
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyperporphyrin effects extended into a J-aggregate supramolecular structure in water†
Adrián
Zurita
,
Anna
Duran
,
Josep M.
Ribó
 ,
Zoubir
El-Hachemi
* and
Joaquim
Crusats
*
,
Zoubir
El-Hachemi
* and
Joaquim
Crusats
*
Departament de Química Inorgànica i Orgànica, Secció de Química Orgànica, Institut de Ciències del Cosmos (ICC), Universitat de Barcelona, Martí Franquès 1, 08028-Barcelona, Catalonia, Spain. E-mail: zelhachemi@ub.edu; j.crusats@ub.edu
First published on 13th January 2017
Abstract
The relationship between the acid–base chemistry and the supramolecular behavior of 5-(4-aminophenyl)-10,15,20-tris(4-sulfonatophenyl)porphyrin in acidic water is reported. A new species exhibiting a prominently red-shifted Q-absorption band at 742 nm is described which is in accordance with a hyperporphyrin-type spectrum of a J-aggregate in water. UV-vis spectroscopy and peak force microscopy reveal that depending on the pH value of the medium the porphyrin self-assembles into two structurally different mesophases which can be reversibly interconverted at will.
Self-organized materials obtained from the self-assembly of monomers that form J-aggregates1 arranged in a regular fashion are nowadays promising candidates with potential applications in energy and electron transfer processes in nanostructured materials. In this regard, supramolecular homoassociates of water-soluble porphyrins have received much attention.2
Inspired both by the hyperporphyrin aggregates described for the first time by Monsù Scolaro et al.3 using hydroxy-substituted porphyrins and by the more recent report by Wamser et al.4 on the acid–base chemistry of amino-substituted hyperporphyrins in organic media, we set forth to investigate if both approaches could converge using the water-soluble 5-(4-aminophenyl)-10,15,20-tris(4-sulfonatophenyl)porphyrin (TPPS3NH2, Scheme 1) as a suitable monomeric building block with the aim to compare its supramolecular behavior with that of the well-known 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin (TPPS4).5 The results of our investigation are presented in what follows.
 | ||
| Scheme 1 Chemical structure of TPPS3NH2 and its schematic representation used in this communication. | ||
The spectrophotometric titration6 of TPPS3NH2 (species of Fig. 1) could not be conveniently performed in buffered solutions at different pH values owing to an enhanced propensity of this porphyrin to aggregate in solutions of high ionic strength (see below). Instead, a 1.4 × 10−6 M aqueous solution of the free base form was titrated with sulfuric acid without any interference from the presence of aggregated species (ESI†). In spite of sharp isosbestic points in the spectrophotometric titration (ESI†), only a rough average value for (pKa1 + pKa2)/2 = 5.06 ± 0.02 corresponding to the transition from the free base porphyrin to the diprotonated form (protonation at the inner pyrroleninic nitrogen atoms, Fig. 1) could be measured. This was caused by the unusually high presence of the monoprotonated species7 as a consequence of its stabilization originated by the electronic conjugation between the protonated central nitrogen atom the peripheral unprotonated amino group.4‡ However, although the presence of the lateral amino group does indeed seem to stabilize the monoprotonated form as reported,4 it does not have a substantial effect on the basicity of the central core of the porphyrin ring in water.6 It had already been unambiguously established that the inner pyrroleninic nitrogen atoms of the macrocycle are more basic than the peripheral meso-4-aminophenyl group,4,8 as we could corroborate during the titration of TPPS3NH2. Then, the pKa value determined for the meso-anilinium cation was found to be: 2.84 ± 0.02, corresponding to a basicity more than two orders of magnitude lower than that of the central nitrogen atoms. As a consequence, solutions of the monomeric free-base, diprotonated, and triprotonated forms can all be adequately obtained so that their UV-vis spectra can be recorded as individual species (Fig. 1 and Table 1). The spectrum of the free base shows the expected absorption bands for a monosubstituted meso-4-aminophenylporphyrin.4 Upon diprotonation of the inner nitrogen atoms a distinctive hyperporphyrin spectrum showing a split Soret band together with an unusually red-shifted Q-band is recorded, in accordance with the presence of the conjugated amino group.§ Further protonation at the anilinic amino group results again in the typical absorption of the diacidic form of a porphyrin protonated at both inner pyrroleninic nitrogen atoms without any hyperporphyrin features in the spectrum.4
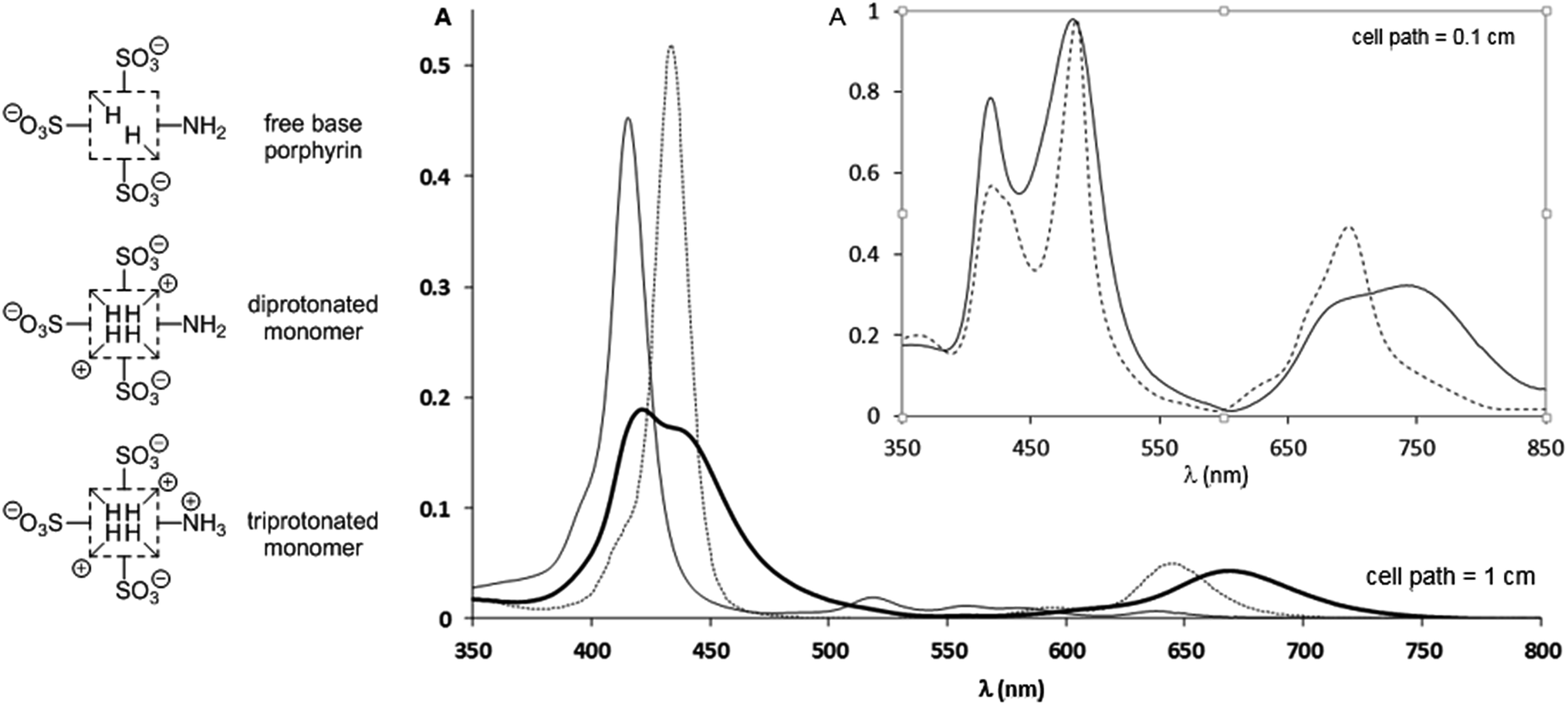 | ||
| Fig. 1 UV-vis spectra of an aqueous 1.40 × 10−6 M solution of TPPS3NH2 at different pH values (H2SO4); continuous line: free base porphyrin monomer (pH = 7.0); bold line: the diprotonated monomer showing a hyperporphyrin-like spectrum (pH = 4.0); dotted line: triprotonated monomer (pH = 1.0). For the whole quantitative spectrophotometric titration of the porphyrin see the ESI.† Inset: supramolecular aggregates of the acidic forms of TPPS3NH2 at different pH values; continuous line: a 6.25 × 10−6 M solution of TPPS3NH2 in an aqueous buffer at pH = 4.0 (AcOH, AcONa 0.1 M) in which the aggregates of the diprotonated form predominate; dotted line: a 1.47 × 10−4 M solution of TPPS3NH2 in aqueous HCl 0.1 M corresponding to the aggregates of the triprotonated zwitterionic form; the presence of residual monomeric units is evident from the shoulder at 433 nm. | ||
| Free base | Diprotonated form | Triprotonated form |
|---|---|---|
| 415 (320.000) | 423 (135.000) | 434 (362.000) |
| 519 (13.400) | 436 (126.400) | 593 (5.700) |
| 560 (9.100) | 610sh (6.900) | 645 (35.000) |
| 579 (7.900) | 669 (33.900) | |
| 639 (4.100) |
We then studied how the protonation state of the peripheral amino group affected the supramolecular behavior of the porphyrin and the electronic properties of the aggregates, in comparison to the currently well-known example of TPPS4.5 In relation to its tetrasulfonated counterpart, TPPS3NH2 was found to have an increased propensity to homoassociate. Already at micromolar concentrations of the porphyrin, an order of magnitude lower than for TPPS4 (ESI†), aggregates could be detected by UV-vis spectroscopy, both in aqueous HCl 0.1 M (triprotonated form, –NH3+) and in AcONa/AcOH 0.1 M buffer at pH = 4.0 (diprotonated