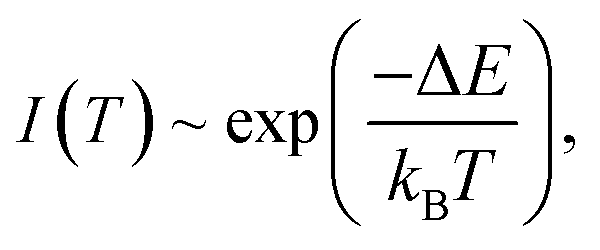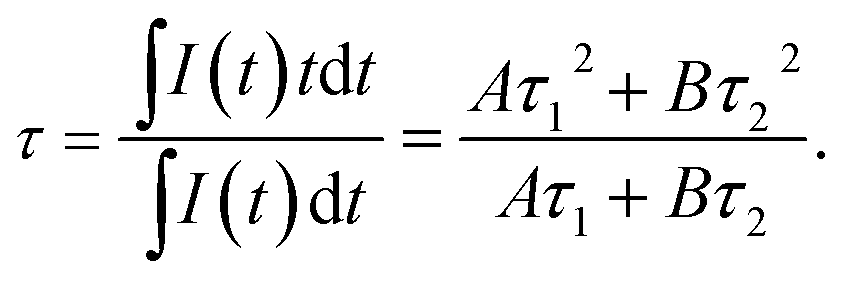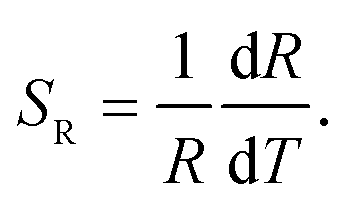DOI:
10.1039/C6RA27971F
(Paper)
RSC Adv., 2017,
7, 10200-10205
Terbium and holmium codoped yttrium phosphate as non-contact optical temperature sensors
Received
9th December 2016
, Accepted 1st February 2017
First published on 6th February 2017
Abstract
Optical thermometry has attracted many studies for non-contact high-resolution real-time temperature sensing. Most promising approaches are based on the ratio of up-converted luminescence intensities of two thermally coupled excited states. Here, we proposed a new strategy utilizing the temperature dependence of the anti-Stokes luminescence by exciting a thermally populated low-lying state to the excited state. Our scheme not only retains the advantage of previous approaches in reducing noise from the Stokes-type stray light, but also has the advantage of high quantum yield as a result of a one-photon excitation process. The temperature-dependent luminescence of Tb3+, Ho3+ codoped YPO4 is employed to demonstrate our scheme. The results show that, under a certain excitation, the emission of Tb3+ enhances dramatically while that of Ho3+ declines with increasing temperature. The sharp temperature-dependent intensity ratio was used to calibrate temperature. A maximum relative sensitivity of 2.51% K−1 at 310 K was obtained, substantially superior to values previously reported for acknowledged optical thermometry phosphors. These results indicate that the YPO4:Tb3+,Ho3+ can be a promising candidate to achieve accurate optical temperature sensing with a high sensitivity, and the mechanism proposed can be used to develop better optical thermometry.
1. Introduction
Non-contact optical temperature sensors have been attracting extensive attention because of their wide applications as molecular probes and nano-thermometry.1–4 Some schemes based on spectral shift, emission bandwidth, absolute intensity, fluorescence lifetime, as well as fluorescence intensity ratio (FIR), have been widely explored.5–9 Among these aspects, FIR of two thermally-coupled energy levels (TCELs) based on up-conversion (UC) luminescence is adopted frequently.10–13 Compared to conventional down-conversion luminescence, UC luminescence can effectively reduce the interference of Stokes-type stray scattering light from sensors and their environment. However, the fluorescence quantum yield of UC luminescence tends to be very low (usually much less than 1% under weak excitation intensity), which means that, to obtain good signal to noise ratio, one has to drastically enhance the laser power, but this often causes heating of the sensor. Besides, the common FIR technique is based on the assumption of transient thermoequilibrium between the two TCELs, which may break down at low temperature when the nonradiative relaxation between the two TCELs is not fast enough, i.e., loss of thermal coupling.
Under the premise of retaining the advantage of UC luminescence and aiming to effectively prompt the fluorescence quantum yield, we consider an alternative mechanism to achieve luminescence temperature sensing. In this work, as the temperature-detection region in our case ranges from room temperature to high temperature (above 700 K), the tetragonal phase yttrium phosphate YPO4 with xenotime structure was chosen as the host material when given its good physical and chemical stability at high temperature.14 Meanwhile, rare-earth ions with TCELs were widely investigated to achieve FIR sensing based on UC luminescence, such as Nd3+, Gd3+, Dy3+, Ho3+, Er3+ and Tm3+.15–21 Trivalent terbium have a 4f7 electronic configuration, and the energy gap between the ground state 7F6 and the first excited state 7F5 is roughly 2100 cm−1, which suggests that the thermal equilibrium between 7F5 and 7F6 can be constructed at a relatively high temperature. In YPO4, Tb3+ and Ho3+ were both expected to substitute for Y3+ in YPO4. Under a certain pulsed laser excitation and by monitoring the characteristic emission of both Ho3+ and Tb3+, we can obtain the FIR between these two emissions. By calculation, we found the sample YPO4:Tb3+,Ho3+ has a high relative sensitivity, which shows the sample can be a potential candidate to achieve high-sensitivity temperature sensing.
