DOI:
10.1039/C6RA27911B
(Paper)
RSC Adv., 2017,
7, 15158-15167
SelW protects against H2O2-induced liver injury in chickens via inhibiting inflammation and apoptosis
Received
8th December 2016
, Accepted 14th February 2017
First published on 7th March 2017
Abstract
Selenium (Se) is recognized as a necessary trace mineral in animal diets. Se deficiency induces a number of diseases and injuries in chickens including liver damage, which is related to oxidative stress. Selenoprotein W (SelW) plays a crucial role in antioxidant defense mechanisms in mammals; however, little is known about the role of SelW and cytokines in H2O2-mediated chicken liver damage since chicken SelW has no cysteine (Cys) at residue 37 (Cys37) that is required for its presumed antioxidant function in mammals. The aim of this study was to evaluate whether chicken SelW possessed the same function as mammal SelW and to investigate the protective role of SelW against H2O2 by influencing the expression of inflammation and apoptosis via building a model of overexpressed or/and knocked down SelW in cultured chicken hepatic cells. The results showed that after the cells were exposed to H2O2, the levels of P53, caspase-3, Bax, Bak, COX-2, PTGEs, TNF-α, iNOS and NF-κB were lower in the SelW overexpressed cells but higher in the SelW knockdown cells compared to the H2O2-only treatment group. However, the result for Bcl-2 was opposite to that of the other apoptosis-promoting genes. Moreover, chickens were fed with a Se-deficient diet containing 0.032 mg kg−1 Se for 15, 25, 35, 45, and 55 days, and their liver tissues were collected and examined for apoptosis and inflammation. The results showed that the levels of P53, Bax, Bak, COX-2, PTGEs, TNF-α, iNOS and NF-κB in the Se-deficient chicken livers were dramatically higher than the levels in the normal control group. In conclusion, our findings suggested that chicken SelW possess an antioxidant function similar to the mammalian homologs despite the lack of Cys37 in the peptide, and SelW regulated related-inflammation and apoptosis during H2O2-dependent chicken liver damage.
Introduction
The essential trace element selenium (Se) plays an important antioxidant role in the biosphere.1 The biological function of Se is primarily implemented through its presence in a family of Se-containing proteins, known as selenoproteins.2 Se deficiency can induce oxidative stress, inflammation, and apoptosis and can also affect the expression of selenoproteins in several tissues.3 The liver, as one of the main organs of Se absorption, is the tissue targeted by Se deficiency.4 Se deficiency can decrease the liver-elimination abilities of toxic substances, which causes liver damage characterized by hepatomegaly or liver atrophy, a rough surface with red and yellow, liver cells having fatty degeneration, cytoplasm disappearance, and diffuse cellular debris.5,6
The production of reactive oxygen species (ROS) is a natural consequence of a variety of essential biochemical reactions; however, excessive ROS is implicated in the pathogenesis of several diseases, such as alcoholic liver diseases,7 non-alcoholic fatty liver disease,8 liver fibrosis9 and hepatitis.10 ROS not only initiates MAPK activation, but also potently regulates the pro-inflammatory transcription factor NF-κB.11 ROS stimulates the production of pro-inflammatory cytokines (tumour necrosis factor alpha (TNF-α)), iNOS/NO, and COX-2/PTGE.12 NF-κB also has been shown to negate the apoptotic pathway by regulating the expression of anti-apoptotic genes, including the caspase-3 inhibitors XIAP and FLIP.13 The Bcl-2 family includes proapoptotic (e.g., Bax, Bak, Bcl-Xs) and anti-apoptotic (e.g., Bcl-2, Bcl-XL) members that coexist harmoniously at rest. Anti-apoptotic Bcl-2 and Bcl-XL inhibit Bax activation by restricting its mitochondrial translocation and hindering its accessibility to pro-apoptotic signals, respectively.14,15 In addition to the Bcl-2 family, P53 also has the ability to regulate and control apoptosis by oxidative stress.16
Se-dependent proteins in human and animal organisms include selenoprotein W, which are one of the selenoproteins having important biochemical functions. SelW binds glutathione17 and has been suggested to act as an antioxidant in vivo.18,19 The expression of mutant SelW in which selenocysteine 13 or cysteine 36 were replaced by serine did not confer resistance to H2O2, implicating the antioxidant activity of SelW in mice.20 Cells that have an overexpression of SelW were found to be more resistant to oxidative stress.21 However, the sequences of SelW among humans, mice, and chickens are different; for instance, the mouse SelW has glutathione bound to it22 and subsequently has been shown to be bound to residue number 37 cysteine, using the primate SelW;23 whereas, the chicken SelW does not contain cysteine at residue number 37 that is necessary for the anti-oxidative function in rats.24 However, whether chicken SelW possesses the same function is still unclear.
This study was conducted to determine whether and how the chicken SelW gene plays a role in protecting the chicken liver against oxidation stress. First, the over-expressed and knockdown SelW hepatocytes of chicken were cultured; furthermore, the extent of the H2O2-induced apoptosis by TUNEL, the abundance of apoptosis and inflammation factors mRNA by q-PCR and proteins by western blot techniques were observed. Second, to further detect the function of SelW, a model of chicken liver damage induced by Se deficiency was built, and then the liver tissue at different time points were collected to detect the mRNA and protein levels of apoptosis and inflammation-related genes. The present study provides some data about the effect of SelW on regulating inflammation and apoptosis in chicken liver induced by oxidative stress.
Materials and methods
Ethics statement
All chicken experiments were approved by the Institutional Animal Care and Use Committee of Northeast Agricultural University, under the approved protocol number SRM-06.
Hepatocyte culture and treatment
Chickens were fed on a commercial stock diet (proteins 20%, lipids 2.5%, carbohydrates 57.5%, minerals 6%, moisture 14%), pre-anesthetized by an intramuscular injection (83 μg kg−1), and anesthetized by an injection of a mixture of natrium thiopental (17.5 mg kg−1) and heparin (1750 U kg−1). The methods of hepatocyte isolation and culture were according to our prior study.25 The liver cells were grown at 37 °C in 5% CO2 atmosphere in William's E supplemented with 10% FBS (Sigma, USA). One-third of the cells comprised the control group, while another one-third of the hepatic cells were transiently transfected with pcDNA3.1/SelW (O-SeW) (Table 1) using X-treme GENE HP DNA transfection reagent (Roche, USA), and the remaining one-third of the cells were knock downed with siRNA (K-SelW) (Table 2) using Lipofectaminet mRNAi MAX (Invitrogen, USA) in accordance with the manufacturer's recommendations. For SelW overexpression experiment, the best transfection optimization condition is that 8 μL transfection reagents add 2 μg pcDNA3.1/GFP and the transfection efficiency is more than 80%. For the SelW knockdown experiment, the transfection optimization condition was 0.9 μL S-RNA + 1.5 μL RNAiMAX and the transfection efficiency was about 75%. After transfection for 24 h (the highest/lowest SelW mRNA levels), the cells were analyzed with an inverted microscope and were treated with 0.2 mM H2O2 (IC50 was 0.2 mM) for 6 h, 12 h, 18 h, and 24 h, respectively, and were then harvested for analysis.
Table 1 Primers for qPCR
| Gene (accession number) |
Primer sequences (5′–3′) |
Product (bp) |
| SelW (GQ919055) |
Forward: TCTAAGCTTAGTGCCATCGCCATGCCGCT |
473 |
| Reverse: CGGAATTCTGGCGTAAAGCAGACCCACACC |
| β-Actin (U20114) |
Forward: GGCTCCCAGCACCATGAA |
185 |
| Reverse: GCCACCGATCCACACAGAGT |
Table 2 S-RNA Sequences for SelW RNA
| S-RNA (sequence location) |
Primer sequences (5′–3′) |
| SelW_stealth (268) |
Forward: CGGCUUCGUGGACACCGACGCCAA |
| Reverse: UUUGGCGUCGGUGUCCACGAAGCCG |
| SelW_stealth (negative control) |
Forward: CGGUCGUGGACACCGACGCCCUAAA |
| Reverse: UUUAGGGCGUCGGUGUCCACGACC |
Animal and treatment
One hundred male chickens (1 day-old; Wei Wei Co. Ltd, Harbin, China) were kept in an air-conditioned animal house with a normal day/night cycle and randomly divided into 10 groups (10 chicken per group): five control groups were fed basal diet and five treatment groups were fed a Se-deficient diet containing 0.032 mg kg−1. The feed and tap water were supplied ad libitum. On 15, 25, 35, 45 and 55 days, following euthanasia with sodium pentobarbital, the liver tissues were quickly collected from the chickens, immediately frozen in liquid nitrogen, and subsequently stored at −80 °C before use.
Analysis of gene expressions by real-time quantitative reverse transcription PCR
Real-time quantitative reverse transcription PCR (q-PCR) was used to detect the expressions of the SelW, P53, caspase-3, Bax, Bak, Bcl-2, COX-2, PTGEs, TNF-α, iNOS, and NF-κB genes. Primer Premier Software (PREMIER Biosoft International, USA) was used to design specific primers for those based on known sequences (Table 3).
