
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic asymmetric conjugate addition of t-butyl nitroacetate to o-quinone methides: synthesis of optically active α-nitro-β,β-diaryl-propionates†
Ri-long Liu‡
,
Xiang-zheng Tang‡,
Xue-jing Zhang,
Ming Yan * and
Albert S. C. Chan
* and
Albert S. C. Chan
Institute of Drug Synthesis and Pharmaceutical Process, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: yanming@mail.sysu.edu.cn
First published on 20th January 2017
Abstract
An asymmetric conjugate addition of t-butyl nitroacetate to in situ generated o-quinone methides had been developed. A chiral squamide derived from 9-amino-9-deoxyepiquinine was found to be the efficient catalyst. α-Nitro-β,β-diaryl-propionates could be obtained in good yields and with excellent enantioselectivities.
o-Quinone methides (o-QMs) are valuable intermediates for organic synthesis. Although most o-QMs are unstable and inseparable, applications of o-QMs in organic synthesis have been developed.1 In recent years, great progress has been made in asymmetric reactions with o-QMs.2 A number of organocatalytic transformations with o-QMs have also been explored. Han and co-workers reported the quinine-catalyzed asymmetric cycloannulation of o-QMs with malononitrile.3 Zhou and co-workers reported asymmetric addition of thiols and malononitrile to o-QMs with bifunctional organocatalysis.4 Fochi, Bernardi and co-workers developed chiral squamide-catalyzed asymmetric addition of Meldrum's acid, malononitrile and 1,3-dicarbonyl compounds to o-QMs.5 Sun, Rueping, Schneider, Shi, List and co-workers reported a series of asymmetric reactions of o-QMs with chiral Brønsted acid catalysts.6 Ye and co-workers developed asymmetric reactions of o-QMs catalyzed by chiral nitrogen heterocyclic carbenes.7 Very recently, Scheidt and co-workers reviewed the related progresses of organocatalytic reactions of in situ generated o-QMs.8
In recent years, we have developed organocatalytic asymmetric conjugate addition of nitroacetates to β,γ-unsaturated-α-ketoesters.9 To expand the application of nitroacetates in organocatalytic transformations, we are interesting in the asymmetric conjugate addition of nitroacetates to o-QMs.10,11 The reaction can provide optically active α-nitro-β,β-diaryl-propionates, which are useful intermediates for the synthesis of chiral drugs and natural products. In this paper, we report an organocatalytic conjugate addition of t-butyl nitroacetate to in situ generated o-QMs. A series of α-nitro-β,β-diaryl-propionates could be obtained in good yields and with excellent enantioselectivities.
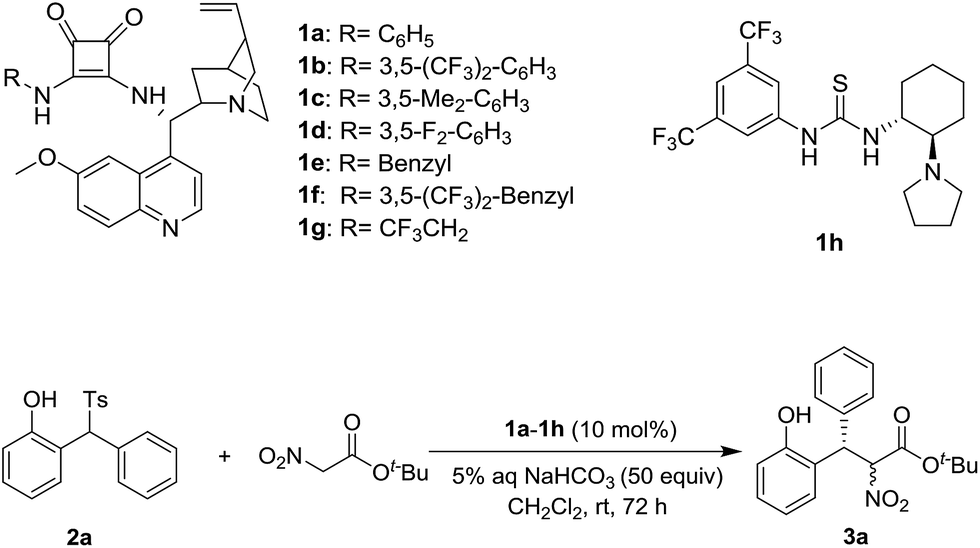
