
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Random copolymer gels of N-isopropylacrylamide and N-ethylacrylamide: effect of synthesis solvent compositions on their properties†
Qiao Wanga,
Chandra Sekhar Biswasab,
Massimiliano Galluzziab,
Yuhang Wua,
Bing Du*a and
Florian. J. Stadler *a
*a
aCollege of Materials Science and Engineering, Shenzhen Key Laboratory of Polymer Science and Technology, Guangdong Research Center for Interfacial Engineering of Functional Materials, Nanshan District Key Lab for Biopolymers and Safety Evaluation, Shenzhen University, Shenzhen 518060, P. R. China. E-mail: dubing@szu.edu.cn; fjstadler@szu.edu.cn
bCollege of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, P. R. China
First published on 30th January 2017
Abstract
Random copolymer gels of N-isopropylacrylamide (NIPAM) and N-ethylacrylamide (NEAM) were synthesized using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 monomer molar ratio in different methanol–water (xm = 0, 0.06, 0.13, 0.21, 0.31 0.43, 0.57, 0.76, where xm = mole fraction of methanol) mixtures. The samples were characterized using different techniques like Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), swelling ratio measurements, deswelling kinetics study, atomic force microscopy (AFM), and rheology. We found that, with the variation of the solvent composition (methanol–water mixtures) the properties of the gels varied significantly. These results can be explained on the basis of the interactions of the two different kinds of monomers with different methanol–water mixtures, their different kinds of thermoresponsiveness and hydrophilicity, and their different cononsolvent properties toward methanol–water mixtures.
:1 monomer molar ratio in different methanol–water (xm = 0, 0.06, 0.13, 0.21, 0.31 0.43, 0.57, 0.76, where xm = mole fraction of methanol) mixtures. The samples were characterized using different techniques like Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), swelling ratio measurements, deswelling kinetics study, atomic force microscopy (AFM), and rheology. We found that, with the variation of the solvent composition (methanol–water mixtures) the properties of the gels varied significantly. These results can be explained on the basis of the interactions of the two different kinds of monomers with different methanol–water mixtures, their different kinds of thermoresponsiveness and hydrophilicity, and their different cononsolvent properties toward methanol–water mixtures.
Introduction
Thermo-responsive water-soluble poly(N-isopropylacrylamide) (PNIPAM) has received significant attention due to its low critical solution temperature (LCST) (around 33 °C), as first reported by Heskins and Guillet.1 PNIPAM undergoes phase separation when increasing the temperature beyond the LCST value, where the polymer chains change from a random coil to a globular state.2–5 Phase separation can also happen when a specific water-miscible organic solvent is mixed with water at a certain range of compositions, for example, methanol,6–9 ethanol,10,11 tetrahydrofuran (THF),12,13 dimethyl sulfoxide (DMSO),14,15 known as cononsolvency,8 a phenomenon where a mixture of two good solvents behaves as a poor solvent for a polymer at particular compositions. Winnik et al. studied cononsolvency of linear PNIPAM chains in different methanol–water mixtures and explored different aspects of cononsolvency of PNIPAM in those solvents.7,8 Tanaka et al. studied cononsolvency of linear PNIAM in methanol–water mixture based on a competitive hydrogen bond model formed between water-polymer and methanol-polymer.16 Hore et al. studied cononsolvency of linear PNIPAM in ethanol-deuterated water mixture.17 Kojima et al. studied cononsolvency of PNIPAM microgels in methanol–water mixture.18 Scherzinger et al. compared cononsolvency of PNIPAM (linear, microgel and macrogel) in methanol–water mixtures.19Hofmeister20 showed that salts in aqueous solution can have a profound effect on solubility as well. In recent years, it was reported that the physico-chemical properties of polymer solutions (spectroscopic properties, viscosity, temperature dependency, …) change upon addition of salts.21–23 While at a first glance the Hofmeister and cononsolvency effects are not related to each other, they have several common points: (1) addition of a second material in the solute leads to worsening (in case of chaotrope materials) of the solubility, (2) the effect depends on concentration and type of this additive. The clear difference between these effects is that salts are not solvents themselves and that they are ions in solution, thus, having particularly strong interactions with ionic materials.
