
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted one-pot synthesis of hybrid nanosystems based on CdS quantum dots functionalized with organic chromophores: effect of the chromophore nature on the nanosystem composition†
Oleg V. Chashchikhin *,
Mikhail F. Budyka,
Tatiana N. Gavrishova and
Vitalii M. Li
*,
Mikhail F. Budyka,
Tatiana N. Gavrishova and
Vitalii M. Li
Institute of Problems of Chemical Physics, Russian Academy of Sciences, pr. Akademika Semenova 1, Chernogolovka, Moscow region 142432, Russian Federation. E-mail: ovchash@icp.ac.ru
First published on 20th December 2016
Abstract
A novel method of one-pot microwave assisted synthesis was employed to prepare hybrid nanosystems (HNSs) with CdS quantum dots (QD) covered by an organic shell containing capping “background” ligand (benzylmercaptan, BM) and functional ligand (FL) with a styrylquinoline (SQ) or merocyanine (MC) chromophore group. An average amount (n) of the FL molecules in the HNS organic shell was measured. We herein report on an unusual dependence of n on the FL relative concentration in the source solution. A non-linear dependence resembling a cooperative effect is found for the SQ-FL, and a linear dependence with critical concentration is found for the MC-FL. The effects are suggested to be associated with the peculiarities of microwave activation of the ligands, which are built into the organic shell of the HNS as a result of a kinetically rather than thermodynamically controlled process. These data shed light on the problem of the formation of the covering organic shell on the QD surface in different conditions.
Introduction
Hybrid organic–inorganic nanosystems (HNSs), wherein semiconductor nanocrystals (quantum dots, QDs) are covered by organic ligand molecules, attract considerable attention due to the possibility to combine useful properties of organic and inorganic subsystems for technological applications in optoelectronics, biology, medicine, for energy production or conversion.1–10 Semiconductor QDs have unique size-dependent electronic and optical properties,11 while organic subsystems can be used to modulate (including reversibly) these properties.1–4,12–14To be correct, special coordinating (capping) organic ligands are often used to cover the QDs for stabilization, these ligands contain special “anchor” groups for attaching to the QD surface.15 Sometimes the stabilizing layer consists of several capping ligands such as trioctylphosphine (TOP) and trioctylphosphine oxide (TOPO); tributylphosphine and TOPO; TOP, TOPO and hexadecylamine;16,17 oleic acid and octadecylphosphonic acid;18 oleic acid and benzophenone;19,20 oleylamine and TOP;21 N-oleylmorpholine and stearic acid;22 stearic acid, TOPO and octadecylamine.23
However, such QDs are not considered as “hybrid nanosystems”. The HNS should contain, apart from covering “inert” (“background”) ligand, a specific functional ligand (FL), for example, organic dye – quencher or sensitizer, photochromic compound, bioactive molecule, etc. Since functional ligand can decompose at high temperatures used upon synthesis of QDs, the HNSs are usually obtained by ligand exchange or self-assembling, an extra procedures after the QD synthesis.3,4,8–10,24 Due to development of microwave-assisted synthesis of QDs,25–29 the possibility has arisen for inserting the FL into the organic shell of the HNS simultaneously with the capping inert ligand at the stage of the QDs synthesis. The number (n) of the FL molecules in the HNS organic shell can be controlled by the relative concentration (x) of the FL in the source solution used for the HNS synthesis, and, at first sight, n should be proportional to x.
We herein report on the two types of dependence of n on x, which were found upon the one-pot microwave-assisted synthesis of the HNSs with the CdS QD covered by organic shell containing the benzyl mercaptan (BM) as an inert capping ligand and an additional FL. Two FLs with the isothiouronium anchor groups (for attaching to the QD surface), which are linked by the nonamethylenic chain to different chromophores, are studied, Scheme 1. The chromophores are the neutral styrylquinoline (SQ) or positively charged N-methyl styrylquinolinium iodide, which is O-alkylated merocyanine (MC). The exponential dependence of n on x was found for the SQ-FL, and the linear dependence with critical concentration was found for the MC-FL.
 | ||
| Scheme 1 Structure of benzyl mercaptan (BM) and functional ligands (FLs) with styrylquinoline (SQ) and O-alkylated merocyanine (MC) chromophore. | ||
It should be noted that the HNSs with the oleic acid stabilized CdS QDs and the SQ-containing ligands have been obtained earlier via the ligand exchange procedures, however, the dependence of n on x has not been studied.30 This dependence has been previously studied for the one-pot microwave-assisted synthesis of the HNSs with the SQ ligands having the thiol anchor group.31 In the present paper we use ligands with another anchor group – isothiouronium instead of the thiol one, and in addition to the neutral SQ chromophore, the positively charged MC chromophore is investigated. The results obtained are discussed below in comparison with the previously published data.