form, –NH2). The UV-vis spectra of these aggregated species are presented on the inset of Fig. 1. The absorption spectrum of the aggregates of the triprotonated form [λmax: 418 nm (H-agg.); 486 nm (J-agg.); 630sh nm; 666sh nm; 696 nm] is very much in line with the typical spectrum of those of TPPS4,5 whereas, remarkably, the spectrum of the aggregates of the diprotonated form is clearly different. In this last case, although the maximum absorption of the J- and H-aggregates is almost unaltered (482 nm and 418 nm respectively), the absorption band of the J-aggregate is clearly wider and there is a most significant red-shift of a Q-band displaced to 742 nm. We attribute these changes in the J-aggregate absorption band to a hyperporphyrin effect caused by the electronic conjugation of the lone pair of the basic peripheral amino group3,4,9 with the exciton band of the aggregate in analogy to what is observed for the corresponding diprotonated monomeric species. It is also significant that when the amino group is not protonated, even at porphyrin concentrations as low as 10 μM the presence of the monomer is not detected. Yet another difference between both types of aggregates is that the triprotonated form (–NH3+) easily flocculates in the solution¶ while the diprotonated form (–NH2) does not. In addition, both different aggregates (–NH3+/–NH2) can be reversibly interconverted merely by changing the pH of the solution once the mesophases are already formed (ESI†). Notice, however, that increasing the acidity of the medium to the extent that the sulfonato groups become protonated (pH ≪ 0) results, as expected, in the total disappearance of the aggregates.
The acid–base reversibility between both aggregates is also evidenced by peak force microscopy (PFM) measurements (Fig. 2). The mesophases deposited onto highly ordered pyrolytic graphite from a solution that shows an electronic spectrum with the hyperporphyrin absorption bands are very regular (aqueous HCl solution of TPPS3NH2 at pH = 4.0 obtained from the addition of a concentrated solution of the sodium salt of the free base porphyrin over the acid and left to aggregate for 24 hours leading to a solution with the expected UV-vis spectrum). In this case, wide plate-like structures consisting of monolayered 2-D aggregates appear on the graphite surface showing regions in which the flat structure is partially folded into a bilayer (3.7 nm of height) in analogy to what has been reported for other porphyrins bearing hydrophobic meso-substituents.10 When an analogous solution of TPPS3NH2 is similarly prepared but at lower pH values so that the amino group is protonated (aqueous HCl solution, pH = 1.3, with a UV-vis spectrum showing the predominance of the aggregates of the triprotonated form), only particles of different heights corresponding to multilayered structured can be detected (ESI†). The reversible interconversion between both types of aggregates was also corroborated by PFM: when a typical solution of TPPS3NH2 at pH = 4.0 showing the folded mesophases is further acidified to pH = 1.0 with HCl, the folded structures do disappear, smaller aggregates form and flocculate, and again only irregularly shaped multilayered structures can be detected on the HOPG surface.