Based on our previous experiences,9 a variety of chiral squamides 1a–1g and thiourea 1h were chosen as the catalysts (Table 1). The reaction of 2-tosylmethylphenol 2a and t-butyl nitroacetate was examined and the results are summarized in Table 1. The in situ generation of o-QM from 2a in the presence of aqueous sodium bicarbonate was reported by Fochi, Bernardi and co-workers.5 To our delight, the expected product 3a was obtained with good yield and enantioselectivity. Almost no diastereoselectivity was achieved in this transformation. The product 3a was obtained as the equivalent mixture of two diastereoisomers. Among the tested squamide catalysts derived from 9-amino-9-deoxyepiquinine, 1a afforded the best yield. The 3,5-bistrifluoromethyl, 3,5-bismethyl and 3,5-difluoro substituted analogs 1b–1d gave lower yields and similar enantioselectivities. Benzyl, 3,5-bistrifluoromethyl benzyl and trifluoroethyl substituted squamides 1e–1g afforded lower enantioselectivities. The thiourea 1h provided the enantiomer of 3a in a moderate yield and with good enantioselectivity.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | Drc | eed (%) |
| a The reactions were carried out with 2a (0.20 mmol), t-butyl nitroacetate (0.4 mmol), 1a–1h (0.02 mmol) in CH2Cl2 (2.5 mL) and 5% aqueous NaHCO3 (16.8 mL) at room temperature for 72 h.b Isolated yields of 3a.c The diastereoisomeric ratios were determined by 1H NMR.d The ee values of 3a were determined by HPLC with a Chiralpak AD-H column. | ||||
| 1 | 1a | 83 | 1/1 | 95/95 |
| 2 | 1b | 76 | 1/1 | 96/96 |
| 3 | 1c | 68 | 1/1 | 95/95 |
| 4 | 1d | 69 | 1/1 | 95/96 |
| 5 | 1e | 61 | 1/1 | 85/84 |
| 6 | 1f | 76 | 1/1 | 86/86 |
| 7 | 1g | 61 | 1/1 | 88/89 |
| 8 | 1h | 66 | 1/1 | −94/−94 |
Using 1a as the catalyst, the effect of the reaction solvent, the base, the catalyst loading and the reaction temperature was examined and the results are summarized in Table 2. Better enantioselectivity and similar yield were achieved in CHCl3. Lower enantioselectivities were observed in other solvents (Table 2, entries 3–7). The replacement of sodium bicarbonate with potassium bicarbonate led to slightly lower yield (Table 2, entry 8). The use of more basic sodium carbonate, potassium carbonate, and cesium carbonate gave poor yields and enantioselectivities (Table 2, entries 9–11). While less basic sodium acetate and potassium fluoride were examined, low yields and good enantioselectivities were observed (Table 2, entries 12 and 13). The decrease of the catalyst loading gave slightly lower yields and enantioselectivities (Table 2, entries 14 and 15). The reaction at 0 °C gave a poor yield and inferior enantioselectivity (Table 2, entry 16). The higher reaction temperature was found to be detrimental for the enantioselectivity (Table 2, entry 17).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | x | Base | T (°C) | Yieldb (%) | eec (%) |
| a The reactions were carried out with 2a (0.20 mmol), t-butyl nitroacetate (0.4 mmol), 1a (0.02 mmol) in solvent (2.5 mL) and 5% aqueous base (50 equiv.) at room temperature for 72 h.b Isolated yields of 3a. The diastereoisomeric ratios were found to be 1/1 in all cases by 1H NMR.c The ee values of 3a were determined by HPLC with a Chiralpak AD-H column.d The reaction was run for 84 h.e The reaction was run for 48 h. | ||||||
