DOI:
10.1039/C6RA27510A
(Paper)
RSC Adv., 2017,
7, 19073-19080
New discrete iodometallates with in situ generated triimidazole derivatives as countercations (Mn+ = Ag+, Pb2+, Bi3+)†
Received
29th November 2016
, Accepted 24th March 2017
First published on 30th March 2017
Abstract
Through employing the solvothermal in situ N-alkylation of organic bases with alcohol molecules, four new iodometallates [L1]2[Ag8I12]I2 (L13+ = 1,1′,1′′-(benzene-1,3,5)tris(3-methyl-1H-imidazol-3-ium)) 1, [L1]2[Ag6I12] 2, [L1]2[Pb3I12]·2H2O 3, and [L1][Bi2I9] 4 were obtained. L13+ originates from the in situ N-alkylation between 1,3,5-tri(1H-imidazol-1-yl)benzene (L2) and CH3OH. X-ray single-crystal diffraction analysis reveals that all of the title compounds possess zero-dimensional (0D) structures. The octanuclear anionic cluster [Ag8I12]4− of 1 possesses a sphere-like structure. An Ag6I6 unit with a hexagram structure occupies the equatorial position, while two AgI4 tetrahedra (sharing point) occupy the axial positions. The anionic cluster, [Ag6I12]6−, of 2 exhibits a hexanuclear structure, which can be described as an aggregation of two incomplete cubanes (each lacking an Ag+ corner). The anionic cluster [Pb3I12]6− of 3 can be described as an aggregation of three PbI6 octahedra (sharing face), whereas two BiI6 octahedra (also sharing face) aggregate to form a dinuclear cluster [Bi2I9]3− of 4.
Introduction
As a new high-efficiency synthetic approach, the in situ ligand preparation approach has seen rapid development in the last two decades.1 The in situ ligand synthesis approach not only simplifies the reactive procedure, but also creates a variety of new organic molecules and complexes. A majority of ligand in situ reactions originate from accidental discovery. Then they are extensively employed in the construction of new organic molecules and complexes. Finally, it becomes a classical reaction. So far, the typical ligand in situ reactions are the cycloaddition of organic nitriles with azide,2 oxidative hydroxylation of aromatic rings,3 dehydrogenative coupling of C–C bonds,4 acylation of multicarboxylic acids with N2H4,5 N-alkylation of heterocyclic ligands with alcohols, and so on.6
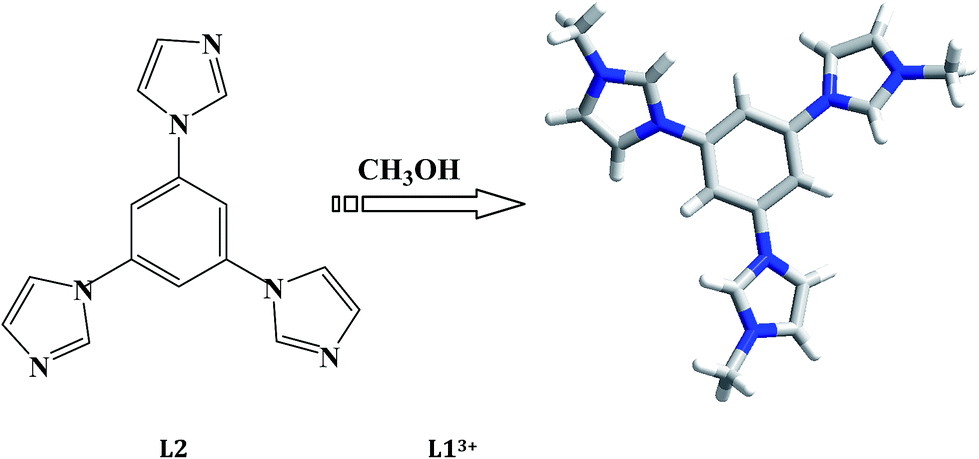
The in situ N-alkylation of organic bases with alcohol molecule was discovered for the first time when preparing compound [CH3CH2S-4-C5H4N–CH2CH3][Cu3I4].7 The N-alkylation between CH3CH2S-4-C5H4N and C2H5OH solvent occurred to generate CH3CH2S-4-C5H4N+–CH2CH3. Latter, the researchers employed this kind of in situ reaction to construct many new hybrid organic–inorganic materials.8 For example, Lang et al. employed the in situ N-alkylation of 4-cynaopyridine (4-cypy) or 4,4′-bipyridine (4,4′-bpy) with various alcohols to prepare a serials of hybrid iodometallates [Mn+ = Cu+, Pb2+, Bi3+].9 Guo et al. employed the in situ N-alkylation of aromatic diamines or 1,4-diazabicyclo[2,2,2]octane (dabco) with diverse alcohols to construct a serials of hybrid iodoplubates.10 Zhang et al. employed the in situ N-alkylation of pyridine (py), 4,4′-bpy, 1,3-bis(4-pyridyl)propane (bpp), or dabco with CH3OH or C2H5OH to create some iodocuprate(I) and bromocuprate(I).11 Wang et al. utilized the N-alkylation of piperazine (pip), pip derivatives, dabco or 3-(aminomethyl)pyridine (ampy) with CH3OH to synthesize a series of metal phosphates and phosphites (Mn+ = Be2+, Ga3+, Zn2+).12 In the past, our group also employed this kind of in situ reaction to construct some iodocuprates(I) and iodoplumbates(II).13 The used organic bases have pip, dabco, 4,4′-bipiperidine (bp), 1,3-bis(4-piperidyl)propane (bpip) and py. Even though the in situ N-alkylation of so many organic bases (cyclic aliphatic organic bases, aromatic bisamine, py/derivatives) have been investigated, the triimidazole molecules are never considered. In this article, the in situ N-alkylation of 1,3,5-tri(1H-imidazol-1-yl)benzene (L2) with alcohols are investigated, and four new iodometallates as the octanuclear [L1]2[Ag8I12]I2 (L13+ = 1,1′,1′′-(benzene-1,3,5)tris(3-methyl-1H-imidazol-3-ium)) 1, hexanuclear [L1]2[Ag6I12] 2, trinuclear [L1]2[Pb3I12]·2H2O 3, and the dinuclear [L1][Bi2I9] 4 were obtained. L13+ originated from the solvothermal in situ N-alkylation of L2 with CH3OH.
Experimental
General
All chemicals are of reagent grade quality, obtained from commercial sources without further purification. Elemental analysis (C, H and N) was performed on a Perkin-Elmer 2400LS II elemental analyzer. Infrared (IR) spectrum was recorded on a Perkin Elmer Spectrum 1 spectrophotometer in 4000–400 cm−1 region using a powdered sample on a KBr plate. Powder X-ray diffraction (XRD) data were collected on a Rigaku/max-2550 diffractometer with Cu-Kα radiation (λ = 1.5418 Å). Thermogravimetric (TG) behavior was investigated on a Perkin-Elmer TGA-7 instrument with a heating rate of 10 °C min−1 in air. Ultraviolet-visible (UV-vis) spectrum was obtained on a Rigaku-UV-3100 spectrophotometer.
Synthesis of title compounds
[L1]2[Ag8I12]I2 1. The yellow columnar crystals of 1 were obtained from a simple solvothermal self-assembly of AgI (12 mg, 0.05 mmol), L2 (28 mg, 0.1 mmol), and HI (45%, 1 mL) in a mixed solvent of CH3CN (4 mL) and CH3OH (2 mL) (pH = 2) at 110 °C for 3 days. Yield: ca. 24% based on Ag(I). Anal. calcd C36H42N12Ag8I14 1: C 13.17, H 1.29, N 5.12. Found: C 13.22, H 1.33, N 5.23%. IR (cm−1): 3086 m, 3051 m, 2925 w, 2849 w, 1614 s, 1575 s, 1419 w, 1386 w, 1239 w, 1190 s, 862 w, 809 s, 747 s, 673 s, 614 s.
[L1]2[Ag6I12] 2. The yellow columnar crystals of 2 were obtained from a simple solvothermal self-assembly of AgBr (19 mg, 0.05 mmol), L2 (28 mg, 0.1 mmol), and HI (45%, 1 mL) in a mixed solvent of CH3CN (6 mL) and CH3OH (2 mL) (pH = 2) at 120 °C for 3 days. Yield: ca. 31% based on Ag(I). Anal. calcd C36H42N12Ag6I12 2: C 15.37, H 1.51, N 5.98. Found: C 15.51, H 1.65, N 5.78%. IR (cm−1): 3063 w, 3034 w, 2920 w, 2854 w, 1637 m, 1618 s, 1386 w, 1194 m, 1135 w, 816 w, 614 s.
[L1]2[Pb3I12]·2H2O 3. The yellow columnar crystals of 3 were obtained from a simple solvothermal self-assembly of PbI2 (46 mg, 0.1 mmol), L2 (14 mg, 0.05 mmol), and HI (45%, 1 mL) in a mixed solvent of CH3CN (8 mL) and CH3OH (2 mL) (pH = 2) at 110 °C for 3 days. Yield: ca. 46% based on Pb(II). Anal. calcd C36H46N12O2Pb3I12 3: C 15.31, H 1.64, N 5.95. Found: C 15.42, H 1.61, N 6.02%. IR (cm−1): 3091 w, 3043 w, 2919 w, 2853 w, 1637 m, 1617 s, 1545 w, 1386 w, 1190 s, 1135 w, 871 w, 818 m, 671 w, 613 s.