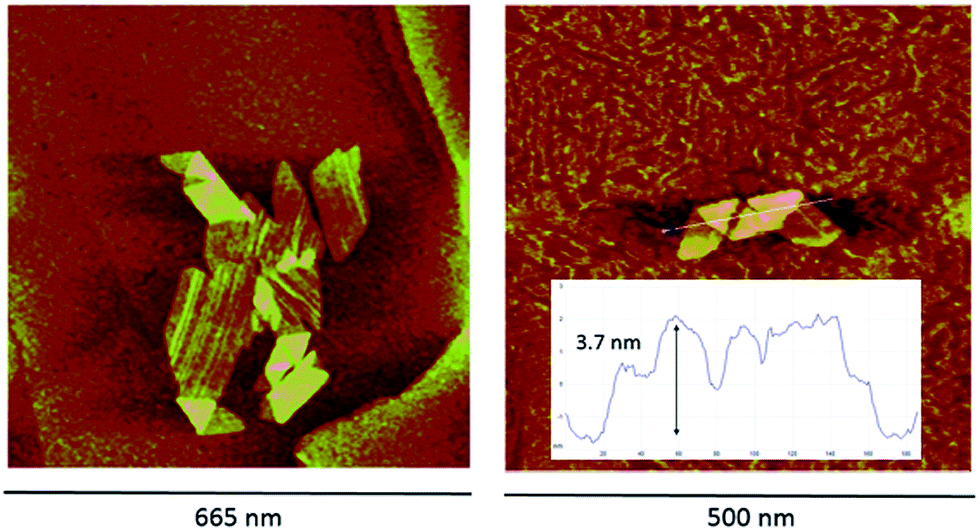 | ||
| Fig. 2 PFM images (topographical map) of TPPS3NH2 aggregates deposited on highly ordered pyrolytic graphite (HOPG) from an 8.8 × 10−5 M aqueous solution of the porphyrin at pH = 4.0 (HCl). The inset shows the height analysis of the section of the spiral structure folded so as to form bilayered regions. | ||
It is now rather well-established how the number and relative position of the anionic sulfonato groups in the para-position of the meso-phenyl groups of a protonated tetraphenylporphyrin determine the shape of their self-assembled mesophases and, hence, some of their properties like chirality expression at the mesoscopic scale.11 The results presented herein based on the tunable amino group (neutral vs. positively charged) at the periphery of the macrocycle show that not only the electronic properties of the J-aggregates can be effectively controlled at will, but also provide further evidence on how the supramolecular architectures of the aggregates can indeed be now predicted to a reasonable extent. The experimental results reported above can be reasonably interpreted as depicted in Scheme 2. Inasmuch as there are two opposite meso-4-sulfonatophenyl groups in TPPS3NH2, the porphyrin is able to form aggregates via the same basic supramolecular pattern than TPPS4.12 In a first stage, oligomeric aggregates of the porphyrin further self-assemble into a two dimensional sheet-like structure consisting of J- and H-aggregates by the interaction of two opposite sulfonato groups of the same molecule with the positively charged central protonated pyrroleninic nitrogen atoms of adjacent units, together with π–π interactions and hydrophobic effects. Thereupon, the out-of-plane 5-(4-aminophenyl) and 15-(4-sulfonatophenyl) groups can give rise to different stereoisomers of the porphyrin aggregates depending on the tacticity of these lateral groups in relation to the 2-D supramolecular sheet.13 Depending on the protonation state of the amino group, the conspicuously different growth of the particles that leads to two distinctive mesophases can be rationalized as follows: when at lower pH values the peripheral amino groups are protonated, multilayered structures can easily grow owing to interlayer coulombic interactions (Scheme 2); this fact, together with the total charge compensation in the zwitterionic nature of the triprotonated species accounts for the increased tendency of the porphyrin to flocculate. The bilayered structures, however, can be exclusively self-assembled when the amino groups are in their basic neutral form and a large enough region of the monolayer rearranges itself into an isotactic pattern with all the 4-aminophenyl groups on the same side of the 2-D structure. In this way, the supramolecular structure as a whole can gain additional thermodynamical stabilization owing to these supplementary interlayer hydrophobic and aromatic π–π interactions provided by folding.
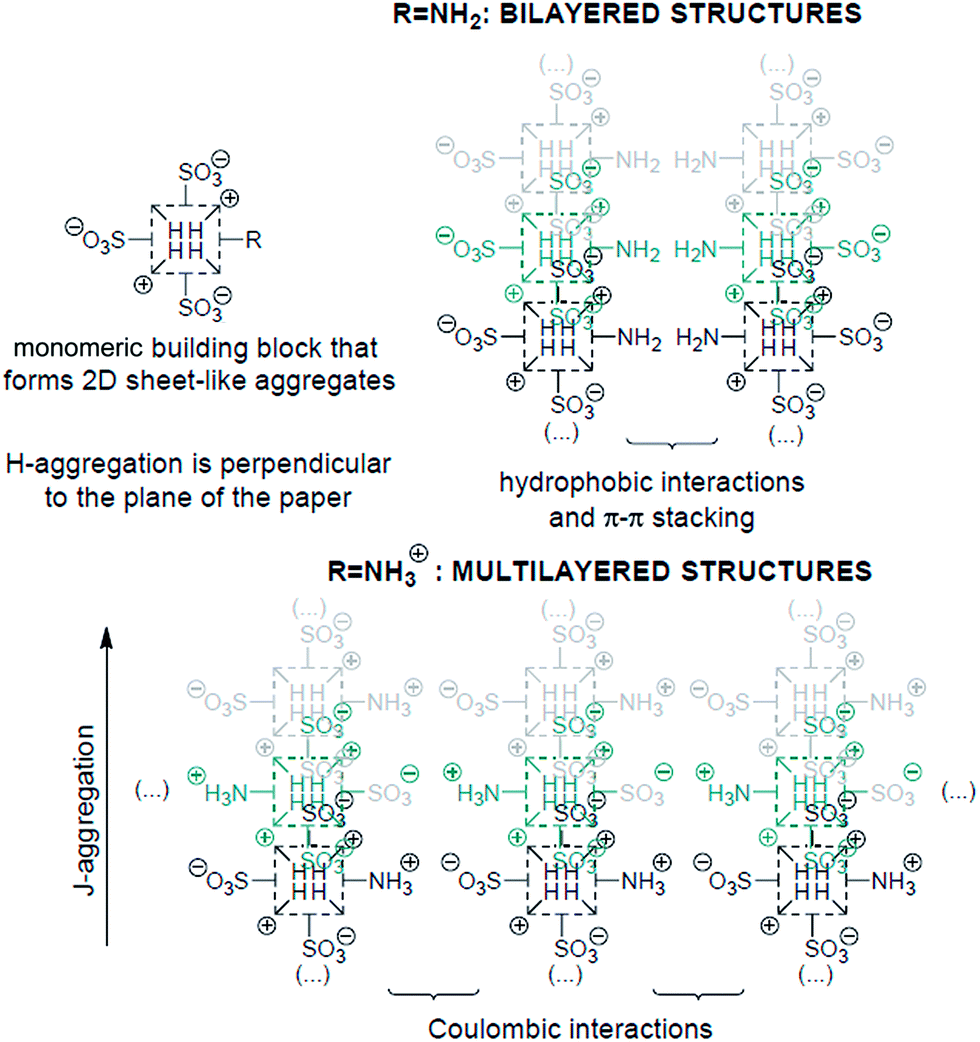 | ||
| Scheme 2 Schematic representation of the different 3D packing motifs of the 2D sheet-like supramolecular structure of the TPPS3NH2 aggregates depending on the protonation state of the peripheral amino group. | ||
In summary, we report a new type of J-aggregate in water that exhibits a pH-activatable hyperporphyrin type absorption spectrum obtained from self-assembly of a meso-sulfonatophenyl-substituted porphyrin. In addition we have shown how the origin of the unusually red-shifted absorption Q-band of the J-aggregate is unambiguously correlated to the protonation state of the peripheral p-aminophenyl group of TPPS3NH2. In this way, the electronic characteristics of the J-aggregates can be reversibly tuned depending on the acidity of the media. This fundamental result may be of potential applicability for the design of novel pH-switchable self-assembled functional materials which optical properties in the near-infrared are based on a programmed response of the porphyrinic J-aggregates.
Acknowledgements
Financial support from the Spanish Ministry of Economy and Competitiveness (MINECO), project CTQ2013-47401-C2-1-P and from Generalitat de Catalunya, project 2014SGR10, is gratefully acknowledged.Notes and references
- (a) J-Aggregates, ed. T. Kobayashi, World Scientific, Singapore, 1996 Search PubMed; (b) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef.
