cycle. The rats were kept with free access to food and water until 12 h prior to the experiments. All procedures and experiments of this study were approved by the Animal Care and Use Committee of Nanjing University of Chinese Medicine, and its approved protocol is “Scientific Protocol (2007) Number 16 of Nanjing University of Chinese Medicine”.LC-MS/MS conditions
The LC-MS/MS system consisted of a Shimadzu LC-20A series liquid chromatograph (Shimadzu) and a QTRAP 5500 mass spectrometer (Applied Bio-systems, AB Sciex) equipped with an electro-spray ionization (ESI) source. The data acquisition and processing were performed using Analyst 1.5.2 software. The separation was performed on a Waters Acquity UHPLC BEH C18 column (100 × 2.1 mm, i.d., 1.7 μm, Waters) protected by a Van Guard BEH C18 column (5 × 2.1 mm, 1.7 μm, Waters) maintained at 40 °C. The mobile phase consisting of a 0.5% formic acid aqueous solution and 0.1% formic acid acetonitrile solution (60:40, v/v) was run at a flow rate of 0.3 mL min−1. The injection volume was 3 μL. The total run time for a LC-MS/MS analysis was 6 min.
An ESI interface in the negative mode was used. The ion spray voltage was set at −4.5 kV and the turbo spray temperature was maintained at 500 °C. Nebulizer gas (gas 1) and heater gas (gas 2) were set at 160 and 260 psi, respectively. The curtain gas was kept at 40 psi and the interface heater was on. Nitrogen gas was used in all cases. Quantification was performed using MRM mode.
Preparation of calibration and quality control (QC) samples
Stock solutions of RA and silibinin (I.S.) were prepared in methanol at a concentration of 0.1122 and 0.1126 mg mL−1, respectively. Stock solutions were then diluted with methanol to produce a series of standard or QC working solutions at desired concentrations. The calibration standards were prepared by adding 10 μL of the series of standard working solutions into 100 μL of blank plasma to provide final concentrations at 8.976, 17.95, 44.88, 89.76, 224.4, 448.8, 1122, 2244, 5610, and 11220 ng mL−1. Low, medium and high levels of QC samples were prepared at 17.95, 448.8 and 5610 ng mL−1, respectively. The I.S. working solution of 2.252 μg mL−1 was also prepared by dilution of the stock solution with methanol. All the solutions were kept at 4 °C and brought to room temperature before use.
Sample preparation
A simple liquid–liquid extraction method was applied to extract RA and I.S. from the rat plasma. In an aliquot of 100 μL of rat plasma, 10 μL of the I.S. and 10 μL of methanol (volume of the corresponding working solution for calibration curve and QC samples) and 16 μL of formic acid/water (1:3, v/v) were added. Then the mixture was vortexed for 1 min and 0.2 mL of acetone/water (70:30, v/v) solution was added, followed by adding 1 mL of ethyl acetate to each tube. Extraction was performed by vortex mixing the tubes for 2 min, followed by centrifugation at 16000 rpm for 3 min. The organic layer was transferred to Eppendorf tubes and dried using a centrifugal concentrator. The residue was reconstituted in 100 μL of water containing 25% acetone. After centrifugation at 16000 rpm for 3 min, aliquots of 3 μL were injected into the UPLC-MS/MS system for analysis.
Method validation
The method was validated for selectivity, linearity, precision, accuracy, extraction recovery and stability according to the FDA guidelines for the validation of bio-analytical methods.27The lower limit of quantification is defined as the lowest concentration on the calibration curve with an acceptable precision (RSD) not exceed 20% and RE (relative error) of accuracy should not deviate by more than 20%.