| 1 | CH2Cl2 | 10 | NaHCO3 | rt | 83 | 95/95 |
| 2 | CHCl3 | 10 | NaHCO3 | rt | 82 | 97/98 |
| 3 | Cl(CH2)2Cl | 10 | NaHCO3 | rt | 64 | 92/92 |
| 4 | Toluene | 10 | NaHCO3 | rt | 51 | 29/35 |
| 5 | EtOAc | 10 | NaHCO3 | rt | 20 | 5/5 |
| 6 | MeOtBu | 10 | NaHCO3 | rt | 36 | 2/2 |
| 7 | EtOH | 10 | NaHCO3 | rt | 80 | 4/4 |
| 8 | CHCl3 | 10 | KHCO3 | rt | 80 | 97/97 |
| 9 | CHCl3 | 10 | Na2CO3 | rt | 23 | 19/16 |
| 10 | CHCl3 | 10 | K2CO3 | rt | 25 | 1/1 |
| 11 | CHCl3 | 10 | Cs2CO3 | rt | 32 | 0/0 |
| 12 | CHCl3 | 10 | NaOAc | rt | 24 | 98/98 |
| 13 | CHCl3 | 10 | KF | rt | 28 | 89/90 |
| 14d | CHCl3 | 5 | NaHCO3 | rt | 80 | 96/96 |
| 15d | CHCl3 | 2 | NaHCO3 | rt | 74 | 94/94 |
| 16 | CHCl3 | 10 | NaHCO3 | 0 | 20 | 87/88 |
| 17e | CHCl3 | 10 | NaHCO3 | 40 | 72 | 95/94 |

The influence of different ester group of nitroacetates was also investigated and the results are summarized in Scheme 1. The reaction of methyl nitroacetate and ethyl nitroacetate did not give the expected products, instead the decarboxylated product 4a was obtained in poor yields and with moderate enantioselectivities. Complicated products were observed in the reaction of i-propyl nitroacetate and i-butyl nitroacetate. The LC-MS analysis showed the existence of a small amount of the expected conjugate addition products, but no pure samples could be isolated and characterized. The hydrolysis of these nitroacetates in aqueous sodium bicarbonate possibly accounted for the results.12 The resistance of t-butyl nitroacetate to the basic hydrolysis ensures the good yield.
 | ||
| Scheme 1 Effect of ester group in nitroacetates. a The reactions were carried out with 2a (0.20 mmol), nitroacetate (0.4 mmol), 1a (0.02 mmol) in CHCl3 (2.5 mL) and 5% aqueous NaHCO3 (16.8 mL) at room temperature for 72 h. b Isolated yields. c The ee values of 4a were determined by chiral HPLC with a Chiralpak OJ-H column. | ||
A variety of 2-tosylmethylphenols 2b–2n were examined in the reaction and the results are summarized in Table 3. The introduction of 4-methyl, 4-t-butyl and 4-methoxyl to the phenyl ring did not affect the yields and enantioselectivities (Table 3, entries 1–3). The substitutions with 5-methoxyl, 6-methoxyl and 4,5-methylenedioxy led to the better yields and excellent enantioselectivities (Table 3, entries 4–6). The 6-methyl substituted substrate 3h gave the inferior yield, but still with excellent enantioselectivity (Table 3, entry 7). The 6-fluoro substitution was tolerated well. The good yield and excellent enantioselectivity were achieved (Table 3, entry 8). Interestingly, the improved diastereoselectivity was observed in this case. Keeping 5-methoxyl substitution at the left phenyl ring, a number of substrates with different substituents at the right phenyl ring of 2a were examined. The substitutions with 2-methyl, 