2. Experimental
2.1. Materials synthesis
Tb3+, Ho3+-codoped YPO4 powder sample was synthesized via hydrothermal method. Firstly, RE(NO3)3 standard solutions (RE = Y, Tb, Ho and Y/Tb/Ho = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.2:0.8) were prepared by dissolving the respective Y2O3, Tb4O7 and Ho2O3 (99.99%) in hot dilute nitric acid with the total metal cations being 2 mmol. After vigorous magnetic stirring for 15 min, NH4H2PO4 was added to the mixed solution. Then, the pH value of the mixture was adjusted to 1 with adding moderate amount of dilute nitric acid. Continuously stirring and several minutes later, the mixture were transferred to the muffle furnace at 160 °C for 18 h. The white precipitate was separated by centrifugation and washed with deionized water and ethanol for several times. And then the precipitate was dried at 60 °C for 2 h and the collected powder was calcined at 850 °C for 4 h in the air. Subsequently after ground, the powder was recalcined at 1200 °C for 8 h under CO reduction atmosphere.
:3.2:0.8) were prepared by dissolving the respective Y2O3, Tb4O7 and Ho2O3 (99.99%) in hot dilute nitric acid with the total metal cations being 2 mmol. After vigorous magnetic stirring for 15 min, NH4H2PO4 was added to the mixed solution. Then, the pH value of the mixture was adjusted to 1 with adding moderate amount of dilute nitric acid. Continuously stirring and several minutes later, the mixture were transferred to the muffle furnace at 160 °C for 18 h. The white precipitate was separated by centrifugation and washed with deionized water and ethanol for several times. And then the precipitate was dried at 60 °C for 2 h and the collected powder was calcined at 850 °C for 4 h in the air. Subsequently after ground, the powder was recalcined at 1200 °C for 8 h under CO reduction atmosphere.
2.2. Materials characterization
The as-prepared phosphors were characterized by an X-ray diffractometer (MAC Science Co., Ltd., MXP18AHF, Tokyo, Japan) using nickel-filtered Cu Kα radiation (λ = 0.15418 nm), with the accelerating voltage and tube current being 40.0 kV and 100.0 mA, respectively. The photoluminescence excitation (PLE) and emission (PL) spectra were measured by a double monochromator (Model Jobin-Yvon HRD-1) equipped with a Hamamatsu R928 photomultiplier. The excitation source was a tunable laser system (Opolette 355 LD system) which has a laser linewidth of 4–7 cm−1 and a pulse duration of 7 ns. The detected signal was analyzed by an EG&G 7265 DSP Lock-in Amplifier and the decay curves were recorded with a Tektronix TDS2024 digital storage oscilloscope (US Patents, Shanghai, China).
3. Results and discussion
3.1. Phase characterization
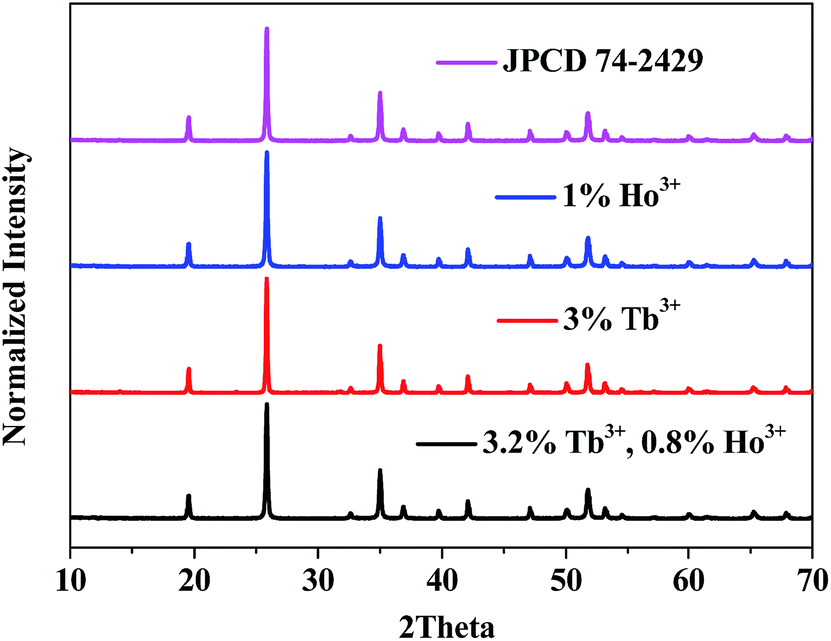
The crystal structure was identified by the XRD patterns in Fig. 1. As shown, all diffraction peaks of the sample fit well with the standard JCPD 74-2479, and no additional peaks are found, indicating no impurity was introduced in our samples.