Table 3 Primers for qPCR
| Gene |
Primer sequences (5′–3′) |
Product (bp) |
| COX-2 |
Forward: TGTCCTTTCACTGCTTTCCAT |
105 |
| Reverse: TTCCATTGCTGTGTTTGAGGT |
| iNOS |
Forward: CCTGGAGGTCCTGGAAGAGT |
82 |
| Reverse: CCTGGGTTTCAGAAGTGGC |
| NF-κB |
Forward: TCAACGCAGGACCTAAAGACAT |
162 |
| Reverse: GCAGATAGCCAAGTTCAGGATG |
| TNF-α |
Forward: GCCCTTCCTGATACCAGATG |
98 |
| Reverse: ACACGACAGCCAAGTCAACG |
| PTGEs |
Forward: GTTCCTGTCATTCGCCTTCTAC |
115 |
| Reverse: CGCATCCTCTGGGTTAGCA |
| BCL-2 |
Forward: ATCGTCCGCCTTCTTCGAGTT |
150 |
| Reverse: ATCCCATCCTCCGTTGTCCT |
| P53 |
Forward: GAGATGCTGAAGGAGATCAATGAG |
145 |
| Reverse: GTGGTCAGTCCGAGGCTTTT |
| Bak |
Forward: ATGGATGCCTGTCTGTCCTGTTC |
106 |
| Reverse: GCAGAGCAGTCCAAAGACACTGA |
| Caspase-3 |
Forward: ACTCTGGAAATTCTGCCTGATGACA |
129 |
| Reverse: CATCTGCATCCGTGCCTGA |
| Bax |
Forward: GTGATGGCATGGGACATAGCTC |
90 |
| Reverse: TGGCGTAGACCTTGCGGATAA |
| SelW |
Forward: CTCCGCGTCACCGTGCTC |
150 |
| Reverse: CACCGTCACCTCGAACCATCCC |
| β-Actin |
Forward: CCGCTCTATGAAGGCTACGC |
128 |
| Reverse: CTCTCGGCTGTGGTGGTGAA |
Total cDNA from hepatic cells and liver tissue were prepared. Quantitative real-time PCR was performed on an ABI PRISM 7500 Detection System (Applied Biosystems, USA). Reactions were performed in a 20 μL reaction mixture containing 10 μL of 2 × SYBR Green I PCR Master Mix (TaKaRa, China), 2 μL of either diluted cDNA, 0.4 μL of each primer (10 μM), 0.4 μL of 50 × ROX reference dye II, and 6.8 μL of PCR-grade water. The PCR procedure for SelW, P53, caspase-3, Bax, Bak, Bcl-2, COX-2, PTGEs, TNF-α, iNOS, NF-κB, and β-actin consisted of 95 °C for 30 s, followed by 40 cycles of 95 °C for 15 s, and finally 60 °C for 30 s. The melting curve analysis showed only one peak for each PCR product. Electrophoresis was performed with the PCR products to verify primer specificity and product purity. A dissociation curve was separated for each plate to confirm the production of a single product. The amplification efficiency for each gene was determined using the DART-PCR program. The mRNA relative abundance was calculated.
Western blot assay
Briefly, protein extracts were subjected to SDS-polyacrylamide gel electrophoresis under reducing conditions on 15% gels. Separated proteins were then transferred to nitrocellulose membranes using a tank transfer for 2 h at 100 mA in tris-glycine buffer containing 20% methanol. Membranes were blocked with 5% skim milk for 1 h and incubated overnight with diluted primary antibodies (SelW, Cox-2, PTGEs, NF-κB, Bax, P53), followed by a horseradish peroxidase conjugated secondary antibody rabbit IgG (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000, Santa Cruz Biotechnology, USA) or goat (1:1000, Santa Cruz Biotechnology). To verify equal loading of samples, the membrane was incubated with a monoclonal GAPDH antibody (1:1000, Santa Cruz Biotechnology), followed by a horseradish peroxidase conjugated goat anti-mouse IgG (1:1000). The signal was detected by an X-ray film (Trans GenBiotech Co., Beijing, China).
:1000, Santa Cruz Biotechnology, USA) or goat (1:1000, Santa Cruz Biotechnology). To verify equal loading of samples, the membrane was incubated with a monoclonal GAPDH antibody (1:1000, Santa Cruz Biotechnology), followed by a horseradish peroxidase conjugated goat anti-mouse IgG (1:1000). The signal was detected by an X-ray film (Trans GenBiotech Co., Beijing, China).
Measurement of apoptosis by TUNEL assay
Apoptotic cells were identified using a terminal deoxyribonucleotide transferase-mediated dUTP nick end labeling (TUNEL) with the In Situ Cell Death Detection kit (TMR red; Roche Applied Science, USA). After treatment with H2O2 for 6 h, cells grown on coverslips (1 × 105 cells per well of a 12-well plate) were washed with PBS and fixed in paraformaldehyde solution (4% in PBS) for 30 min at room temperature. The cells were permeabilized in a solution containing 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice, followed by incubation in freshly prepared TUNEL reaction mixture for 60 min at 37 °C in the dark. The coverslips were washed with PBS. Afterwards, the DAPI stain slides were then mounted and evaluated under a fluorescence microscope.
Statistical analysis
Statistical analysis was performed using SPSS statistical software (version 13; SPSS Inc., Chicago, IL, USA) and significant values (p < 0.05) were obtained by one-way analysis of normal distribution and equal variance. All the data showed a normal distribution and passed equal variance testing. Differences between the means were assessed by Tukey's honest significant difference test for post hoc multiple comparisons. Data are expressed herein as the mean ± standard deviation. Bars that do not share a common letter were considered to be significantly different to each other (p < 0.05).
Results
Efficiency of SelW overexpression and knockdown
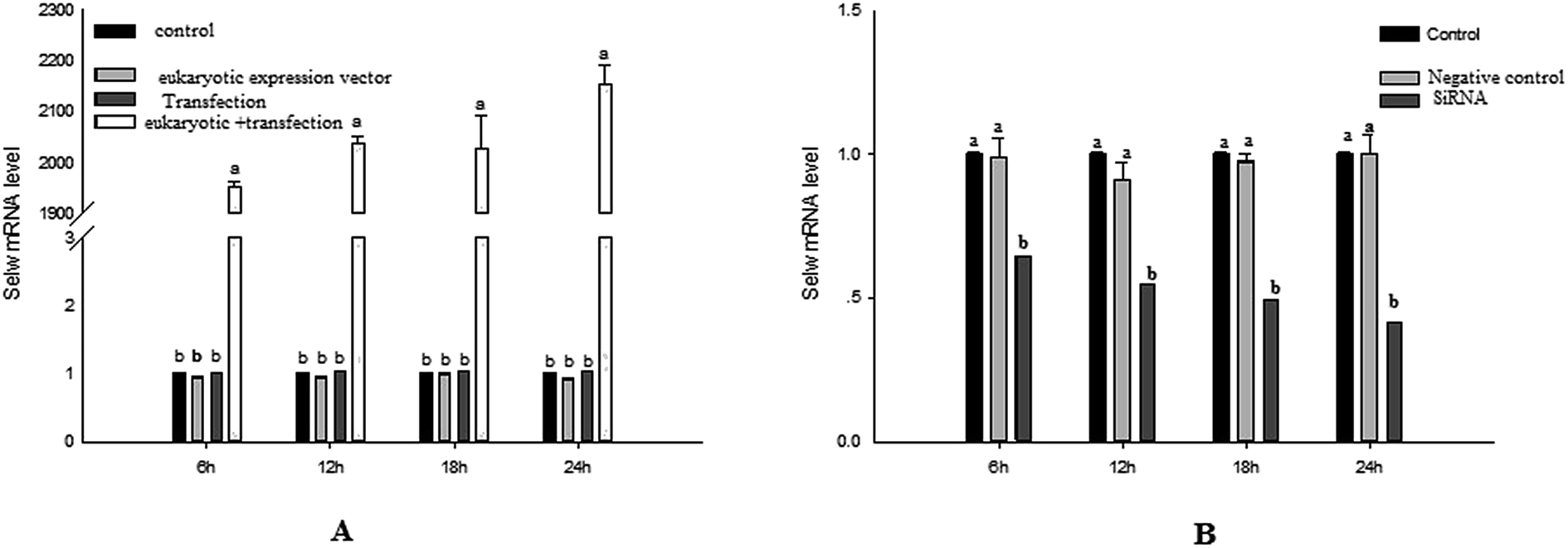
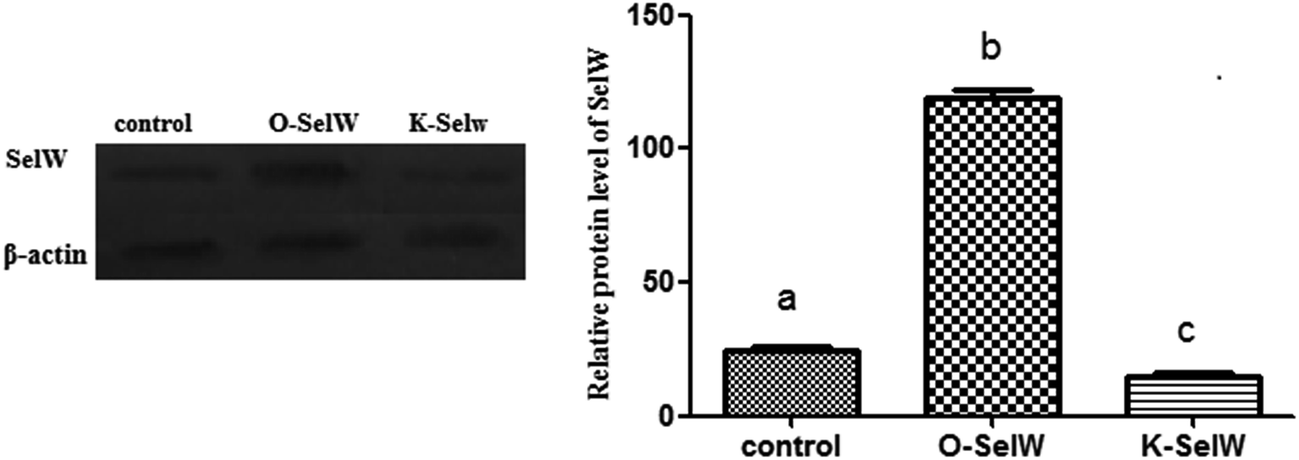
To estimate the levels of SelW in hepatic cell after overexpression and knockdown, the levels of SelW mRNA were tested by q-PCR, respectively. The results showed that the expression of SelW mRNA was much higher than the other groups in the SelW overexpression group (O-SelW), and it was highest on the 24 h time point (Fig. 1A). Moreover, it was shown that the mRNA level of SelW was lower than the other groups in the SelW knockdown group (K-SelW), and it was lowest after treatment for 24 h (Fig. 1B). Moreover, the relative level of SelW protein is shown in Fig. 2, where it can be seen it increased (p < 0.05) in the O-SelW group and decreased (p < 0.05) in the K-SelW group compared to the control group (Fig. 2).