[L1][Bi2I9] 4. The red columnar crystals of 4 were obtained from a simple solvothermal self-assembly of BiCl3 (32 mg, 0.05 mmol), L2 (28 mg, 0.1 mmol), and HI (45%, 1 mL) in a mixed solution of CH3CN (6 mL) and CH3OH (2 mL) (pH = 2) at 110 °C for 3 days. Yield: ca. 39% based on Bi(III). Anal. calcd C18H21N6Bi2I9 4: C 14.49, H 1.13, N 4.47. Found: C 14.35, H 1.01, N 4.58%. IR (cm−1): 3100 w, 3071 w, 2929 w, 2853 w, 1623 s, 1587 s, 1548 s, 1418 w, 1385 w, 1348 w, 1208 s, 1110 w, 1087 m, 855 m, 813 s, 768 m, 680 w, 611 s.The title four hybrids are stable in air, and soluble in H2O, methanol, ethanol, acetone and acetonitrile.
X-ray crystallography
The data were collected with Mo-Kα radiation (λ = 0.71073 Å) on a Rigaku R-AXIS RAPID IP diffractometer for 1, 3 and 4, and on a Siemens SMART CCD diffractometer for 2. With SHELXTL program, the structures of 1–4 were solved using direct methods.14 The non-hydrogen atoms were assigned anisotropic displacement parameters in the refinement, and the hydrogen atoms were treated using a riding model. The hydrogen atoms on the water molecule in 3 are not located. The structures were then refined on F2 using SHELXL-97.14 CCDC numbers are 1519637–1519640 for 1–4, respectively. The crystallographic data for 1–4 are summarized in Table 1.
Table 1 Crystal data of title compounds
| |
1 |
2 |
3 |
4 |
| Formula |
C36H42N12Ag8I14 |
C36H42N12Ag6I12 |
C36H46N12O2Pb3I12 |
C18H21N6Bi2I9 |
| M |
3282.38 |
2812.84 |
2823.22 |
1881.47 |
| T (K) |
293(2) |
293(2) |
293(2) |
293(2) |
| Crystal system |
Rhombohedral |
Monoclinic |
Monoclinic |
Orthorhombic |
| Space group |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
P21/c |
P21/n |
P212121 |
| a (Å) |
14.090(2) |
11.2094(3) |
12.496(3) |
12.485(3) |
| b (Å) |
14.090(2) |
13.2371(4) |
20.569(4) |
13.080(3) |
| c (Å) |
31.201(6) |
21.8384(5) |
12.634(3) |
23.566(5) |
| α (°) |
90 |
90 |
90 |
90 |
| β (°) |
90 |
92.965(2) |
100.16(3) |
90 |
| γ (°) |
120 |
90 |
90 |
90 |
| V (Å3) |
5364.4(15) |
3236.04(15) |
3196.3(11) |
3848.4(13) |
| Z |
3 |
2 |
2 |
4 |
| Dc (g cm−3) |
3.048 |
2.887 |
2.933 |
3.247 |
| μ (mm−1) |
8.219 |
7.540 |
13.716 |
16.371 |
| Reflections collected |
17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 338 338 |
18068 |
30245 |
37053 |
| Unique reflections |
2739 |
5716 |
7292 |
8789 |
| Rint |
0.0504 |
0.0460 |
0.0870 |
0.0698 |
| Gof |
1.094 |
0.979 |
1.050 |
1.040 |
| R1, I > 2σ(I) |
0.0417 |
0.0343 |
0.0492 |
0.0382 |
| wR2, all data |
0.0954 |
0.0810 |
0.0926 |
0.0705 |
Results and discussion
Synthetic analysis
All of the title compounds were obtained under the solvothermal conditions. The reactions of metal halides (Mn+ = Ag+, Pb2+, Bi3+), L2 and HI in a mixed solvent (containing CH3OH) at a strong acidic condition created the title compounds 1–4. The N-alkylation of L2 with CH3OH occurred to generate L13+ (see Scheme 1). The equations below show a potential reactive process: (i) CH3OH + HI = CH3I + H2O; (ii) L2 + 3CH3I = L1I3. The second-step reaction can occur at an acidic or neutral environment, but the formation of CH3I for the first-step reaction maybe needs a strong acidic environment. The other alcohol molecules as C2H5OH and C3H7OH were used to be in place of CH3OH, but the IR spectrum analyses for the obtained solid samples reveal that the N-alkylation of L2 with these alcohols did not occur. The reaction using CuI as the metal resource was also carried out, but unfortunately, the crystal data of the product [L1]2[Cu6I12] did not pass the cif-checking examination. The reactions without the CH3OH solvent were also investigated, and only a Pb2+ hybrid [H3L2][PbI4] was obtained.
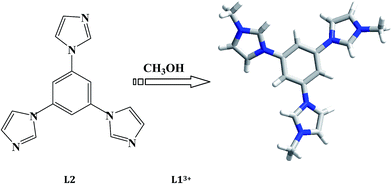 |
| | Scheme 1 In situ N-alkylation of L2 with CH3OH into L13+. | |
Structural description
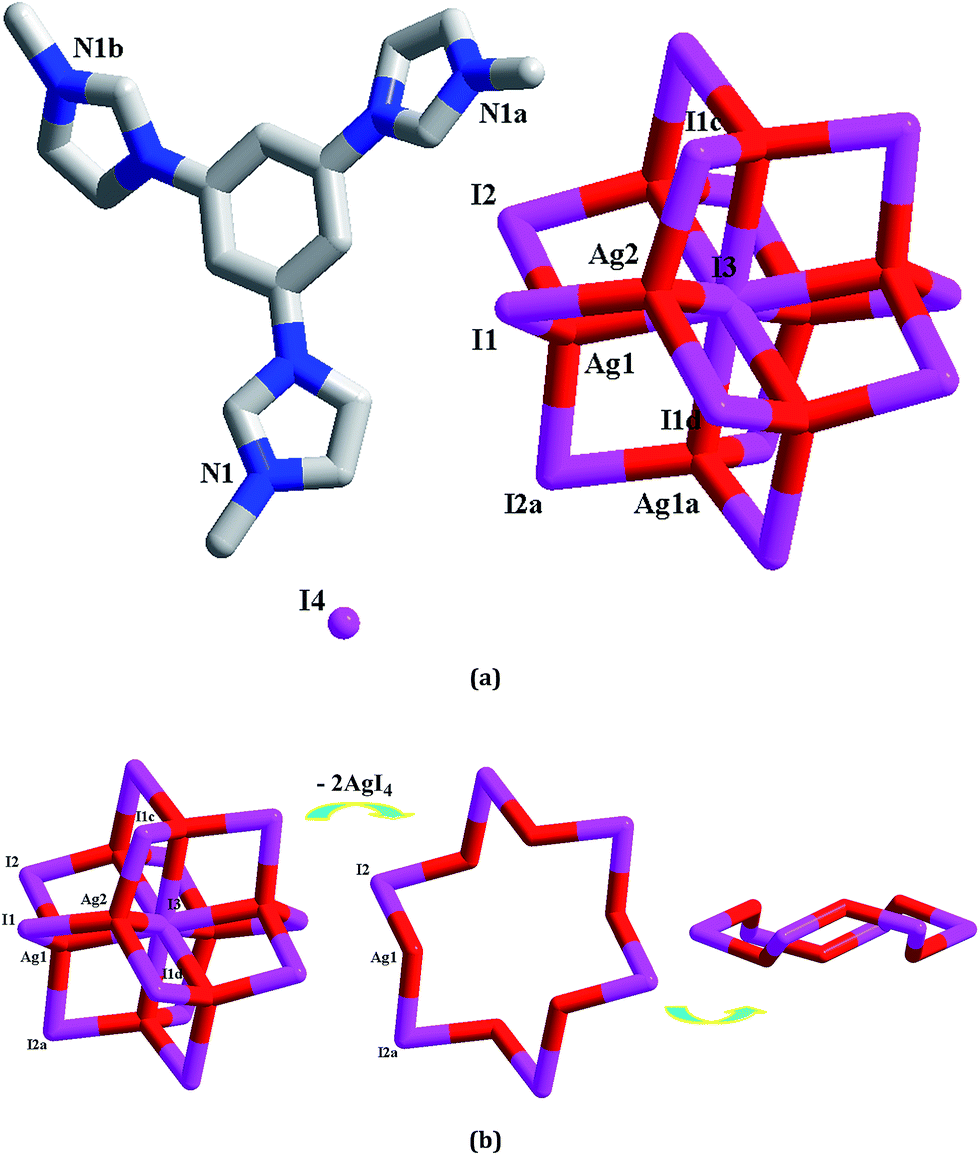
[L1]2[Ag8I12]I2 1. X-ray single-crystal diffraction analysis reveals that 1 is an octanuclear iodoargentate with L13+ as the countercation (see Fig. 1a). The asymmetric unit of 1 is found to be composed of two types of Ag+ ions (Ag1, Ag2), four types of I− ions (I1, I2, I3, I4), and one L13+ molecule. Ag1 and Ag2 are both in a tetrahedral site with four I− ions as the donor atoms (I1, I2, I2a and I3 for Ag1; I1, I1c, I1d and I3 for Ag2). The Ag–I distances span a quite wide range from 2.7704(6) Å to 3.1694(15) Å. For four types of I− ions, I4 exists in a free form, while the other three I− ions act as the bridging ligands. I1 and I2 adopt a simple double-bridged coordination mode, whereas I3 adopts a special μ8 coordination mode. With L13+ as the counteraction, Ag+ and I− aggregate to form an octanuclear cluster. This cluster possesses a sphere-like structure (see Fig. 1b). Six symmetry-related I2 ions bridge six symmetry-related Ag1 centers to form a hexagram, occupying the equatorial position of the sphere. Note that this hexagram is not planar. Six I− ions are nearly co-planar with a minor mean deviation of 0.0852 Å. Three Ag+ ions are above the six iodine plane, while the other three Ag+ ions lie below the six iodine plane. Two Ag(2)I4 tetrahedra occupy the axial positions. These two tetrahedra share the I3 ion. I3 lies at the center of the sphere. Via the Ag1–I1 and Ag1–I3 interactions, the Ag6I6 hexagram and two AgI4 tetrahedral aggregate together to form the octanuclear anionic cluster of 1. The shortest Ag⋯Ag separation in 1 is Ag1⋯Ag1a = 3.4455(10) Å. To the formation of this octanuclear cluster, the free I4 ion should play a key role.