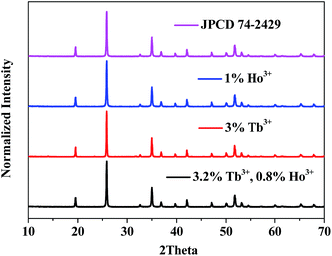 |
| | Fig. 1 XRD patterns of (a) YPO4:4%Tb3+, (b) YPO4:1%Ho3+ and (c) YPO4:3.2%Tb3+,0.8%Ho3+ powder samples. The standard JPCD card (no. 74-2429) is presented as a reference. | |
3.2. Room-temperature luminescence properties
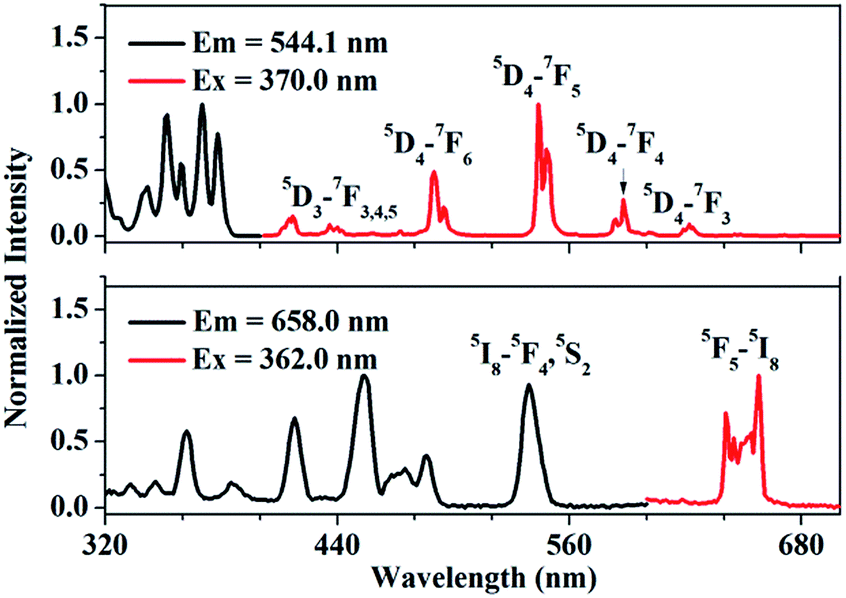
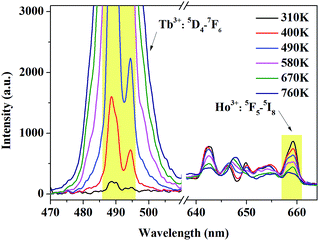
Fig. 2(a) shows the photoluminescence excitation (PLE) and emission (PL) spectra of Tb3+ singly-doped YPO4. As presented, the PLE spectrum of YPO4:Tb3+ consists of several excitation peaks located at 345.0, 355.0, 362.0, 370.0 and 381.0 nm, respectively. Under 370.0 nm excitation, the PL spectrum consists of several characteristic emission peaks which can be attributed to 5D3,4–7FJ transition of Tb3+. The transitions we care about are 5D4–7F5 (543.9 nm) and 5D4–7F6 (488.0 nm). Fig. 2(b) shows the PLE and PL spectrum of Ho3+ singly-doped YPO4. Under 362.0 nm excitation, some characteristic emission peaks centered at 641.0, 645.0 and 658.0 nm can be observed, which of all can be ascribed to the 5F5–5I8 transition of Ho3+.22 We can observe from Fig. 2 that, Tb3+ and Ho3+ have similar characteristic peaks at around 540.0 nm, which provides the possibility to simultaneously excite Tb3+ and Ho3+ under a certain excitation wavelength.
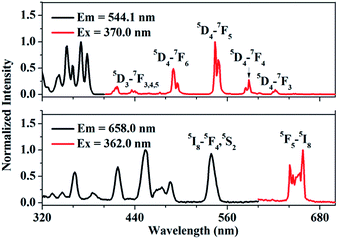 |
| | Fig. 2 Photoluminescence excitation (PLE) and emission spectra (PL) of (a) Tb3+-singly doped YPO4 and (b) Ho3+-singly doped YPO4 at room-temperature. | |
3.3. Temperature-dependent luminescence properties of YPO4:Tb3+,Ho3+
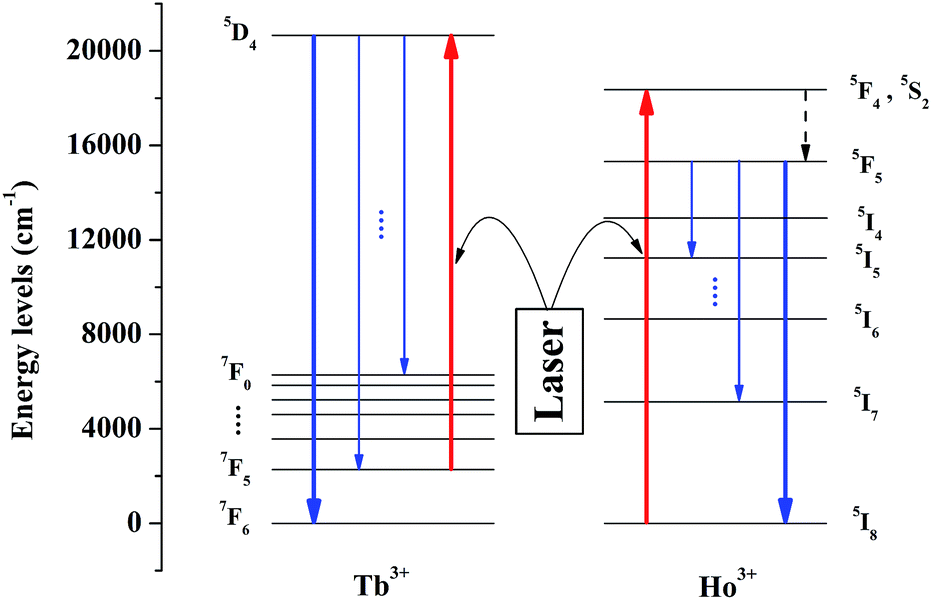
The schematic diagram of the mechanism for temperature sensing is depicted in Fig. 3. Tb3+ and Ho3+ can be simultaneously excited under a certain excitation, and the transitions we concern about are 7F5–5D4 of Tb3+ and 5I8–5F4 (5S2) of Ho3+. The thermally-activated Tb3+ which locates at the lowest excited state stark level of 7F5 multiplets can be excited to 5D4 state via resonance absorption, followed by the radiative transitions from 5D4 to 7FJ states, and we detect the emission between 5D4 to 7F6. In theory, as the system is in thermal equilibrium, the population of Tb3+ situated at 7F5 and 7F6 follows the Boltzmann distribution. Therefore, as the temperature rises, the number of Tb3+ being excited to 5D4 increases correspondingly and so does the emission intensity, which means that this emission can be utilized to measure the temperature variation. Meanwhile, Ho3+ was doped as a reference to ensure the accuracy of the luminescence intensity of Tb3+. Under the same excitation, Ho3+ situated in the ground state 5I8 can be excited to 5F4 (5S2) and due to the non-radiative relaxation from 5F4 (4S2) to 5F5, the emission of 5F5–5I8 transition can be observed.