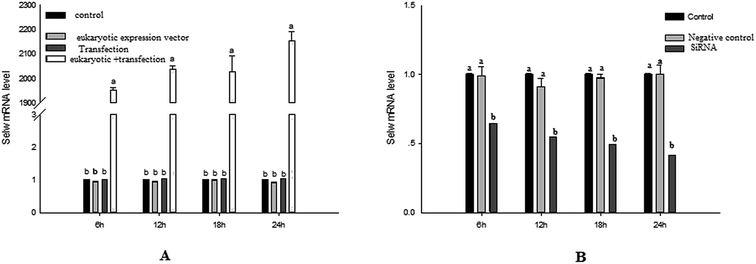 |
| | Fig. 1 The expression of SelW mRNA in chicken hepatic cells with SelW overexpression and knockdown. (A) The overexpression group, divided into four treatment groups: control group, pCDNA3.1 group, transfected SelW group, and transfected pCDNA3.1/SelW group. (B) The knockdown group, divided into three treatment groups: control group, negative control group, and transfected SelW RNAi group. Bars with different letters were significantly different (p < 0.05). The data are represented as the mean ± SD, n = 6. | |
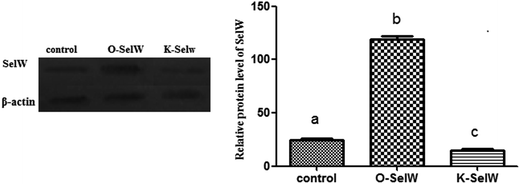 |
| | Fig. 2 The expression of SelW protein in chicken hepatic cells with SelW overexpression and knockdown. Control means hepatic cells without treatment, O-SelW means hepatic cells transfected with pCDNA3.1/SelW, and K-SelW means hepatic cells transfected with SelW RNAi. Bars with different letters were significantly different (p < 0.05). The data are represented as the mean ± SD, n = 6. | |
Effects of SelW on the expression of TNF-α, NF-κB, PTGEs, COX-2, and iNOS in chicken hepatic cells
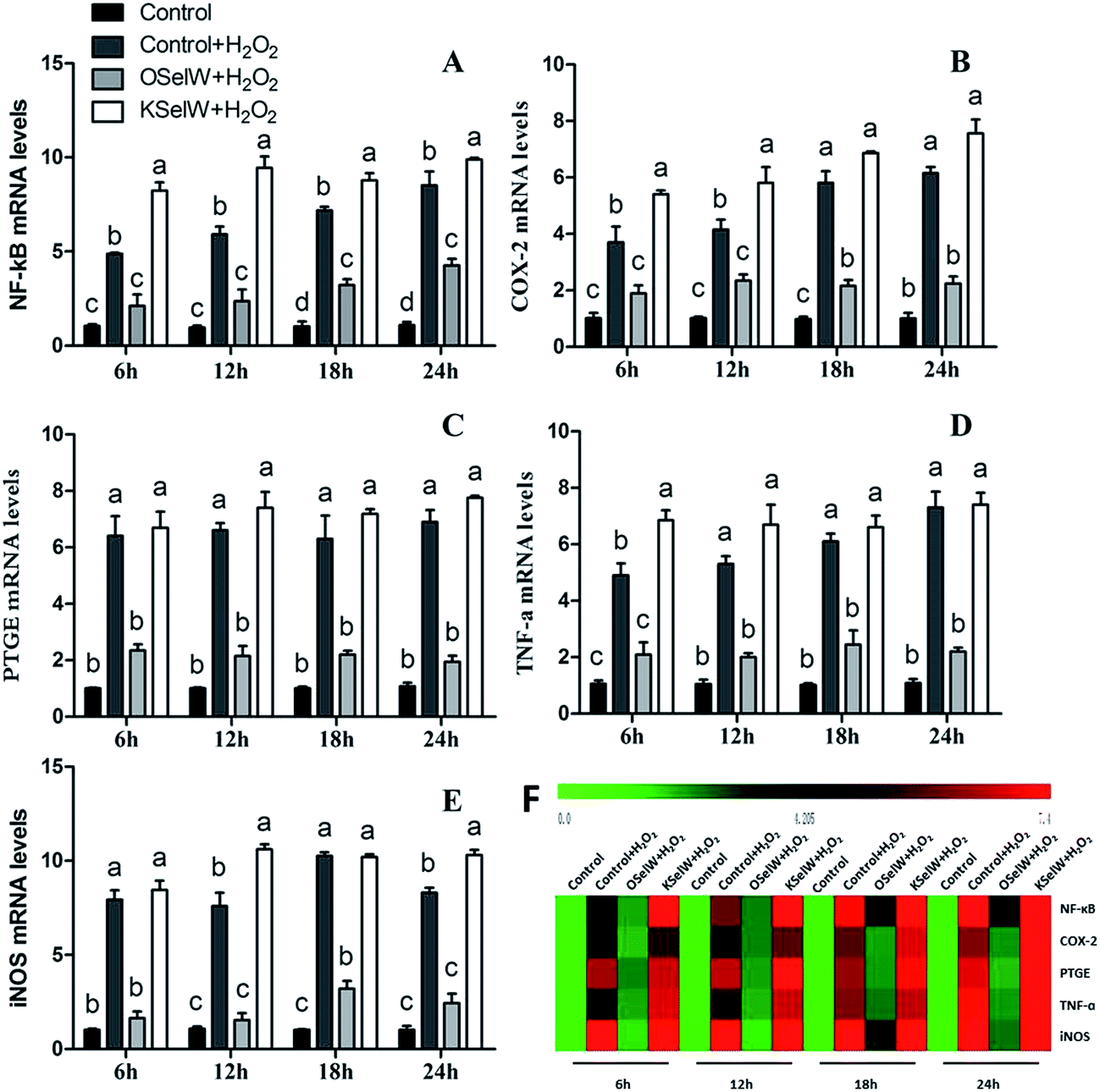
To assess whether SelW could inhibit inflammation factors expression in the liver of chickens, the mRNA levels of TNF-α, NF-κB, PTGE, COX-2, and iNOS were monitored by q-PCR. As shown in Fig. 3, the mRNA levels of TNF-α, NF-κB, PTGEs, COX-2, and iNOS were significantly lower in chicken hepatic cells in the O-SelW group (p < 0.05), whereas they were much higher in cells with K-SelW than in the normal control group at 6 h, 12 h, 18 h, and 24 h after H2O2 treatment. Treatment with O-SelW significantly decreased the levels of TNF-α, NF-κB, PTGEs, COX-2, and iNOS in the hepatic cells by more than 2-fold (Fig. 3). To explicitly present the voluminous mRNA dataset comprising all the significantly induced mRNA expression in each group and at each time point, a heat map analysis was applied based on the RT-qPCR results. The results demonstrate a clear visualization of the induction patterns of all the identified mRNAs according to the abundance differences (Fig. 3). It can be seen that K-SelW significantly increases the expression levels of the inflammation factors, while O-SelW remarkably decreases those levels.