- (a) R. F. Pasternack, P. R. Huber, P. Boyd, G. Engasser, L. Francesconi, E. Gibbs, P. Fasella, G. C. Venturo and L. D. C. Hinds, J. Am. Chem. Soc., 1972, 94, 4511 CrossRef CAS; (b) O. Ohno, Y. Kaizu and J. Kobayashi, J. Chem. Phys., 1993, 99, 4128 CrossRef CAS; (c) D. L. Akins, H.-R. Zhu and C. Guo, J. Phys. Chem., 1994, 98, 3612 CrossRef CAS; (d) N. C. Maiti, M. Ravikanth, S. Mazumdar and N. Periasamy, J. Phys. Chem., 1995, 99, 17192 CrossRef CAS; (e) R. Rubires, J. Crusats, Z. El-Hachemi, T. Jaramillo, M. Lopez, E. Valls, J.-A. Farrera and J. M. Ribó, New J. Chem., 1999, 23, 189 RSC; (f) N. Micali, F. Mallamace, A. Romeo, R. Purrello and L. Monsù Scolaro, J. Phys. Chem. B, 2000, 104, 5897 CrossRef CAS; (g) R. Rubires, J. A. Farrera and J. M. Ribó, Chem.–Eur. J., 2001, 7, 436 CrossRef CASM. A. Castriciano, A. Romeo, V. Villari, N. Micali and L. Monsù Scolaro, J. Phys. Chem. B, 2003, 107, 8765 CrossRef CAS; (h) A. S. R. Koti, J. Taneja and N. Periasamy, Chem. Phys. Lett., 2003, 375, 171 CrossRef CAS; (i) R. Rotomskis, R. Augulis, V. Snitka, R. Valiokas and B. Liedberg, J. Phys. Chem. B, 2004, 108, 2833 CrossRef CAS; (j) M. de Napoli, S. Nardis, R. Paolese, M. G. H. Vicente and R. Purrello, J. Am. Chem. Soc., 2004, 126, 5934 CrossRef CAS PubMed; (k) A. L. Yeats, A. D. Schwab, B. Massare, D. E. Johnston, A. T. Johnson, J. C. De Paula and W. Smith, J. Phys. Chem. C, 2008, 112, 2170 CrossRef CAS; (l) L. Zhang and M. Liu, J. Phys. Chem. B, 2009, 113, 14015 CrossRef CAS PubMed; (m) M. A. Castriciano, A. Romeo, G. De Luca, V. Villari, L. M. Scolaro and N. Micali, J. Am. Chem. Soc., 2011, 133, 765 CrossRef CAS; (n) A. Sorrenti, Z. El-Hachemi, J. Crusats and J. M. Ribó, Chem. Commun., 2011, 47, 8551 RSC.
- G. De Luca, A. Romeo and L. Monsù Scolaro, J. Phys. Chem. B, 2006, 110, 14135 CrossRef CAS PubMed.
- A. B. Rudine, B. D. DelFatti and C. Wamser, J. Org. Chem., 2013, 78, 6040 CrossRef CAS.
- J. M. Ribó, J. Crusats, J.-A. Farrera and M. L. Valero, Chem. Commun., 1994, 30, 681 RSC.
- (a) P. Hambright, Porphyrins and Metalloporphyrins, ed. K. M. Smith, Elsevier Scientific Publishing Company, Amsterdam–Oxford–New York, 1975, pp. 235–238 Search PubMed and references cited therein; (b) T. P. G. Sutte, R. Rahimi, P. J. Hambright, C. Bommer, M. Kumar and P. Neta, J. Chem. Soc., Faraday Trans., 1993, 85, 495 RSC; (c) T. Gensch, C. Viappiani and S. E. Braslavsky, J. Am. Chem. Soc., 1999, 121, 10573 CrossRef CAS.
- (a) F. Hibbert and K. P. P. Hunte, J. Chem. Soc., Perkin Trans. 2, 1977, 1624 RSC; (b) B. Cunderlikova, O. Kaalhus, R. Cunderlik, A. Mateasik, J. Moan and M. Kongshaug, Photochem. Photobiol., 2004, 79, 242 CrossRef CAS.
- (a) E. C. A. Ojadi, H. Linschitz, M. Gouterman, R. I. Walter, J. S. Lindsey, R. W. Wagner, P. R. Droupadi and W. Wang, J. Phys. Chem., 1993, 97, 13192 CrossRef CAS; (b) I. Hatay, B. Su, M. A. Méndez, C. Corminboeuf, T. Khoury, C. P. Gros, M. Bourdillon, M. Meyer, J. M. Barbe, M. Ersoz, S. Záliš, Z. Samec and H. H. Girault, J. Am. Chem. Soc., 2010, 132, 13733 CrossRef CAS.