Due to its unique thermoresponsive properties, PNIPAM is the most important thermo-responsive polymer. For this reason, it became very important in applied fields like, bio-separation,24–26 controlled release,27–29 water capturing,30 sensors,31,32 etc. There are several reports available on PNIPAM gels and homopolymers. Wu and Zhou reported the swelling properties of PNIPAM microgels in water,33 and phase transition in water of swollen microgels.34 Shirota et al. reported the effect of deuterium isotope on phase transition temperature of PNIPAM gels prepared in water.35 Kratz, et al. prepared PNIPAM microgels in water with different cross-linker density and investigated their different behavior at swollen and collapsed state in water by various methods.36 Lynch and Dawson investigated the effect of polymeric additives on pore size distribution and deswelling kinetics of PNIPAM hydrogel in water.37 Liu et al. prepared PNIPAM hydrogels by reversible addition fragmentation chain transfer (RAFT) polymerizations, and investigated the swelling properties in water.38 As porous structures are very important for swelling properties and their applications, lots of efforts have been given to synthesize them in water miscible organic solvents (cononsolvent medium), like methanol,9,39 ethanol,40,41 acetone,40,42 1,4-dioxane,10 THF,12 or DMSO.42
Recently, we reported the synthesis and characterization of the PNIPAM hydrogels by tuning the stereoregularity of the PNIPAM gels by using the rare earth Lewis acid Y(OTf)3 in different methanol–water mixtures.39,43
Atomic force microscopy (AFM) is an effective instrument for the characterization of mechanical properties of gels. There are few studies regarding measurement of mechanical properties of PNIPAM using AFM technique. Matzelle et al.44 used AFM technique to study the effect of cross-linking density on elastic properties in water of PNIPAM and polyacrylamide (PAM) hydrogels at different temperatures. Goodman et al. reported AFM measurement of polymer brushes of PNIPAM, poly(N,N-dimethylacrylamide) (PDMA), or poly(methoxyethylacrylamide) (PMEA) grafted on polystyrene (PS) seed in sodium chloride solution and at pH = 7.45 Sui et al. used AFM technique to understand the effect of both cononsolvency in methanol–water and grafting density on the collapse dynamics of PNIPAM brushes.46
Rheology is another powerful tool for characterization of mechanical and solution properties of polymers and gels. For example, we can get the idea about their mechanical properties from compression test. At the same time, we also get the information about LCST by measuring the temperature sweep experiment.47–49 Puleo et al. investigated rheological properties of PNIPAM crosslinked hydrogel in water.50 One of our group studied rheological properties of supramolecular gel based on NIPAM and catechol acrylamide copolymer in water.51
PNEAM is another important thermoresponsive polymer in the family of N-alkylacrylamides, which shows an LCST in water in a wide range of temperatures, e.g. in between 62 °C and 82 °C (there is some literature disagreement on the exact LCST and possibly there are additional factors influencing the LCST of PNEAM, which have not been discovered so far).50,52 Although it is not as studied as PNIPAM so far, still there are many researchers are interested about it due to its growing importance nowadays. Lowe et al. investigated physio-chemical properties of PNEAM microgels in water in absence or presence of sodium chloride in water.53 Xue et al. investigated swelling behaviors, polymer–solvent interaction parameters and elastic moduli of PNEAM hydrogels in water with different cross-linking density.54 Cai and Gupta studied the properties of PNEAM hydrogels synthesized in absence or presence of acrylic acid (AA) and NEAM copolymer microgel particles and studied their application for lignin separation.55 Hirano et al. synthesized NEAM and N-n-propylacrylamide (NnPAM) copolymers with different stereoregularity in toluene to investigate its effect on LCST.56 Savoji et al. synthesized block random copolymers using NEAM and NnPAM with different chemical composition, and investigated the phase transition behavior.57 They also reported the formation of micelles in water with dually-responsive diblock random copolymers consists of NEAM, NnPAM and 2-(diethylamino)ethyl methacrylate.58 Nichifor and Zhu copolymerized styrene with NEAM and studied their LCSTs with respect to chemical composition, molecular weight and polymer concentration in water.59 Recently, the effect of stereoregularity on thermoresponsive properties was investigated for PNEAM gels prepared in methanol–water mixtures,60 and the effect of synthesis solvent compositions on stereoregularity of PNEAM gels in presence of Y(OTf)3 in methanol–water mixtures.61
So far to the best of our knowledge, there is no report concerning synthesis of random copolymer gels using 1:1 NIPAM and NEAM molar ratio in different compositions of methanol–water mixtures. In this study, we synthesized a series of such random copolymer gels and studied their swelling (deswelling, cononsolvency, swelling ratio, etc.), morphological, mechanical, and rheological properties in detail.