Bioavailability and pharmacokinetics of RA in rats
The validated method was applied to analyze the plasma concentrations of RA in rats after oral and intravenous administration of RA. Twenty-four male rats were randomly divided into four groups with six in each. Three groups were given RA at a single oral dose of 12.5, 25 and 50 mg kg−1, and one group received a single dose of RA of 0.625 mg kg−1 by intravenous injection via caudal vein after an overnight fasting period. The rats were fasted for the first 2 h with free access to water after dosing. Serial blood samples (approximately 0.2 mL) were collected from the eyeground vein into 1.5 mL heparinized tubes at 0, 2, 5, 10, 20, 30, 45, 60, 90, 120, 180, 240, 360, 480 and 720 min after oral administration and at 0, 2, 5, 10, 20, 30, 45, 60, 75, 90, 120, 180, 240 and 360 min after intravenous injection. Plasma was isolated from the blood samples by cooling them for 2 h at 4 °C, followed by centrifugation at 3000 rpm for 10 min. The collected plasma was transferred into 1.5 mL Eppendorf tubes and then stored at −20 °C for analysis.Pharmacokinetic parameters were estimated using Drug and Statistics 3.0 (DAS 3.0, Mathematical Pharmacology Professional Committee of China) employing the noncompartmental model. The oral bioavailability (F) of RA is calculated by comparing the respective AUC after oral and intravenous administration according to the following equation:
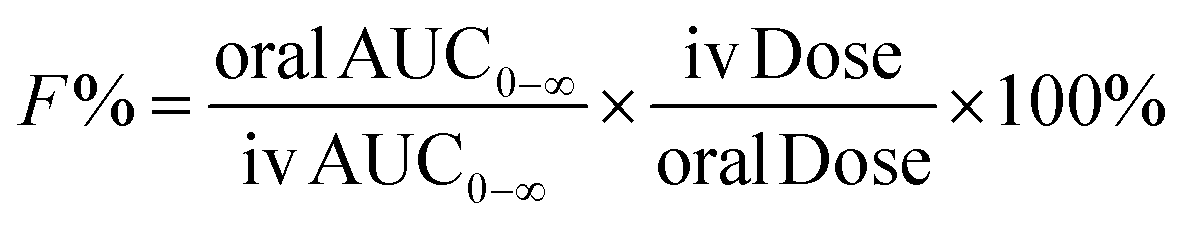 | (1) |
Statistical analyses
Descriptive statistics were used to summarize the pharmacokinetic parameters for RA. Statistical comparisons of AUC0–t, AUC0–∞, Cmax between the three doses were performed using an analysis of variance (ANOVA). Multivariable regression analysis was used to test associations between AUC0–t, AUC0–∞, Cmax and the dose. Dose proportionality for AUC0–t, AUC0–∞ and Cmax was assessed by linear regression of ln-transformed parameters on the natural ln-transformed dose [ln(PK) = β0 + β1 × ln(dose)]. Values of the proportionality constant, β1, and its corresponding 90% CI were estimated with the SPSS 19.0 software. The primary methodology used to assess dose proportionality from 12.5 to 50 mg kg−1 was a comparison of the 90% CI of the slopes with the modified acceptance range [1 + (ln(θL)/ln(r)), 1 + (ln(θH)/ln(r))] based on a power model,28,29 where θ L and θ H are the lower and upper limits of the confidence interval and r is the maximal dose ratio for the study. Dose proportionality was assumed if the plot of pharmacokinetic parameter vs. dose indicated linearity and the 90% CI for the slope fell within the modified acceptance range.Results and discussions
LC-MS/MS development
In the full scan mass spectra, the deprotonated molecular ions [M − H]− of RA and silibinin (I.S.) (m/z at 359.0 and 480.6) were stable and exhibited higher abundance. Quantitation was performed using multiple reaction monitoring (MRM) of the transitions of m/z 359.0 → 160.9 for RA and 480.6 → 301.0 for I.S., respectively. Declustering potential, collision energy and cell exit potential of RA and I.S. were optimized as −70, −22, −15 V and −210, −26, −21 V, respectively. The optimized mass spectrometric parameters had a higher signal for both precursor ions and product ions mentioned above.Selection of the LC conditions was key in order to obtain chromatograms with better peak responses. We found that acetonitrile–water can achieve higher peak responses and shorter analysis times for the target compounds in chromatograms. It was found that the peak shapes and responses of analytes were improved with an eluent consisting of water containing 0.5% formic acid and acetonitrile containing 0.1% formic acid (60:40, v/v). Satisfactory separation was achieved in 6 min by isocratic elution using the LC conditions described above.