2-methoxyl, 4-methyl and 4-methoxyl provided the products with excellent yields and enantioselectivities (Table 3, entries 9–12). While R2 was changed to a methyl group, excellent yield and moderate enantioselectivity were achieved (Table 3, entry 13). It should be noted that the introduction of 5-methoxyl substitution at the left phenyl ring significantly increases the reactivity of the substrates. Shorten reaction time was required in these cases. 1-(Phenyl(tosyl)methyl)naphthalen-2-ol was prepared and examined under the standard conditions, but no expected product was obtained. The big steric hindrance of this substrate possibly prohibited the reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Yieldb (%) | Drc | eed (%) |
| a The reactions were carried out with 2b–2n (0.20 mmol), t-butyl nitroacetate (0.4 mmol), catalyst 1a (0.02 mmol) in CHCl3 (2.5 mL) and 5% aqueous NaHCO3 (16.8 mL) at room temperature for 72 h.b Isolated yields.c The diastereoisomeric ratios were determined by 1H NMR.d The ee values were determined by HPLC with a Chiralpak AD-H column.e The reaction was run for 24 h.f The reaction was run for 40 h. | |||||
| 1 | 4-Me | C6H5 | 3b, 73 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
97/98 |
| 2 | 4-tBu | C6H5 | 3c, 80 | 1:1 |
96/92 |
| 3 | 4-MeO | C6H5 | 3d, 80 | 1:1 |
97/97 |
| 4e | 5-MeO | C6H5 | 3e, 98 | 1:1 |
98/98 |
| 5 | 6-MeO | C6H5 | 3f, 95 | 1:1 |
96/98 |
| 6 | 4,5-OCH2O– | C6H5 | 3g, 92 | 1:1 |
99/99 |
| 7 | 6-Me | C6H5 | 3h, 52 | 1:1 |
97/95 |
| 8 | 6-F | C6H5 | 3i, 81 | 5:1 |
97/97 |
| 9e | 5-MeO | 2-Me–C6H4 | 3j, 98 | 1:1 |
99/99 |
| 10e | 5-MeO | 2-MeO–C6H4 | 3k, 90 | 1:1 |
97/97 |
| 11e | 5-MeO | 4-Me–C6H4 | 3l, 98 | 1:1 |
96/98 |
| 12e | 5-MeO | 4-MeO–C6H4 | 3m, 99 | 1:1 |
99/98 |
| 13f | 5-MeO | Me | 3n, 94 | 1.5:1 |
83/86 |

The transformation of the product 3e and 3g was explored (Scheme 2).13 The ester exchange was achieved by the treatment of 3e/3g with TFA. The lactones 5e/5g were obtained with good yields and diastereoselectivities.14 The basic hydrolysis of 5e/5g gave the decarboxylation products 4e/4g. The absolute configuration of 4g was determined as S by the comparison of the optical rotation with the reported data.15 The reduction of 4e/4g and the protection of the amino group with (Boc)2O provided 6e/6g in good yields and without the erosion of optical purity.
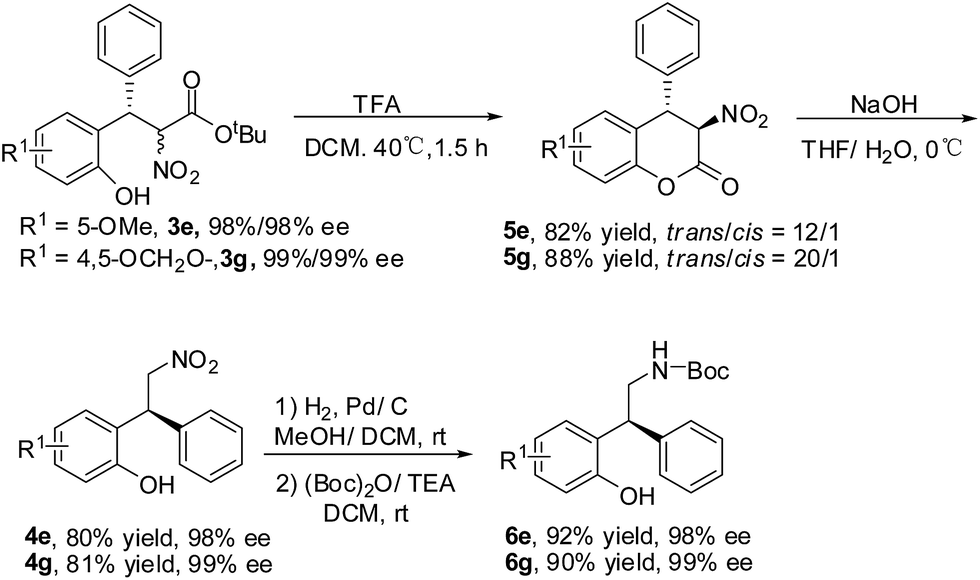 | ||
| Scheme 2 Transformation of the product 3e and 3g. | ||
A tentative reaction mechanism is suggested in Scheme 3. o-QM is generated from 2-tosylmethylphenol 2a in the presence of aqueous sodium bicarbonate. The double hydrogen bonding of o-QM with catalyst 1a increases its electrophilic reactivity. In the meantime, 1a abstracts a proton from t-butyl nitroacetate to give the nitro enolate, which is drawn close to o-QM by the hydrogen bonding with the ammonium cation. The subsequent attack from si-face of o-QM provides the product 3a.