Experimental section
Materials
NIPAM (98%) was recrystallized from n-hexane. NEAM (Sigma-Aldrich, 99%) was purified by passing through column (neutral aluminum oxide filled). Methanol was dried and distilled over calcium oxide. N,N,N′,N′-Tetramethylethylenediamine (TEMED, Aladdin, 98%), N,N′-methylenebisacrylamide (BIS) and ammonium persulfate (APS) were used as received. All chemicals are of analytical grade, and are purchased from Macklin, Shanghai, China, unless specifically mentioned. Double distilled water was used for all experiments.Synthesis of copolymer gels
A slightly modified procedure was used based on literature:9,39 three stock solutions were prepared: (i) an aqueous solution of TEMED (107 mmol l−1); (ii) a methanolic solution of TEMED (107 mmol l−1); and (iii) an aqueous solution of APS (84 mmol l−1) was used to synthesize copolymer gels. First, required amount of NIPAM, NEAM, BIS, water, methanol, TEMED solution (as specified in Table 1) were added to a 30 ml glass vial fitted with a rubber septum. The solutions were purged with nitrogen for 30 min and then immersed in a thermal bath maintained at 10 °C. The APS solution in water (maintained at 10 °C) was also purged with N2 for 30 min and then added to the pre-gel mixture with degassed syringe, mixed them well immediately by tilting the vials up and down and allowed them to react at 10 °C for 12 hours. The prepared gels were cut into small square types pieces (8 × 8 mm2) having approximately same size followed by dialysis in deionized water for 7 days to remove all unreacted chemicals. After the dialysis, the gels were dried under vacuum at 50 °C for 72 h. The conversion of the obtained gels was determined gravimetrically.:1) in the presence of different compositions of methanol–water mixturea
| Run ID | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| X′0 | X′′0 | X0 | X0.06 | X0.13 | X0.21 | X0.31 | X0.43 | X0.57 | X0.76 | |
| a Polymerization temperature = 5 °C, polymerization time = 12 h.b Determined gravimetrically after drying under vacuum at 50 °C for 72 h after dialysis.c Determined by rheology. | ||||||||||
| NIPAM (mg) | 1200 | 0 | 640 | 640 | 640 | 640 | 640 | 640 | 640 | 640 |
| NEAM (mg) | 0 | 1120 | 560 | 560 | 560 | 560 | 560 | 560 | 560 | 560 |
| BIS (mg) | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 |
| MeOH (ml) | 0 | 0 | 0 | 1.875 | 3.75 | 5.625 | 7.5 | 5.625 | 7.5 | 9.375 |
| Water (ml) | 9.375 | 9.375 | 9.375 | 7.5 | 5.625 | 3.75 | 1.875 | 3.75 | 1.875 | 0 |
| Solution of TEMED (107 mmol dm−3) in water (ml) | 3.75 | 3.75 | 3.75 | 3.75 | 3.75 | 3.75 | 3.75 | 0 | 0 | 0 |
| Solution of TEMED (107 mmol dm−3) in methanol (ml) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3.75 | 3.75 | 3.75 |
| Solution of APS (84 mmol dm−3) in water (ml) | 1.875 | 1.875 | 1.875 | 1.875 | 1.875 | 1.875 | 1.875 | 1.875 | 1.875 | 1.875 |
| Conversionb (%) | 97 | 98 | 99 | 99 | 97 | 98 | 91 | 94 | 82 | 65 |