Method validation
220 ng mL−1. A typical standard curve was y = 0.0089x − 0.3320 (r2 = 0.9980), where y represents the peak area ratios of RA to the I.S. and x represents the plasma concentrations of RA.The linearity and lower limit of quantification (LLOQ) of RA in rat plasma was 8.976 ng mL−1 with an intra- and inter-day relative standard deviation (RSD) of less than 3.85%, and an intra- and inter-day relative error (RE) of −8.74% and −7.88%, which was sufficient for the pharmacokinetic studies of RA in rats.
| Analytes | Concentration (ng mL−1) | Matrix effects | Extraction recoveries | ||
|---|---|---|---|---|---|
| Mean ± SD (%) | RSD (%) | Mean ± SD (%) | RSD (%) | ||
| RA | 17.95 | 95.47 ± 7.14 | 7.48 | 78.19 ± 8.42 | 10.76 |
| 448.8 | 87.30 ± 5.29 | 6.06 | 80.18 ± 11.84 | 14.77 | |
| 5610 | 94.71 ± 4.88 | 5.15 | 72.03 ± 6.18 | 8.59 | |
| Silibinin (I.S.) | 225.2 | 96.83 ± 4.43 | 4.58 | 84.58 ± 6.79 | 8.03 |
Pharmacokinetic parameters and bioavailability study
The validated analytical method was successfully applied to a pharmacokinetic study of RA in rats. The mean plasma concentration–time curves of RA after oral administration of 12.5, 25, 50 mg kg−1 and intravenous administration of 0.625 mg kg−1 to rats are presented in Fig. 2. The corresponding pharmacokinetic parameters calculated using non-compartmental analysis are listed as mean ± SD in Table 2.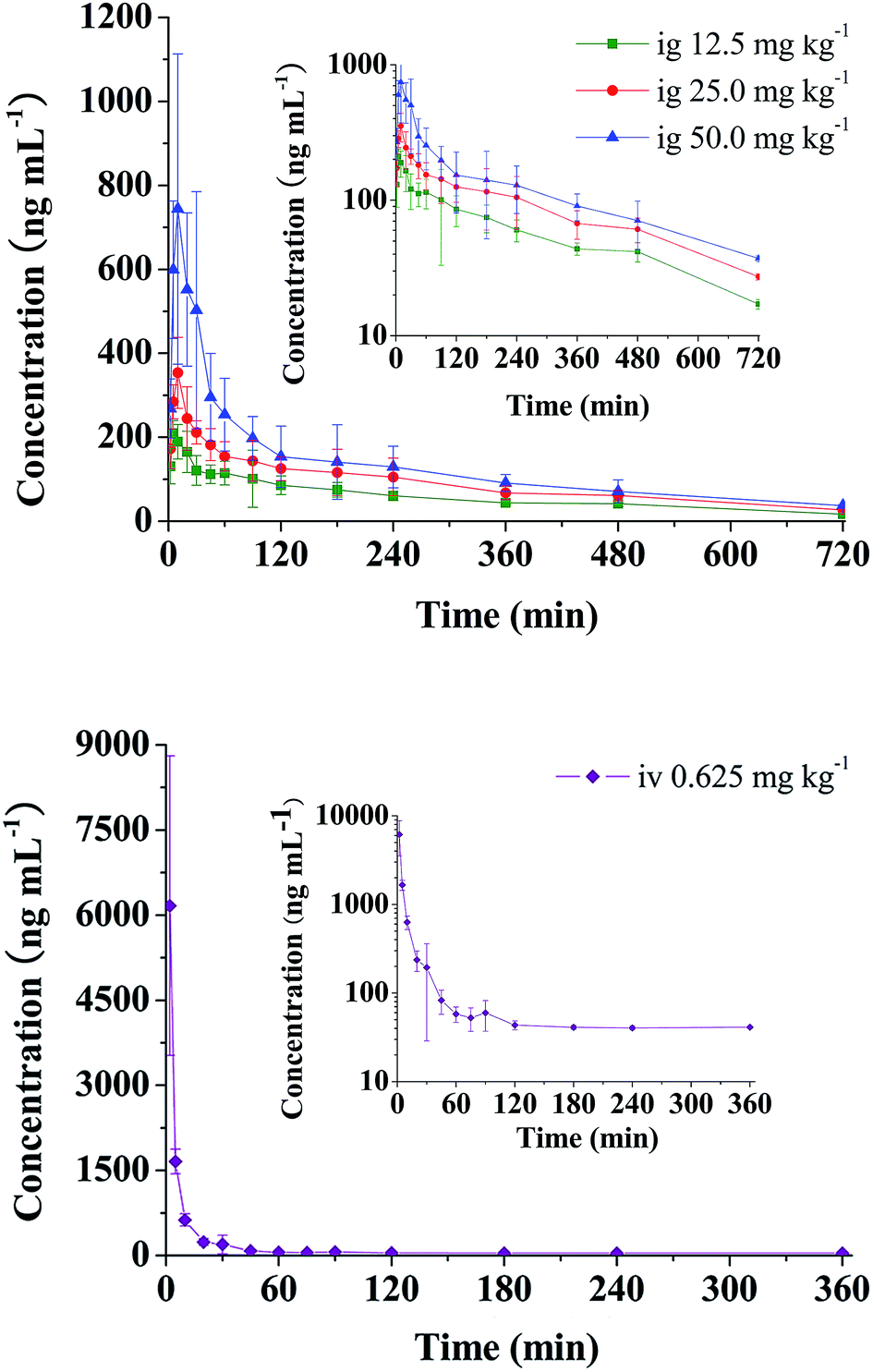 | ||
| Fig. 2 Mean plasma concentration–time curves of RA in rats after administration of single oral (12.5, 25 and 50 mg kg−1) and intravenous (0.625 mg kg−1) doses. | ||
| Parameters | Oral (12.5 mg kg−1) | Oral (25 mg kg−1) | Oral (50 mg kg−1) | Intravenous (0.625 mg kg−1) |