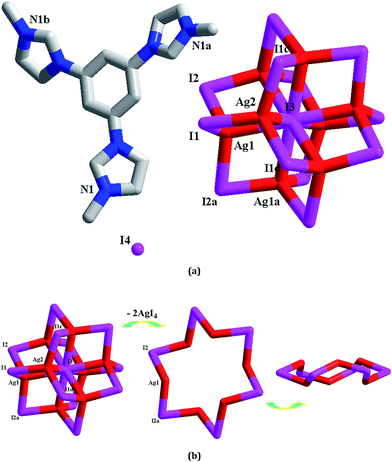 |
| | Fig. 1 Molecular structure (a) and anionic cluster structure (b) for 1 (a: y − 1/3, −x + y + 1/3, −z + 1/3; b: x − y + 2/3, x + 1/3, −z + 1/3; c: −x + y, −x + 1, z; d: −y + 1, x − y + 1, z). | |
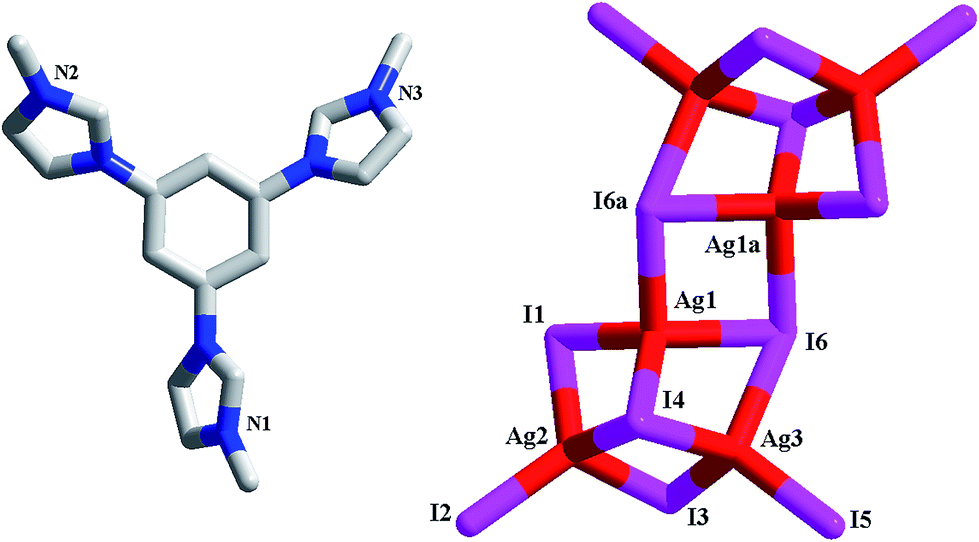
[L1]2[Ag6I12] 2. Without the existence of the free I− ion, Ag+ and I− aggregate into a hexanuclear cluster. Here L13+ still serves as the countercation in 2 (see Fig. 2). The asymmetric unit of 2 is found to be composed of three types of Ag+ ions (Ag1, Ag2, Ag3), six types of I− ions (I1, I2, I3, I4, I5, I6), and one L13+ molecule. Three Ag+ centers all exhibit a tetrahedral geometric configuration with an AgI4 donor set. The Ag–I bond length range is 2.7512(9)–2.9597(9) Å. Six I− ions adopt three types of coordination modes: terminal mode for I2 and I5; double-bridged mode for I1 and I3; triple-bridged mode for I4 and I6. The Ag–I–Ag angles for linear I1 and I3 are 67.93(2)° (for I1) and 78.41(3)° (for I3), respectively. Both I4 and I6 adopt a triple pyramidal geometrical configuration. However, the triple pyramid for I4 are regular, while the triple pyramid for I6 suffers from a severe distortion. It looks more like a slice of the rectangular pyramid (losing an Ag+ corner). The distances from the apical I− ions to the three Ag+ planes are 2.33 Å (for I4) and 1.04 Å (for I6), respectively. Templated by L13+, a hexanuclear iodoargentate forms. Three I− ions (I1, I3 and I6) first bridge alternately three Ag+ centers to form a trinuclear cluster. The I4 ion caps these three Ag+ centers, making the trinuclear cluster more stable. Note that this trinuclear cluster can be described as a incomplete cubane (lacking a tetrahedral Ag+ corner). Then via the interactions between Ag1 and I6a, two such trinuclear clusters further aggregate to form the title hexanuclear [Ag6I12]6− cluster. The terminal I2 and I5 are used to complete the tetrahedral coordination of Ag2 and Ag4. The shortest Ag⋯Ag separation in 2 is Ag1⋯Ag3 = 3.0833(10) Å.
 |
| | Fig. 2 Molecular structure of 2 (a: −x + 2, −y + 1, −z). | |
[L1]2[Pb3I12]·2H2O 3. 3 is a trinuclear iodoplumbate(II) with L13+ as the countercation (see Fig. 3). The asymmetric unit of 3 is found to be composed of two types of Pb2+ ions (Pb1, Pb2), six types of I− ions (I1, I2, I3, I4, I5, I6), one L13+ molecules, and one lattice water molecule (Ow1). For six ions, I1, I2 and I3 adopt a terminal mode, whereas I4, I5 and I6 adopt a linear bridging mode. The Pb–I–Pb angle range is 76.83(2)–80.37(3)°. Pb1 and Pb2 are both in an octahedral site, coordinated by six I− ions. The Pb–I bond length range is 3.0481(9)–3.2565(11) Å. Templated by L13+, Pb2+ and I− aggregate into a trinuclear cluster. This trinuclear cluster can be described as a linear arrangement of three PbI6 octahedra via sharing the face. Pb2 is located at a special position, serving as an inversion center. The contact distance between Pb1 and Pb2 is 4.174 Å. In the reported metal(II) halides (M2+ = Pb2+, Cd2+), a kind of chain structure is often observed, which can be described as a linear array of MX6 octahedra via sharing the face.15 Although the trinuclear structure in 3 is seldom found, it is actually a segment of this type of chain.