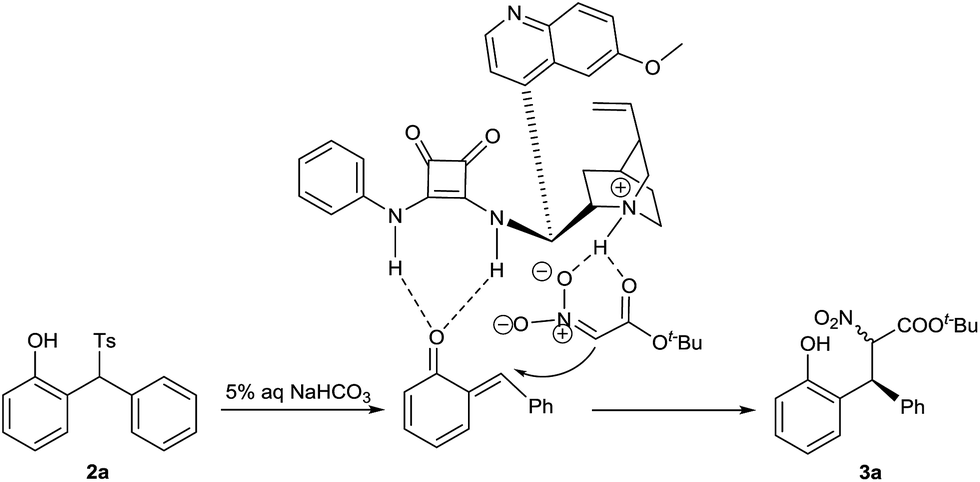 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed an organocatalytic asymmetric reaction of o-QMs with t-butyl nitroacetate. 2-Tosylmethylphenols could be used as the suitable precursors of o-QMs in the presence of aqueous sodium bicarbonate. A chiral squamide derived from 9-amino-9-deoxyepiquinine was identified as the superior catalyst. A variety of t-butyl α-nitro-β,β-diaryl-propionates were prepared in good yields and with excellent enantioselectivities. The further elaboration of the products provided the useful chiral intermediates for the synthesis of drugs and natural products.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21172270, 21472248) for the financial support of this study.Notes and references
- (a) R. W. V. D. Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367 CrossRef; (b) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655 CrossRef CAS PubMed; (c) M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924 RSC.
- (a) Z.-B. Wang and J.-W. Sun, Synthesis, 2015, 47, 3629 CrossRef; (b) L. Caruana, M. Fochi and L. Bernardi, Molecules, 2015, 20, 11733 CrossRef CAS PubMed.
- A. Adili, Z.-L. Tao, D.-F. Chen and Z.-Y. Han, Org. Biomol. Chem., 2015, 13, 2247 CAS.
- (a) W.-G. Guo, B. Wu, X. Zhou, P. Chen, X. Wang, Y.-G. Zhou, Y. Liu and C. Li, Angew. Chem., Int. Ed., 2015, 54, 4522 CrossRef CAS PubMed; (b) B. Wu, X. Gao, Z. Yan, W.-X. Huang and Y.-G. Zhou, Tetrahedron Lett., 2015, 56, 4334 CrossRef CAS; (c) B. Wu, X. Gao, Z. Yan, M.-W. Chen and Y.-G. Zhou, Org. Lett., 2015, 17, 6134 CrossRef CAS PubMed.
- L. Caruana, M. Mondatori, V. Corti, S. Morales, A. Mazzanti, M. Fochi and L. Bernardi, Chem.–Eur. J., 2015, 21, 6037 CrossRef CAS PubMed.