|---|---|---|---|---|
| AUC0–t (min ng mL−1) | 41789.84 ± 4673.43 |
64220.00 ± 9287.91 |
96070.00 ± 22151.45 |
62412.99 ± 14262.99 |
| AUC0–∞ (min ng mL−1) | 51990.55 ± 10945.85 |
78517.08 ± 8497.73 |
111994.79 ± 21458.01 |
153368.64 ± 83621.74 |
| Cmax (ng mL−1) | 215.21 ± 29.32 | 361.57 ± 81.01 | 790.96 ± 358.41 | 6166.89 ± 2641.58 |
| Tmax (min) | 8.33 ± 6.06 | 10.83 ± 4.92 | 18.33 ± 9.83 | — |
| t1/2 (min) | 332.34 ± 168.77 | 314.13 ± 185.84 | 295.32 ± 72.29 | 42.90 ± 10.41 |
| MRT0–t (min) | 244.41 ± 16.69 | 243.20 ± 18.91 | 228.85 ± 21.05 | 50.12 ± 14.94 |
| MRT0–∞ (min) | 439.13 ± 190.70 | 434.88 ± 211.45 | 366.64 ± 56.78 | 1747.51 ± 1436.59 |
| CL (L min−1 kg−1) | 0.25 ± 0.05 | 0.32 ± 0.03 | 0.46 ± 0.08 | 0.01 ± 0.00 |
| V (L kg−1) | 111.83 ± 31.14 | 141.81 ± 69.63 | 197.65 ± 63.60 | 9.41 ± 2.86 |
| F (%) | 1.69 ± 0.36 | 1.28 ± 0.14 | 0.91 ± 0.17 | — |
As seen from Table 2 and Fig. 2, it was found that after single oral administration (12.5, 25 and 50 mg kg−1), RA could be absorbed rapidly into blood, and reached its peak concentration in plasma within 19 min. After that RA eliminated slowly with t1/2 of 295.32–332.34 min and CL between 0.248 and 0.460 L min−1 kg−1. Cmax values were estimated to be 215.21, 361.57 and 790.96 ng mL−1, respectively. The AUC increased with increasing doses, and the AUC0–t values were 41789.84, 64220.00 and 96070.00 (ng min mL−1), receptively. In addition, no significant difference was observed in t1/2 and MRT (p > 0.05) at three dosages. The absolute oral bioavailabilities of RA in rats were quite low with values being 1.69 ± 0.36%, 1.28 ± 0.14% and 0.91 ± 0.17%, receptively.