As mentioned above, reported here HNSs contain FLs with chromophoric groups, which undergo reversible trans–cis photoisomerisation.32 Thus, HNSs could be applied as photoswitches. Investigation of photochemical properties of the MC-FL containing HNSs is underway. The HNSs with the SQ ligands having the thiol anchor group31 were used for the studies of photolysis kinetics of the FL (in the free form and attached to the QD's surface).33
Results and discussion
The obtained nanoparticles characterisation by (HR)TEM, SAED and XRD techniques can be found in the ESI.† According the TEM images (Fig. SI-1a†) nanoparticles have almost spherical shape and good size distribution (Fig. SI-1c†) centered at 2.4 nm and ranged between 2.1 and 2.9 nm. SAED image (Fig. SI-1a,† left inset) and XRD pattern (Fig. SI-1b†) indicate hexagonal CdS crystal structure of nanoparticles.The absorption spectra of the unlinked (“free”) SQ-FL, free QD and HNSs with the SQ ligands in dimethylformamide (DMF) are shown in Fig. 1a. Free SQ-FL and QD have absorption maximums at 360 nm and 333 nm, respectively. The size of nanoparticles was calculated from the first excitonic peak position in the absorption spectra (333 nm) following the formula (1)
| E = Ebulk + h2/8R2(1/me + 1/mh) − 1.8e2/4πε0εR, | (1) |
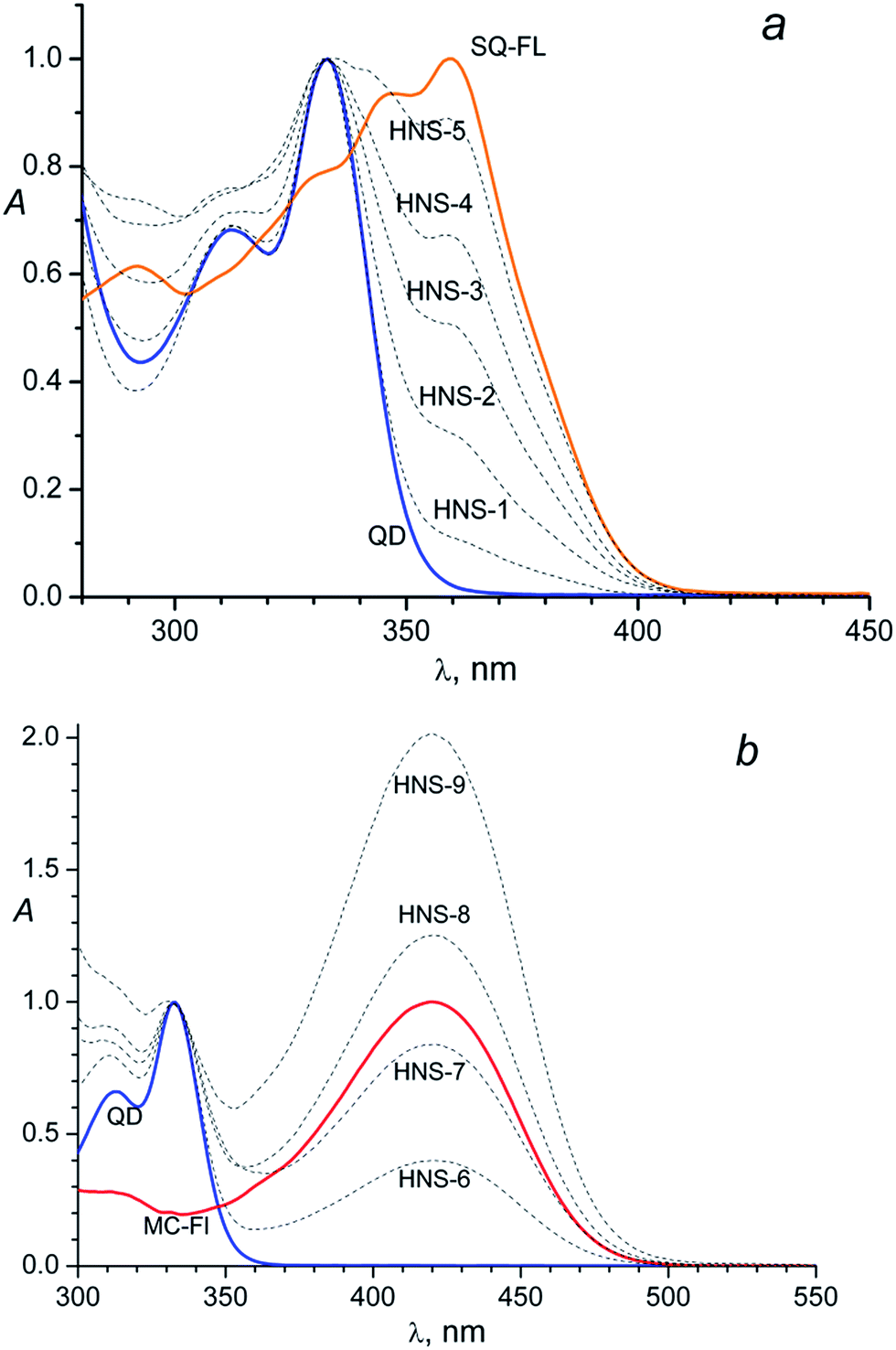 | ||
| Fig. 1 (a) Normalized (at 333 nm) absorption spectra of the CdS QDs (diameter of 2.4 nm), the functional ligand SQ-FL, and hybrid nanosystems HNS-1–HNS-5; (b) normalized absorption spectra of the CdS QDs (diameter of 2.4 nm), N-methyl-2-(4-[9-mercaptononoxy]styryl)quinolinium iodide – the functional ligand MC-FL, and hybrid nanosystems HNS-6–HNS-9. The QD and HNSs spectra are normalized at 333 nm, the MC-FL spectrum is normalized at 420 nm. | ||
So, the presence of the FL in the HNS organic shell can be clearly detected due to an additional absorbance at 360 nm: a shoulder at the long-wavelength tail of the QD absorption band in the HNS-1–HNS-3 spectra, or separate long-wavelength absorption band in the HNS-4 and HNS-5 spectra, Fig. 1a.
