
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular sub-monomeric cell-penetrating guanidine-rich peptoids – synthesis, assembly and biological evaluation†
Etienne Marouseaua,
Albane Neckebroecka,
Heidi Larkina,
Antoine Le Rouxa,
Leonid Volkovb,
Christine L. Lavoiea and
Éric Marsault *a
*a
aInstitut de Pharmacologie de Sherbrooke, Department of Pharmacology and Physiology, Université de Sherbrooke, 3001, 12e Avenue Nord, J1H 5N4, Sherbrooke, QC, Canada. E-mail: Eric.Marsault@usherbrooke.ca
bBiophotonics Core Facility, Centre de Recherche du Centre Hospitalier de l’Université de Sherbrooke, 3001, 12e Avenue Nord, J1H 5N4, Sherbrooke, QC, Canada
First published on 18th January 2017
Abstract
Guanidinium-rich transporters are promising agents to deliver a wide range of molecules through cell membranes. This article reports the synthesis of a modular, conformationally restricted guanidinium display scaffold, designed with the goal to study the impact of conformational restrictions on the resulting properties of the transporters and ultimately probe the anionic chemical space of the cellular membrane. The scaffold was assembled on solid phase as a peptoid, and its subsequent biological evaluation showed very good cellular penetration and low cytotoxicity.
Introduction
A wide range of drug discovery targets are intracellular, such as protein–protein interactions, which makes them difficult to reach.1 On the other hand, peptides, ribonucleic acids (RNAs), and more broadly high molecular weight or highly polar molecules, often represent promising leads in vitro for intracellular targets,2 yet fail to reach efficacy in cells or in vivo for their inability to cross cell membranes.3 There is thus a need for cellular penetration agents for these two chemical spaces to meet.The discovery of the protein transduction domain of Antennapedia homeodomain and the transactivated transcription (Tat) protein of HIV-1 changed this paradigm,4,5 demonstrating that some large peptides can cross cell membranes. From these pioneering works and based on the fundamentals of these observations, cell-penetrating peptides (CPPs) and CPP-like transporters have been further developed and diversified.6 Promising proofs of concepts were made, demonstrating the feasibility to deliver otherwise impermeable cargoes to various cell types and tissues, including the brain, using such transporters.7–9 CPPs were originally classified in three categories: cationic, amphipathic and lipophilic.10,11
Amongst cationic CPPs, the guanidinium-rich transporters (GRTs) emerged after the observation that polylysines and polyarginines are sufficient and efficient peptides to carry cargos inside the cells, and that peptidomimetic analogs, owing to their higher stability, entered cells more readily.12,13 Several research groups have subsequently studied GRTs featuring various backbones.13–18
Additionally, it has also been shown that the interaction between sulfated (anionic) glycosaminoglycans (GAGs) and GRTs was critical for cellular entry.19,20 However, how precisely these interactions allow cationic CPPs to enter cells is still a matter of debate and several parameters21,22 and mechanisms23,24 have been proposed and studied. The lack of a toolset to interrogate the cellular membrane chemical space, however, limits this exploration.25 In order to better understand this poorly characterized chemical space, we and others reported the use of macrocyclisation as a way to study the impact of GRT conformational restrictions on cellular permeation.26–30
In the present study, we designed novel peptoidic GRTs bearing modular scaffolds, assembled via a sub-monomer approach, to display a conformationally restricted pattern of 2–3 guanidines per sub-monomer. This modular design enables structural variations to sample distances between guanidine moieties, with the goal to explore potential interactions with the negatively charged groups displayed on GAGs.14,31 In other words, this design provides a diversifiable cationic chemical space as an exploratory tool to better understand the anionic chemical space of the cellular membrane.