Oral bioavailability is a complicated parameter that involves a number of chemical and physical processes. Poor bioavailability of RA may result from several different causes such as its intestinal absorption and metabolism. Its high hydrophilicity makes RA exhibit poor permeation across intestinal epithelial cells.30
Besides, in rats orally administered with RA, the polyphenol was absorbed, degraded, and/or conjugated as m-hydroxyphenylpropionic acid, m-coumaric acid, and sulphated forms of caffeic acid and ferulic acid,20 which were then absorbed by a specific intestinal monocarboxylic acid transporter-mediated active process,21 before being excreted gradually in the urine.
Considering the low absolute bioavailability of RA and its derivatives' efficacy, the effects of RA may be explained not solely by the established absorption of RA, but also by the absorption of its microbial phenolic acid metabolites. The assigned effect of RA and its metabolites is a need for further research.31–34
Dose proportionality
According to One-Way ANOVA analysis, the results of homogeneity of variance test statistics indicated that the variances of Cmax, AUC0–t and AUC0–∞ were not homogenous. The Cmax, AUC0–t and AUC0–∞ need to be ln-transformed. The results are summarized in Table 3. After curve estimation analysis with different models, relationships between lnCmax and dose, lnAUC0–t and dose, lnAUC0–∞ and dose could be well fitted with power model, as showed in Table 4. Plots of the fitted function for the power model with associated 90% confidence intervals are presented for AUC0–t, AUC0–∞ and Cmax in Fig. 3. Results of the assessment based on the power model are presented in Table 5. The slopes (90% confidence intervals (CIs)) were 0.586 (0.466, 0.707) for AUC0–t, 0.553 (0.433, 0.673) for AUC0–∞ and 0.887 (0.679, 1.095) for Cmax. The corresponding 90% CIs for AUC0–t, AUC0–∞ and Cmax spanned the acceptance interval (0.839, 1.161) defined by the equations for r = 50/12.5, θL = 0.8 and θH = 1.25, indicating inconclusive results.
| Parameters | Cmax | lnCmax |
AUC0–t | lnAUC0–t |
AUC0–∞ | lnAUC0–∞ |
| Sig. | 0.006 | 0.123 | 0.010 | 0.215 | 0.038 | 0.415 |
| Equation | R square | ||
|---|---|---|---|
| lnCmax |
lnAUC0–t |
lnAUC0–∞ |
|
| Linear | 0.766 | 0.771 | 0.749 |
| Logarithmic | 0.756 | 0.804 | 0.792 |
| Inverse | 0.694 | 0.780 | 0.778 |
| Quadratic | 0.769 | 0.805 | 0.794 |
| Cubic | 0.769 | 0.805 | 0.794 |
| Compound | 0.779 | 0.771 | 0.745 |
| Power | 0.778 | 0.808 | 0.791 |
| Growth | 0.779 | 0.771 | 0.745 |
| Exponential | 0.779 | 0.771 | 0.745 |
| Logistic | 0.779 | 0.771 | 0.745 |
 | ||
| Fig. 3 Relationship between the extent of systemic exposure and dose following oral administration of RA ranged from 12.5 to 50 mg kg−1 in rats. The empty circles are the individual observed values; the solid lines are the fitted values based on the power model, and the dashed lines are the 90% CIs. (A), AUC0–t; (B), AUC0–∞; (C), Cmax. | ||
| Parameters | Acceptance range | RA | |