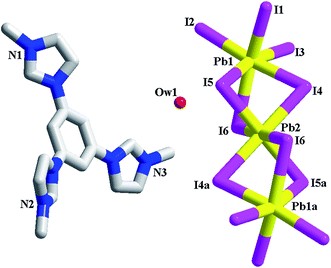 |
| | Fig. 3 Molecular structure of 3 (a: −x, −y, −z + 1). | |
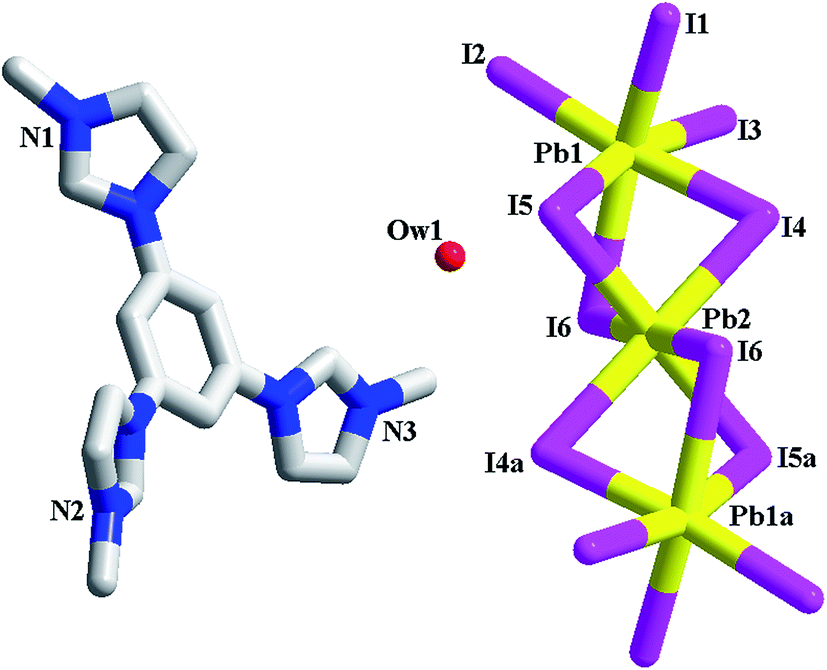
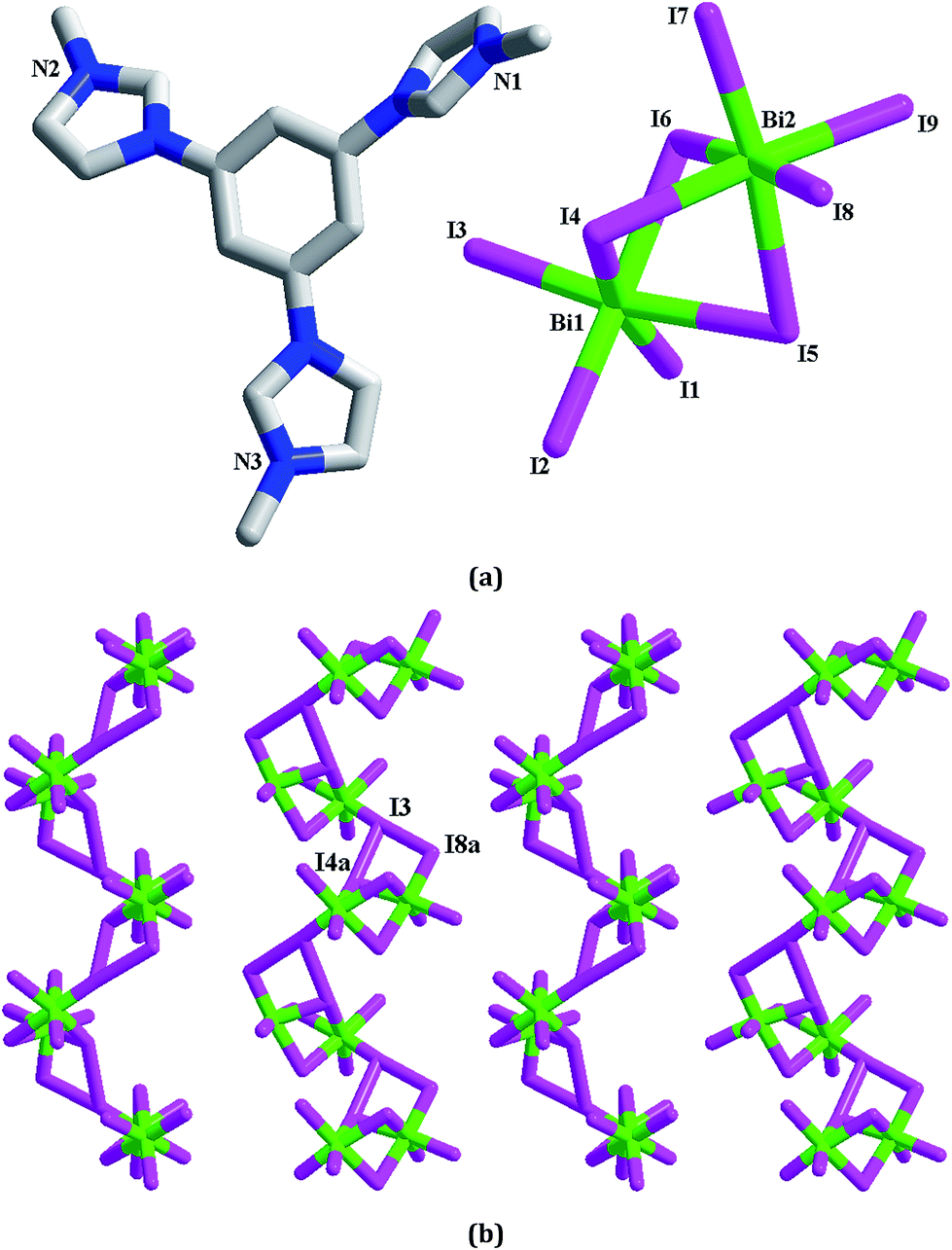
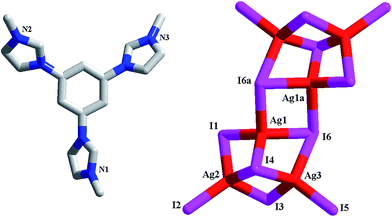
[L1][Bi2I9] 4. 4 is a dinuclear iodobismuthate(III) with L13+ as the countercation (see Fig. 4a). The asymmetric unit of 4 is found to be composed of two types of Bi3+ ions (Bi1, Bi2), nine types of I− ions (I1–I9), and one L13+ cation. Although nine types of I− ions are found in 4, the inorganic anion of 4 only exhibits a simple dinuclear structure. Six I− ions (I1–I3, I7–I9) act as the terminal ligands, completing the octahedral coordination of Bi1 and Bi2. The remaining three I− ions (I4–I6) adopt a μ2 coordination mode, triply bridging two Bi3+ centers into a dinuclear cluster. Two Bi3+ centers are both in an octahedral site, surrounded by six I− ions. The Bi–I bond length range is 2.8692(10)–3.3736(10) Å. In fact, this dinuclear cluster can be viewed as a segment of the trinuclear cluster observed in 3. The formation of the different cluster structures for 3 and 4 should be relative to the metal center. The difference on the charge and the size for Pb2+ and Bi3+ directly influence the formation of the cluster structures. The Bi1⋯Bi2 distance is 4.232 Å. In 4, there exist the weak I⋯I interactions (I3⋯I4a = 3.997 Å, I3⋯I8a = 3.904 Å), via which, the neighboring dinuclear units are linked together into a 1-D supramolecular chain (see Fig. 4b).
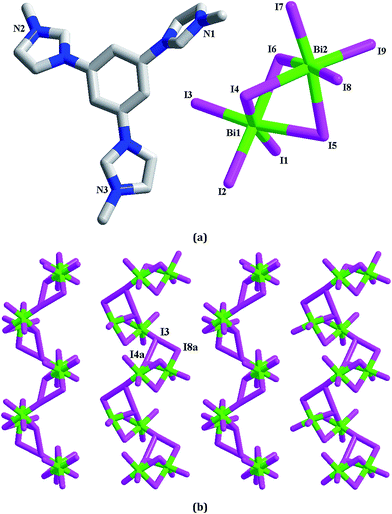 |
| | Fig. 4 Molecular structure (a) and I⋯I interactions between inorganic dinuclear clusters (b) in 4 (a: −x + 1, y + 1/2, −z − 1/2). | |
Structural discussion
The title four compounds are proved to be four new organically modified iodometallate. Although the cations are all the L13+ molecule, the iodometallates exhibits the distinct structures. This should be mainly related to the metal center. By the size (characterized by ionic radius) and the charge, the metal center controls the structure of the iodometallate. In 1 and 2, the iodoargentates show the different structures, which should be due to the existence of the free I− ion in 1. As expected, the triimidazole molecule L2 is N-alkylated by CH3OH to form L13+. The N-alkylation of the L2 molecule causes two important changes: (i) changing the size of the organic cation. The size of the organic molecule directly affects the structure of the inorganic iodometallate. In general, the larger organic cation leads to the formation of the discrete iodometallate, while the smaller organic cation corresponds to the formation of the infinite iodometallate. Although the exception is frequently observed in the reported hybrids, in the title four compounds the especial situation does not occur. With the larger L13+ as the countercation, all of the iodometallates in 1–4 display the discrete structures; (ii) changing the weak interactions between the organic molecule and inorganic anionic moiety. The N–H⋯X hydrogen-bonded interaction has been confirmed to play a non-ignorable role in determining the structure of inorganic anionic moiety. Once the N atom is alkylated, this kind of weak interaction will disappear.