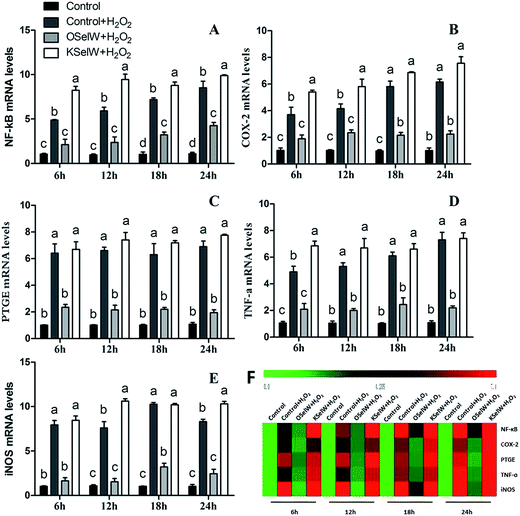 |
| | Fig. 3 Effects of SelW on the mRNA expression of NF-κB, COX-2, PTGEs, TNF-α, and iNOS in chicken hepatic cells. Control, hepatic cells without exposure to H2O2; control + H2O2, hepatic cells exposed to 0.2 mM H2O2; control + OSelW + H2O2, hepatic cells transfected pCDNA3.1/SelW before exposure to 0.2 mM H2O2, control + KSelW + H2O2, hepatic cells transfected SelW RNAi before exposure to 0.2 mM H2O2. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
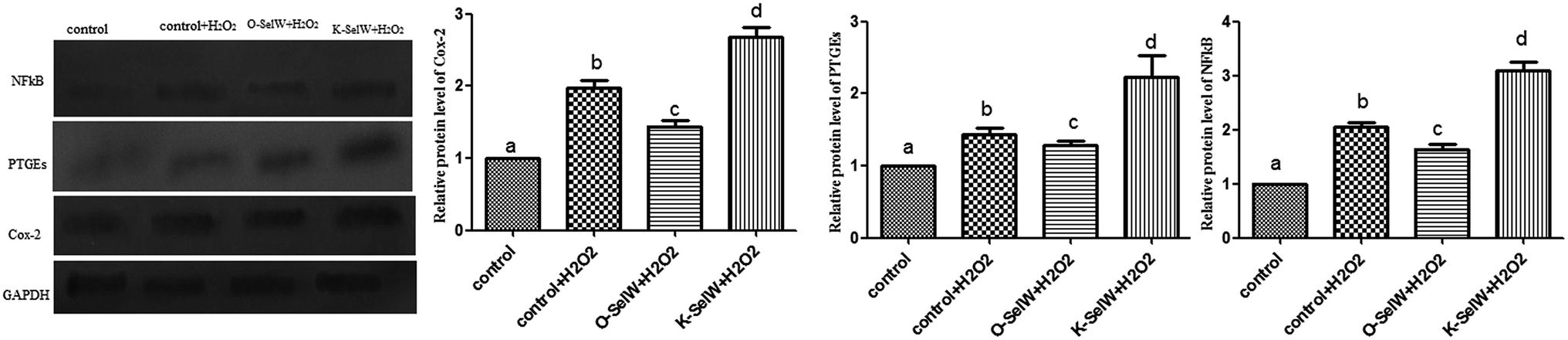
Moreover, the relative protein levels of NF-κB, PTGEs, and COX-2 are shown in Fig. 4, and it can be seen that they were also increased (p < 0.05) in the O-SelW group and decreased (p < 0.05) in the K-SelW group compared to that in the H2O2-only treatment group (Fig. 4).
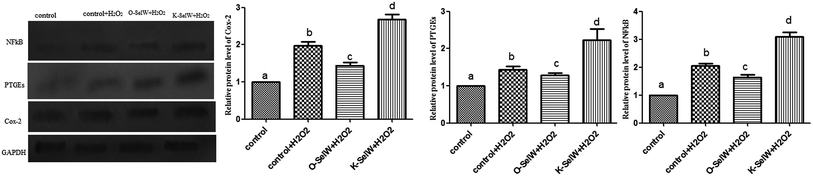 |
| | Fig. 4 Effects of SelW on the protein expression of NF-κB, COX-2, and PTGEs in chicken hepatic cells. Control, hepatic cells without exposure to H2O2; control + H2O2, hepatic cells exposed to 0.2 mM H2O2; control + O-SelW + H2O2, hepatic cells transfected pCDNA3.1/SelW before exposure to 0.2 mM H2O2, control + K-SelW + H2O2, hepatic cells transfected SelW RNAi before exposure to 0.2 mM H2O2. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
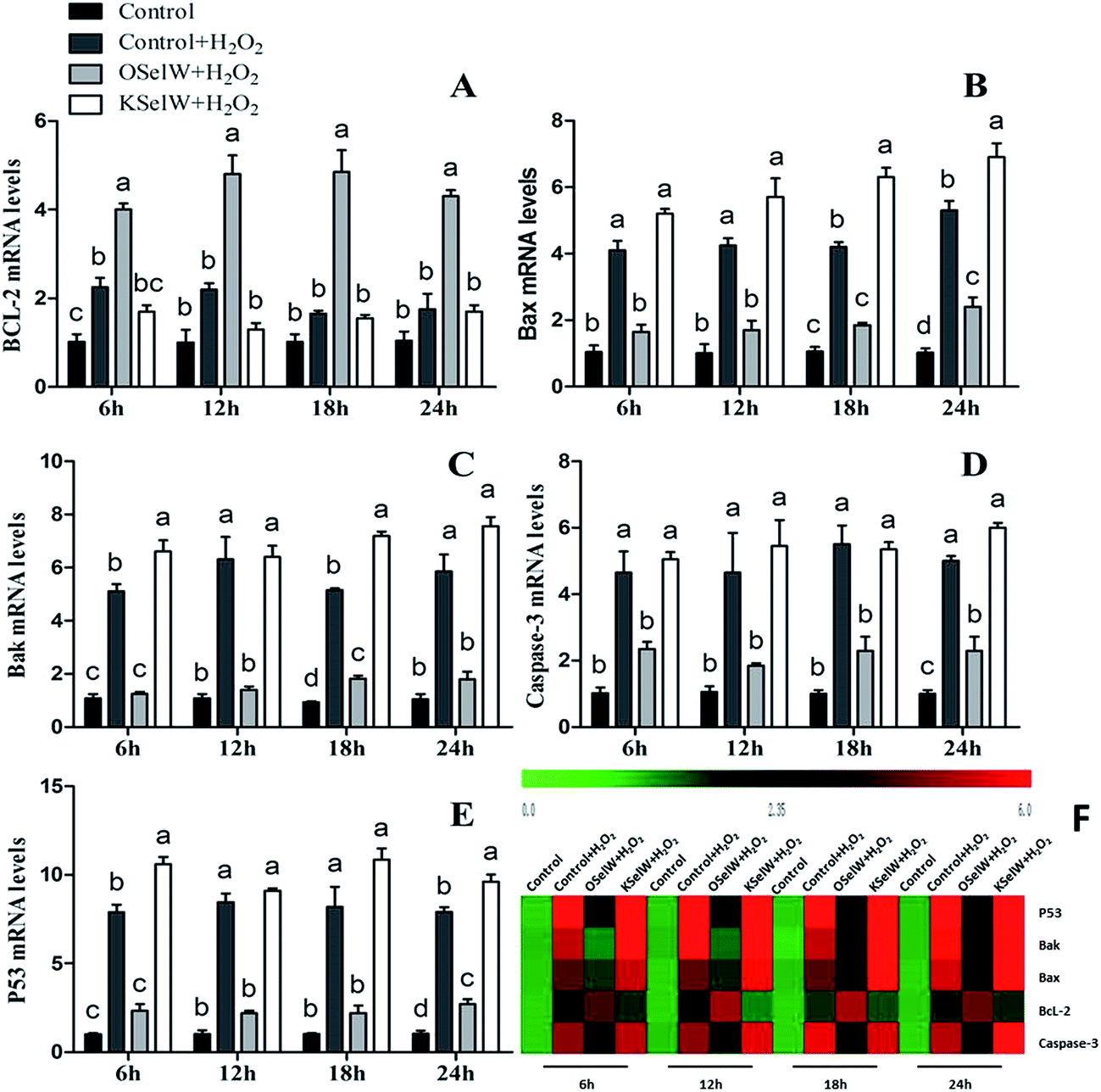
Effects of SelW on the expression of Bcl-2 family, caspase-3, and P53 in chicken hepatic cells
To determine whether SelW could affect apoptosis in chicken hepatic cells, the mRNA levels of Bcl-2, Bax, Bak, caspase-3, and P53 were tested by q-PCR. As shown in Fig. 5, there were higher mRNA levels of caspase-3, Bak, Bax, and P53 in the K-SelW treatment group and lower in O-SelW when compared with those in the corresponding control group (p < 0.05). While the mRNA level of Bcl-2 in O-SelW was higher than in K-SelW, with the increase in H2O2 treatment time, the mRNA levels of these apoptosis-related genes showed an increased trend. In the heat map, it can be seen that K-SelW could significantly increase the expression levels of the apoptosis-related genes (except Bcl-2), while O-SelW remarkably decreased the mRNA level of those genes (except for Bcl-2).