- M. Gouterman, The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 3, p. 1 Search PubMed.
- C. Escudero, J. Crusats, I. Díez-Pérez, Z. El-Hachemi and J. M. Ribó, Angew. Chem., Int. Ed., 2006, 45, 8032 CrossRef CAS.
- (a) J. Crusats, J. Claret, I. Díez-Pérez, Z. El-Hachemi, H. García-Ortega, R. Rubires, F. Sagués and J. M. Ribó, Chem. Commun., 2003, 1588 RSC; (b) A. Cabrer, J. M. Ribó, Z. El-Hachemi and J. Crusats, J. Porphyrins Phthalocyanines, 2015, 19, 852 CrossRef CAS.
- (a) Z. El-Hachemi, C. Escudero, F. Acosta-Reyes, M. T. Casas, V. Altoe, S. Aloni, G. Oncins, A. Sorrenti, J. Crusats, J. L. Campos and J. M. Ribó, J. Mater. Chem. C, 2013, 1, 3337 RSC; (b) M. J. Short, J. A. Berriman, C. Kübel, Z. El-Hachemi, J. V. Naubron and T. S. Balaban, ChemPhysChem, 2013, 14, 3209 CrossRef; (c) J. V. Hollingsworth, A. J. Richard, M. G. H. Vicente and P. S. Russo, Biomacromolecules, 2012, 13, 60 CrossRef CASS. C. M. Gandini, E. L. Gelamo, R. Itri and M. Tabak, Biophys. J., 2003, 85, 1259 CrossRef CAS.
- Z. El-Hachemi, T. S. Balaban, J. L. Campos, S. Cespedes, J. Crusats, C. Escudero, C. S. Kamma-Lorger, J. Llorens, M. Malfois, G. R. Mitchell, A. P. Tojeira and J. M. Ribó, Chem.–Eur. J., 2016, 22, 9740 CrossRef CAS PubMed.
- M. Vitasovic, M. Gouterman and H. Linschitz, J. Porphyrins Phthalocyanines, 2011, 5, 191 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: For materials and methods, synthetic procedures, HPLC analysis, acid–base titrations, aggregation studies and further PFM images. See DOI: 10.1039/c6ra27441b |
| ‡ When the titrations of TPPS3NH2 were performed with methanesulfonic acid in DMSO the results paralleled those reported for 5-(4-aminophenyl)-10,15,20-tris(4-carbomethoxyphenyl)porphyrin to the minor detail (ref. 4). However, when the titration was performed in water with sulfuric acid, although a significant presence of the porphyrin monocation could still be inferred, an increase of the intensity of the Soret band similar to that reported in ref. 4 was not observed (ESI†). |
| § The split Soret band by 13 nm together with the red-shifted Q bands and the decreased ε value depicted in Fig. 1 remain unaltered at the concentration limit of the UV-vis technique (TPPS3NH2 ∼10−7 M); in addition, we were unable to detect any resonance light scattering signal in this solution. The typical hyperporphyrin electronic spectra arise in transition metalloporphyrins as a consequence of the interaction between the porphyrin molecular orbitals and the p or d orbitals of the central metal atom (ref. 9). In the case of meso-aminophenyl substituted porphyrins with four hydrogen atoms in the core of the macrocycle, the same type of hyperporphyrin electronic spectra arise through the interaction of the peripheral meso-substituent with the molecular orbitals of the porphyrin ring. Further protonation on the amino group at the phenyl substituent results in the disappearance of the hyperporphyrin feature in the spectrum, as reported also herein (ref. 14). |
| ¶ We have observed a similar behavior in all the members of a series of 5-(aryl-or-heteroaryl)-10,15,20-tris(4-sulfonatophenyl)porphyrins in which there are three negatively charged peripheral sulfonate groups plus a fourth substituent bearing a positive charge. Unexpectedly, none of these porphyrins lead to solutions with detectable CD-signals during their supramolecular homoassociation process in acidic water, in contrast with what is commonly observed with TPPS4 and its partially sulfonated counterparts. The results will be published elsewhere in due course. |
| This journal is © The Royal Society of Chemistry 2017 |