Characterization
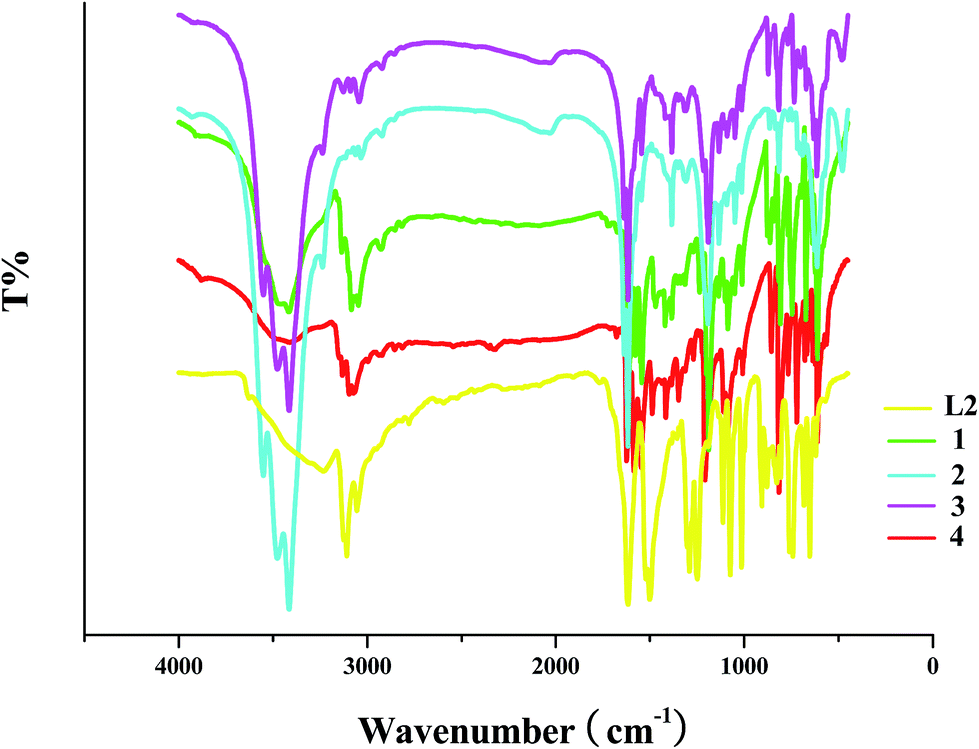
The obvious difference between L13+ and L2 is the existence of the –CH3 group in the L13+ molecule (see Scheme 1). In the IR spectra of the as-synthesized hybrids, once the characteristic peaks of the –CH3 group appear, this means that the N-alkylation of the L2 molecule with CH3OH has occurred. For the –CH3 group, the stretching vibration peak of C–H (νC–H) generally appears in the wavenumber range of 2925–2845 cm−1, and the in-plane bending vibration peak of C–H (βC–H) is generally observed around 1380 cm−1. In the IR spectra of 1–4 (see Fig. 5), these characteristic peaks are found, suggesting that L2 has been methylated by CH3OH to form the L13+ molecule. νC–H(–CH3): 2925, 2849 cm−1 for 1; 2920, 2854 cm−1 for 2; 2919, 2853 cm−1 for 3; 2929, 2853 cm−1 for 4. βC–H(–CH3): 1419, 1386 cm−1 for 1; 1386 cm−1 for 2; 1386 cm−1 for 3; 1418, 1385 cm−1 for 4. Note that νC–H(–CH3) is a kind of weak peak, while βC–H(–CH3) is a kind of sharp peak. In the IR spectrum of L2, the characteristic C–H peaks for the –CH3 group are not observed (also see Fig. 5). Moreover, νC–H(ring): 3086, 3051 cm−1 for 1; 3063, 3034 cm−1 for 2; 3091, 3043 cm−1 for 3; 3100, 3071 cm−1 for 4. νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN(ring): 1614, 1575, 1543 cm−1 for 1; 1637, 1618 cm−1 for 2; 1637, 1617, 1545 cm−1 for 3; 1623, 1587, 1548 cm−1 for 4. νC–N(ring): 1239, 1190 cm−1 for 1; 1194, 1135 cm−1 for 2; 1190, 1135 cm−1 for 3; 1348, 1208, 1110, 1087 cm−1 for 4. γC–H(ring; γ: out-of-plane bending vibration): 862, 809, 747 cm−1 for 1; 816 cm−1 for 2; 871, 818 cm−1 for 3; 855, 813, 768 cm−1 for 4.
C/CN(ring): 1614, 1575, 1543 cm−1 for 1; 1637, 1618 cm−1 for 2; 1637, 1617, 1545 cm−1 for 3; 1623, 1587, 1548 cm−1 for 4. νC–N(ring): 1239, 1190 cm−1 for 1; 1194, 1135 cm−1 for 2; 1190, 1135 cm−1 for 3; 1348, 1208, 1110, 1087 cm−1 for 4. γC–H(ring; γ: out-of-plane bending vibration): 862, 809, 747 cm−1 for 1; 816 cm−1 for 2; 871, 818 cm−1 for 3; 855, 813, 768 cm−1 for 4.
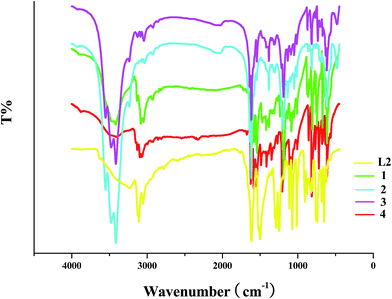 |
| | Fig. 5 IR spectra of title compounds. | |
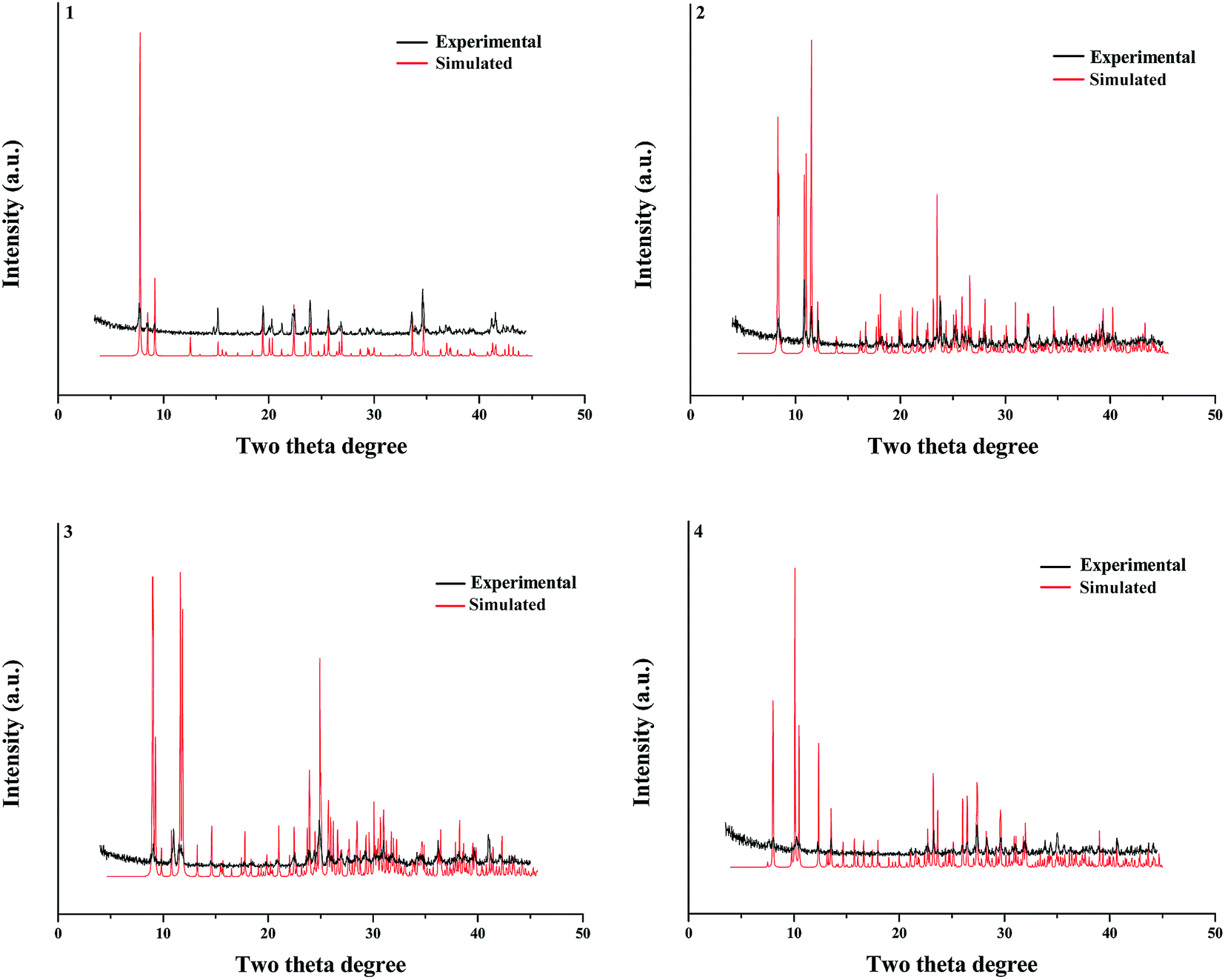
Powder XRD diffraction
Fig. 6 presents the experimental and simulated powder XRD patterns of 1–4. The experimental powder XRD pattern for each compound is in accord with the simulated one generated on the basis of structural data, confirming that the as-synthesized product is pure phase.