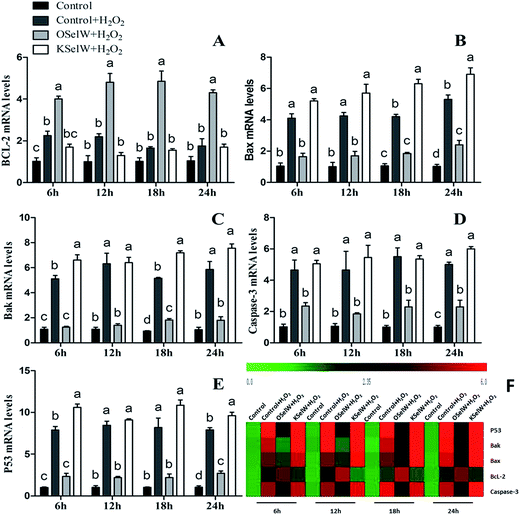 |
| | Fig. 5 Effects of SelW on the mRNA expression of Bcl-2, Bax, Bak, caspase-3, and P53 in chicken hepatic cells. Control, hepatic cells without exposure to H2O2; control + H2O2, hepatic cells exposed to 0.2 mM H2O2; control + O-SelW + H2O2, hepatic cells transfected pCDNA3.1/SelW before exposure to 0.2 mM H2O2, control + K-SelW + H2O2, hepatic cells transfected SelW RNAi before exposure to 0.2 mM H2O2. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
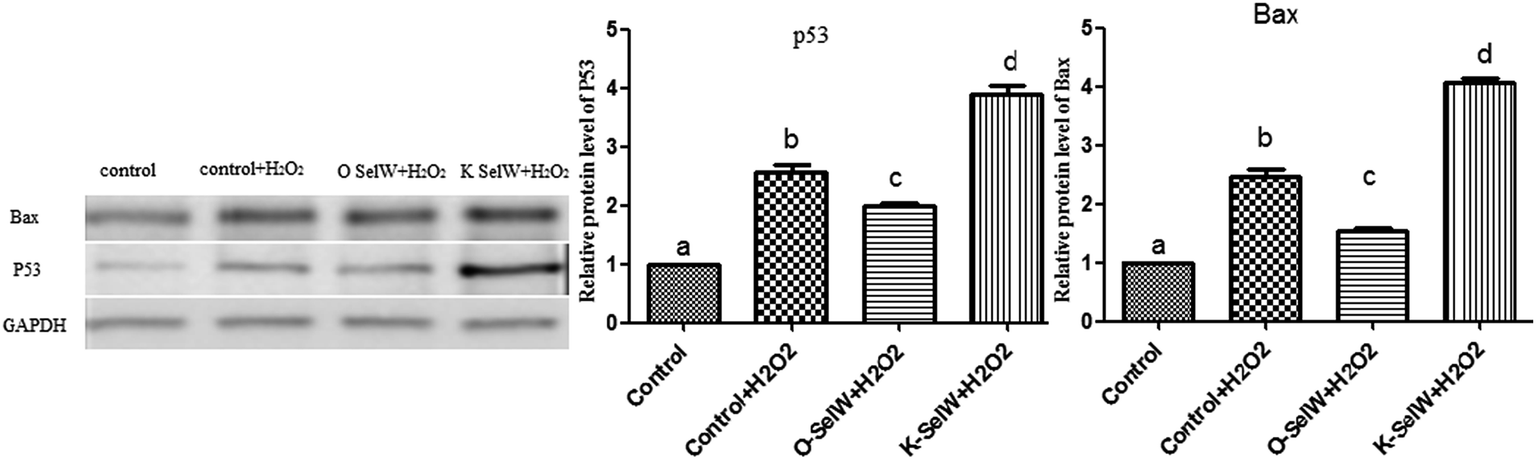
Moreover, the relative protein levels of Bax and P53 are shown in Fig. 6, and they were also increased (p < 0.05) in the O-SelW group and decreased (p < 0.05) in the K-SelW group compared to those in the H2O2-only treatment group (Fig. 6).
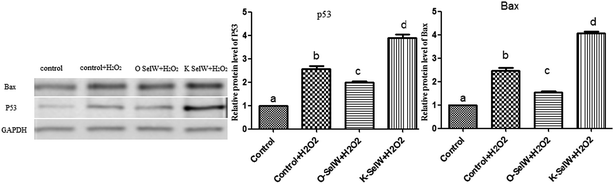 |
| | Fig. 6 Effects of SelW on the protein expression of Bax and P53 in chicken hepatic cells. Control, hepatic cells without exposure to H2O2; control + H2O2, hepatic cells exposed to 0.2 mM H2O2; control + O-SelW + H2O2, hepatic cells transfected pCDNA3.1/SelW before exposure to 0.2 mM H2O2, control + K-SelW + H2O2, hepatic cells transfected SelW RNAi before exposure to 0.2 mM H2O2. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
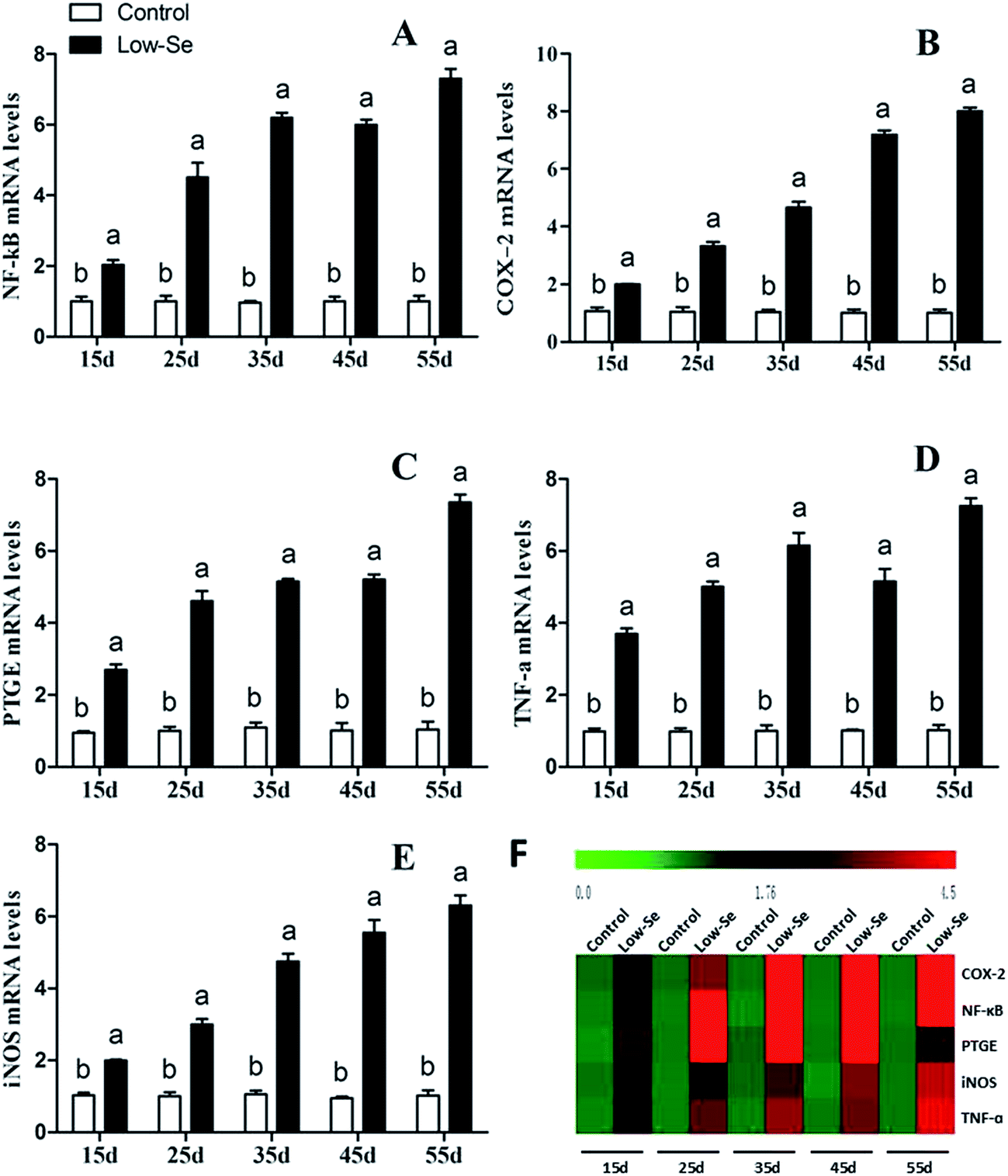
Effects of Se deficiency on the expression of TNF-α, NF-κB, PTGEs, COX-2, and iNOS in chicken liver tissue
The effects of Se deficiency on the mRNA levels of TNF-α, NF-κB, PTGEs, COX-2, and iNOS genes in chicken liver tissue are shown in Fig. 7, respectively. The levels were significantly increased over time in the low-Se treatment groups compared to that in the control group (p < 0.05). With the increase in the Se deficiency time, the mRNA levels of these inflammatory factors showed an increased tendency. In the heat map, low-Se treatment could significantly increase the expression levels of inflammatory factors.