| Appearance | Transparent | Transparent | Transparent | Transparent | Translucent | Translucent | Translucent | Translucent | Transparent | Transparent |
| Swelling ratio at 20 °C (Ws/Wd) | 13.1 | 15.1 | 15.9 | 17.4 | 19.2 | 23.0 | 23.4 | 24.2 | 24.2 | 23.5 |
| Swelling ratio at 85 °C (Ws/Wd) | 1.3 | 3.4 | 1.5 | 1.5 | 1.5 | 1.5 | 1.5 | 1.6 | 1.6 | 1.6 |
| LCSTc (±0.5) (°C) | 33.0 | 81.0 | 68.1 | 68.4 | 68.8 | 68.4 | 67.7 | 67.5 | 67.2 | 66.5 |
FTIR characterization
FTIR spectra of the gels were taken in the range of 400–4000 cm−1 range by making pellet with KBr.Surface morphology
Gels were first swollen in deionized water at 20 °C for 24 h to reach the equilibrium swelling conditions. They were then frozen in liquid nitrogen and freeze-dried in vacuo. The surface morphology of these freeze-dried hydrogels were analyzed by field emission scanning electron microscopy (FESEM, HITACHI-SU-70, JAPAN) at 5 kV voltage.Cononsolvency study
Cononsolvency study of different gels at different temperatures were done by dipping the samples in different methanol–water mixtures (xm = 0, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30, 0.35, 0.40, 0.45, 0.50, 0.55, 0.60, 0.80, and 1; where xm = mole fraction of methanol) for 24 h to get equilibrium swelling conditions were measured gravimetrically. The swelling ratio (Ws/Wd) was taken as the ratio of the weight of the equilibrium swollen gel (Ws) to that of the dried gel (Wd).Temperature dependence of swelling ratio in water
Swelling ratios of the different hydrogels at 20, 25, 30, 35, 40, 43, 46, 50, 55, 60, 70, 80, and 85 °C were measured gravimetrically. The pre-weighed dry gels (Wd) were immersed into deionized water at the desired temperatures for 24 h to reach its equilibrium state, their weights (Ws) were taken after removing surface water with moistened filter paper. Swelling ratio (SR) was calculated using the ratio of Ws to Wd as:| SR = Ws/Wd | (1) |
Temperature dependent deswelling kinetics at 85 °C
Deswelling kinetics of the gels in water at 85 °C obtained after immersing in water at 20 °C for 24 h were measured gravimetrically. The pre-weighed equilibrium swollen gel in water at 20 °C were quickly transferred into water at 85 °C. At definite time intervals, the gels were taken out, wiped the surface water out with moistened filter paper, weighed the gels (Wt), and immerse the gels back in the water at 85 °C. Percentage of water retention (WR) was calculated using following equations:| % of WR = 100 × (Wt − Wd)/(Ws − Wd) | (2) |
Deswelling rate (DR) was calculated using eqn (3):
| Rate of water release = (100 − WR)/t | (3) |
Measurements of mechanical properties by AFM