The same picture is observed for the MC containing nanosystems, Fig. 1b. In this case, owing to the larger difference in the absorption maximums of the two subsystems (333 nm for the QD and 420 nm for the MC-FL), the presence of an organic chromophore is detected by the separate long-wavelength absorption band in the spectra of all HNSs, Fig. 1b.
Comparison of Fig. 1a and b with Fig. 1 in the previous paper31 testifies that the HNS absorption spectrum is determined by the nature of the photochromic fragment and does not depend on the nature of the anchor group (thiol in ref. 31 and isothiouronium in the present study). However, the anchor group has an influence on the ability of the FL to bind to the QD surface, see below.
Qualitatively, in both cases (Fig. 1a and b) one can see identical dependence: the higher the FL relative concentration (x) in the source solution, the higher the relative absorbance of this ligand in the HNS; here x = CFL/(CFL + CBM)sol, where Ci is the concentration of component i in the solution.
Photoluminescence characterization of the HNSs and constituting QD and FLs can be found in the ESI, Fig. SI-2† for SQ-FL containing HNSs and Fig. SI-3† for MC-FL containing HNSs. Photoluminescence and photoluminescence excitation spectra of HNSs contain bands both of the QD and the corresponding FL.
Presence of the FL in HNS is confirmed by FTIR spectroscopy. Fig. 2 shows spectra of the pure QD, SQ-FL, MC-FL and some representative HNSs. In the spectra of all HNSs, the CdS characteristic bands (698, 1648 cm−1) are observed. Additionally, in the spectra of the first series of the HNSs (Fig. 2a) one can see the characteristic bands of the SQ-FL (the most intensive bands are 823, 1174, and 1511 cm−1), whereas in the spectra of the second series of the HNSs (Fig. 2b), the characteristic bands of the MC-FL are observed (837, 1169, 1594 cm−1).