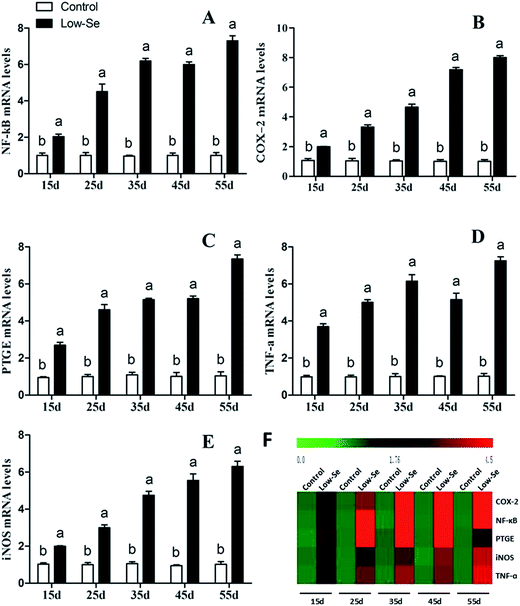 |
| | Fig. 7 Effects of the Se-deficient diet on the mRNA expression of NF-κB, COX-2, PTGEs, TNF-α, and iNOS in chicken liver tissue. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
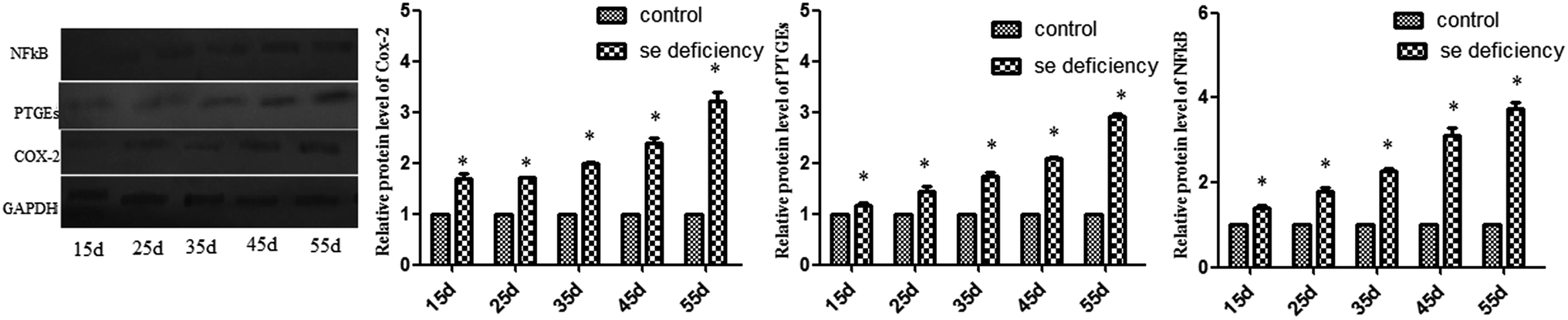
In addition, the relative protein levels of PTGEs, NF-κB, and COX-2 are shown in Fig. 8, where it can be seen that they were increased (p < 0.05) in the Se-deficient groups.
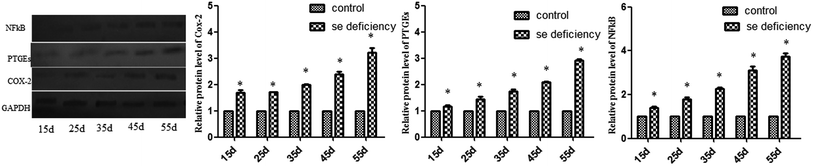 |
| | Fig. 8 Effects of the Se-deficient diet on the protein expression of COX-2, NF-κB, and PTGEs in chicken liver tissue. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
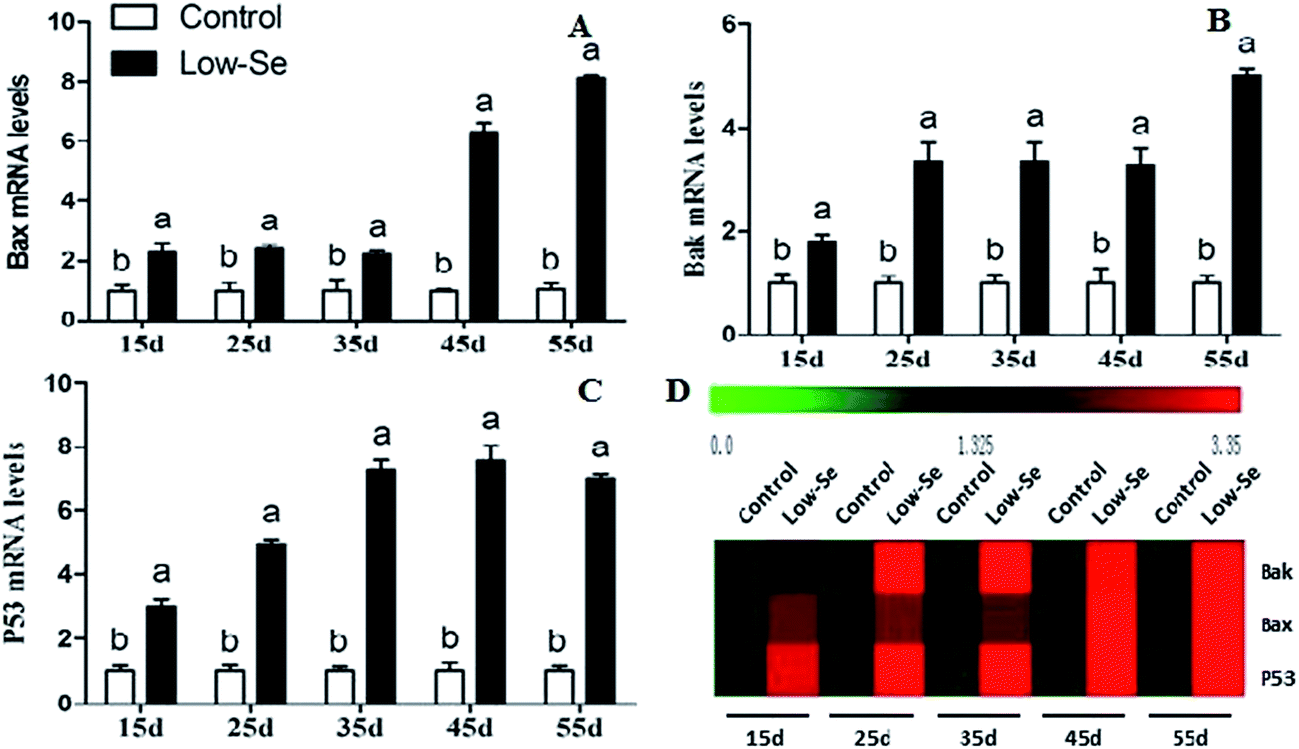
Effects of Se deficiency on the expression of Bak, Bax, and P53 in chicken liver tissue
The effects of Se deficiency on the mRNA levels of the related apoptosis gene in chicken liver are shown in Fig. 9. The mRNA levels of Bak, Bax, and P53 were significantly increased in the low-Se groups compared to the control group from 15 to 55 days. In the heat map, it can be seen that low-Se treatment could significantly increase the expression levels of the pro-apoptosis factors.
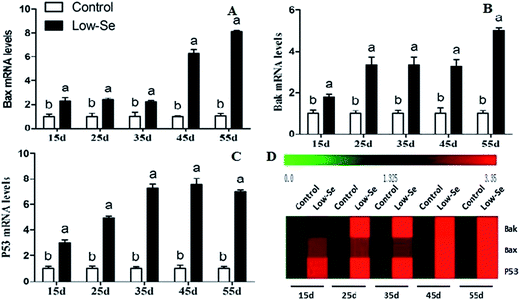 |
| | Fig. 9 Effects of the Se-deficient diet on the mRNA expression of Bax, Bak, and P53 in chicken liver tissue. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
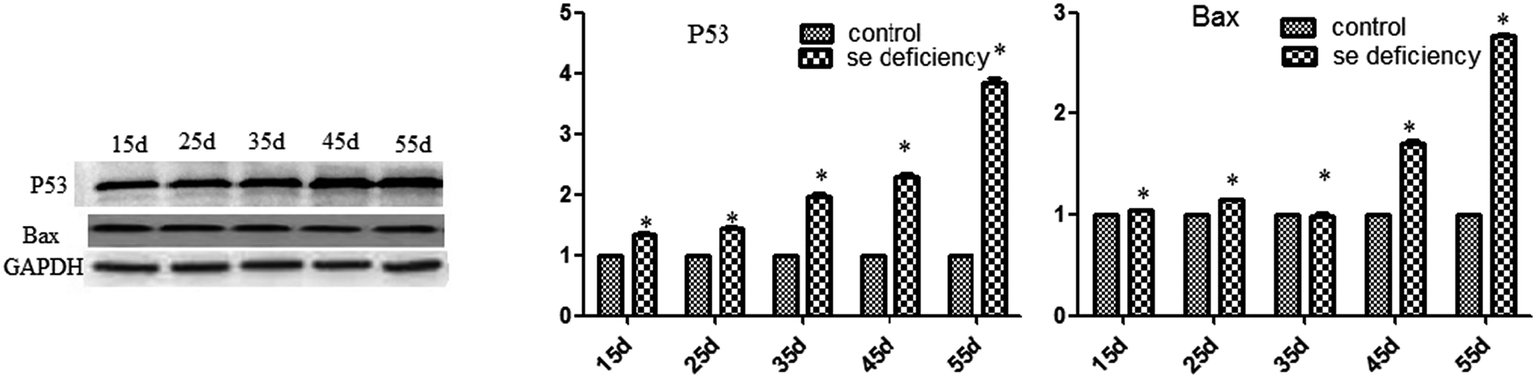
In addition, the relative protein levels of Bax and P53 are shown in Fig. 10, where it can be seen that they were increased (p < 0.05) in the Se-deficient groups, which is consistent with the mRNA level.