- (a) Z.-W. Lai, Z.-B. Wang and J.-W. Sun, Org. Lett., 2015, 17, 6058 CrossRef CAS PubMed; (b) Z.-B. Wang, F.-J. Ai, Z. Wang, W.-X. Zhao, G.-Y. Zhu, Z.-Y. Lin and J.-W. Sun, J. Am. Chem. Soc., 2015, 137, 383 CrossRef CAS PubMed; (c) C.-C. Hsiao, S. Raja, H.-H. Liao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 5762 CrossRef CAS PubMed; (d) C.-C. Hsiao, H.-H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258 CrossRef CAS PubMed; (e) S. Saha and C. Schneider, Chem.–Eur. J., 2015, 21, 2348 CrossRef CAS PubMed; (f) G. C. Tsui, L.-P. Liu and B. List, Angew. Chem., Int. Ed., 2015, 54, 7703 CrossRef CAS PubMed; (g) J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460 CrossRef CAS PubMed; (h) J.-J. Zhao, Y.-C. Zhang, M.-M. Xu, M. Tang and F. Shi, J. Org. Chem., 2015, 80, 10016 CrossRef CAS PubMed; (i) Y.-C. Zhang, Q.-N. Zhu, X. Yang, L.-J. Zhou and F. Shi, J. Org. Chem., 2016, 81, 1681 CrossRef CAS PubMed; (j) Q. Wu, J.-J. Zhao, S.-B. Sun, M.-S. Tu and F. Shi, Acta Chim. Sin., 2016, 74, 576 CrossRef CAS.
- (a) H. Lv, L. You and S. Ye, Adv. Synth. Catal., 2009, 351, 2822 CrossRef CAS; (b) H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS PubMed.
- A. A. Jaworski and K. A. Scheidt, J. Org. Chem., 2016, 81, 10145 CrossRef CAS PubMed.
- (a) R.-J. Lu, W.-T. Wei, J.-J. Wang, S.-Z. Nie, X.-J. Zhang and M. Yan, Tetrahedron, 2012, 68, 9397 CrossRef CAS; (b) R.-L. Liu, Y.-Y. Yan, T. Zhang, X.-J. Zhang and M. Yan, Tetrahedron: Asymmetry, 2015, 26, 1416 CrossRef CAS.
- For the reviews of nitroacetates in organic synthesis, see: (a) L. F. Tietze and C. Schneider, e-EROS Encyclopaedia of Reagents for Organic Synthesis, Ethyl Nitroacetate, 2001 Search PubMed; (b) M. T. Ship chandler, Synthesis, 1979, 9, 666 CrossRef.
- For recent reports of organocatalytic asymmetric reactions with nitroacetates, see: (a) C. Liu and Y.-X. Lu, Org. Lett., 2010, 12, 2278 CrossRef CAS PubMed; (b) Y.-R. Zhou, Q. Liu and Y.-F. Gong, Tetrahedron Lett., 2013, 54, 3011 CrossRef CAS; (c) M.-Y. Han, Y. Zhang, H.-Z. Wang, W.-K. An, B.-C. Ma, Y. Zhang and W. Wang, Adv. Synth. Catal., 2012, 354, 2635 CrossRef CAS; (d) S. Shirakawa, S. J. Terao, R.-J. He and K. Maruoka, Chem. Commun., 2011, 47, 10557 RSC; (e) Y.-Z. Li, F. Li, P. Tian and G.-Q. Lin, Eur. J. Org. Chem., 2013, 1558 CrossRef CAS; (f) J. Kwiatkowski and Y.-X. Lu, Org. Biomol. Chem., 2015, 13, 2350 RSC.
- The reaction of 2a with nitromethane under the standard conditions is not successful. No product 4a was observed in the reaction mixture. The lower reactivity of nitromethane comparing with nitroacetates possibly accounts for the result.
- The direct reduction of nitro group of 3e and 3g was not successful via general reduction methods (such as the catalytic hydrogenation over Pd/C, Pt/C, RANEY®-Ni, and the reduction with Zn, Sn, Fe powder). The big steric hindrance of 3e and 3g may prohibit the reaction of the nitro group.
- The reduction of 5e/5g was not successful despite extensive efforts were made (the catalytic hydrogenation over Pd/C, Pt/C, RANEY®-Ni, and the reduction with Zn, Sn, Fe powder). We think the big steric hindrance of 5e and 5g hampers the reduction.
- Y. Sohtome, B. Shin, N. Horitsugi, R. Takagi, K. Noguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 7299 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of the products, copies of 1H, 13C NMR spectra and HPLC chromatograms. See DOI: 10.1039/c6ra28068d |
| ‡ These two authors contribute equally to this study. |
| This journal is © The Royal Society of Chemistry 2017 |