AFM measurements of the gels were performed using a commercial AFM Dimension Icon (Bruker, USA) in Force Volume (FV) mechanical imaging mode.56,62 The mechanical analysis on hydrogels series were performed with medium resolution during imaging (64 × 64) but higher resolution in force curves (4096 points), which allows for much clearer, quantitative evidence. All samples were imaged, while being immersed in deionized water at room temperature (T = 23 °C). Spherical colloidal probes (Novascan) with 2500 nm radius and spring constant k ≈ 0.08 N m−1 were used for all measurements. The exact geometry of every tip used in experiments was analyzed by scanning electron microscopy. The elastic spring constant was calibrated for each tip in air using thermal tuning method.Rheological properties
Rheological properties of the gels are measured by using an Anton Paar MCR 302 rheometer using 8 mm parallel plates with a Peltier temperature control device. Two types of measurements were performed on the gels; temperature sweep and compression test. For both measurements, the gels were swollen first to obtain equilibrium conditions at respective temperatures. For temperature sweep, the gels were glued between the 8 mm parallel plates of the rheometer by superglue. Next, the measurement vessel was filled with water, followed by the temperature sweep from 1 °C to 90 °C and 90 °C to 1 °C at a heating rate q = 1 K min−1. For the compression tests at 20 °C, i.e. well below the LCST of the gels, the gels were placed between two 8 mm plates and compressed from an initial height of 3–4 mm until a final height of 0.1 mm at a speed of 0.01 mm s−1. The experiment was stopped automatically, if the force exceeded 18 N, to avoid damage to the air bearing of the rheometer. However, the force required for breaking all samples was significantly below this threshold.Result and discussion
Synthesis
The random copolymer gels of PNIPAM and PNEAM gel synthesis conditions and their characterization data are given in Table 1. The observed yields (%) were varied in between 65 and 98%. Conversion is low for the gels synthesized in the presence of maximum methanol amount (xm = 0.76, run X0.76, Table 1). This may be due to the lower solubility of the initiator APS in methanol-rich solvents. Appearance of the as prepared hydrogels changed from transparent to translucent depending on the solvent compositions. The observed transparency of the gels X′0 (pure PNIPAM gel synthesized in water), X0 (copolymer gel synthesized in water), X0.06 (copolymer gel synthesized in 0.06 mole fraction of methanol), X0.57 (copolymer gel synthesized in 0.57 mole fraction of methanol), and X0.76 (copolymer gel synthesized in 0.76 mole fraction of methanol), is due to the homogeneous and highly solvated coiled conformation of PNIPAM and PNEAM chains in the gel owing to the strong interaction of polymer chains with the solvent. The observed translucency of the gels X0.13 (copolymer gel synthesized in 0.16 mole fraction of methanol), X0.21 (copolymer gel synthesized in 0.21 mole fraction of methanol), X0.31 (copolymer gel synthesized in 0.31 mole fraction of methanol), and X0.43 (copolymer gel synthesized in 0.43 mole fraction of methanol), is due to the effect of cononsolvency of both PNIPAM and PNEAM towards