Results and discussion
Our first objective was to develop a modular synthesis (Fig. 1), hinging around protected sub-monomers S1 assembled in solution, assembly of multimers followed by solid phase guanidinylation. This general strategy is expected to enable modifications to the peptoid sequence and the introduction of additional scaffolds. Thus, Rink amide resin was chosen for solid phase synthesis and a β-alanine spacer between the GRT and the cargo completes the construction.32 Fluorescein (FL) was chosen as the first cargo to assess transporter permeability by flux cytometry.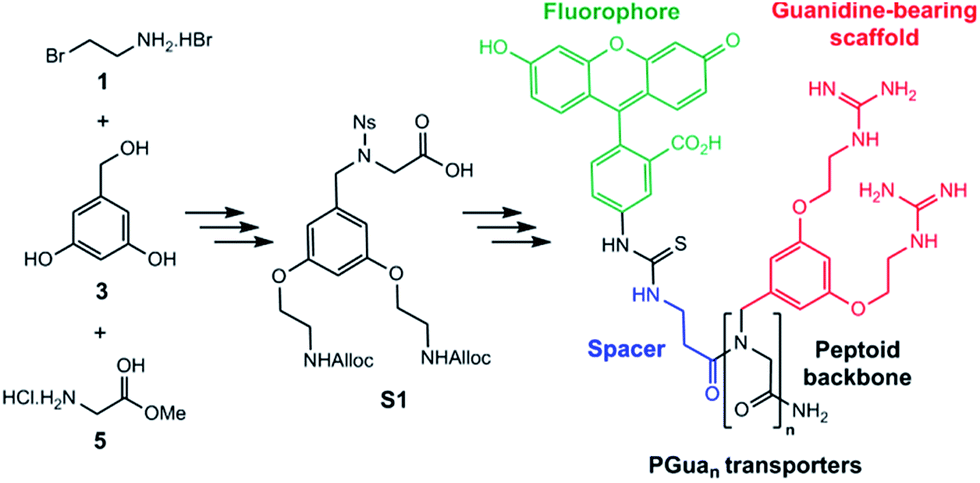 | ||
| Fig. 1 Synthetic route for PGuan transporters. | ||
Construction of building block S1 (Scheme 1) started with Alloc protection of 2-bromoethylamine hydrobromide 1 in excellent yield to provide 2. Displacement of the bromide of 2 with the phenol groups of 3,5-dihydroxybenzyl alcohol 3 yielded dialkylated intermediate 4 efficiently. Meanwhile, activation of the amine of glycine methyl ester hydrochloride 5 with a 2-nitrobenzene sulfonyl (nosyl) group produced intermediate 6, isolated by precipitation in excellent yield. Intermediates 4 and 6 were then combined using the Fukuyama–Mitsunobu reaction33 to obtain intermediate 7 in good yield, which was hydrolyzed in excellent yield to S1 for an overall yield of 40%.
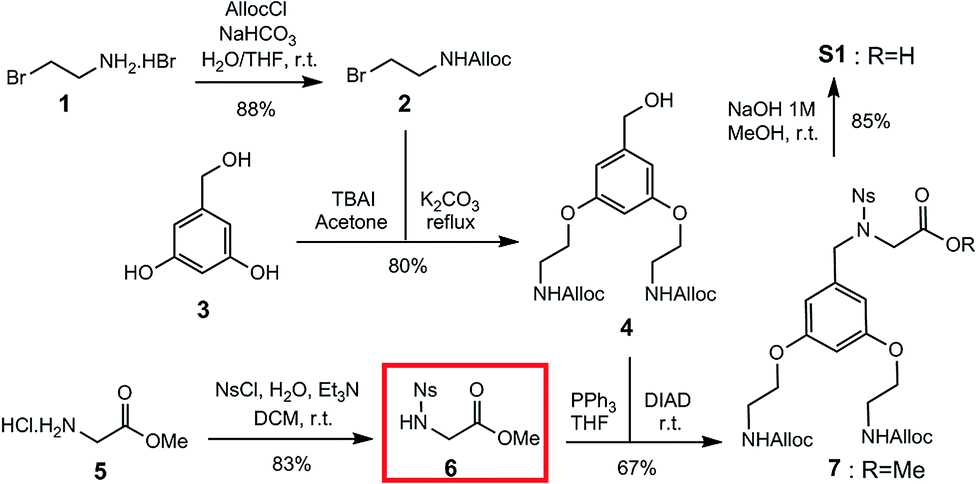 | ||
| Scheme 1 Solution synthesis of sub-monomer S1. | ||
The next step was to build the corresponding GRT (PGuan, Fig. 2), which was accomplished using submonomer assembly. In order to assess the respective role of backbone and guanidine on properties, the corresponding benzyl and aminated analogues (PBnn and PAmn, respectively, Fig. 2) were built as controls, as well as the corresponding polyarginines (Rn, Fig. 2). Accordingly, dimers to hexamers were synthesized to assess the impact of guanidine number on permeability.
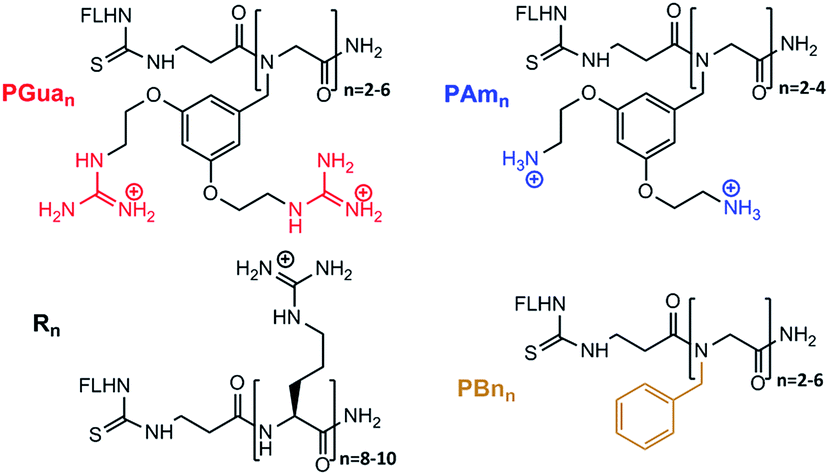 | ||
| Fig. 2 Structures of the transporters. | ||
The first focus was to optimize conditions to obtain aminated and guanidinylated compounds (Scheme 2). A nosylamide alkylation strategy was applied by sequential addition of S1 followed by nosyl deprotection. The coupling step with hexafluorophosphate azabenzotriazole tetramethyluronium (HATU) and DIPEA to obtain 9 worked best with 3 eq. of S1 and was performed overnight to get 10. Nosyl group deprotection was performed using mercaptoethanol and DBU.34
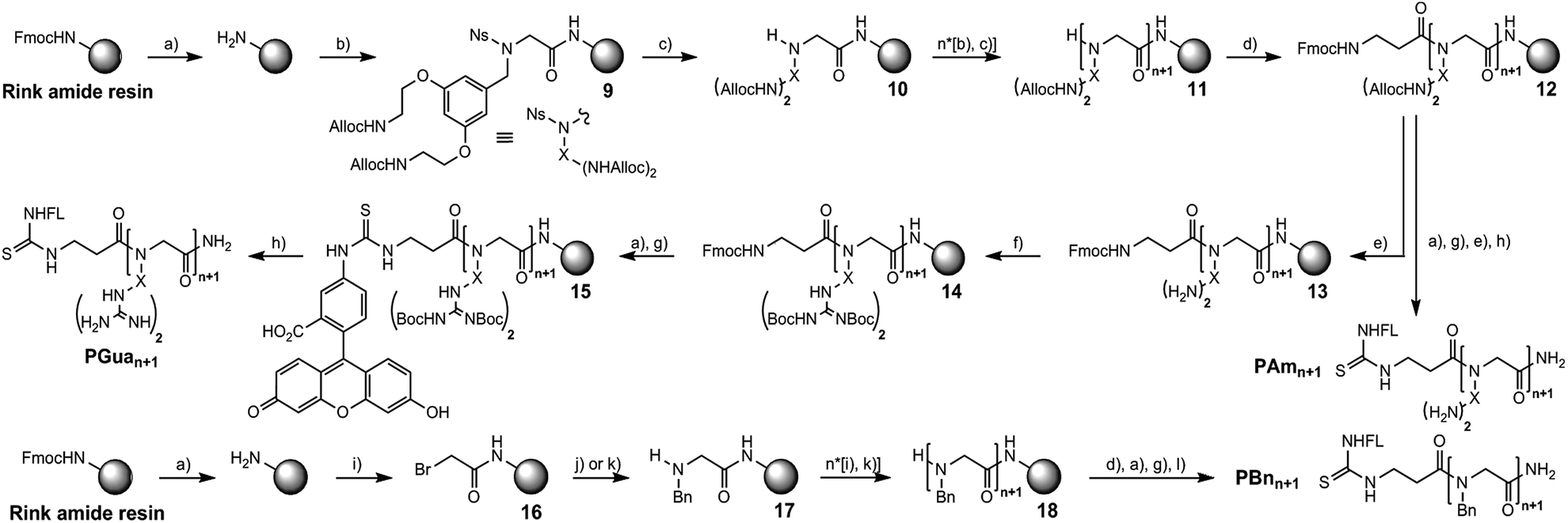 | ||
Scheme 2 Solid phase synthetic routes of PGuan, PAmn and PBnn. Reagents and conditions: (a) 20% pip in DMF, rt, 20 min; (b) 3 eq. S1, 3 eq. HATU, 6 eq. DIPEA, DMF, rt, o/n; (c) 10 eq. mercaptoethanol, 5 eq. DBU, DMF, rt, 2 × 30 min; (d) 5 eq. Fmoc-βAla-OH, 5 eq. HATU, 6 eq. DIPEA, DMF, rt, 2 × 1 h; (e) 0.5 eq./Alloc Pd(PPh3)4, 2 eq./Alloc DMBA, DCM, rt, 2 h; (f) 2 eq./amine Goodman's reagent, 2 eq./amine DIPEA, DCM, rt, 4 h; (g) 2 eq. FITC, 4 eq. DIPEA, DCM/Pyr (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), rt, 30 min; (h) TFA/TIPS/water (95:2.5:2.5), rt, 2 h; (i) 5 eq. bromoacetic acid, 2.5 eq. DIC, DCM, rt, 15 min; (j) 2 eq. benzylamine, 6 eq. DIPEA, DMF, rt, o/n; (k) 20 eq. benzylamine, DMF, rt, 1 h; (l) TFA, rt, 1 h. :3), rt, 30 min; (h) TFA/TIPS/water (95:2.5:2.5), rt, 2 h; (i) 5 eq. bromoacetic acid, 2.5 eq. DIC, DCM, rt, 15 min; (j) 2 eq. benzylamine, 6 eq. DIPEA, DMF, rt, o/n; (k) 20 eq. benzylamine, DMF, rt, 1 h; (l) TFA, rt, 1 h. | ||
After assembling the desired number of submonomers, intermediate 11 was obtained, and β-alanine was coupled to obtain intermediate 12. Among several conditions to deprotect the allyl group with palladium(0) tetrakis triphenylphosphine (phenylsilane,35 dimedone, HCl/morpholine and dimethylbarbituric acid (DMBA)),36 DMBA gave the best results to generate polyamine 13. The latter was then guanidinylated with Goodman's reagent (1,3-Di-Boc-2-(trifluoromethylsulfonyl)guanidine) successfully to deliver intermediate 14.15,37 Deprotection of the Fmoc protecting group followed by addition of fluorescein isothiocyanate (FITC) yielded intermediate 15.
A mixture of TFA/water/triisopropylsilane (TIPS) simultaneously cleaved the peptoid and deprotected Boc groups to give PGuan.
For aminated analogs, the order of steps was changed, starting from 12, to conjugate fluorescein first. Deprotection of the Alloc groups worked well here too, and cleavage from the resin gave PAmn transporters. Increasing the size of PGuan or PAmn was uneventful. Overall yield remained close to 10% after purification. To assess potential contributions of the aromatic backbone on cellular penetration, FL-βAla-(NPhe)n-NH2 (PBnn) were chosen as non-functionalised controls. The bromoacetic acid strategy was applied to these syntheses (Scheme 2),38 in overall good yields (up to 55%) after purification. Finally, control polyarginine transporters (Rn) were synthesized using standard Fmoc chemistry with Fmoc-Arg(Pbf)-OH.
As a primary biological assay, to validate which compounds qualified for further analyses, permeation was evaluated with flow cytometry in HeLa cells.39 Uptake was measured by the displacement of the mean cell fluorescence after incubation with 5 μM fluorescein-labelled compounds in culture medium compared to a blank. To ensure extracellular fluorescence quenching, cells were incubated with trypsin before analysis,19,40 and a buffer containing trypan blue was added (see ESI†).41 The first results (Fig. 3) validated several hypotheses: (1) non-functionalized control analogs (Fig. 3A) showed no significant difference compared to blank, proving backbone is not responsible for uptake; (2) aminated analogs (Fig. 3B) did not show any significant cellular penetration with up to 8 amine moieties; (3) guanidinylated compounds (Fig. 3C) start entering cells with 6 guanidines and showed a maximal uptake of 80% of the reference R9 for 8 guanidines, consistent with previous literature.42
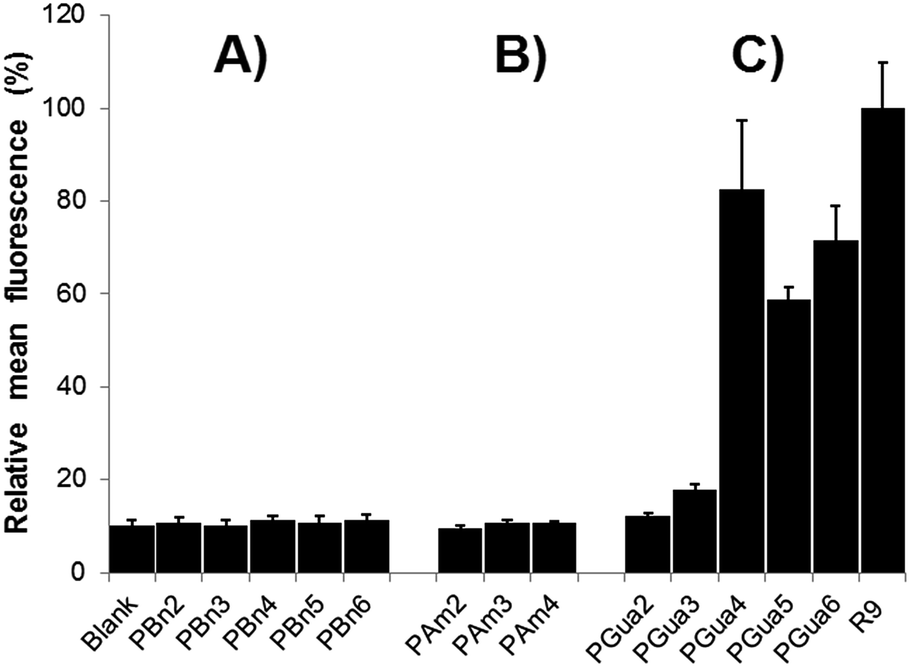 | ||
| Fig. 3 Cellular penetration of (A) backbone-only analogs; (B) aminated analogs; (C) guanidinylated analogs and nonaarginine in HeLa cells as determined by flow cytometry. Standard deviation was calculated according to the number of experiments; PBn4 n = 15; blank n = 12; PBn2,5,6, PGua5,6 and R9 n = 8; PBn3 and PGua4 n = 7; PAm2,3,4 and PGua2,3 n = 6. Mean fluorescence was normalized with respect to control R9. | ||
GAGs have been shown to be key for cellular selectivity.14,43,44 and a potentially important avenue to cancer-targeted delivery.45 Hence, the second experiment was intended to assess the dependence of cellular uptake upon GAGs. Thus, GAG-depleted Chinese hamster ovarian (CHO) cells (pgsA-745),46 and wild type cells (CHO-K1), were incubated in the same conditions as described for HeLa cells, and flow cytometry analysis was performed (Fig. 4). In CHO-K1 cells, the uptake of PGuan compounds was found to be significantly lower compared to polyarginines, with diminution ranging from 28% (PGua4) to 58% (PGua6) compared to HeLa cells. This suggests that, as opposed to Hela cells, CHO-K1 cells discriminate polyarginines compared to this new GRT scaffold. This also suggests that a structure-permeability relationship is achievable with the various cell lines, similarly to our reports with macrocyclic GRTs.29
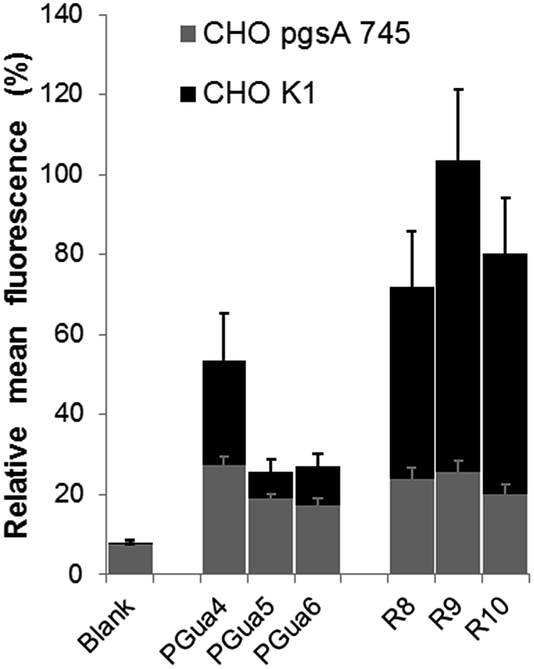 | ||
| Fig. 4 Cellular penetration of guanidinylated analogs and polyarginines in CHO-K1 (black) and CHO-pgsA-745 (grey) as determined by flow cytometry. Each value represents an average of 8 experiments, with the standard deviation. Mean fluorescence was normalized with respect to control R9. | ||