|---|---|---|---|
| β1 | 90% confidence interval | ||
| AUC0–t | 0.839–1.161 | 0.586 | 0.466–0.707 |
| AUC0–∞ | 0.839–1.161 | 0.553 | 0.433–0.673 |
| Cmax | 0.839–1.161 | 0.887 | 0.679–1.095 |
Conclusions
In summary, it has been firstly carried out to comprehensively investigate absolute bioavailability, pharmacokinetics, and dose proportionality on the pharmacokinetics of single oral dose of RA. The absolute bioavailability of RA in rats was estimated as 1.69%, 1.28% and 0.91% after oral administration of RA at the doses of 12.5, 25 and 50 mg kg−1. The relationships between pharmacokinetic parameters (Cmax, AUC0–t and AUC0–∞) of RA and dose could be well fitted with power model. Pharmacokinetic properties of RA in rats after oral administration were characterized as rapid absorption, middle-speed elimination and poor absolute bioavailability. Analysis by power model demonstrated that systemic exposure exhibited lack of dose proportionality over the dose ranged from 12.5 to 50 mg kg−1. The data presented in this study could provide useful information for its rational clinical application and optimal dosage form design.Declaration of interest
The authors declare no conflict of interest.Acknowledgements
This study was financially supported by the Priority Academic Program Development (PAPD) fund of Jiangsu Higher Education Institution and The Natural Science Foundation of Jiangsu Province (No. BK20141465).References
- M. R. Al-Sereiti, K. M. Abu-Amer and P. Sen, Indian J. Exp. Biol., 1999, 37, 124–130 CAS.
- M. Heinrich, J. Kufer, M. Leonti and M. Pardo-de-Santayana, J. Ethnopharmacol., 2006, 107, 157–160 CrossRef PubMed.
- P. Ramirez, F. J. Senoran, E. Ibanez and G. Reglero, J. Chromatogr. A, 2004, 1057, 241–245 CrossRef CAS PubMed.
- Y. Peng, J. Yuan, F. Liu and J. Ye, J. Pharm. Biomed. Anal., 2005, 39, 431–437 CrossRef CAS PubMed.
- M. E. González-Trujano, E. I. Peña, A. L. Martínez, J. Moreno, P. Guevara-Fefer, M. Déciga-Campos and F. J. López-Muñoz, J. Ethnopharmacol., 2007, 111, 476–482 CrossRef PubMed.
- X. Chu, X. Ci, J. He, L. Jiang, M. Wei and Q. Cao, Molecules, 2012, 17, 3586–3598 CrossRef CAS PubMed.
- S. K. Ku, E. J. Yang, K. S. Song and J. S. Bae, Food Chem. Toxicol., 2013, 59, 311–315 CrossRef CAS PubMed.
- R. Domitrović, M. Skoda, M. V. Vasiljev, O. Cvijanović, P. E. Pernjak and M. B. Stefan, Food Chem. Toxicol., 2013, 51, 370–378 CrossRef PubMed.
- D. O. Moon, M. O. Kim, J. D. Lee, Y. H. Choi and G. Y. Kim, Cancer Lett., 2010, 288, 183–191 CrossRef CAS PubMed.
- K. A. Scheckel, S. C. Degner and D. F. Romagnolo, J. Nutr., 2008, 138, 2098–2105 CrossRef CAS PubMed.
- M. J. Jordán, V. Lax, M. C. Rota, S. Lorán and J. A. Sotomayor, J. Agric. Food Chem., 2012, 60, 9603–9608 CrossRef PubMed.
- G. S. Li, W. L Jiang, J. W. Tian, G. W. Qu, H. B. Zhu and F. H. Fu, Phytomedicine, 2010, 17, 282–888 CrossRef CAS PubMed.
- H. P. Dong, S. J. Park, J. M. Kim, W. Y. Jung and J. H. Ryu, Fitoterapia, 2010, 81, 644–648 CrossRef PubMed.