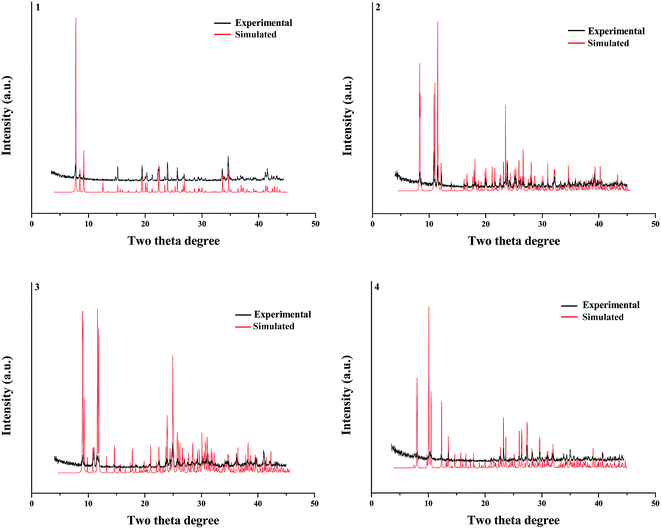 |
| | Fig. 6 Powder XRD patterns of title compounds. | |
TG analysis
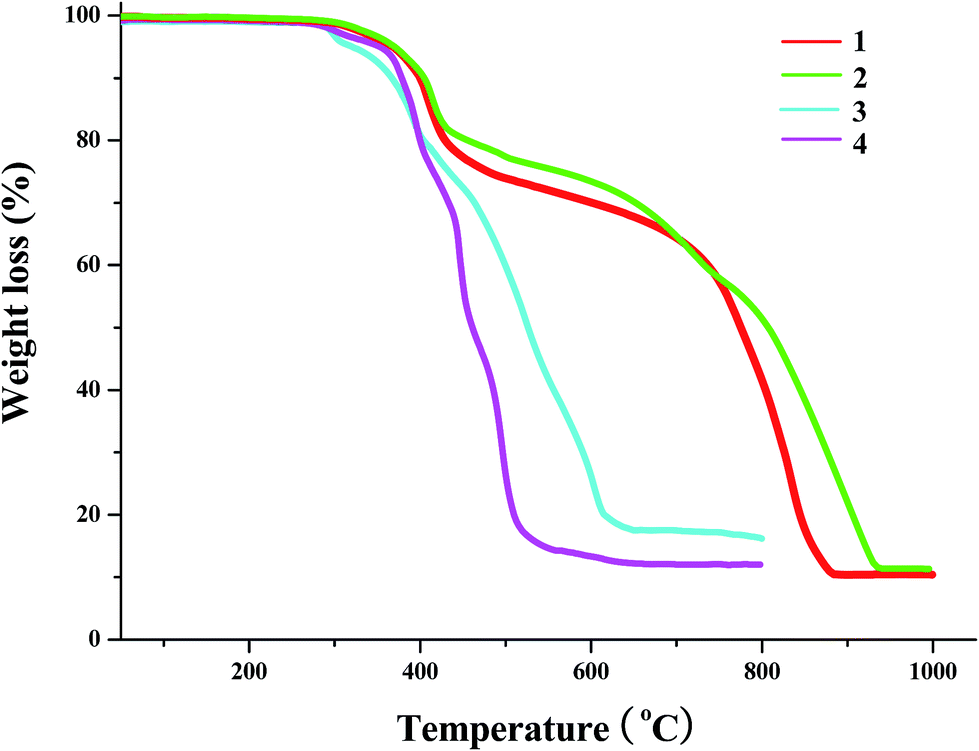
The TG behaviors of the title compounds (temperature range: 30–1000 °C for 1 and 2; 30–800 °C for 3 and 4) are investigated (see Fig. 7). Based on the TG curves, we can know that (i) 1, 2 and 4 possess the better thermal stability, and can be thermal stable up to ca. 275 °C; (ii) two iodoargentates 1 and 2 show the similar TG behaviors. Both underwent the two steps of weight loss. The first step of weight loss (found: ca. 30% for 1, ca. 35% for 2) corresponds to the loss of organic cation in the form of L1I3 (calcd: 31.2% for 1, 36.4% for 2). The second step should be a transforming process from AgI to Ag2O. The observed residue content (ca. 10.6% for 1, ca. 11.4% for 2) is lower than that of the calculated (28.2% for 1; 24.7% for 2), suggesting that the evaporation of AgI also occurred in this step; (iii) in the temperature range of 275–800 °C, 3 and 4 only underwent the one step of weight loss. In this step, the organic cation first removed. Then PbI2 or BiI3 transformed into PbO or Bi2O3. Synchronously, part of AgI evaporated, revealed by the low residue content (found: 17.5% for 3, 12.3% for 4; calcd: 23.7% for 3, 24.8% for 4); (iii) since the content of the water molecule in 3 is minor (1.28%), it is difficult to be observed for the removal of the water molecule. Fig. S1† gives the DSC (differential scanning calorimeter) curves of 1–4.
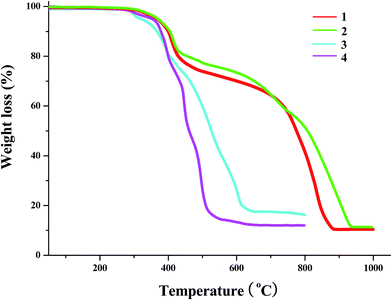 |
| | Fig. 7 TG curves of 1–4. | |
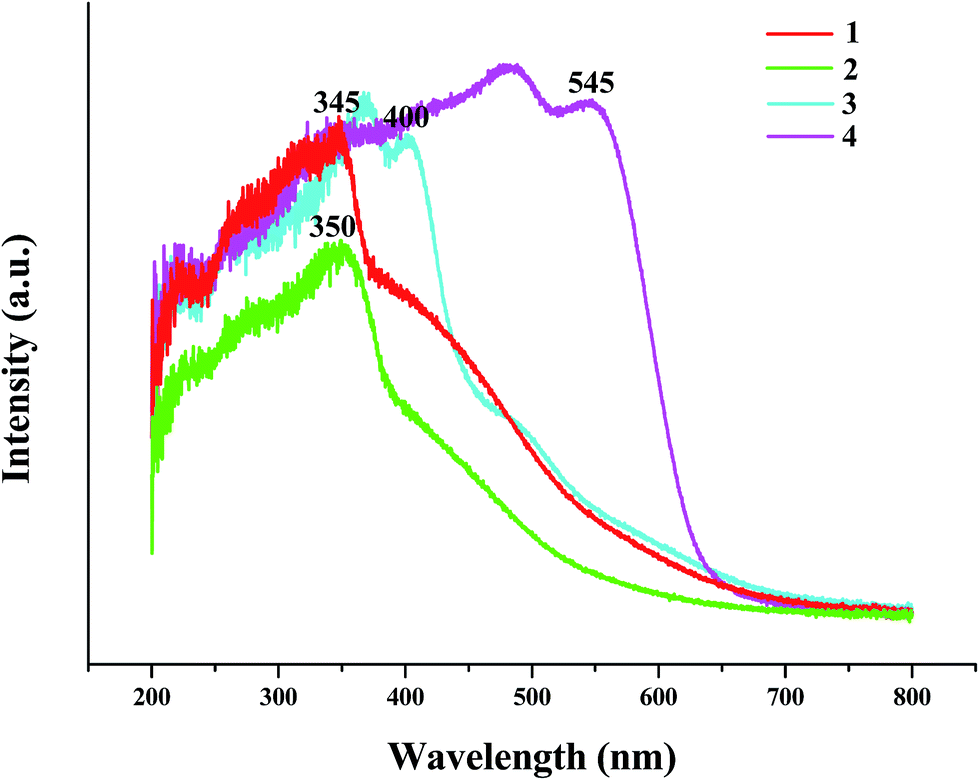
UV-vis spectrum analysis
The solid-state UV-vis spectra of the title compounds are also investigated. As shown in Fig. 8, the adsorption edges at 345 nm for 1, 350 nm for 2, 400 nm for 3, and 545 nm for 4, suggests that the energy gaps for four hybrids are 3.59 eV for 1, 3.54 eV for 2, 3.10 eV for 3, and 2.28 eV for 4, respectively. Compared with the energy gaps of AgI (2.81 eV),16 PbI2 (2.53 eV),10d and BiI3 (ca. 1.7–2.0 eV),17 the energy gaps for the title four hybrids all become wide (0.78 eV for 1, 0.73 eV for 2, 0.57 eV for 3, and 0.28–0.58 eV for 4), which should be due to the hybrid.