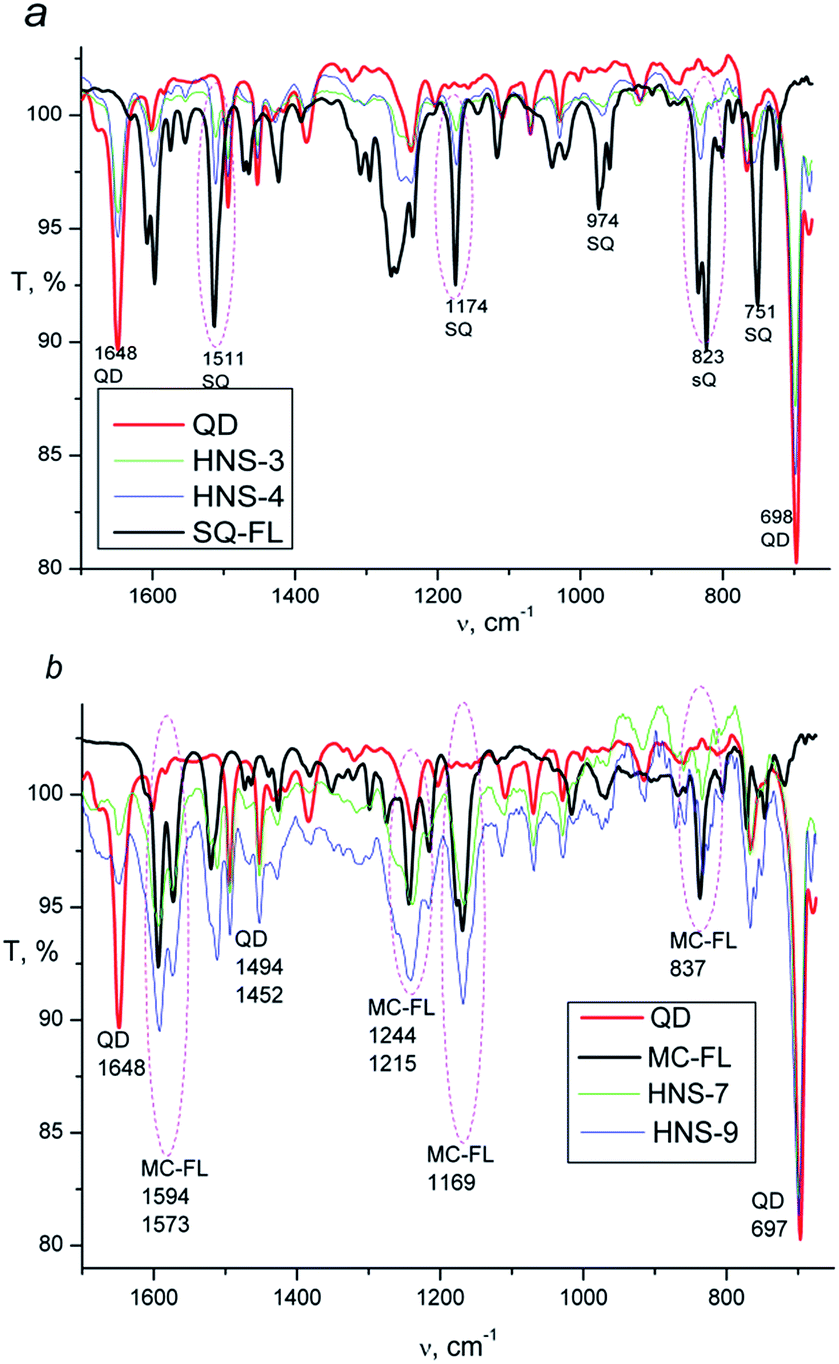 | ||
| Fig. 2 ATR-FTIR spectra: (a) QD, SQ-FL, HNS-3 and HNS-4; (b) QD, MC-FL, HNS-7 and HNS-9. | ||
For the quantitative analysis of the HNS composition we used electronic absorption spectra. Provided the spectra of the QD and FL remain unchanged upon binding of these components to HNS, the absorbance of the latter (AHNS) is defined as
| AHNS = (εQD + n × εFL) × CHNS × l, | (2) |
Fig. 3 shows the dependence of n on x for the different functional ligands studied. One can see that, quantitatively, completely different dependences are observed.