In CHO-pgsA-745 cells, the uptake of guanidinylated compounds (PGuan) and polyarginines were both abrogated. Interestingly, the remaining levels of uptake compared to CHO-K1 cells, irrespective of compounds, remained around 20% (Fig. 4) with very low standard deviation, independently of their structures or number of guanidines. This suggests the presence of a dual uptake mechanism (one dependent on GAGs and modular, the other independent of GAGs and non-modular).
Finally, cytotoxicity was assessed for a series of analogous compounds (PBn4, PAm4, PGua4 and R8) using the lactate dehydrogenase (LDH) assay.47
As shown in Fig. 5, no significant cytotoxicity was observed for the compounds tested, except for PGua4, which induced 25% mortality at 50 μM concentration only.
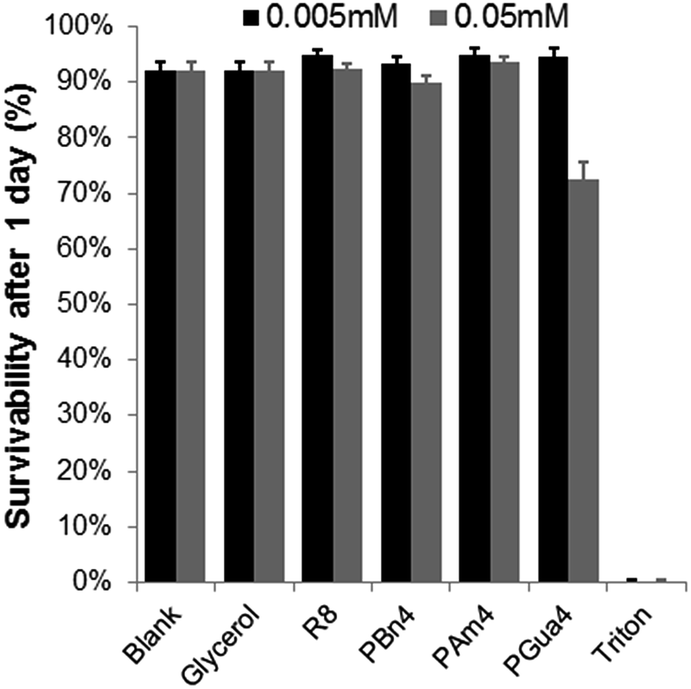 | ||
| Fig. 5 Cellular toxicity of PGua4 and its analogs in HeLa cells. Each value represents an average of 6 experiments, with its standard deviation shown. Triton was used as a negative control. | ||
Conclusion
A new type of sub-monomer was created to synthesize GRTs based on a peptoid backbone. Solid phase synthesis worked well and is applicable to subsequent backbone diversification. Biological results confirm that this scaffold is conducive of good cellular uptake. The best compound in this class so far proved to be PGua4, bearing 8 guanidine groups, which was superior to similar scaffolds bearing 10 or 12 guanidines. Flow cytometry suggests the potential presence of dual mechanisms of uptake of the compounds in CHO cells. Finally, cell survival assay showed promising results with a low cytotoxicity for PGua4. This demonstrates that the proposed methodology is promising and that further investigations could identify new GRT structures to support the development of structure-cellular penetration relationship. This toolset provides the first step of a broader project aimed at providing a broad cationic chemical space as a complement to interrogate the anionic chemical space defined by the cell membrane.Acknowledgements
We would like to thank the Natural Sciences and Engineering Research Council of Canada and the Canadian Foundation for Innovation. The FRQNT-funded PROTEO Network and the Institut de Pharmacologie de Sherbrooke are acknowledged for graduate scholarship to E. Marouseau. Pr. Steve Bourgault (Université du Québec A Montréal, QC, Canada) is gratefully acknowledged for providing CHO-K1 and CHO-pgsA-745 cell lines.Notes and references
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS PubMed
.
- C. Corbi-Verge and P. M. Kim, Cell Commun. Signaling, 2016, 14, 1–12 CrossRef PubMed
- G. L. Verdine and L. D. Walensky, Clin. Cancer Res., 2007, 13, 7264–7270 CrossRef CAS PubMed
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CAS
- E. Vivès, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef
- S. Stalmans, E. Wynendaele, N. Bracke, B. Gevaert, M. D'Hondt, K. Peremans, C. Burvenich and B. De Spiegeleer, PLoS One, 2013, 8, e71752 CAS
- M. Pooga, U. Soomets, M. Hällbrink, a. Valkna, K. Saar, K. Rezaei, U. Kahl, J. X. Hao, X. J. Xu, Z. Wiesenfeld-Hallin, T. Hökfelt, T. Bartfai and U. Langel, Nat. Biotechnol., 1998, 16, 857–861 CrossRef CAS PubMed
- A. Srimanee, J. Regberg and Ü. Langel, in Cell-Penetrating Peptides: Methods and Protocols, ed. Ü. Langel, Springer New York, New York, NY, 2nd edn, 2015, pp. 349–356 Search PubMed
- J. E. Dixon, G. Osman, G. E. Morris, H. Markides, M. Rotherham, Z. Bayoussef, A. J. El Haj, C. Denning and K. M. Shakesheff, Proc. Natl. Acad. Sci., 2016, 113, E291–E299 CrossRef CAS PubMed
- R. Fischer, M. Fotin-Mleczek, H. Hufnagel and R. Brock, ChemBioChem, 2005, 6, 2126–2142 CrossRef CAS PubMed
- K. Kurrikoff, M. Gestin and Ü. Langel, Expert Opin. Drug Delivery, 2016, 13, 373–387 CrossRef CAS PubMed
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS PubMed
- W. Huang, J. Seo, J. S. Lin and A. E. Barron, Mol. Biosyst., 2012, 8, 2626–2628 RSC
- L. Elson-Schwab, O. B. Garner, M. Schuksz, B. E. Crawford, J. D. Esko and Y. Tor, J. Biol. Chem., 2007, 282, 13585–13591 CrossRef CAS PubMed
- T. Schröder, N. Niemeier, S. Afonin, A. S. Ulrich, H. F. Krug and S. Bräse, J. Med. Chem., 2008, 51, 376–379 CrossRef PubMed
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed
- Y. Demizu, M. Oba, K. Okitsu, H. Yamashita, T. Misawa, M. Tanaka, M. Kurihara and S. H. Gellman, Org. Biomol. Chem., 2015, 13, 5617–5620 CAS
- M. Nepal, S. Thangamani, M. N. Seleem and J. Chmielewski, Org. Biomol. Chem., 2015, 13, 5930–5936 CAS
- J. P. Richard, K. Melikov, H. Brooks, P. Prevot, B. Lebleu and L. V. Chernomordik, J. Biol. Chem., 2005, 280, 15300–15306 CrossRef CAS PubMed
- L. E. Prevette, N. C. Benish, A. R. Schoenecker and K. J. Braden, Biophys. Chem., 2015, 207, 40–50 CrossRef CAS PubMed
- J. B. Rothbard, T. C. Jessop and P. A. Wender, Adv. Drug Delivery Rev., 2005, 57, 495–504 CrossRef CAS PubMed
- R. Wallbrecher, W. P. R. Verdurmen, S. Schmidt, P. H. Bovee-Geurts, F. Broecker, A. Reinhardt, T. H. Van Kuppevelt, P. H. Seeberger and R. Brock, Cell. Mol. Life Sci., 2014, 71, 2717–2729 CAS
- P. A. Wender, W. C. Galliher, E. A. Goun, L. R. Jones and T. H. Pillow, Adv. Drug Delivery Rev., 2008, 60, 452–472 CrossRef CAS PubMed
- A. Tchoumi Neree, P. T. Nguyen and S. Bourgault, Int. J. Mol. Sci., 2015, 16, 27391–27400 CrossRef PubMed