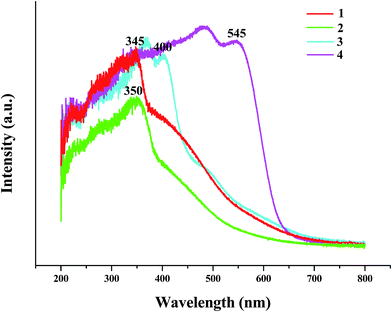 |
| | Fig. 8 Solid-state UV-vis spectra of title hybrids. | |
Conclusion
In summary, we employed the in situ N-alkylation of organic bases with alcohols to construct four new iodometallates (Mn+ = Ag+, Pb2+, Bi3+) with L13+ as the countercation. L13+ derived from the in situ N-alkylation of a kind of triimidazole molecule with CH3OH. Note that the in situ N-alkylation of triimidazole with alcohols was investigated for the first time. Synthetically, a strong acidic environment is quite important. This chiefly affects the formation of the important intermediate CH3I. The N-methylation of L2 not only changes the size of the organic cation, but also makes another potential factor in influencing the cluster structure (N–H⋯I hydrogen bond) disappear. With the large bulk L13+ as the counteraction, the inorganic anionic moieties all show the discrete structures. By the size and the charge, the metal center controls the structure of the inorganic anionic cluster. The incorporated I− anion also plays a key role in controlling the cluster structure. Based on the IR spectrum, we can preliminarily judge whether the triimidazole molecule has been N-alkylated by alcohols. The UV-vis spectrum analysis indicates the energy gaps of the title four hybrids.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21271083).
References
-
(a) X. M. Zhang, Coord. Chem. Rev., 2005, 249, 1201–1219 CrossRef CAS;
(b) X. M. Chen and M. L. Tong, Acc. Chem. Res., 2007, 40, 162–170 CrossRef CAS PubMed;
(c) H. Zhao, Z. R. Qu, H. Y. Ye and R. G. Xiong, Chem. Soc. Rev., 2008, 37, 84–100 RSC;
(d) H. B. Zhu and S. H. Gou, Coord. Chem. Rev., 2011, 255, 318–338 CrossRef CAS.
-
(a) D. C. Zhong, Y. Q. Wen, J. H. Deng, X. Z. Luo, Y. N. Gong and T. B. Lu, Angew. Chem., Int. Ed., 2015, 54, 11795–11799 CrossRef CAS PubMed;
(b) Y. Z. Tang, J. B. Xiong, J. X. Gao, Y. H. Tan, Q. Xu and H. R. Wen, Inorg. Chem., 2015, 54, 5462–5466 CrossRef CAS PubMed;
(c) T. Wen, D. X. Zhang, Q. R. Ding, H. B. Zhang and J. Zhang, Inorg. Chem. Front., 2014, 1, 389–392 RSC;
(d) J. D. Tsai and C. I. Yang, Dalton Trans., 2014, 43, 15576–15582 RSC;
(e) Z. F. Liu, M. F. Wu, F. K. Zheng, S. H. Wang, J. Xiao, G. C. Guo and A. Q. Wu, CrystEngComm, 2013, 15, 7038–7047 RSC;
(f) T. Jin, F. Kitahara, S. Kamijo and Y. Yamamoto, Chem.–Asian J., 2008, 3, 1575–1580 CrossRef CAS PubMed;
(g) X. M. Zhang, Y. F. Zhao, H. S. Wu, S. R. Batten and S. W. Ng, Dalton Trans., 2006, 3170–3178 RSC.
-
(a) X. M. Zhang, M. L. Tong and X. M. Chen, Angew. Chem., Int. Ed., 2002, 41, 1029–1031 CrossRef CAS;
(b) X. M. Zhang, M. L. Tong, M. L. Gong, H. K. Lee, L. Luo, K. F. Li, Y. X. Tong and X. M. Chen, Chem.–Eur. J., 2002, 8, 3187–3194 CrossRef CAS PubMed;
(c) S. L. Zheng, J. P. Zhang, W. T. Wong and X. M. Chen, J. Am. Chem. Soc., 2003, 125, 6882–6883 CrossRef CAS PubMed;
(d) J. Tao, Y. Zhang, M. L. Tong, X. M. Chen, T. Yuen, C. L. Lin, X. Y. Huang and J. Li, Chem. Commun., 2002, 1342–1343 RSC.
-
(a) A. J. Black, N. R. Champness, S. S. M. Chung, W. S. Li and M. Schröder, Chem. Commun., 1997, 1675–1676 Search PubMed;
(b) C. M. Liu, S. Gao and H. Z. Kou, Chem. Commun., 2001, 1670–1671 RSC;
(c) N. Zheng, X. Bu and P. Feng, J. Am. Chem. Soc., 2002, 124, 9688–9689 CrossRef CAS PubMed;
(d) O. R. Evens and W. Lin, Cryst. Growth Des., 2001, 1, 9–11 CrossRef;
(e) Q. H. Wei, L. Y. Zhang, G. Q. Yin, L. X. Shi and Z. N. Chen, J. Am. Chem. Soc., 2004, 126, 9940–9941 CrossRef CAS PubMed;
(f) J. Zhang, T. Wu, P. Y. Feng and X. H. Bu, Chem. Mater., 2008, 20, 5457–5459 CrossRef CAS PubMed.
-
(a) X. X. Hu, J. Q. Xu, P. Cheng, X. Y. Chen, X. B. Cui, J. F. Song, G. D. Yang and T. G. Wang, Inorg. Chem., 2004, 43, 2261–2266 CrossRef CAS PubMed;
(b) Y. N. Wang, Q. S. Huo, P. Zhang, J. H. Yu and J. Q. Xu, Spectrochim. Acta, Part A, 2016, 167, 33–40 CrossRef CAS PubMed;
(c) J. H. Yu, Y. C. Zhu, D. Wu, Y. Yu, Q. Hou and J. Q. Xu, Dalton Trans., 2009, 8248–8256 RSC;
(d) Y. N. Wang, G. H. Li, F. Q. Bai, J. H. Yu and J. Q. Xu, Dalton Trans., 2014, 43, 15617–15627 RSC;
(e) J. Jin, F. Q. Bai, M. J. Jia, Y. Peng, J. H. Yu and J. Q. Xu, Dalton Trans., 2012, 41, 2382–2392 RSC;
(f) Y. N. Wang, Q. F. Yang, G. H. Li, P. Zhang, J. H. Yu and J. Q. Xu, Dalton Trans., 2014, 43, 11646–11657 RSC;
(g) Y. N. Wang, J. H. Yu and J. Q. Xu, Inorg. Chem. Front., 2014, 1, 673–681 RSC;
(h) Y. N. Wang, J. H. Yu and J. Q. Xu, Polyhedron, 2014, 83, 220–227 CrossRef CAS.
-
(a) C. Feng, D. Zhang, Z. Chu and H. Zhao, Polyhedron, 2016, 115, 288–296 CrossRef CAS;
(b) Y. W. Li, H. Y. Ma, S. N. Wang, J. Xu, D. C. Li, J. M. Dou and X. H. Bu, RSC Adv., 2015, 5, 88809–88815 RSC;
(c) H. L. Jia, Z. Shi, Q. F. Yang, J. H. Yu and J. Q. Xu, Dalton Trans., 2014, 43, 5806–5814 RSC;
(d) R. Sultana, T. S. Lobana and A. Castineriras, RSC Adv., 2015, 5, 100579–100588 RSC;
(e) S. Zou, Q. Li and S. Du, RSC Adv., 2015, 5, 34936–34941 RSC;
(f) X. Hang, S. Wang, X. Zhu, H. Han and W. Liao, CrystEngComm, 2016, 18, 4938–4943 RSC;
(g) R. Y. Huang, H. Jiang, C. H. Zhu and H. Xu, RSC Adv., 2016, 6, 3341–3349 RSC;
(h) L. Fan, L. Li, B. Xu, M. Qian, J. Li and H. Hou, Inorg. Chim. Acta, 2014, 423, 46–51 CrossRef CAS;
(i) B. Ji, D. Deng, J. Ma, C. Sun and B. Zhao, RSC Adv., 2015, 5, 2239–2248 RSC;
(j) H. Zhao, S. Y. Zhou, C. Feng, N. X. Wei and G. G. Wang, Inorg. Chim. Acta, 2014, 421, 169–175 CrossRef CAS;
(k) D. C. Zhong, H. B. Guo, J. H. Deng, Q. Chen and X. Z. Luo, CrystEngComm, 2015, 17, 3519–3525 RSC;
(l) Y. W. Li, Y. Tao and T. L. Hu, Solid State Sci., 2012, 14, 1117–1125 CrossRef CAS;
(m) C. Xu, L. Li, Y. Wang, Q. Guo, X. Wang, H. Hou and Y. Fan, Cryst. Growth Des., 2011, 11, 4667–4675 CrossRef CAS;
(n) S. R. Zheng, S. L. Cai, J. Fan, T. T. Xiao and W. G. Zhang, Inorg. Chem. Commun., 2011, 14, 1097–1101 CrossRef CAS;
(o) C. C. Ji, J. Li, Y. Z. Li, Z. J. Guo and H. G. Zheng, CrystEngComm, 2011, 13, 459–466 RSC.
- J. K. Cheng, Y. G. Yao, J. Zhang, Z. J. Li, Z. W. Cai, X. Y. Zhang, Z. N. Chen, Y. B. Chen, Y. Kang, Y. Y. Qin and Y. H. Wen, J. Am. Chem. Soc., 2004, 126, 7796–7797 CrossRef CAS PubMed.