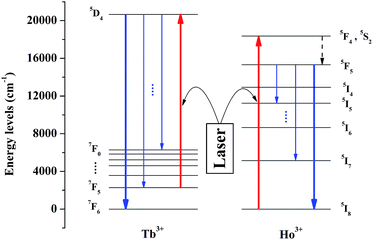 |
| | Fig. 3 Schematic energy level diagram of Tb3+ and Ho3+ in YPO4. Under a certain excitation, the excitation we care about are 7F6–5D4 (Tb3+) and 5I8–5F4, 5S2 (Ho3+), and the emission we concern are 5D4–7F6 (Tb3+) and 5F5–5I8 (Ho3+). | |
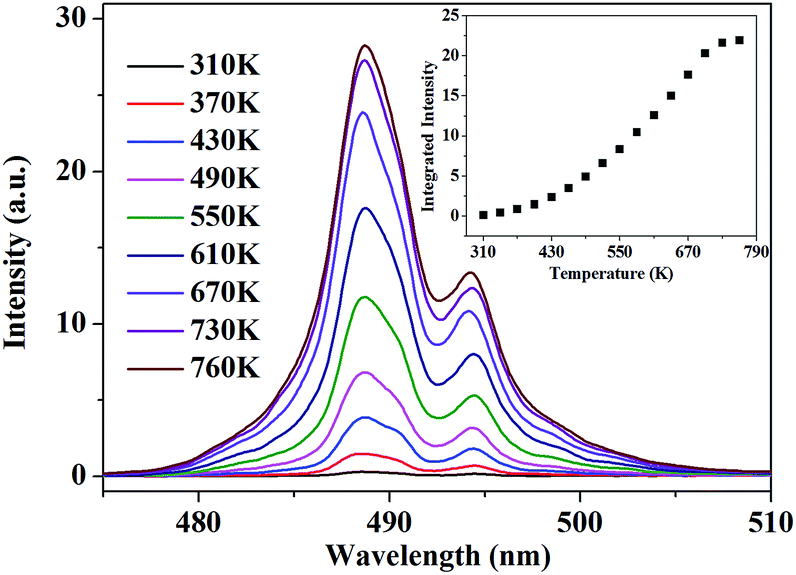
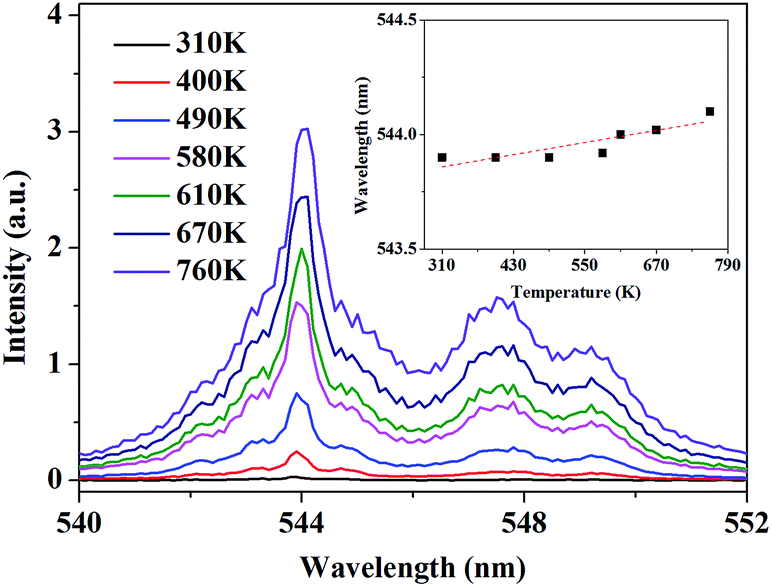
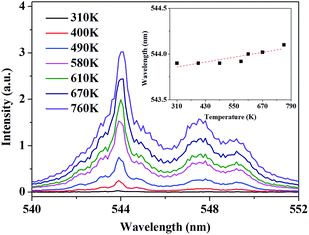
According to this inspiration, we investigated the temperature-dependent emission of 5D4–7F6 transition of Tb3+, as well as 5F5–5I8 transition of Ho3+ in our as-prepared YPO4:Tb3+,Ho3+ powder sample under excitation of 543.9 nm, which suits well to the resonance absorption of Tb3+ at room temperature. In order to ensure strong emission intensity of Tb3+ and effectively reduce the energy transfer between Tb3+ and Ho3+, we choose 3.2% and 0.8% as the doping concentration of Tb3+ and Ho3+, respectively. The emission spectra were measured at various temperatures between 310 K and 760 K, ranging from 470.0 to 510.0 nm and from 630.0 to 670.0 nm which corresponds to the transition of 5D4–7F6 of Tb3+ and 5F5–5I8 of Ho3+, respectively. As shown in Fig. 4, under 543.9 nm excitation, the absolute luminescence intensity of Tb3+ enhances monotonously with increasing temperature until 730 K and nearly keeps invariant when the temperature further rises to 760 K.