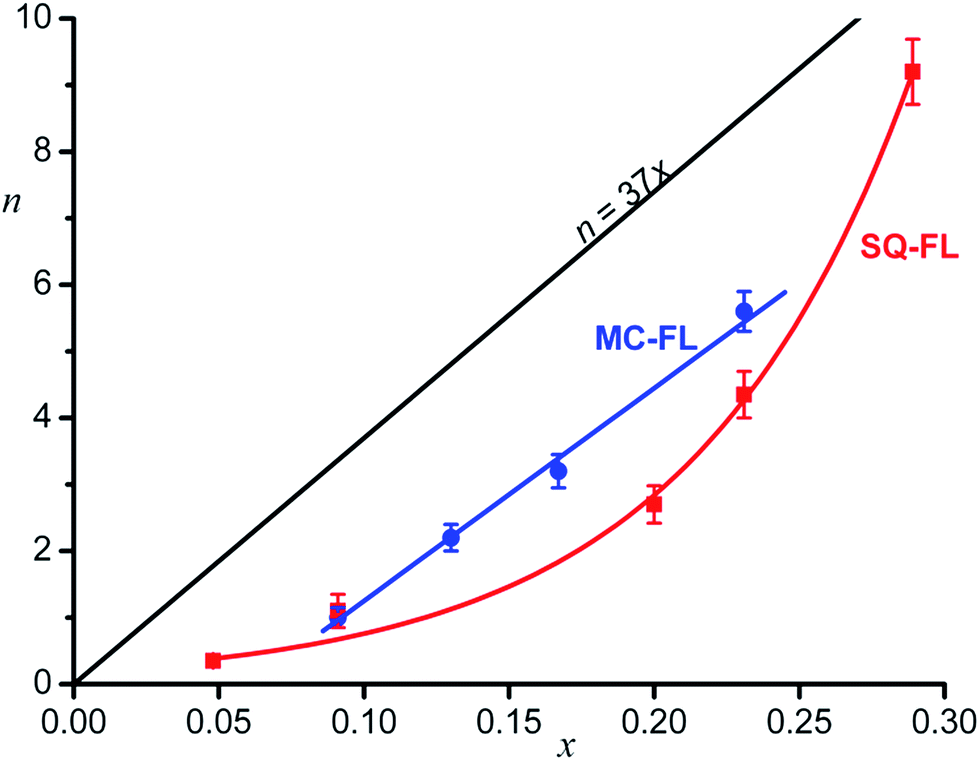 | ||
| Fig. 3 Average number of the functional ligand molecules (n) in the hybrid nanosystem shell as a function of the relative concentration (x) of this ligand in the source solution, experimental points and approximating curves, see text. | ||
Theoretically calculated dependence of n = 37x under the condition of equally probable adsorption of the inert and functional ligands and the densest ligand packing on the QD surface (see below) is also shown.
In the case of the SQ-FL, the experimental points are approximated by the exponential
| n = (0.20 ± 0.03)exp((13.2 ± 0.7)x), | (3) |
| n = (32.0 ± 1.5)x − (1.95 ± 0.21), R = 0.998. | (4) |
Before discussing the dependences obtained, it is worthwhile to consider an expected theoretical dependence of n on x. We assume that all ligands compete for the same coordination centres on the QD surface, the number of such centres being determined by the size of the QD and the nature of the facets. We assume also a cylindrical model for the ligands, the cross section being circle with area of S = 0.385 nm2. So, we can estimate the upper limit of the number of ligand molecules in a monolayer of the HNS organic shell as the maximal number (Nmax) of the circles with area S that can occupy the surface of a sphere of radius r,
| Nmax = 4πηr2/S. | (5) |
Here r is the QD radius (assuming spherical QD), and η is a packing density. For the densest packing of the circles on a sphere, η = π/4 = 0.785, and since r = 1.2 nm, we obtain for the QDs studied Nmax = 37. This number is less than the earlier estimated value (86), which has been obtained using the understated cross-sectional area of BM (0.21 nm2) and without considering the packing density.31
If monolayer of the organic ligands in the HNS is formed in a dynamic equilibrium of attachment and detachment of different molecules to the QD surface, one could expect the proportionality between the relative concentration of the FL on the HNS surface (n/Nmax) and in the source solution (x), n/Nmax = αx, where α is a proportionality coefficient. In the simplest case, α = 1, wherefrom n = Nmaxx = 37x. The expected theoretical dependence is shown in Fig. 3.
One can see that both experimental plots curves are below the theoretical line. Obviously, α < 1, since the number of really accessible binding sites is lower than Nmax, which was estimated as an upper limit according to simple geometrical model provided the entire QD surface is occupied by the ligands. Really, full filling of the QD surface doesn't occur, the number of the binding sites depends on the nanocrystal facet type.6
The second remarkable feature of the experimental plot for the SQ-FL in Fig. 3 is non-linearity. The upward deviation of the plot suggests that the addition of each next SQ-FL molecule to the QD occurs easier than that of preceding one, and this non-proportional growth resembles a peculiar cooperative effect. During self-association of dyes, nucleic bases, and surfactants, the cooperative effect is characterised by the fact that, after the formation of a dimer, the addition of following molecules to a growing aggregate is facilitated, due to specific interactions of the substrates. If such aggregate is attached to the growing QD, one could anticipate increasing relative proportion of the FL in the HNS organic shell. For the ligands studied, at spectroscopic concentrations (∼10–5 M), we did not observe the aggregate formation. However, in syntheses, the ligand concentration was higher by two orders of magnitude, so we could not exclude formation of dimers, trimers, etc. Indeed, slight changes (broadening and red shift) in the absorbance spectra were observed for the SQ-FL studied at the synthetic concentrations (∼10–3 M) in the quartz cells with an optical path length of l = 0.1 mm. For the MC-FL there was no difference in the absorbance spectra recorded at various concentrations.
The exponential dependence with significantly higher coefficients has been found earlier for the SQ-FL with the thiol anchor group,31 indicating the higher binding ability of the thiol group in comparison with the isothiouronium one.
As compared to the SQ-FL, in the case of the MC-FL, the experimental plot is almost parallel to the theoretical line and crosses the abscissa axis at x = 0.061, Fig. 3. Before the MC-FL relative concentration reaches this critical value, the ligand is not built into the HNS organic shell. This behaviour resembles a critical micelle concentration (CMC), when micelles only form above the CMC. It is interesting that at x = 0.061, theoretical value of n is equal to 2.3, i.e. only when the quantity of the MC-FL in solution exceeds two molecules of the ligand per the QD, the latter begins to adsorb the former.
The microwave-assisted synthesis has some peculiarities, which are discussed in the literature.27 These “specific MW effects” are related to a direct interaction of the MW electric field with molecules in the reaction mixture. The BM is a polar molecule, the FLs are salts and exist in the DMF solution as ion pairs, moreover, the SQ chromophore is neutral whereas the MC one is positively charged. These three ligands interact differently with the MW electric field, and activated differently. Therefore, they can interact with the QD binding sites with different rates. In equilibrium, these differences do not matter; however, if the HNSs are formed in non-equilibrium conditions, these differences are reflected in the HNS composition. We can conclude that, during the MW-assisted synthesis, the hybrid nanosystems are formed in kinetically rather than thermodynamically controlled process that gives rise to the effects observed.
The developed herein one-pot microwave assisted synthesis of HNSs can be compared with the reported previously ligand exchange method.30 In the one-pot synthesis, up to 80% of the FLs, dissolved in the reaction mixture, are connected to nanoparticles, whereas only ca. 40% of the FLs in the case of the exchange method. Being simpler in the procedure, the one-pot method additionally saves a notable quantity of the FL, which is often expensive or synthetically hard-to-reach.
Experimental
Chemicals
Cadmium acetate dihydrate (Sigma-Aldrich, >98%), thiourea (reagent grade), benzyl mercaptan (Sigma-Aldrich, 99%), dimethylformamide (reagent grade) and chloroform (reagent grade) were used as received.Synthesis
The precursor of the SQ functional ligand, 2-(E)-(4-(9-bromononyl-1-oxy)styryl)quinoline was synthesised according to ref. 30. Synthesis of the precursor of the MC functional ligand, N-methyl-2-(E)-(4-(9-iodononyl-1-oxy)styryl)quinolinium iodide is described in the ESI.†Hybrid nanosystems were synthesised similarly to the previously described procedure,31 slightly modified in order to obtain in situ the functional ligand via nucleophilic substitution of halogen by thiourea. The corresponding precursor (0.0025–0.017 mmol) and equimolar quantity of thiourea, as well additional 0.05 mmol of thiourea were dissolved in DMF (5 mL) and underwent microwave (MW) irradiation for 10 min using experimental setup, which has been described previously.29 In brief, the setup is equipped with 920 W 2.45 GHz magnetron (Daewoo 2M218 HF) and a cylindrical working chamber. Then cadmium acetate dihydrate (0.05 mmol) and benzyl mercaptan (0.05 mmol) were added and the solution underwent microwave irradiation for 40 s. Thus, in all cases, the concentrations of cadmium acetate (10 mM), thiourea (10 mM) and benzyl mercaptan (10 mM) were kept constant, whereas concentrations of the FLs varied, Table 1. Since all ligands used are soluble in ethanol, to separate the nanosystems from the unbound ligands, fourfold excess of ethanol was added, the precipitate centrifuged at 3000 rpm, and supernatant was decanted. Then precipitate was dissolved in small amount of DMF and re-precipitated with excess of ethanol, this procedure was repeated twice. Final precipitate was washed with ethanol (the absorption spectrum of the supernatant did not detect the presence of the FL) and dried under vacuum. Table 1 shows a list of samples prepared using different concentrations of the functional ligands, samples HNS-1–HNS-5 with the SQ-FL and samples HNS-6–HNS-9 with the MC-FL.
| Sample | CSQ-FL, mM | Sample | CMC-FL, mM |
|---|---|---|---|
| HNS-1 | 0.5 | HNS-6 | 1 |
| HNS-2 | 1 | HNS-7 | 1.5 |
| HNS-3 | 2.5 | HNS-8 | 2 |
| HNS-4 | 3 | HNS-9 | 3 |
| HNS-5 | 3.45 |
Measurement and characterisation
Conclusions
Hybrid nanosystems with CdS quantum dot (QD) as an inorganic core coated by organic shell containing benzyl mercaptan as an inert (background) ligand and a functional ligand (FL) were prepared by one-pot microwave-assisted synthesis. Two FLs were studied, both containing chromophores, styrylquinoline (SQ) or O-alkylated merocyanine (MC). Owing to the presence of a chromophore, the average number (n) of the FL molecules in the nanosystem organic shell was determined by UV-Vis spectroscopy. The dependence of n on the relative concentration (x) of the FL in source solution is determined by the nature of the chromophoric group in the FL: the n depends on x exponentially for the SQ ligand and linearly with a critical point for the MC ligand. The effects observed are suggested to be associated with non-equilibrium conditions of the hybrid nanosystem formation. Because of the peculiarities of microwave activation of the ligands, the product composition is controlled by reaction kinetic rather than thermodynamic.Notes and references
- L. Zhu, M. Zhu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2005, 127, 8968 CrossRef CAS PubMed.
- L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17 RSC.
- Z. Erno, I. Yildiz, B. Gorodetsky, F. M. Raymo and N. R. Branda, Photochem. Photobiol. Sci., 2010, 9, 249 CAS.
- A. Credi, New J. Chem., 2012, 36, 1925 RSC.
- I. V. Martynenko, A. O. Orlova, V. G. Maslov, A. V. Baranov, A. V. Fedorov and M. Artemyev, Beilstein J. Nanotechnol., 2013, 4, 895 CrossRef CAS PubMed.
- J. Y. Kim, O. Voznyy, D. Zhitomirsky and E. H. Sargent, Adv. Mater., 2013, 25, 4986 CrossRef CAS PubMed.
- R. Ladj, A. Bitar, M. Eissa, Y. Mugnier, R. Le Dantec, H. Fessi and A. Elaissari, J. Mater. Chem. B, 2013, 1, 1381 RSC.
- R. Freeman and I. Willner, Chem. Soc. Rev., 2012, 41, 4067, 10.1039/c2cs15357b.
- S. B. Brichkin and E. V. Chernykh, High Energy Chem., 2011, 45, 1 CrossRef CAS.
- T. Avellini, C. Lincheneau, F. Vera, S. Silvi and A. Credi, Coord. Chem. Rev., 2014, 263–264, 151 CrossRef CAS.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS PubMed.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859 RSC.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203 RSC.
- T. J. Fukaminato, J. Photochem. Photobiol., C, 2011, 12, 177 CrossRef CAS.
- N. V. Hullavarad, S. S. Hullavarad and P. C. Karulkar, J. Nanosci. Nanotechnol., 2008, 8, 3272 CrossRef CAS PubMed.
- C. De Mello Donega, S. G. Hickey, S. F. Wuister, D. Vanmaekelbergh and A. Meijerink, J. Phys. Chem. B, 2003, 107, 489 CrossRef CAS.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207 CrossRef CAS.
- A. Rahmi, A. Umar, M. Salleh, B. Majlis and M. Oyama, Mater. Sci. Forum, 2011, 663–665, 25 CAS.
- D. G. Wu, M. E. Kordesch and P. G. Van Patten, Chem. Mater., 2005, 17, 6436 CrossRef CAS.
- C. Wang, Y. Jiang, Z. Zhang, G. Li, L. Chen and J. Jie, J. Nanosci. Nanotechnol., 2009, 9, 4735 CrossRef CAS PubMed.
- J. R. Dethlefsen and A. Dossing, Nano Lett., 2011, 11, 1964 CrossRef CAS PubMed.
- X. Liu, Y. Jiang, X. Lan, Y. Zhang, S. Li, J. Li, T. Han, B. Wang and H. Zhong, Mater. Chem. Phys., 2011, 130, 909 CrossRef CAS.
- J. J. Li, Y. A. Wang, W. Z. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. G. Peng, J. Am. Chem. Soc., 2003, 125, 12567 CrossRef CAS PubMed.
- N. Kaur, N. Singh, B. McCaughan and J. F. Callan, Sens. Actuators, B, 2010, 144, 88, DOI:10.1016/j.snb.2009.09.066.
- Y. Wada, H. Kuramoto, J. Anand, T. Kitamura, T. Sakata, H. Mori and S. Yanagida, J. Mater. Chem., 2001, 11, 1936 RSC.
- M. Majumder, S. Karan and B. Mallik, J. Lumin., 2011, 131, 2792 CrossRef CAS.
- M. Baghbanzadeh, L. Carbone, P. D. Cozzoli and C. O. Kappe, Angew. Chem., Int. Ed., 2011, 50, 11312 CrossRef CAS PubMed.
- T. Xuan, X. Wang, J. Liu, H. Li, L. Pan and Z. Sun, J. Mater. Chem. C, 2013, 1, 4550, 10.1039/c3tc30680a.
- M. F. Budyka, O. V. Chaschikhin and P. A. Nikulin, Nanotechnol. Russ., 2015, 10, 13 CrossRef CAS.
- M. F. Budyka, O. V. Chashchikhin, T. N. Gavrishova, M. G. Spirin and S. B. Brichkin, Nanotechnol. Russ., 2014, 9, 116 CrossRef CAS.
- M. F. Budyka, O. V. Chaschikhin and P. A. Nikulin, Nanotechnol. Russ., 2016, 11, 78 CrossRef CAS.
- M. F. Budyka, T. N. Gavrishova, N. I. Potashova and O. V. Chashchikhin, High Energy Chem., 2015, 49, 237 CrossRef CAS.
- M. F. Budyka and O. V. Chashchikhin, High Energy Chem., 2016, 50, 349 CrossRef CAS.
- C. Unni, D. Philip, S. L. Smitha, K. M. Nissamudeen and K. G. Gopchandran, Spectrochim. Acta, Part A, 2009, 72, 827 CrossRef CAS PubMed.
- Y. J. Xi, Z. G. Ling and H. G. Rong, Microelectron. Eng., 2003, 66, 115 CrossRef.
- L. E. Brus, J. Chem. Phys., 1984, 80, 4403 CrossRef CAS.
- M. Majumder, S. Karan, A. K. Chakraborty and B. Mallik, Spectrochim. Acta, Part A, 2010, 76, 115 CrossRef PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, XRD, TEM, PL. See DOI: 10.1039/c6ra27577j |
| This journal is © The Royal Society of Chemistry 2017 |