-
(a) B. Xin, Y. Li, G. Zeng, Y. Peng, G. Li, Z. Shi and S. Feng, Z. Anorg. Allg. Chem., 2013, 639, 611–617 CrossRef CAS;
(b) G. N. Niu, J. R. Shi, X. J. Han, X. Zhang, K. Li, J. Li, T. Zhang, Q. S. Liu, Z. W. Zhang and C. Li, Dalton Trans., 2015, 44, 12561–12575 RSC;
(c) J. Wu, Y. Yan, B. Liu, X. Wang, J. Li and J. Yu, Chem. Commun., 2013, 49, 4995–4997 RSC;
(d) G. Zeng, S. Xing, X. Han, B. Xin, Y. Yang, X. Wang, G. Li, Z. Shi and S. Feng, RSC Adv., 2015, 5, 40792–40797 RSC;
(e) C. Zhang, J. Shen, Q. Guan, T. Yu and Y. Fu, Solid State Sci., 2015, 46, 14–18 CrossRef CAS;
(f) G. N. Liu, L. L. Liu, Y. N. Chu, Y. Q. Sun, Z. W. Zhang and C. Li, Eur. J. Inorg. Chem., 2015, 478–487 CrossRef CAS;
(g) G. N. Liu, X. M. Jiang, Q. S. Fan, M. B. Hussain, K. Li, H. Sun, X. Y. Li, W. Q. Liu and C. Li, Inorg. Chem., 2017, 56, 1906–1918 CrossRef CAS PubMed;
(h) T. Yu, L. An, L. Zhang, J. Shen, Y. Fu and Y. Fu, Cryst. Growth Des., 2014, 14, 3875–3879 CrossRef CAS;
(i) J. Shen, C. Zhang, T. Yu, L. An and Y. Fu, Cryst. Growth Des., 2014, 14, 6337–6342 CrossRef CAS.
-
(a) J. Y. Xue, J. C. Li, H. X. Li, H. Y. Li and J. P. Lang, Tetrahedron, 2016, 72, 7014–7020 CrossRef CAS;
(b) H. Chan, Y. Chen, M. Dai, C. N. Lü, H. F. Wang, Z. G. Ren, Z. J. Huang, C. Y. Ni and J. P. Lang, CrystEngComm, 2012, 14, 466–473 RSC;
(c) Y. Chen, Z. Yang, C. X. Guo, C. Y. Ni, H. X. Li, Z. G. Ren and J. P. Lang, CrystEngComm, 2011, 13, 243–250 RSC;
(d) Y. Chen, Z. Yang, C. X. Guo, C. Y. Ni, Z. G. Ren, H. X. Li and J. P. Lang, Eur. J. Inorg. Chem., 2010, 5326–5333 CrossRef CAS.
-
(a) G. E. Wang, M. S. Wang, X. M. Jiang, Z. F. Liu, R. G. Lin, L. Z. Cai, G. C. Guo and J. S. Huang, Inorg. Chem. Commun., 2011, 14, 1957–1961 CrossRef CAS;
(b) G. E. Wang, X. M. Jiang, M. J. Zhang, H. F. Chen, B. W. Liu, M. S. Wang and G. C. Guo, CrystEngComm, 2013, 15, 10399–10404 RSC;
(c) G. E. Wang, G. Xu, M. S. Wang, L. Z. Cai, W. H. Li and G. C. Guo, Chem. Sci., 2015, 6, 7222–7226 RSC;
(d) Z. J. Zhang, S. C. Xiang, G. C. Guo, G. Xu, M. S. Wang, J. P. Zou, S. P. Guo and J. S. Huang, Angew. Chem., Int. Ed., 2008, 47, 4149–4152 CrossRef CAS PubMed;
(e) G. E. Wang, G. Xu, B. W. Liu, M. S. Wang, M. S. Yao and G. C. Guo, Angew. Chem., Int. Ed., 2016, 55, 514–518 CrossRef CAS PubMed.
-
(a) J. J. Hou, C. H. Guo and X. M. Zhang, Inorg. Chim. Acta, 2006, 359, 3991–3995 CrossRef CAS;
(b) J. J. Hou, S. L. Li, C. R. Li and X. M. Zhang, Dalton Trans., 2010, 39, 2701–2707 RSC;
(c) S. L. Li and X. M. Zhang, Inorg. Chem., 2014, 53, 8376–8383 CrossRef CAS PubMed.
-
(a) G. M. Wang, J. Q. Jiao, X. Zhang, X. M. Zhao, X. Yin, Z. H. Wang, Y. X. Wang and J. H. Lin, Inorg. Chem. Commun., 2014, 39, 94–98 CrossRef CAS;
(b) G. M. Wang, J. H. Lin, J. Q. Jiao, X. Zhang, X. M. Zhao, X. Yin, J. S. Huang, Y. X. Wang and J. H. Lin, Inorg. Chem. Commun., 2014, 43, 105–109 CrossRef CAS;
(c) G. M. Wang, J. H. Li, X. Zhang, J. Q. Jiao, Z. Z. Bao, X. M. Zhao, W. W. Jiang, Y. X. Wang and J. H. Lin, Inorg. Chem. Commun., 2014, 46, 295–300 CrossRef CAS;
(d) G. Wang, Z. Ding, J. Li, X. Lv, X. Zhang, X. Zhao, Z. Wang, Y. Wang and J. Lin, CrystEngComm, 2014, 16, 3296–3304 RSC;
(e) G. Wang, J. Li, X. Zhang, P. Wang, B. B. Pang, Z. Wang, Y. Wang, J. Lin and C. Pan, Dalton Trans., 2013, 42, 13084–13091 RSC;
(f) G. M. Wang, J. H. Li, L. Wei, X. Zhang and Z. Z. Bao, RSC Adv., 2016, 5, 74811–74820 RSC.
-
(a) Q. Hou, F. Q. Bai, M. J. Jia, J. Jin, J. H. Yu and J. Q. Xu, CrystEngComm, 2012, 14, 4000–4007 RSC;
(b) J. J. Zhao, X. Zhang, Y. N. Wang, H. L. Jia, J. H. Yu and J. Q. Xu, J. Solid State Chem., 2013, 207, 152–157 CrossRef CAS;
(c) Q. Hou, J. J. Zhao, T. Q. Zhao, J. Jin, J. H. Yu and J. Q. Xu, J. Solid State Chem., 2011, 184, 1756–1760 CrossRef CAS;
(d) Q. Hou, J. N. Xu, J. H. Yu, T. G. Wang, Q. F. Yang and J. Q. Xu, J. Solid State Chem., 2010, 183, 1561–1566 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
(a) B. Guo, X. Zhang, Y. N. Wang, J. J. Huang, J. H. Yu and J. Q. Xu, Dalton Trans., 2015, 44, 5095–5105 RSC;
(b) G. Q. Mei and W. Q. Liao, J. Mater. Chem. C, 2015, 3, 8535–8541 RSC;
(c) Y. J. She, S. P. Zhao, Z. F. Tian and X. M. Ren, Inorg. Chem. Commun., 2014, 46, 29–32 CrossRef CAS;
(d) S. Eppel, N. Fridman and G. Frey, Cryst. Growth Des., 2015, 15, 4363–4371 CrossRef CAS;
(e) H. H. Li, Z. R. Chen, L. C. Cheng, Y. J. Wang, M. Feng and M. Wang, Dalton Trans., 2010, 39, 11000–11007 RSC;
(f) J. J. Liu, Y. F. Guan, C. Jiao, M. J. Lin, C. C. Huang and W. X. Dai, Dalton Trans., 2015, 44, 5957–5960 RSC;
(g) C. H. Wang, H. J. Du, Y. Li, Y. Y. Niu and H. W. Hou, New J. Chem., 2015, 39, 7372–7378 RSC;
(h) H. L. Jia, G. H. Li, H. Ding, Z. M. Gao, G. Zeng, J. H. Yu and J. Q. Xu, RSC Adv., 2013, 3, 16416–16425 RSC.
-
(a) T. Yu, Y. Fu, Y. Wang, P. Hao, J. Shen and Y. Fu, CrystEngComm, 2015, 17, 8752–8761 RSC;
(b) T. L. Yu, P. F. Hao, J. J. Shen, H. H. Li and Y. L. Fu, Dalton Trans., 2016, 45, 16505–16510 RSC;
(c) C. Zhang, J. Shen, Q. Guan, T. Yu and Y. Fu, Solid State Sci., 2015, 46, 14–18 CrossRef CAS.
-
(a) A. M. Goforth, M. A. Tershansy, M. D. Smith, L. Peterson Jr, J. G. Kelley, W. J. I. DeBenedetti and H.-Z. zur Loye, J. Am. Chem. Soc., 2011, 133, 603–612 CrossRef CAS PubMed;
(b) L. M. Wu, X. T. Wu and L. Chen, Coord. Chem. Rev., 2009, 253, 2787–2804 CrossRef CAS;
(c) J. Heine, Dalton Trans., 2015, 44, 10069–10077 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1519637–1519640. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27510a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Qi-Sheng Huoa,
Jie-Hui Yu
*b,
Qi-Sheng Huoa,
Jie-Hui Yu