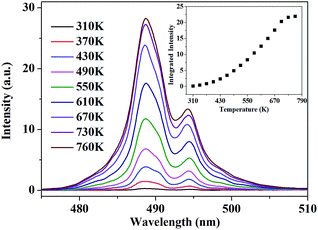 |
| | Fig. 4 PL spectra of 5D4–7F6 transition under 543.9 nm excitation at various temperature. The inset shows the variation of the integrated fluorescence intensity with temperature. | |
To quantitatively present the temperature-dependent variation, we obtained the integrated fluorescence intensity ranging from 470.0 nm to 510.0 nm of 5D4–7F6 transition of Tb3+. The experimental data are fitted with the Arrhenius formula:1
| |
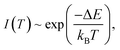 | (1) |
where
kB is Boltzmann constant. An effective energy difference Δ
E = 1846 cm
−1 is obtained. This value is smaller than the energy gap between
7F
5 and
7F
6 of Tb
3+, which is estimated to be 2100 cm
−1 from the emission spectrum.
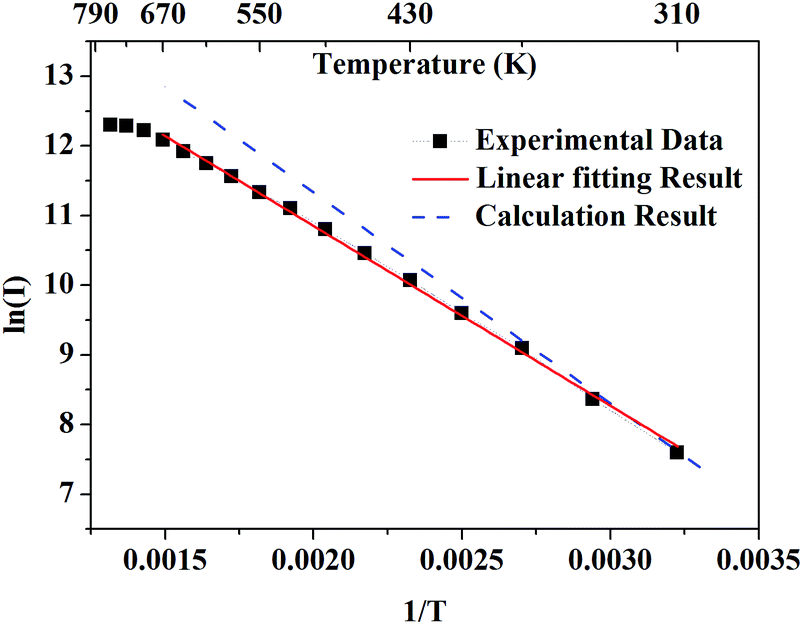
Fig. 5 presents the logarithmic integrated fluorescence intensity in response to the inverse of temperature. An obvious deviation from the calculated energy gap can be explicitly observed. This is due to the fact that apart from the variation of population of
7F
5 of Tb
3+, some other factors such as variations with temperature of absorption efficiency and luminescent quantum efficiency need to be taken into account.
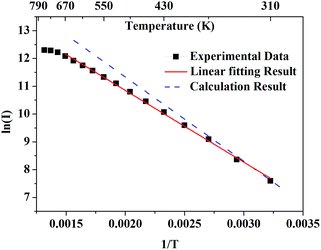 |
| | Fig. 5 The black solid rectangles present the logarithmic integrated fluorescence intensity in response to the inverse of temperature. The red solid line presents the linear fitting result of experimental data. The blue dashed line presents the theoretical results calculated from the emission spectrum. | |
The obvious deviation is mainly linked to the variation of excitation efficiency at various temperature. Fig. 6 shows the PLE spectra at various temperature. The inset presents that the peak position of PLE spectrum witnesses a slight red-shift (about 2 Å) as temperature increases. Due to the sharp spectral shape in PLE spectra, the proportion of 543.9 nm in the whole excitation range can be largely influenced by the shift of temperature-dependent excitation peaks, leading to the declining excitation efficiency contributing to the 5D4–7F6 emission of Tb3+.
 |
| | Fig. 6 The PLE spectra monitored at 488.0 nm at various temperature. The Inset shows the shift of peak position with increasing temperature. | |
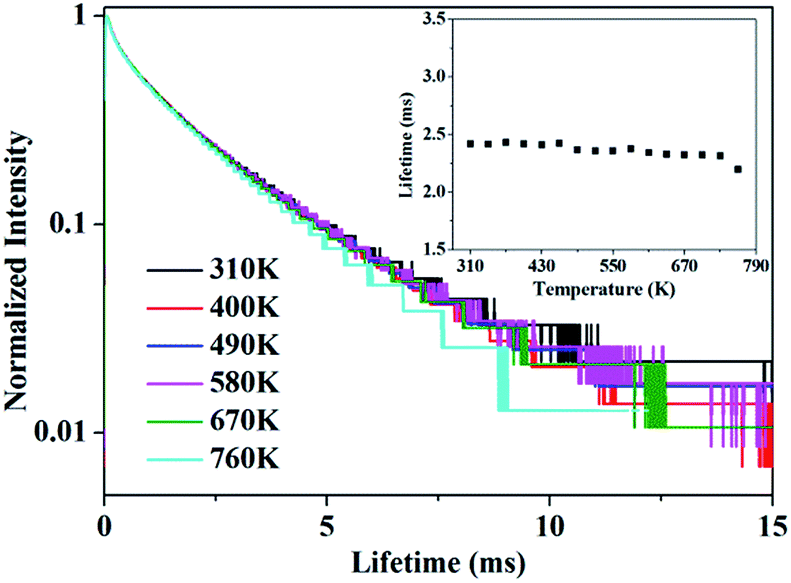
In addition, the decay curves depending on temperature is given in Fig. 7. The fluorescence lifetime of 5D4 of Tb3+ can be determined from the decay curves by monitoring the emission of 5D4–7F5 at 543.9 nm under the excitation of 7F6–5D4 at 488.0 nm. Fig. 7 shows the normalized decay curves at a series of temperatures, which presents that the lifetime of 5D4 nearly keeps invariant over the whole temperature range concerned, indicating there is little thermal quenching of Tb3+ and weak energy transfer between Tb3+ and Ho3+. The decay curve can be well fitted by double exponential equation:
| | |
I(t) = Aexp(−t/τ1) + Bexp(−t/τ2),
| (2) |
where
I is the luminescence intensity,
A and
B are constants,
t is the time,
τ1 and
τ2 are the decay time for the two exponential components, respectively. The result shows that the fluorescence decay curve consists of fast and slow decay components, which may due to the different doping concentration distribution in our sample.
23 The average lifetime can be calculated by the following formula:
| |
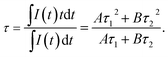 | (3) |
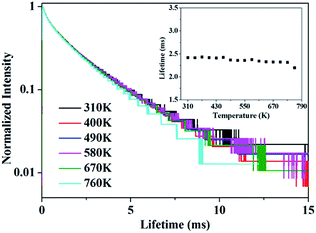 |
| | Fig. 7 The decay curve of 5D4 energy level of Tb3+ when monitored at 543.9 nm under 488.0 nm excitation. The inset shows the variation of luminescence lifetime of 5D4 with various temperature. | |
The calculated average lifetime is obtained to be ∼2.40 ms at the temperature of 310–760 K.
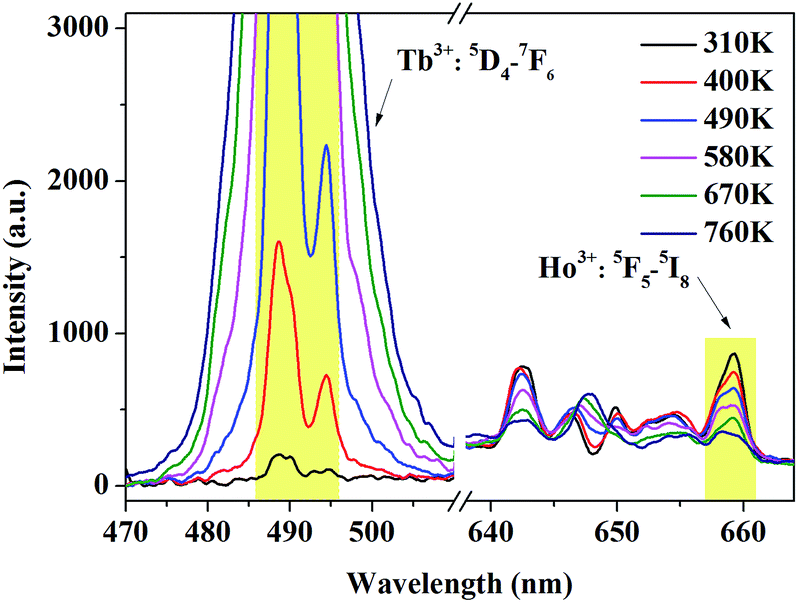
It can be concluded from Fig. 6 that, more dramatic variation of the emission of Tb3+ which contributes to larger FIR value will be obtained if we adjust the excitation wavelength from 543.9 nm to 544.1 nm. Under 544.1 nm excitation, the emission spectra ranging from 470.0 to 510.0 nm and 630 to 670 nm were measured at various temperatures ranging from 310 K to 760 K corresponding to 5D4–7F6 transition of Tb3+ and 5F5–5I8 transition of Ho3+, respectively. From Fig. 8, it can be explicitly observed that the luminescence intensity of Tb3+ witnesses a monotonous increase while the tendency of Ho3+ gradually decrease with rising temperature. As temperature increases, the enhancing luminescence intensity of Tb3+ is mainly linked with the larger population in 7F5, while the declining intensity of Ho3+ is appropriately accounted by the common multiple-photon relaxation process between 5F5 and 5I4 when given the large phonon energy of YPO4 (about 1080 cm−1) and small energy difference between 5F5 and 5I4 (less than 2400 cm−1).24
 |
| | Fig. 8 Temperature-dependent absolute intensity of 5D4–7F6 transition of Tb3+ and 5F5–5I8 transition of Ho3+ under 544.1 nm excitation. The integrated range to obtain FIR is selected as 486.0–496.0 nm for Tb3+ and 657.0–661.0 nm for Ho3+. | |
To effectively reduce the effect of noise and avoid the overlap of emission between Tb3+ and Ho3+, we choose the integrated area of a certain range within 486.0–496.0 nm of Tb3+ and 657.0–661.0 nm of Ho3+ rather than a whole range. The integrated fluorescence intensity from these two ranges were calculated to quantitatively depict the temperature-dependent variation, and the FIR between Tb3+ and Ho3+ depending on temperature is presented in Fig. 9. To better understand the temperature sensing performance, it is of great significance to investigate the sensing sensitivity for an optical thermometer. The relative sensitivity SR is a key parameter to evaluate the property of the sensors, which is defined as the relative change of the FIR (denoted as R) in response to the variation of temperature, which can be obtained by the following equation:
| |
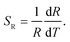 | (4) |
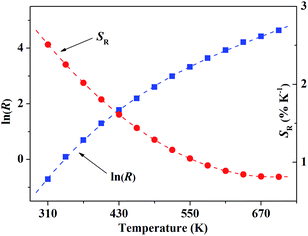 |
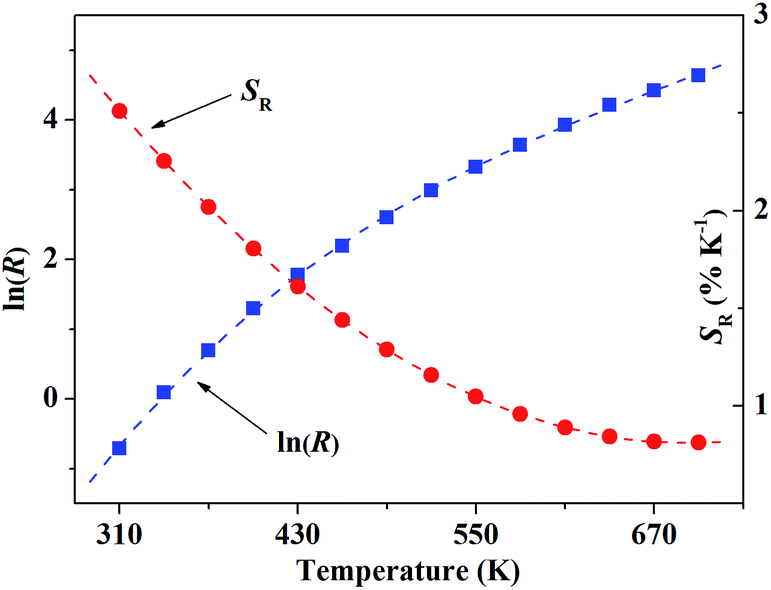
| | Fig. 9 The blue solid rectangles present ln(R) in response to the temperature. The red dashed line is the polynomial fitting curve, which just describe the variation tendency of the data. | |
Fig. 9 shows the relationship of ln(R) and SR in response to the temperature T. As many factors have to be considered in our case, the variation of FIR cannot be quantitatively described by a simple model, so the data was dealt with polynomial fitting just to present the variation tendency of ln(R) in response to T.
According to formula (4), the corresponding relative sensitivity for our sample is given within the temperature range of 310–700 K, which is displayed in Fig. 8 as well. The result of polynomial fitting shows the SR can be described approximately with
| | |
SR = 0.06378 − (16.07 × 10−5)T + (11.60 × 10−8)T2
| (5) |
The calculated SR value reaches around 2.60% K−1 at T = 300 K. Several typical temperature sensors based on FIR technique and their relative sensitivity were displayed in Table 1. Compared with other previously reported rare-earth ions doped temperature sensors, the sensitivity of our sample is much higher, especially at room temperature.15–20,25,26 The result indicates that YPO4:Tb3+,Ho3+ can be a potentially excellent candidate for optical temperature sensors with high relative sensitivity.
Table 1 Compared with other previously reported rare-earth ions doped temperature sensors, the sensitivity of our sample is much higher, especially at room temperature.15–20,25,26 The result indicates that YPO4:Tb3+, Ho3+ can be a potentially excellent candidate for optical temperature sensors with high relative sensitivity
| Materials |
The range of T (K) |
SR max (% K−1) |
SR at 300 K (% K−1) |
| PKBAN glass:Nd3+ (ref. 15) |
300–450 |
1.50 |
1.50 |
| NaLuF4:Yb3+,Tm3+,Gd3+ (ref. 16) |
300–460 |
1.26 |
0.74 |
| Y4Al2O9:Dy3+ (ref. 17) |
300–420 |
1.60 |
1.60 |
| Y2O3:Yb3+,Ho3+ (ref. 18) |
299–419 |
1.19 |
1.19 |
| AgLa(MoO4)2:Yb3+,Er3+ (ref. 19) |
298–458 |
1.26 |
1.26 |
| NaYF4:Yb3+,Tm3+,Pr3+ (ref. 20) |
300–460 |
1.53 |
0.42 |
| LiAl5O3:Cr3+ (ref. 25) |
300–480 |
0.83 |
0.21 |
| NaGd(MoO4)2:Tb3+,Pr3+ (ref. 26) |
303–483 |
2.05 |
0.61 |
| YPO4:Tb3+,Ho3+ |
310–550 |
2.60 |
2.60 |
4. Conclusion
We proposed a scheme for optical temperature sensing and implemented it with the material YPO4:3.2%Tb3+,0.8%Ho3+. In our sample, the relative sensitivity SR can reach the value of 2.60% at 300 K, which is superior to many other common temperature sensing materials, which indicates that YPO4:Tb3+,Ho3+ can be a potential candidate as optical temperature sensor. The sample YPO4:Tb3+,Ho3+ based on the FIR between 5D4–7F6 of Tb3+ and 5F5–5I8 of Ho3+ serves as a way of temperature sensing not only bearing the advantages of both UC luminescence and FIR techniques but also dramatically enhancing the fluorescence quantum yield, which presents excellent temperature-dependent luminescence properties with a high sensing sensitivity. Furthermore, as the scheme is mainly based on the energy structure of Tb3+ and Ho3+ in our case, host materials and doping ions can be further explored to optimize the luminescence properties and sensing sensitivity.