- Y. Q. Liu, Q. Cai, C. Liu, F. W. Bao and Z. Q. Zhang, J. Anal. Methods Chem., 2014, 2014, 617367 Search PubMed , 9 pages.
- X. Li, Y. Chen, Y. Cai, G. Liu, J. Jia and Y. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 820, 41–47 CrossRef CAS PubMed.
- X. Y. Wang, X. H. Ma, W. Li, Y. Chu, J. H. Guo, S. M. Li, J. M. Wang, H. C. Zhang, S. P. Zhou and Y. H. Zhu, J. Pharm. Biomed. Anal., 2013, 86, 82–91 CrossRef CAS PubMed.
- X. J. Lai, L. Zhang, J. S. Li, H. Q. Liu, X. H. Liu and L. Q. Di, Fitoterapia, 2011, 82, 883–888 CrossRef CAS PubMed.
- J. Y. Jia, Y. L. Lu, X. C. Li, G. Y. Liu, S. J. Li, Y. Liu, Y. M. Liu, C. Yu and Y. P. Wang, Curr. Ther. Res., 2010, 71, 260–271 CrossRef PubMed.
- X. Li, C. Yu, Y. Lu, Y. Gu, J. Lu, W. Xu, L. Xua and Y. Wang, Drug Metab. Dispos., 2007, 35, 234–239 CrossRef CAS PubMed.
- S. Baba, N. Osakabe, M. Natsume and J. Terao, Life Sci., 2004, 75, 165–178 CrossRef CAS PubMed.
- Y. Konishi, Y. Hitomi, M. Yoshida and E. Yoshioka, J. Agric. Food Chem., 2005, 53, 4740–4746 CrossRef CAS PubMed.
- J. Sun, L. Zhang, J. Song, S. Tian, C. Huang, Z. Feng, Y. Lv and G. Du, J. Ethnopharmacol., 2013, 148, 617–623 CrossRef PubMed.
- Y. T. Wu, Y. F. Chen, Y. J. Hsieh, I. Jaw, M. S. Shiao and T. H. Tsai, Int. J. Pharm., 2006, 326, 25–31 CrossRef CAS PubMed.
- Y. Konishi and S. Kobayashi, Biosci., Biotechnol., Biochem., 2005, 69, 583–591 CrossRef CAS PubMed.
- Y. Konishi, Y. Hitomi and E. Yoshioka, J. Agric. Food Chem., 2004, 52, 2527–2532 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, D. Li, C. T. Ho, J. Li and X. Wan, Food Funct., 2016, 7, 1273–1281 CAS.
- http://www.fda.gov/downloads/drugs/, accessed October 2013.
- B. P. Smith, F. R. Vandenhende, K. A. Desante, N. A. Farid, P. A. Welch and J. T. Callaghan, Pharm. Res., 2000, 17, 1278–1283 CrossRef CAS.
- V. S. Sethuraman, S. Leonov, L. Squassante, T. R. Mitchell and M. D. Hale, Pharm Stat., 2007, 6, 35–41 CrossRef PubMed.
- Z. Qiang, Z. Ye, C. Hauck, P. A. Murphy, J. A. McCoy, M. P. Widrlechner, M. B. Reddy and S. Hendrich, J. Ethnopharmacol., 2011, 137, 1107–1112 CrossRef CAS PubMed.
- T. Yasuda, A. Takasawa, T. Nakazawa, J. Ueda and K. Ohsawa, J. Pharm. Pharmacol., 2003, 55, 239–244 CrossRef CAS PubMed.
- I. M. Chung, J. J. Lim, M. S. Ahn, H. N. Jeong, T. J. An and S. H. Kim, J. Ginseng Res., 2016, 40, 68–75 CrossRef CAS PubMed.
- S. F. Nabavi, K. P. Devi, D. S. Malar, A. Sureda, M. Daglia and S. M. Nabavi, Mini-Rev. Med. Chem., 2015, 15, 776–788 CrossRef CAS PubMed.
- T. Silva, C. Oliveira and F. Borges, Expert Opin. Ther. Pat., 2014, 24, 1257–1270 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |