
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross C–S coupling reaction catalyzed by copper(I) N-heterocyclic carbene complexes†
Wei-Kai Huangab,
Wei-Ting Chenc,
I.-Jui Hsu*c,
Chien-Chung Han *b and
Shin-Guang Shyu*a
*b and
Shin-Guang Shyu*a
aInstitute of Chemistry, Academia Sinica, Taipei 11529, Taiwan, Republic of China. E-mail: sgshyu@chem.sinica.edu.tw
bDepartment of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan, Republic of China. E-mail: cchan@mx.nthu.edu.tw
cDepartment of Molecular Science and Engineering, National Taipei University of Technology, Taipei 10608, Taiwan, Republic of China. E-mail: ijuihsu@ntut.edu.tw
First published on 17th January 2017
Abstract
Copper(I) N-heterocyclic carbene has a good activity towards aryl halides and was used as a model complex to study the catalytic cycle of Cu(I) to catalyze the cross C–S coupling reaction because the N-heterocyclic carbene has a strong electron donating property, and ligand dissociation can be avoided. Free radical scavenger cumene does not retard the yield of the reaction indicating that the catalytic reaction goes through a non free radical path. Switching the solvent from toluene to DMF lowered the yield of the reaction. DFT calculation shows that the activation of aryl halide is the rate determining step, and the activation energy is higher for the reaction in DMF than in toluene. A plausible catalytic cycle is proposed with the support of DFT calculation.
Introduction
Ligands can enhance the Ullmann type Cu(I) catalyzed C–S cross coupling reaction,1,2 and phen-based (phen = phenanthroline) ligands have been studied intensively in the last decade.3 A Cu(I) phen complex is generally considered as an intermediate in the catalytic cycle,3a,c,4 and DFT calculation has been carried out to support this hypothesis.5 Several studies indicate that the mechanism is more complicated than first thought.6 Actually, phen does not coordinate with the Cu(I) firmly. An equilibrium between (phen)CuSAr, [Cu(phen)2]+ and [Cu(SAr)2]1− was proposed and confirmed by the observations from in situ ESI-MS studies.6e [K(phen)][Cu(SAr)2] was also proposed as an intermediate in the catalytic reaction based on in situ ESI-MS studies. All these results echo the labile coordination property of the phen ligand towards K1+ and Cu(I).6e In addition, aggregation of CuSR can occur in the reaction.6a–d,f,g In fact, [K(Me2phen)3]2, [Cu4(SCH2Ph)6]2− and [phenCuSAr]2 have been synthesized (through the respective reaction between CuCl and KSR, and that between CuOtBu and HSAr, in the presence of phen)6f and studied to elucidate the possible involvements of [Cu4(SCH2Ph)6]2− or [phenCuSAr]2 in the catalytic cycle. All these diverse observations indicate that the mechanism of C–S cross coupling reaction catalyzed by Cu(I)/ligand has not been settled yet.N-Heterocyclic carbene has a strong electron donating property and ligand dissociation can be avoided.7 Thus, LCu(SAr) (L = N-heterocyclic carbene ligand) can maintain as a monomer in the solution.8 We conceive that this highly stable LCu(SAr) may serve as the dominant intermediate in the catalytic cycle, which could help simplify the elucidation on the corresponding catalytic activity. Herein, we report the synthesis of Cu(I) N-heterocyclic carbenes, and the studies on their catalytic activities in the C–S cross coupling reaction between aryl iodide and thiophenol. DFT studies were carried out and a catalytic cycle based on experimental results and calculation is proposed.
Experimental
General procedures
All reagents were purchased from commercial sources and used without further purification. Copper iodide(I), iodobenzene, 1-iodo-naphthalene, 2-iodotoluene, 3-iodotoluene, 4-iodotoluene, 4-iodoanisole, 4-iodobenzonitrile, lithium tert-butoxide, sodium tert-butoxide, potassium tert-butoxide, thiophenol, and 2,2,6,6-tetramethyl-1-piperidinyloxy were purchased from ACROS. 1,4-Di-tert-butylbenzene was purchased from Alfa Aesar. Isopropylbenzene and 4-iodobenzotrifluoride were purchased from Aldrich. p-Xylene, toluene and DMF (dried) were purchased from Merck. All reagents were transferred to the reaction vessel (Pyrex tube with a Teflon screw cap) in a glove box. 1H and 13C NMR spectra were recorded in CDCl3 or C6D6 on Bruker AV 400 or Bruker AMX 400 instruments. GC experiments were performed on Agilent 6890N gas chromatograph equipped with a 30 m × 0.53 mm × 3.0 μm HP-1 or 30 m × 0.25 mm × 0.25 μm DB-5MS capillary columns and a FID detector.Preparation of N-heterocyclic carbene
1H NMR (CDCl3): δ 2.09 (s, 12H), 2.33 (s, 6H), 6.98 (s, 4H), 7.03 (s, 2H); 13C{1H} NMR (CDCl3): δ 17.9, 21.3, 122.4, 129.6, 134.7, 135.3, 139.7.
1H NMR (CDCl3): δ 1.34 (d, 12H, J = 6.9 Hz), 1.41 (d, 12H, J = 6.9 Hz), 2.65–2.70 (m, 4H), 7.37 (s, 2H), 7.41 (d, 4H, J = 7.8 Hz), 7.60 (t, 2H, J = 7.5 Hz); 13C{1H} NMR (CDCl3): δ 24.1, 25.0, 28.9, 123.3, 124.4, 124.9, 130.8, 154.8.
1H NMR (C6D6): δ 1.90 (s, 12H), 2.11 (s, 6H), 5.97 (s, 2H), 6.68 (s, 4H), 6.89–6.90 (overlapping multiplets, 3H), 7.34 (br m, 2H); 13C{1H} NMR (C6D6): δ 18.1, 21.5, 121.5, 127.9, 129.9, 134.0, 135.4, 136.2, 139.2, 147.3.
Typical procedure for copper(I)-catalyzed C–S coupling
In a glove box, copper catalyst (0.1 mmol, 10 mol%), ligand (0.1 mmol, 10 mol%), aryl iodide (1.0 mmol), base (1.5 mmol), and solvent (3.0 mL) were transferred to a Pyrex tube fitted with a Teflon screw-cap. Thiophenol (113.0 μL, 1.1 mmol) was added under nitrogen outside the glove box. The mixture was then heated at 120 °C in oil bath for 6 h and then cooled to room temperature. Ethyl acetate (10.0 mL) was added to the reaction mixture and the resultant suspension was filtered through Celite. The filtrate was characterized by GC with 1,4-di-tert-butylbenzene as the internal standard.Reaction of copper(I) catalyzed C–S coupling with different N-heterocyclic carbenes
The same procedure, amount of reagents and reaction conditions as in the typical procedure stated above was applied in the reaction. For reaction using IMes-Cu-Cl as the copper catalyst, 40.0 mg, 0.1 mmol of IMes-Cu-Cl was used. For reaction using IMes-Cu-SPh as the copper catalyst, 47.7 mg, 0.1 mmol of IMes-Cu-SPh was used. For reaction using IPr-Cu-Cl as the copper catalyst, 47.0 mg, 0.1 mmol of IPr-Cu-Cl was used.Reaction between iodobenzene, thiophenol and NaOtBu with IMes-Cu-Cl: GC yield: 88% (response factor for diphenyl sulfide: 1.11).
Reaction between iodobenzene, thiophenol and KOtBu with IMes-Cu-Cl: GC yield: 84% (response factor for diphenyl sulfide: 1.11).
Reaction between iodobenzene, thiophenol and LiOtBu with IMes-Cu-SPh: GC yield: 83% (response factor for diphenyl sulfide: 1.11).
Reaction between iodobenzene, thiophenol and LiOtBu with IPr-Cu-Cl: GC yield: 48% (response factor for diphenyl sulfide: 1.11).
Reaction of copper(I) catalyzed C–S coupling with CuI
In a glove box, CuI (19.0 mg, 0.1 mmol), LiOtBu (120.0 mg, 1.5 mmol), iodobenzene (114.0 μL, 1.0 mmol), and toluene (3.0 mL) were transferred to a Pyrex tube fitted with a Teflon screw-cap. Thiophenol (113.0 μL, 1.1 mmol) was added under nitrogen outside the glove box. The mixture was then heated at 120 °C in oil bath for 6 h and then cooled to room temperature. Ethyl acetate (10.0 mL) was added to the reaction mixture and the resultant suspension was filtered through Celite. The GC yield of diphenyl sulfide was 24% (response factor for diphenyl sulfide: 1.11).Reaction of copper(I) catalyzed C–S coupling with radical scavengers
In a glove box, IMes-Cu-Cl (40.0 mg, 0.1 mmol), LiOtBu (120.0 mg, 1.5 mmol), iodobenzene (114.0 μL, 1.0 mmol), toluene (3.0 mL), and radical scavengers (TEMPO, 156.0 mg, 1.0 mmol or cumene, 142.0 μL, 1.0 mmol) were transferred to a Pyrex tube fitted with a Teflon screw-cap. Thiophenol (113.0 μL, 1.1 mmol) was added under nitrogen outside the glove box. The mixture was then heated at 120 °C in oil bath for 6 h and then cooled to room temperature. Ethyl acetate (10.0 mL) was added to the reaction mixture and the resultant suspension was filtered through Celite. The GC yield of diphenyl sulfide were 86% (TEMPO); 78% (cumene) (response factor for diphenyl sulfide: 1.11).Reaction of IMes-Cu-Cl catalyzed C–S coupling with different aryl iodide
In a glove box, IMes-Cu-Cl (40.0 mg, 0.1 mmol), LiOtBu (120.0 mg, 1.5 mmol), aryl iodide (1.0 mmol), and solvent (3.0 mL) were transferred to a Pyrex tube fitted with a Teflon screw-cap. Thiophenol (113.0 μL, 1.1 mmol) was added under nitrogen outside the glove box. The mixture was then heated at 120 °C in oil bath for 6 h and then cooled to room temperature. Ethyl acetate (10.0 mL) was added to the reaction mixture and the resultant suspension was filtered through Celite. The GC yields were obtained via quantitative GC analysis with 1,4-di-tert-butylbenzene as the internal standard.The same procedure, amount of reagents and reaction conditions was applied for obtaining the isolated yields. The products were separated by column chromatography on silica gel with hexane as the eluent. Aryl sulfides were characterized by NMR and were consistent with the literature data.10
GC yield in DMF: 55% (response factor for diphenyl sulfide: 0.99).
GC yield in DMF: 61% (response factor for 2-methylphenyl phenyl sulfide: 0.96).
GC yield in DMF: 60% (response factor for 3-methylphenyl phenyl sulfide: 0.96).
GC yield in DMF: 50% (response factor for 4-methylphenyl phenyl sulfide: 0.96).
Kinetic experiments
In a glove box, 1.0 mmol of copper catalyst (amount of catalyst used: IMes-Cu-Cl, 400.0 mg; IMes-Cu-SPh, 477.0 mg), LiOtBu (1.2 g, 15.0 mmol), iodobenzene (1.14 mL, 10.0 mmol), and p-xylene (30.0 mL) were transferred to a round-bottom flask. Thiophenol (1.13 mL, 11.0 mmol) was added under nitrogen outside the glove box. The mixture was heated at reflux under nitrogen. We followed the reaction by quantitative GC analysis (1,4-di-tert-butylbenzene as the internal standard) every ten-minute for the first two hours and then every 30 min for the next four hours.Computation methods
All calculations were carried out by Gaussian 09 (ver. C.01).11 Based on the previous DFT calculations of Ullmann-type reactions,5,12 the exchange functional of B3LYP5,12a,b,d,m,13 was used. In order to find the more efficient basis set for our studies, the basis sets of LanL2DZ(f)14 and SDD15 for Cu in combination with 6-31+G(d), 6-31G(d), 6-311G(d,p), and 6-311+G(d,p) for all other atoms were used to optimize structures and compare the experimental geometries. The detailed combinations of different basis sets were listed in ESI.† We noticed that the basis set of 6-31+G(d) for main group atoms provided the consistent results as that of triple-ζ level basis sets. Thus, the basis sets of SDD and 6-31+G(d) were employed for heavy atoms (Cu and I) and main group atoms (H, C, N, O, S, Cl, Li and Na), respectively. This basis set is labeled as BS-1. For those single-point energy calculations, the basis set of SDD for heavy atoms (Cu and I) and 6-311+G(d,p) for main group atoms (H, C, N, O, S, Cl, Li and Na) were used and labeled as BS-2. The tighter integration grid is specified by “Int = UltraFine” keyword. Frequency calculations were performed to verify the real minima (no imaginary frequency) of stationary points or transition state (one imaginary frequency) and also to provide those free energies at 298.15 K and 1 atm. The intrinsic reaction coordinate (IRC)16 was used to confirm that the transition state of Cu-carbene complex reaction system is connected to correct reactant and product. Solvent effect (toluene, ε = 2.37; DMF, ε = 37.22) was calculated by self-consistent reaction filed with CPCM solvation model,17 and these calculations were carried out on the optimized gas-phase geometries. In addition, the energy barriers were also calculated by PBE0, MPWB1K, B3LYP-D2, and ωB97XD to evaluate the effects of exchange functionals and dispersion corrections in this study.Results and discussion
Reactivity of Cu(I) N-heterocyclic carbenes
Cu(I) N-heterocyclic carbenes IMes-Cu-Cl and IPr-Cu-Cl were synthesized according to literature method,9 and their structures are shown in Scheme 1. We follow the reaction procedure reported in the literature to investigate the influence of N-heterocyclic carbene ligand in the copper(I)-catalyzed C–S coupling reaction by using LCu-Cl as the copper source.1a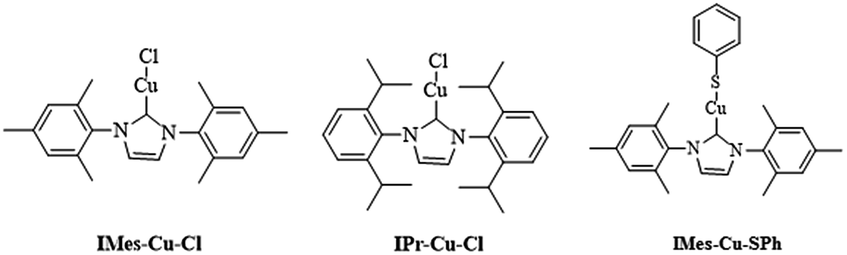 | ||
| Scheme 1 The structures of N-heterocyclic carbene. | ||
A mixture of LCuCl (0.1 mmol), iodobenzene (1.0 mmol), thiophenol (1.1 mmol) and 1.5 equiv. (1.5 mmol) of LiOtBu in toluene was stirred at 120 °C for 6 h. Reactions using only CuI, were also carried out under the same reaction condition for comparison. All the results are listed in Table 1.
|
|||
|---|---|---|---|
| Entry | Catalyst | Base | Yielda (%) |
| a GC yield with 1,4-di-tert-butylbenzene as internal standard.b Reaction run using 100 mol% TEMPO as radical scavenger.c 100 mol% cumene was used as radical scavenger.d GC yield with DMF (3.0 mL) as the solvent. | |||
| 1 | CuI | LiOtBu | 24 |
| 2 | IMes-Cu-Cl | LiOtBu | 81 (86)b, (78)c |
| 3 | IPr-Cu-Cl | LiOtBu | 48 (39)d |
| 4 | IMes-Cu-Cl | — | 14 |
| 5 | IMes-Cu-Cl | NaOtBu | 89 |
| 6 | IMes-Cu-Cl | KOtBu | 84 |
| 7 | IMes-Cu-SPh | LiOtBu | 83 |
Using both Cu(I) carbene complexes resulted in much better yields than using CuI alone (entries 1 to 3). The fact that IPr-Cu-Cl catalyst gave a lower yield than that of IMes-Cu-Cl (entries 2 to 3) may be caused by the higher steric hindrance of the bulky IPr ligand. Thus, IMes-Cu-Cl was used as the catalyst for other iodo-substrates, which all resulted in good yields under the same reaction condition (Table 2), as expected.2 The results are consistent with the general phenomena that ligand can enhance the Ullmann-type C–S coupling reaction.1–3,4b–g
|
|||
|---|---|---|---|
| Entry | Halide | Product | Yield (%) GCa/isolatedb |
| a GC yield with 1,4-di-tert-butylbenzene as internal standard.b Hexane was used as the eluent in silica gel column chromatography.c GC yield with DMF (3.0 mL) as the solvent. | |||
| 1 |  |
 |
81(55)c/82 |
| 2 | 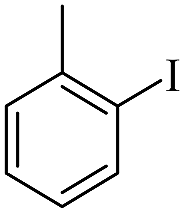 |
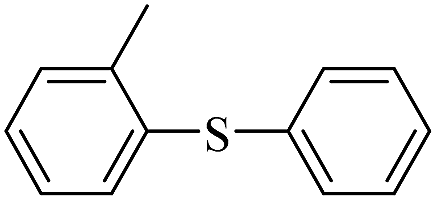 |
94(61)c/96 |
| 3 | 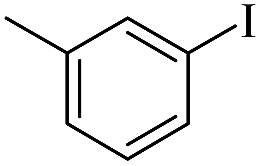 |
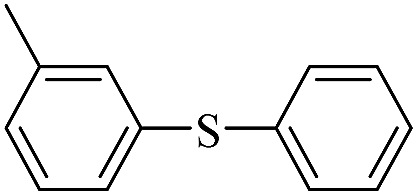 |
69(60)c/71 |
| 4 | 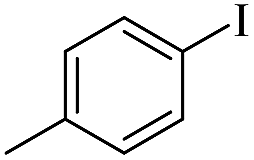 |
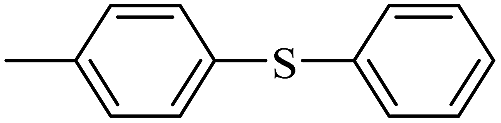 |
75(50)c/73 |
| 5 | 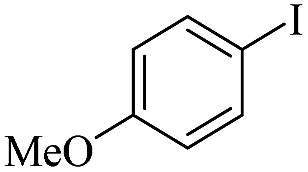 |
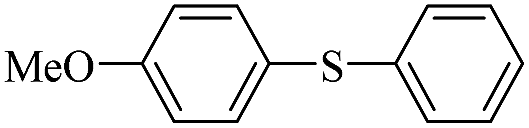 |
93/89 |
| 6 | 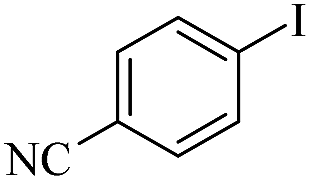 |
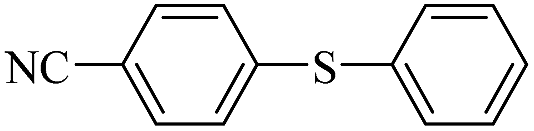 |
99/96 |
| 7 | 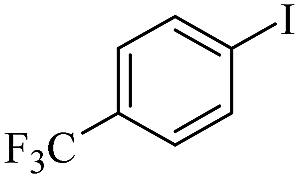 |
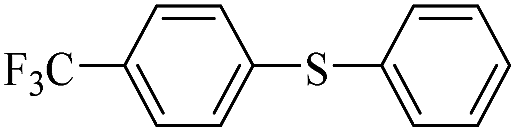 |
99/94 |
| 8 | 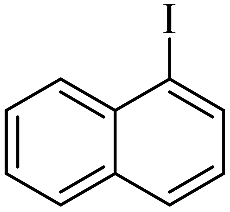 |
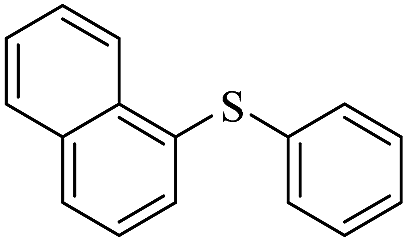 |
90/91 |
Both L-Cu-Cl and L-Cu-SPh (L = IMes, IPr) remain as a monomeric structure as shown by their single crystal X-ray diffraction studies.8,9b,18 In order to ensure the carbene complex is monomeric in solution as in the solid state, diffusion-ordered NMR spectroscopy (DOSY) of IMes-Cu-SPh in d8-toluene was carried out. The estimated hydrodynamic radius of IMes-Cu-SPh is 5.04 Å – comparable to the corresponding computed radii of 5.23 Å.19 Addition of cumene or TEMPO as radical scavenger did not actually reduce the reaction yield, indicating that the contribution of free radical reaction path is limited if there is any (Table 1, entry 2).
Proposed catalytic cycle and LCu-SPh kinetically competent
As generally suggested, the reaction between LCu-Cl and MSPh (M = Li, Na or K) – produced from the deprotonation of HSPh by MOtBu – generates LCu-SPh.4 Oxidative addition of ArI to Lcu-SPh produces LCu(SPh)(Ar)I which further produce PhSAr and LCu-I through reductive elimination. LCu-I can react with MSPh to regenerate LCu-SPh to complete the catalytic cycle (Fig. 1).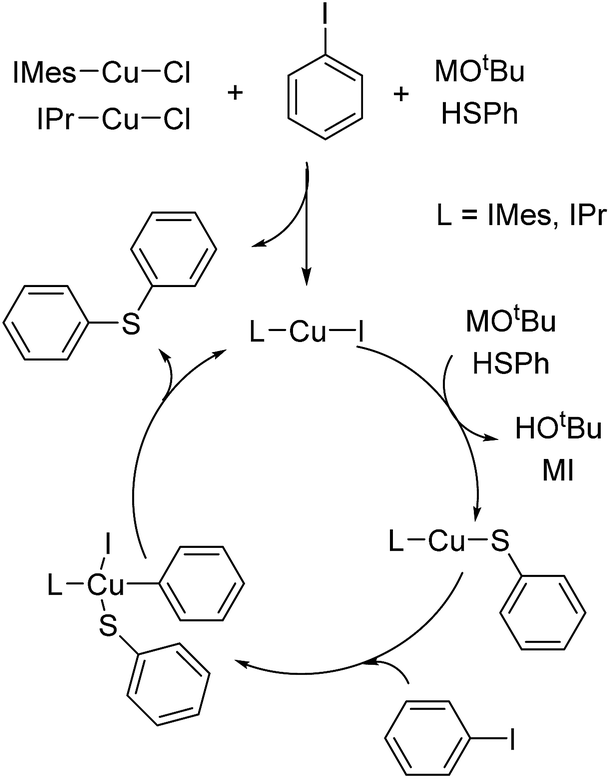 | ||
| Fig. 1 The proposed catalytic cycle by oxidative addition/reductive elimination reaction. | ||
In order to ensure that LCu-SPh is the catalyst or the dominant intermediate in the catalytic cycle, IMes-Cu-SPh was synthesized by the reaction between IMes-Cu-Cl and NaSPh.8 Similar kinetic data were obtained for both reactions using IMes-Cu-Cl and IMes-Cu-SPh indicating that IMes-Cu-SPh is kinetically competent. (Fig. 2).
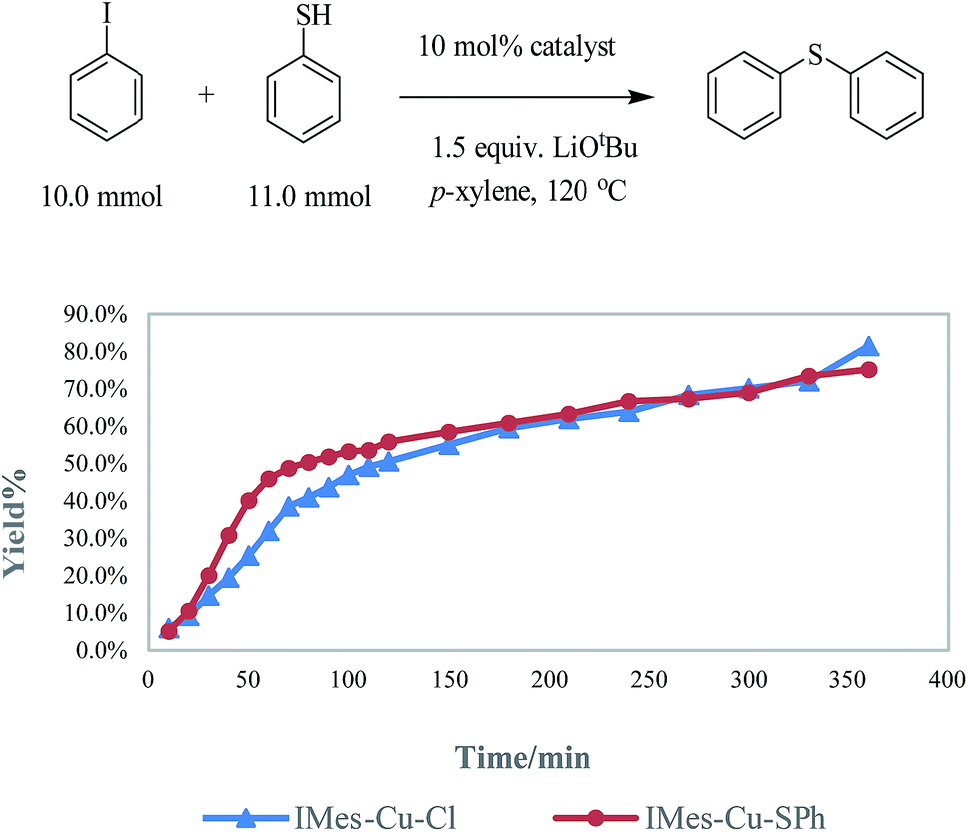 | ||
| Fig. 2 The kinetic experiments of C–S cross coupling with different catalyst. | ||
DFT calculations
Four possible mechanisms of Ullmann type reaction – oxidative addition/reductive elimination (OA/RE), σ-bond metathesis (σBM), single electron transfer (SET) and halogen atom transfer (HAT) – have been proposed.3a,c,4,5,6e,g,h,12,20,21 The first two are usually non-free radical reactions. However, a singlet biradical transition state for σBM is also reported implying the possibility of free radical path of σBM.12j The others (SET and HAT) are free radical paths.Moreover, in Cu-catalyzed C–X (X = C, O, N, S, P) cross coupling reactions, whether the reactions go through non-free radical or free radical paths still debatable.5b,d,e,g,6e,g,h,12b,d,e,20 Adding radical scavenger experiment in this study indicates that the contribution of free radical reaction path is limited if there is any. Therefore, our DFT study focus on non-free radical paths, and possible non free radical mechanisms are proposed (Scheme 2).
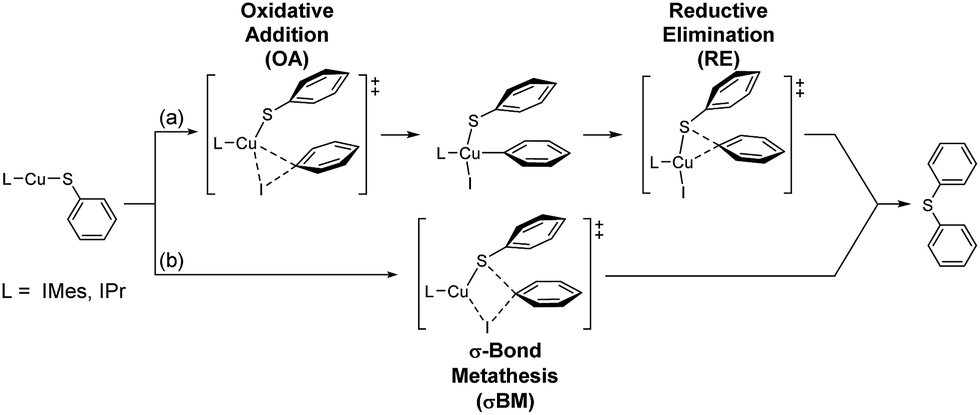 | ||
| Scheme 2 The possible non free radical mechanisms. | ||
The results of DOSY and kinetic experiment indicates that IMes-CuSPh dominates and is kinetically competent with IMes-Cu-Cl in toluene. This observation is consistent with the reported theoretical study (1,10-phenanthrolines as ligand)5e which states that L-CuSPh may transforms to other Cu species through disproportionation, dimerization and ligand dissociation. These transformations are greatly endergonic with free energy differences 14–69 kcal mol−1 (see ESI†) implying that L-CuSPh dominates in toluene also. In addition, comparing the binding capacities between phen and carbene ligands by the dissociation of LCuSPh to form CuSPh (27.08 kcal mol−1 for L = phen, 40.19 kcal mol−1 for L = IMes and 40.25 kcal mol−1 for L = IPr), the N-heterocyclic carbene ligands have stronger binding capacity than that of phen and explains the ligand labile issue.
The energy profiles of L-CuSPh-catalyzed OA/RE and σBM paths are shown in Fig. 3a. As expected, the rate-determining-step is the oxidative addition of PhI to LCuSPh to produce LCu(SAr)(Ph)I. The reaction with IMes-CuSPh catalyst has a lower activation energy (40.61 kcal mol−1) than that of IPr-Cu-SPh (46.01 kcal mol−1) implying that IMes-CuSPh gave a higher reaction rate than that of IPr-Cu-SPh. In addition to the OA/RE reaction path, the reaction may go through the hypothetical σBM mechanism. DFT calculation with B3LYP functional reveals that the path of σBM has a higher activation energy (43.18 kcal mol−1) than that of the oxidative addition step (40.61 kcal mol−1) for L = IMes (Fig. 3), but, for L = IPr, both reaction paths have similar activation energy (46.01 vs. 46.70 kcal mol−1).
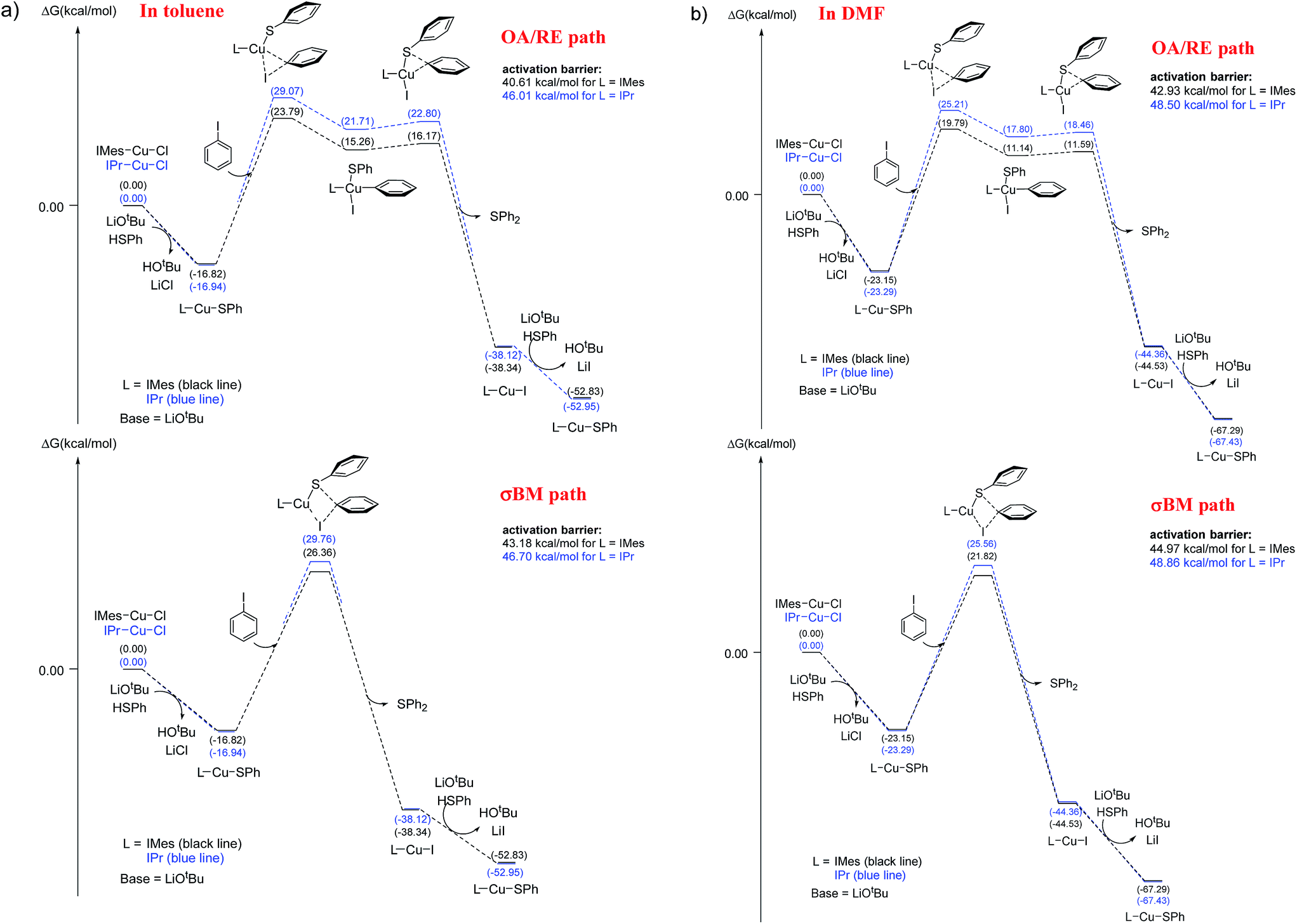 | ||
| Fig. 3 Free energy profile (kcal mol−1) for oxidative addition/reductive elimination paths and σ-bond metathesis paths. (a) In toluene, (b) in DMF. | ||
Base on such small energy differences, it is ambiguous to select the reaction path accordingly. However, ∼1 kcal mol−1 energy difference is used to explain the selection between C–N and C–O coupling,12c and both three-center and four-center oxidative pathway must be envisaged due to the ∼1.19 kcal mol−1 energy difference was also reported.12e
In C–N coupling reaction,5b the energy barrier is 29 kcal mol−1 determined from the experimental kinetic data at 120 °C. However, the DFT calculation results indicate that the energy barrier is 38 kcal mol−1 (by using B3LYP functional). Similar overestimation of energy barrier by using B3LYP functional was also reported in the study of Liu et al.12d Some theoretical studies show that the energy barrier is about 28–35 kcal mol−1 for those experiments carried out around 90–120 °C.5d,e,12c It seems the overestimated energy barrier is a common phenomenon in theoretical studies. Moreover, PBE0 and MPWB1K were also used to study Ullmann type reaction.12c,e–g. In order to investigate the effects of exchange functionals and dispersion corrections in the energy barrier, PBE0 (hybrid GGA), MPWB1K (hybrid meta GGA) and dispersion-corrected functionals B3LYP-D2 and ωB97XD are selected to calculate energy barriers. The larger energy gap (5.37–9.37 kcal mol−1 for L = IMes; 3.22–8.67 kcal mol−1 for L = IPr) between OA/RE and σBM paths were provided by PBE0, MPWB1K, B3LYP-D2 and ωB97XD (see Table 3). Using PBE0, MPWB1K, B3LYP-D2 and ωB97XD can obtain lower energy barrier than that of B3LYP, especially the results obtained by dispersion-corrected DFT methods (lower 6.79–20.66 kcal mol−1 than that of B3LYP at BS-2 basis set). On the other hand, the basis set effect is limited, and especially the results obtained by BS-1 and BS-2 are very close. These results suggest that the catalytic cycle may predominantly go through the OA/RE sequence. Moreover, all DFT methods report a consistent result that IMes-CuSPh gave a higher reaction rate than IPr-Cu-SPh (see Table 3). Thus, B3LYP can achieve qualitative analysis even it reports higher energy barriers.
| OA/RE L = IMes/L = IPr | σBM L = IMes/L = IPr | |
|---|---|---|
| a The free energy unit is kcal mol−1.b The basis set in combination with SDD (for Cu and I) and 6-31G(d) (for others) is noted as BS-small.c BS-1 was used to carry out geometry optimization. | ||
| B3LYP/BS-smallb | 38.34/43.48 | 40.92/44.68 |
| PBE0/BS-smallb | 32.54/37.35 | 37.91/40.57 |
| MPWB1K/BS-smallb | 34.80/36.61 | 40.91/42.80 |
| B3LYP/BS-1 | 40.89/46.12 | 43.30/46.88 |
| B3LYP/BS-2c | 40.61/46.01 | 43.18/46.70 |
| B3LYP-D2/BS-2c | 22.32/25.35 | 29.59/32.54 |
| ωB97XD/BS-2c | 25.72/31.24 | 34.89/39.91 |
The effect of aprotic solvent
The low solubility of base in toluene generates mass transfer effects which affect the deprotonation rate so thus the overall rate of the reaction.21 Using aprotic solvent can dissolve the base and avoid these factors. We use DMF as the solvent to dissolve the base to evaluate the above mentioned solvent effect. Both reaction with IMes-Cu-Cl and IPr-Cu-Cl as the catalyst gave better yield in toluene than in DMF (Table 1, entry 2; Table 2, entries 1 to 4). These observations reveal that DMF retards instead of increases the reaction rate even the mass transfer effect is removed.The presence of DMF may affect the rate determination step (the aryl iodide activation step in the catalytic cycle) if the reaction mechanism remains the same as that of using toluene as the reaction media. The B3LYP calculation was carried out again to evaluate the feasibility of the proposed mechanism. The results are shown in Fig. 3b and Table 4. The rate-determining-step, the oxidative addition of PhI to LCu-SPh to produce LCu(SAr)(Ph)I, is the same as in the toluene system. Both reactions having a higher activation energy (42.93 and 48.50 kcal mol−1 for IMes-Cu-Cl and IPr-Cu-Cl respectively) in DMF than that in toluene (40.61 and 46.01 kcal mol−1) is consistent with the results that both carbene complexes gave a higher yield in toluene than in DMF under the same reaction conditions. Similarly, in DMF, the reaction with IPr-Cu-Cl having a higher activation energy (48.50 kcal mol−1) than that of IMes-Cu-Cl (42.93 kcal mol−1) is consistent with the observation that IMes-Cu-Cl gave a higher yield under the same reaction conditions. In addition, the results obtained by B3LYP-D2 and ωB97XD (see Table 4) are also consistent with above-mentioned observation.
Conclusions
N-Heterocyclic carbene complexes were used to catalyse Ullmann type C–S coupling reaction with good yields. The non-labile carbene ligand enables the carbene complex to maintain its structure in the system and enter the catalytic cycle. Kinetic study showed that IMes-Cu-SPh is kinetically competent indicating IMes-Cu-SPh is an intermediate in the catalytic cycle. The monomeric structure of carbene complex was supported by single crystal X-ray structure in solid state and DOSY analysis in solution. Radical scavenger did not retard the catalytic reaction indicating non-free radical path dominates in the reaction cycle. A catalytic cycle with the aryl iodide activation step as the rate determining step is proposed based on DFT calculation. A lower product yield in DMF indicates that DMF retard the reaction even the mass transfer effect was removed. DFT calculation shows that the reaction in DMF has a higher activation energy in the rate determining step than that in toluene further supports the observation. In addition, using smaller basis set 6-31+G(d) for main group atoms (H, C, N, O, S, Cl, Li and Na) obtains consistent results as that of triple-ζ basis sets. Therefore, we suggest this basis set for Cu-catalysed C–S coupling studies to reduce computational cost. In this study, using dispersion-corrected functionals B3LYP-D2 and ωB97XD can reduce energy barrier effectively. This result indicates that dispersion correction is significant for the C–S coupling reaction catalysed by Cu(I) carbene complex.Acknowledgements
We are grateful to the Ministry of Science and Technology, Republic of China and Academia Sinica for financial support of this work.Notes and references
- (a) C. G. Bates, R. K. Gujadhur and D. Venkataraman, Org. Lett., 2002, 4, 2803 CrossRef CAS PubMed; (b) W. Deng, Y. Zou, Y.-F. Wang, L. Liu and Q.-X. Guo, Synlett, 2004, 15, 1254 Search PubMed; (c) Y.-J. Chen and H.-H. Chen, Org. Lett., 2006, 8, 5609 CrossRef CAS PubMed; (d) C. Enguehard-Gueiffier, I. Thery, A. Gueiffier and S. L. Buchwald, Tetrahedron, 2006, 62, 6042 CrossRef CAS; (e) D. Zhu, L. Xu, F. Wu and B. Wan, Tetrahedron Lett., 2006, 47, 5781 CrossRef CAS; (f) X. Lv and W. Bao, J. Org. Chem., 2007, 72, 3863 CrossRef CAS PubMed; (g) H. Zhang, W. Cao and D. Ma, Synth. Commun., 2007, 37, 25 CrossRef CAS; (h) N. R. Jogdand, B. B. Shingate and M. S. Shingare, Tetrahedron Lett., 2009, 50, 6092 CrossRef CAS; (i) D. Prasad, A. B. Naidu and G. Sekar, Tetrahedron Lett., 2009, 50, 1411 CrossRef CAS; (j) Y. Li, X. Li, H. Wang, T. Chen and Y. Xie, Synthesis, 2010, 3602 CrossRef CAS; (k) S. Roy and P. Phukan, Tetrahedron Lett., 2015, 56, 2426 CrossRef CAS.
- J. P. Wu, A. K. Saha, N. Haddad, C. A. Busacca, J. C. Lorenz, H. Lee and C. H. Senanayake, Adv. Synth. Catal., 2016, 358, 1924 CrossRef CAS . In this paper, aryl iodide with electron withdrawing group and thiophenol with electron donating group were used, and good yields were obtained. However, aryl iodide with electron withdrawing group and thiophenol with electron donating group in C–S cross coupling reaction gives good yields without adding catalyst ( Y. Zhang, K. C. Ngeow and J. Y. Ying, Org. Lett., 2007, 9, 3495 CrossRef PubMed ). We therefore carried out the cross coupling reaction with aryl iodide with electron donating group, which need Cu(I) catalyst to proceed the reaction, to evaluate the catalytic activity of Cu(I) carbene.
- (a) C. G. Bates, P. Saejueng, M. Q. Doherty and D. Venkataraman, Org. Lett., 2004, 6, 5005 CrossRef CAS PubMed; (b) B. Jiang, H. Tian, Z.-G. Huang and M. Xu, Org. Lett., 2008, 10, 2737 CrossRef CAS PubMed; (c) Y. Feng, X. Zhao, J. Wang, F. Zheng and H. Xu, Chin. J. Chem., 2009, 27, 2423 CrossRef CAS; (d) P.-S. Luo, F. Wang, J.-H. Li, R.-Y. Tang and P. Zhong, Synthesis, 2009, 921 CAS; (e) L.-F. Niu, Y. Cai, C. Liang, X.-P. Hui and P.-F. Xu, Tetrahedron, 2011, 67, 2878 CrossRef CAS; (f) Y.-A. Chen, S. S. Badsara, W.-T. Tsai and C.-F. Lee, Synthesis, 2015, 47, 181 CAS.
- (a) R. A. Altman, E. D. Koval and S. L. Buchwald, J. Org. Chem., 2007, 72, 6190 CrossRef CAS PubMed; (b) A. K. Verma, J. Singh and R. Chaudhary, Tetrahedron Lett., 2007, 48, 7199 CrossRef CAS; (c) L. Rout, P. Saha, S. Jammi and T. Punniyamurthy, Eur. J. Org. Chem., 2008, 2008, 640 CrossRef; (d) Y. Feng, H. Wang, F. Sun, Y. Li, X. Fu and K. Jin, Tetrahedron, 2009, 65, 9737 CrossRef CAS; (e) E. Haldón, E. Alvarez, M. C. Nicasio and P. J. Pérez, Organometallics, 2009, 28, 3815 CrossRef; (f) H.-J. Xu, X.-Y. Zhao, J. Deng, Y. Fu and Y.-S. Feng, Tetrahedron Lett., 2009, 50, 434 CrossRef CAS; (g) A. M. Thomas, S. Asha, K. Sindhu and G. Anilkumar, Tetrahedron Lett., 2015, 56, 6560 CrossRef CAS.
- (a) S.-L. Zhang, L. Liu, Y. Fu and Q.-X. Guo, Organometallics, 2007, 26, 4546 CrossRef CAS; (b) J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971 CrossRef CAS PubMed; (c) S. Zhang and Y. Ding, Organometallics, 2011, 30, 633 CrossRef CAS; (d) S. Zhang, Z. Zhu and Y. Ding, Dalton Trans., 2012, 41, 13832 RSC; (e) S.-L. Zhang and H.-J. Fan, Organometallics, 2013, 32, 4944 CrossRef CAS; (f) L.-C. Li, F.-F. Sun, R. Pang and D.-Y. Wu, Comput. Theor. Chem., 2014, 1038, 40 CrossRef CAS; (g) S.-L. Zhang, W.-F. Bie and L. Huang, Organometallics, 2014, 33, 5263 CrossRef CAS.