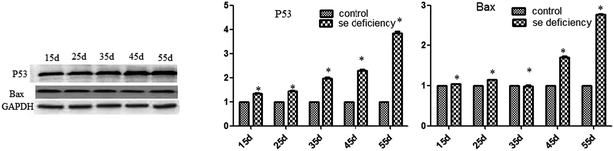 |
| | Fig. 10 Effects of the Se-deficient diet on the protein expression of Bax and P53 in chicken liver tissue. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
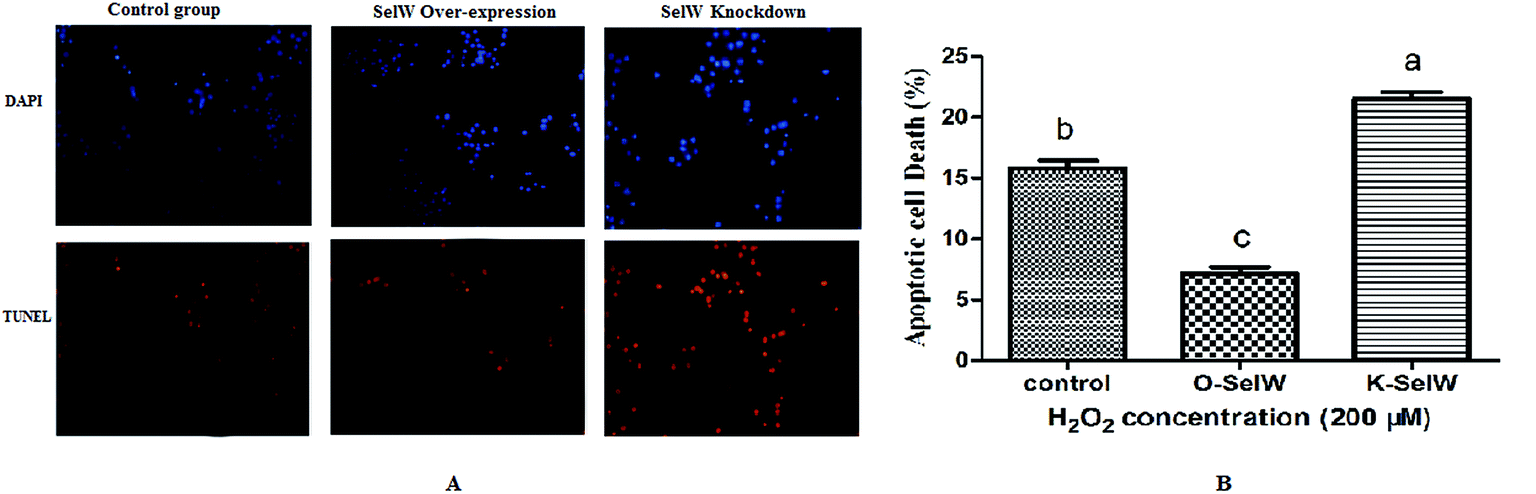
Measurement of cellular apoptosis using the TUNEL assay
We performed the TUNEL assay in chicken hepatic cells induced by H2O2 to confirm the occurrence of apoptosis. Apoptotic cells had red-stained nuclei, while living cells had blue-stained nuclei. Hepatic cells from the K-SelW group had an increased number of apoptotic cells compared to the control group and O-SelW group, which further demonstrated that SelW could protect chicken livers against apoptosis induced by H2O2 (Fig. 11). The effects of SelW on the percentage of apoptotic cells in hepatic cells are shown in Fig. 11, where it can be seen that the percentage of apoptotic cells in hepatic cells was decreased (p < 0.05) in the O-SelW group compared to that in the control group.
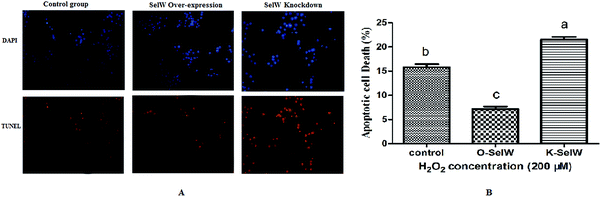 |
| | Fig. 11 The results of tunnel assay by fluorescence microscopy. Control group, hepatic cells exposed to 0.2 mM H2O2; O-SelW group, hepatic cells transfected pCDNA3.1/SelW before exposure to 0.2 mM H2O2, K-SelW, hepatic cells transfected SelW RNAi before exposure to 0.2 mM H2O2. DAPI (blue) means living cells, TUNEL (red) means apoptotic cells. Bars with different letters were significantly different (p < 0.05), and those sharing same letter were not significantly different (p > 0.05). The data are represented as the mean ± SD, n = 6. | |
Discussion
SelW serves as an antioxidant in mammals.26 Rat SelW contains Cys at residues 10 and 37, but chicken SelW has only a Cys at residue 10.24,27,28 Han utilized the chicken SelW cDNA and expressed it in CHO-K1 cells and found that an overexpression of SelW cells resulted in a significant decrease in sensitivity to oxidative stress caused by H2O2 compared with the control cells, and that the overexpression of SelW cells also had lower mRNA levels of caspase-3 and fas.29 In proliferating myoblasts exposed to H2O2, SelW caused an immediate response, suggesting that it protected the developing myoblasts from oxidative stress. Following deletion of SelW by RNA interference in the mouse skeletal muscle cell line C2C12 (C3H), the viability of the SelW siRNA-transfected group decreased significantly by 21.5% compared with non-SelW-depleted cells.30 In the current study, we successfully constructed the models for the overexpression of SelW and knockdown of SelW in chicken hepatic cells. We used an efficient overexpressed SelW sequence and siRNA sequence to successfully overexpress and suppress the gene and protein expression of chicken SelW in chicken's hepatic cells by more than 2100-fold and less than 40%, respectively. These models were the foundation for the follow-up study.
Oxidative stress could induce cell apoptosis and inflammation. H2O2, one of the key oxidative stress factors, can promote the cell inflammation and apoptosis process. The mechanisms by which H2O2 (ROS/RNS) causes or regulates apoptosis typically include caspase activation, Bcl-2 family gene and proteins.13,31 Apoptosis was associated with both intrinsic and extrinsic apoptosis pathways under the regulation of Bcl-2 family proteins and caspase-3 activation during the process.32 Oxidative stresses, such as an addition of extracellular hydrogen peroxide, could induce NF-κB nuclear translocation in several cell lines. NF-κB was distinctly induced in H2O2-treated hepatocytes.33 In addition, inhibition of the production of NO, PGE2, TNF-α, and IL-1β has been proposed to be a useful approach for the treatment of various inflammatory diseases.34,35 In the present study, we detected the inflammation- and apoptosis-related factors to clarify the functions of SelW response to oxidation stress damage. The mRNA levels of COX-2, NF-κB, TNF-α, PTGEs, iNOS Bak, Bax, caspase-3, and P53 in the O-SelW group were significant lower than in the control group, while in the K-SelW group they were higher than those in the control group. On the contrary, the upregulation of Bcl-2 mRNA expression resulted in the O-SelW group compared with the control group, while downregulation of the Bcl-2 mRNA level was observed in the K-SelW group. Silencing of the myoblast SelW gene can increase their apoptosis rate and susceptibility to H2O2 upregulated Bax and caspase-3 and downregulated Bcl-2.21 The siRNA-induced suppression of the SelW gene in cultured chicken splenic lymphocyte resulted in a downregulation in Bcl-2 expression and an upregulation in Bax, Bak, caspase-3, and P53 genes.36 These results indicated that SelW prevents H2O2-induced liver damage from downregulation of the pro-inflammation and pro-apoptosis factors and upregulation of the anti-apoptosis factors.
In our previous study, we found that SelW expression was lower in the liver of chickens fed a Se-deficient diet than in those fed a normal diet,25 and that SelW mRNA expression was significantly increased in a dose-dependent manner in cells treated with 10−8, 10−7, and 10−6 mol L−1 of Se.36 In poultry, Se deficiency reduces growth performance and results in exudative diathesis,37,38 and inhibits the function of neutrophils, NK cells, B cells, and T cells.39 The supplementation of Se leads to enhanced comparative metabolic and immune response of chickens40 and inhibits cells apoptosis being induced.41 However, Se excess could stimulate cell apoptosis and aggravate cell apoptosis induced by other inducers,42 and has shown downregulation for Bcl-2 and upregulation of Bax and p53 in cultured cortical neurons.43 In this study, we fed chicken with a Se-deficient diet to decrease the SelW intake and found Se deficiency can upregulate the expression of NF-κB, TNF-α, iNOS, COX-2, PTGEs, Bax, Bak, and P53. This indicated that Se deficiency can upregulate the inflammatory and apoptosis factors.