- M. E. Favretto, R. Wallbrecher, S. Schmidt, R. Van De Putte and R. Brock, J. Controlled Release, 2014, 180, 81–90 CrossRef CAS PubMed
- D. Oh, S. A. Darwish, A. N. Shirazi, R. K. Tiwari and K. Parang, ChemMedChem, 2014, 92618, 2449–2453 CrossRef PubMed
- R. Wallbrecher, L. Depré, W. P. R. Verdurmen, P. H. Bovée-Geurts, R. H. Van Duinkerken, M. J. Zekveld, P. Timmerman and R. Brock, Bioconjugate Chem., 2014, 25, 955–964 CrossRef CAS PubMed
- N. Nischan, H. D. Herce, F. Natale, N. Bohlke, N. Budisa, M. C. Cardoso and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 1950–1953 CrossRef CAS PubMed
- H. Traboulsi, H. Larkin, M. A. Bonin, L. Volkov, C. L. Lavoie and É. Marsault, Bioconjugate Chem., 2015, 26, 405–411 CrossRef CAS PubMed
- Z. Qian, A. Martyna, R. L. Hard, J. Wang, G. Appiah-Kubi, C. Coss, M. A. Phelps, J. S. Rossman and D. Pei, Biochemistry, 2016, 55, 2601–2612 CrossRef CAS PubMed
- M. Okuyama, H. Laman, S. R. Kingsbury, C. Visintin, E. Leo, K. L. Eward, K. Stoeber, C. Boshoff, G. H. Williams and D. L. Selwood, Nat. Methods, 2007, 4, 153–159 CrossRef CAS PubMed
- M. Jullian, A. Hernandez, A. Maurras, K. Puget, M. Amblard, J. Martinez and G. Subra, Tetrahedron Lett., 2009, 50, 260–263 CrossRef CAS
- T. Fukuyama, C. K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373–6374 CrossRef CAS
- E. Biron, J. Chatterjee and H. Kessler, J. Pept. Sci., 2006, 12, 213–219 CrossRef CAS PubMed
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC
- D. Fernández-Forner, G. Casals, E. Navarro, H. Ryder and F. Albericio, Tetrahedron Lett., 2001, 42, 4471–4474 CrossRef
- K. Feichtinger, H. L. Sings, T. J. Baker, K. Matthews and M. Goodman, J. Org. Chem., 1998, 63, 8432–8439 CrossRef CAS
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS
- A. Gräslund, F. Madani, S. Lindberg, Ü. Langel and S. Futaki, J. Biophys., 2011, 2011, 414729 Search PubMed
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, J. Biol. Chem., 2003, 278, 585–590 CrossRef CAS PubMed
- J. Hed, G. Hallden, S. G. O. Johansson and P. Larsson, J. Immunol. Methods, 1987, 101, 119–125 CrossRef CAS PubMed
- H. Silva, F. Frézard, E. J. Peterson, P. Kabolizadeh, J. J. Ryan and N. P. Farrell, Mol. Pharmacol., 2012, 9, 1795–1802 CrossRef CAS PubMed
- K. J. Lim, B. H. Sung, J. R. Shin, Y. W. Lee, D. J. Kim, K. S. Yang and S. C. Kim, PLoS One, 2013, 8, e66084 CAS
- S. Stalmans, N. Bracke, E. Wynendaele, B. Gevaert, K. Peremans, C. Burvenich, I. Polis and B. De Spiegeleer, PLoS One, 2015, 10, e0139652 Search PubMed
- I. Geisler and J. Chmielewski, Chem. Biol. Drug Des., 2009, 73, 39–45 CAS
- X. Bai, G. Wei, A. Sinha and J. D. Esko, J. Biol. Chem., 1999, 274, 13017–13024 CrossRef CAS PubMed
- E. Amador, L. E. Dorfman and W. E. Wacker, Clin. Chem., 1963, 12, 391–399 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27898a |
| This journal is © The Royal Society of Chemistry 2017 |