- (a) I. Dance and J. Calabrese, Inorg. Chim. Acta, 1976, 19, L41 CrossRef; (b) M. Baumgartner, W. Bensch, P. Hug and E. Dubler, Inorg. Chim. Acta, 1987, 136, 139 CrossRef CAS; (c) S. Shi, X. Zhang and X. Shi, J. Phys. Chem., 1995, 99, 14911 CrossRef CAS; (d) P. Ahte, P. Palumaa and T. Tamm, J. Phys. Chem. A, 2009, 113, 9157 CrossRef CAS PubMed; (e) S.-W. Cheng, M.-C. Tseng, K.-H. Lii, C.-R. Lee and S.-G. Shyu, Chem. Commun., 2011, 47, 5599 RSC; (f) C. Chen, Z. Weng and J. F. Hartwig, Organometallics, 2012, 31, 8031 CrossRef CAS PubMed; (g) C. Uyeda, Y. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548 CrossRef CAS PubMed; (h) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525 RSC; (i) E. Guzmán-Percástegui, D. J. Hernández and I. Castillo, Chem. Commun., 2016, 52, 3111 RSC.
- (a) N. Fröhlich, U. Pidun, M. Stahl and G. Frenking, Organometallics, 1997, 16, 442 CrossRef; (b) J. Huang, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 2370 CrossRef CAS; (c) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (d) A. R. Chianese, X. Li, M. C. Janzen, J. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663 CrossRef CAS; (e) R. Dorta, E. D. Stevens, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 10490 CrossRef CAS PubMed; (f) A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322 CrossRef CAS; (g) M.-T. Lee and C.-H. Hu, Organometallics, 2004, 23, 976 CrossRef CAS; (h) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407 CrossRef CAS; (i) R. H. Crabtree, J. Organomet. Chem., 2005, 690, 5451 CrossRef CAS.
- S. A. Delp, C. Munro-Leighton, L. A. Goj, M. A. Ramírez, T. B. Gunnoe, J. L. Petersen and P. D. Boyle, Inorg. Chem., 2007, 46, 2365 CrossRef CAS PubMed.
- (a) M. H. Voges, C. Rømming and M. Tilset, Organometallics, 1999, 18, 529 CrossRef CAS; (b) A. P. McLean, E. A. Neuhardt, J. P. S. John, M. Findlater and C. D. Abernethy, Transition Met. Chem., 2010, 35, 415 CrossRef CAS.
- (a) J. Yi, Y. Fu, B. Xiao, W.-C. Cui and Q.-X. Guo, Tetrahedron Lett., 2011, 52, 205 CrossRef CAS; (b) H.-J. Xu, Y.-Q. Zhao, T. Feng and Y.-S. Feng, J. Org. Chem., 2012, 77, 2878 CrossRef CAS PubMed; (c) P. Gogoi, S. Hazarika, M. J. Sarma, K. Sarma and P. Barman, Tetrahedron, 2014, 70, 7484 CrossRef CAS; (d) A. R. Martin, D. J. Nelson, S. Meiries, A. M. Slawin and S. P. Nolan, Eur. J. Org. Chem., 2014, 2014, 3127 CrossRef CAS.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- (a) E. R. Strieter, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2008, 131, 78 CrossRef PubMed; (b) R. Giri and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 15860 CrossRef CAS PubMed; (c) G. O. Jones, P. Liu, K. Houk and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 6205 CrossRef CAS PubMed; (d) H.-Z. Yu, Y.-Y. Jiang, Y. Fu and L. Liu, J. Am. Chem. Soc., 2010, 132, 18078 CrossRef CAS PubMed; (e) G. Lefèvre, G. g. Franc, C. Adamo, A. Jutand and I. Ciofini, Organometallics, 2012, 31, 914 CrossRef; (f) G. Lefevre, G. Franc, A. Tlili, C. Adamo, M. Taillefer, I. Ciofini and A. Jutand, Organometallics, 2012, 31, 7694 CrossRef CAS; (g) G. Lefèvre, A. Tlili, M. Taillefer, C. Adamo, I. Ciofini and A. Jutand, Dalton Trans., 2013, 42, 5348 RSC; (h) A. I. Konovalov, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 13410 CrossRef CAS PubMed; (i) P. F. Larsson, C. J. Wallentin and P. O. Norrby, ChemCatChem, 2014, 6, 1277 CAS; (j) T. Chen, M. Yan, C. Zheng, J. Yuan, S. Xu, R. Chen and W. Huang, Chin. J. Chem., 2015, 33, 961 CrossRef CAS; (k) D.-H. Yu, J.-N. Shao, R.-X. He and M. Li, Chin. Chem. Lett., 2015, 26, 564 CrossRef CAS; (l) X. Ge, X. Chen, C. Qian and S. Zhou, RSC Adv., 2016, 6, 29638 RSC; (m) L. C. Li, L. Zhang, W. Wang, R. Pan, S. Mao and A. M. Tian, Int. J. Chem. Kinet., 2016, 48, 11 CrossRef CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS.
- (a) M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS; (b) G. Igel-Mann, H. Stoll and H. Preuss, Mol. Phys., 1988, 65, 1321 CrossRef CAS; (c) A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431 CrossRef CAS.
- (a) K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS.
- (a) C. J. Cramer and D. G. Truhlar, Chem. Rev., 1999, 99, 2161 CrossRef CAS PubMed; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed; (c) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- H. Kaur, F. K. Zinn, E. D. Stevens and S. P. Nolan, Organometallics, 2004, 23, 1157 CrossRef CAS.
- For details, see the ESI.†.
- (a) C.-K. Tseng, M.-C. Tseng, C.-C. Han and S.-G. Shyu, Chem. Commun., 2011, 47, 6686 RSC; (b) C. K. Tseng, C. R. Lee, C. C. Han and S. G. Shyu, Chem.–Eur. J., 2011, 17, 2716 CrossRef CAS PubMed; (c) C.-K. Tseng, C.-R. Lee, M.-C. Tseng, C.-C. Han and S.-G. Shyu, Dalton Trans., 2014, 43, 7020 RSC; (d) H.-J. Chen, I.-J. Hsu, M.-C. Tseng and S.-G. Shyu, Dalton Trans., 2014, 43, 11410 RSC; (e) H.-J. Chen, M.-C. Tseng, I.-J. Hsu, W.-T. Chen, C.-C. Han and S.-G. Shyu, Dalton Trans., 2015, 44, 12086 RSC.
- S. Sung, D. Sale, D. C. Braddock, A. Armstrong, C. Brennan and R. P. Davies, ACS Catal., 2016, 6, 3965 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, DFT calculation data and spectral data. See DOI: 10.1039/c6ra27757h |
| This journal is © The Royal Society of Chemistry 2017 |