In summary, our results indicated that chicken SelW resembles its mammalian homologs in protecting hepatic cells against ROS-mediated inflammation and apoptosis, despite the lack of Cys37 in the peptide.
Acknowledgements
This study was supported by the International (Regional) Cooperation and Exchange Projects of the National Natural Science Foundation of China (31320103920), the National Natural Science Foundation of China (31272626), the National Natural Science Foundation of China (31502134), University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province, (UNPYSCT-2016002), and “Young Talents” project of Northeast Agricultural University (No. 14QC18).
References
- M. Mseddi, R. Ben Mansour, F. Mnif, B. Gargouri, M. Abid, F. Guermazi, H. Attia and S. Lassoued, Ther. Adv. Endocrinol. Metab., 2015, 6, 181–188 CrossRef CAS PubMed.
- D. Behne and A. Kyriakopoulos, Mammalian selenium-containing proteins, Annu. Rev. Nutr., 2001, 21, 453–473 CrossRef CAS PubMed.
- J. Yu, H. Yao, X. Gao, Z. Zhang, J. F. Wang and S. W. Xu, Biol. Trace Elem. Res., 2015, 163, 144–153 CrossRef CAS PubMed.
- C. P. Liu, J. Fu, S. L. Lin, X. S. Wang and S. Li, Biol. Trace Elem. Res., 2014, 159, 192–198 CrossRef CAS PubMed.
- A. Al-Bader, H. Abul, T. Hussain, M. Al-Moosawi, T. C. Mathew and H. Dashti, Mol. Cell. Biochem., 1998, 185, 1–6 CrossRef CAS PubMed.
- Z. Zhang, M. Bi, Q. Liu, J. Yang and S. Xu, Oncotarget, 2016, 7, 77110–77116 Search PubMed.
- Z. Jiang, C. Chen, J. Wang, W. Xie, M. Wang, X. Li and X. Zhang, J. Nat. Med., 2016, 1, 45–53 CrossRef PubMed.
- G. H. Koek, P. R. Liedorp and A. Bast, Clin. Chim. Acta, 2011, 15, 1297–1305 CrossRef PubMed.
- J. Y. Ann, H. Eo and Y. Lim, Genes Nutr., 2015, 10, 46 CrossRef PubMed.
- S. Nanba, F. Ikeda, N. Baba, K. Takaguchi, T. Senoh, T. Nagano, H. Seki, Y. Takeuchi, Y. Moritou, T. Yasunaka, H. Ohnishi, Y. Miyake, A. Takaki, K. Nouso, Y. Iwasaki and K. Yamamoto, J. Clin. Pathol., 2016, 3, 226–233 CrossRef PubMed.
- L. Flohé, R. Brigelius-Flohé, C. Saliou, M. G. Traber and L. Packer, Free Radical Biol. Med., 1997, 22, 1115–1126 CrossRef.
- H. H. Tsai, W. R. Lee, P. H. Wang, K. T. Cheng, Y. C. Chen and S. C. Shen, J. Dermatol. Sci., 2013, 2, 122–131 CrossRef PubMed.
- S. W. Ryter, H. P. Kim, A. Hoetzel, J. W. Park, K. Nakahira, X. Wang and A. M. Choi, Antioxid. Redox Signaling, 2007, 9, 49–89 CrossRef CAS PubMed.
- E. H. Cheng, M. C. Wei, S. Weiler, R. A. Flavell, T. W. Mak, T. Lindsten and S. J. Korsmeyer, Mol. Cell, 2001, 705–711 CrossRef CAS.
- D. M. Finucane, E. Bossy-Wetzel, N. J. Waterhouse, T. G. Cotter and D. R. Green, J. Biol. Chem., 1999, 274, 2225–2233 CrossRef CAS PubMed.
- R. S. Morrison, Y. Kinoshita, M. D. Johnson, W. Guo and G. A. Garden, Neurochem. Res., 2003, 28, 15–27 CrossRef CAS PubMed.
- J. Loflin, N. Lopez, P. D. Whanger and C. Kioussi, J. Inorg. Biochem., 2006, 10, 1679–1684 CrossRef PubMed.
- V. Pagmantidis, G. Bermano, S. Villette, I. Broom, J. Arthur and J. Hesketh, FEBS Lett., 2005, 579, 792–796 CrossRef CAS PubMed.
- J. Dw, T. S. Kim, Y. W. Chung, B. J. Lee and I. Y. Kim, FEBS Lett., 2002, 517, 225–228 CrossRef.
- Y. W. Chung, D. Jeong, O. J. Noh, Y. H. Park, S. I. Kang, M. G. Lee, T. H. Lee, M. B. Yim and I. Y. Kim, Mol. Cells, 2009, 27, 609–613 CrossRef CAS PubMed.
- H. D. Yao, Q. Wu, Z. W. Zhang, S. Li, X. L. Wang, X. G. Lei and S. W. Xu, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3112–3120 CrossRef CAS PubMed.
- M. A. Beilstein, S. C. Vendeland, E. Barofsky, O. N. Jensen and P. D. Whanger, J. Inorg. Biochem., 1996, 61, 117–124 CrossRef CAS PubMed.
- Q. P. Gu, M. A. Beilstein, E. Barofsky, W. Ream and P. D. Whanger, Arch. Biochem. Biophys., 1999, 361, 25–33 CrossRef CAS PubMed.
- P. D. Whanger, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1448–1452 CrossRef CAS PubMed.
- Z. H. Jiang, P. A. Khoso, H. D. Yao, Z. W. Zhang, X. Y. Zhang and S. W. Xu, RSC Adv., 2015, 5, 37896–37905 RSC.
- Y. Sun, O. P. Gu, P. D. Whanger, Y. Sun, Q. P. Gu and P. D. Whanger, J. Inorg. Biochem., 2001, 84, 151–156 CrossRef CAS PubMed.
- S. C. Vendeland, M. A. Beilstein, C. L. Chen, O. N. Jensen, E. Barofsky and P. D. Whanger, J. Biol. Chem., 1993, 268, 17103–17107 CAS.
- H. D. Yao, Q. Wu, Z. W. Zhang, J. L. Zhang, S. Li, J. Q. Huang, F. Z. Ren, S. W. Xu, X. L. Wang and X. G. Lei, J. Nutr., 2013, 143, 613–619 CrossRef CAS PubMed.
- Y. H. Han, Z. W. Zhang, J. Su, B. Zhang, S. Li and S. W. Xu, Biol. Trace Elem. Res., 2012, 147, 395–402 CrossRef CAS PubMed.
- W. Xiao-Long, Y. Chuan-Ping, X. Kai and Q. Ou-Jv, Biochemistry, 2010, 75, 201–207 Search PubMed.
- X. F. Liu, L. M. Zhang, H. N. Guan, Z. W. Zhang and S. W. Xu, Food Chem. Toxicol., 2013, 60, 168–176 CrossRef CAS PubMed.
- L. Chen, M. W. Gong, Z. F. Peng, T. Zhou, M. G. Ying, Q. H. Zheng, Q. Y. Liu and Q. Q. Zhang, Mar. Drugs, 2014, 12, 1939–1958 CrossRef PubMed.
- R. Schreck and P. A. Baeuerle, Trends Cell Biol., 1991, 1, 39–42 CrossRef CAS PubMed.
- S. Kawanishi, Y. Hiraku, S. Pinlaor and N. Ma, Biol. Chem., 2006, 387, 365–372 CrossRef CAS PubMed.
- L. Goossens, N. Pommery and J. P. Henichart, Curr. Top. Med. Chem., 2007, 7, 283–296 CrossRef CAS PubMed.
- D. Yu, Z. W. Zhang, H. D. Yao, S. Li and S. W. Xu, BioMetals, 2014, 27, 277–291 CrossRef CAS PubMed.
- A. Bartholomew, D. Latshaw and D. E. Swayne, Biol. Trace Elem. Res., 1998, 62, 7–16 CrossRef CAS PubMed.
- H. Yao, R. Fan, X. Zhao, W. Zhao, W. Liu, J. Yang, H. Sattar, J. Zhao, Z. Zhang and S. Xu, Oncotarget, 2016, 7, 57618–57632 Search PubMed.
- L. Kiremidjian-Schumacher, M. Roy, H. I. Wishe, M. W. Cohen and G. Stotzky, Biol. Trace Elem. Res., 1992, 33, 23–35 CrossRef CAS PubMed.
- S. V. Rao, B. Prakash, M. V. Raju, A. K. Panda, R. K. Kumari and E. P. Reddy, Biol. Trace Elem. Res., 2016, 172, 511–520 CrossRef CAS PubMed.
- Y. J. Zhou, S. P. Zhang, C. W. Liu and Y. Q. Cai, Toxicol. In Vitro, 2009, 23, 288–294 CrossRef CAS PubMed.
- L. Guan, B. Han, Z. Li, F. Hua, F. Huang, W. Wei, Y. Yang and C. Xu, Apoptosis, 2009, 14, 218–225 CrossRef CAS PubMed.
- R. Xiao, J. T. Qiao, H. F. Zhao, J. Liang, H. L. Yu, J. Liu, A. M. Guo and W. Wang, Neurotoxicology, 2006, 27, 478–484 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Shi-Wen Xu*a
*b and
Shi-Wen Xu*a