Acknowledgements
The authors are thankful for the financial support of National Natural Science Foundation of China (Grants no. 11374291, 11204292, 11274299, 11574298, 11404320 and 11604037) and the National Key Research and Development Program of China (2016YFB0701001).
Notes and references
- X. D. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- S. L. Shinde and K. K. Nanda, Angew. Chem., Int. Ed., 2013, 52, 11325–11328 CrossRef CAS PubMed.
- S. H. Zheng, W. B. Chen, D. Z. Tan, J. J. Zhou, Q. B. Guo, W. Jiang, C. Xu, X. F. Liu and J. R. Qiu, Nanoscale, 2014, 6, 5675–5679 RSC.
- X. J. Xue, F. Wang and X. G. Liu, J. Mater. Chem., 2011, 21, 13107–13127 RSC.
- O. A. Savchuk, G. P. Haro, J. J. Carvajal, D. Jaque, J. Massons, O. M. Aguil and F. Díaz, Nanoscale, 2014, 6, 9727–9733 RSC.
- E. H. Song, S. Ding, M. Wu, S. Ye, F. Xiao, G. P. Dong and Q. Y. Zhang, J. Mater. Chem. C, 2013, 1, 4209–4215 RSC.
- S. S. Zhou, X. Y. Li, X. T. Wei, C. K. Duan and M. Yin, Sens. Actuators, B, 2016, 231, 641–645 CrossRef CAS.
- M. K. Mahata, K. Kumar and V. K. Rai, Sens. Actuators, B, 2015, 209, 775–780 CrossRef CAS.
- D. Q. Chen, Z. Y. Wan, Y. Zhou, X. Z. Zhou, Y. L. Yu, J. S. Zhong, M. Y. Ding and Z. G. Ji, ACS Appl. Mater. Interfaces, 2015, 7, 19484–19493 CAS.
- S. A. Wade, S. F. Collins and G. W. Baxter, J. Appl. Phys., 2003, 94, 4743–4756 CrossRef CAS.
- O. A. Savchuk, J. J. Carvajal, C. Cascales, M. Aguilo and F. Díaz, ACS Appl. Mater. Interfaces, 2016, 8, 7266–7273 CAS.
- K. Z. Zheng, Z. Y. Liu, C. J. Lv and W. P. Qin, J. Mater. Chem. C, 2013, 1, 5502–5507 RSC.
- A. Pandey, V. K. Rai, V. Kumar and H. C. Swart, Sens. Actuators, B, 2015, 209, 352–358 CrossRef CAS.
- Z. L. Xiu, Z. S. Yang, M. K. Lu, S. W. Liu, H. P. Zhang and G. J. Zhou, Opt. Mater., 2006, 29, 431–434 CrossRef CAS.
- P. Rodríguez, L. L. Martín, S. F. L. Luis, I. R. Martín, K. K. Kumar and C. K. Jayasankar, Sens. Actuators, B, 2014, 195, 324–331 CrossRef.
- K. Zheng, Z. Liu, C. Lv and W. Qin, J. Mater. Chem. C, 2013, 1, 5502–5507 RSC.
- Z. Boruc, M. Kaczkan, B. Fetlinski, S. Turczynski and M. Malinowski, Opt. Lett., 2012, 37, 5214–5216 CrossRef CAS PubMed.
- A. Pandey and V. K. Rai, Dalton Trans., 2013, 42, 11005–11011 RSC.
- T. Li, C. F. Guo, S. S. Zhou, C. K. Duan and M. Yin, J. Am. Ceram. Soc., 2015, 1–5 Search PubMed.
- S. Zhou, G. Jiang, X. Li, S. Jiang, X. Wei, Y. Chen, M. Yin and C. Duan, Opt. Lett., 2014, 39, 6687–6690 CrossRef CAS PubMed.
- J. Cao, X. Li, Z. Wang, Y. Wei, L. Chen and H. Guo, Sens. Actuators, B, 2016, 224, 507–513 CrossRef CAS.
- R. Capelletti, A. Baraldi, E. Buffagni, M. Mazzera, N. Magnani, E. M. Rodriguez, J. G. Solé and M. Bettinelli, Spectrosc. Lett., 2010, 43, 382–388 CrossRef CAS.
- J. Y. Sun, X. Y. Zhang, Z. G. Xia and H. Y. Du, J. Appl. Phys., 2012, 111, 013101 CrossRef.
- O. K. Moune, M. D. Faucher and N. Edelstein, J. Lumin., 2002, 96, 51–68 CrossRef CAS.
- X. Y. Li, G. C. Jiang, S. S. Zhou, X. T. Wei, Y. H. Chen and C. K. Duan, Sens. Actuators, B, 2014, 202, 1065–1069 CrossRef CAS.
- Y. Gao, F. Huang, L. Hang, J. Zhou, J. Xu and Y. S. Wang, Adv. Funct. Mater., 2016, 26, 3139–3145 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Min Yin
*a and
Min Yin