these synthesis solvent compositions.In Fig. 1, the FTIR spectra of all the gels are shown. From the FTIR-spectra, we see that all the spectra are almost identical, indicating that all the gels have almost the same chemical compositions. To confirm it further, we have also done elemental analysis of the gels and it proves that all gels have identical chemical compositions within the experimental accuracy, as proven by the elements contents of C, N, O and H. The C to N ratio is also identical within the experimental accuracy in all cases and are very close to the expected value. The figure is shown in Fig. SI1.†
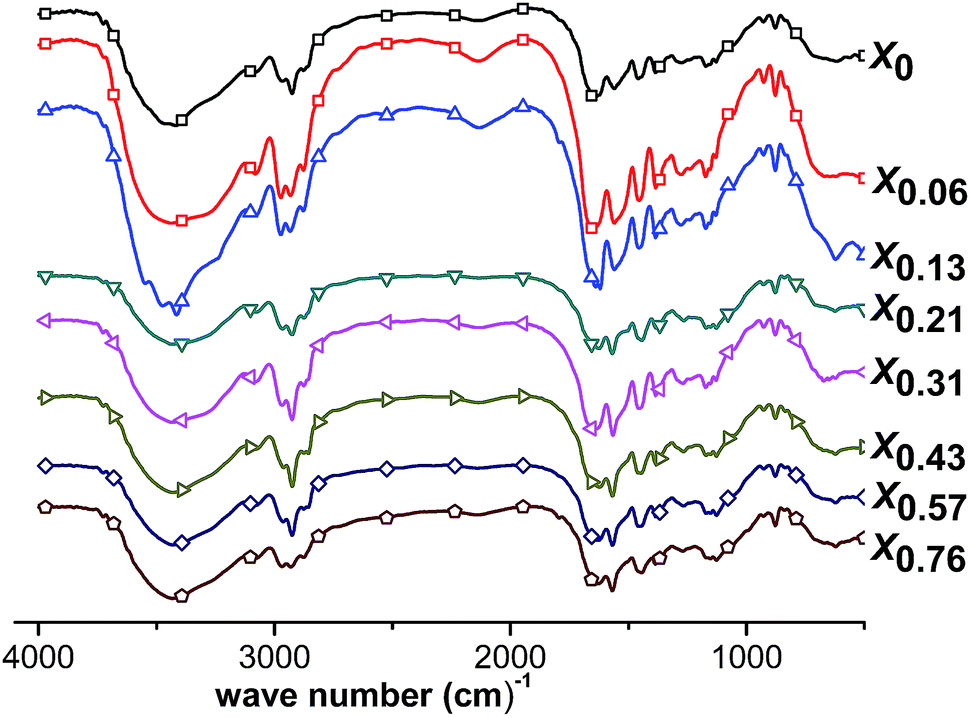 | ||
| Fig. 1 FTIR spectra of all the gels. | ||
Surface morphology and swelling at room temperature
The surface morphology of the gels is shown in Fig. 2. From the morphology images it is evident that, the gel prepared in water using pure NIPAM [Fig. 2(a), X0] does not show any visible pores, as the synthesis is done in highly homogeneous solvent. The gel synthesized using 1:1 NIPAM–NEAM (molar ratio) in pure water (xm = 0) is slightly porous [Fig. 2(b), X0] due to presence of NEAM owing to slightly different interactions with the solvent. The gel synthesized at xm = 0.06 is also slightly porous due to the presence of methanol in the synthesis solvent upon addition to PNEAM [Fig. 2(c), X0.06], due to cononsolvency on PNEAM and PNIPAM alike. The gels, synthesized at xm = 0.13, 0.21 and 0.31 [Fig. 2(d), run X0.13; Fig. 2(e), run X0.21; and Fig. 2(f), run X0.31 respectively] show considerable porosity due the heterogeneity of the solvent media during synthesis owing to the cononsolvency effect. Cononsolvency is prominent both for PNIPAM and PNEAM in these solvent compositions.9,58 This will be described in the cononsolvency discussion part in more detail. With the further increase in the xm value from 0.43 through 0.57 to 0.76 [run X0.43, Fig. 2(g); run X0.57, Fig. 2(h); and run X0.76 (figure not shown here)] cononsolvency does not play a significant role during the synthesis specially for X0.57 and X0.76. As a result, the porosity of the gels is not significant in this region. Hence, the porosity of the gels can be facilely tailored by changing the synthesis solvent compositions.
 | ||
| Fig. 2 Surface morphology of the gels: (a) X′0, (b) X0, (c) X0.06, (d) X0.13, (e) X0.21, (f) X0.31, (g) X0.43, and (h) X0.57. | ||
The swelling ratios of the gels prepared using 1:1 monomer ratio in different methanol–water mixture at 20 °C (Table 1) are shown in Fig. 3 as a function of synthesis solvent composition [here mole fraction of methanol (xm)]. It, the swelling ratio mainly depends on the porosity of the gels and molecular volume of synthesis solvent. The higher the porosity, and hydrophilicity of the gels are, the higher are their swelling ratios. Higher molecular volume of the synthesis media is also favorable for high swelling ratio.63 As the molar ratio of both PNEAM and PNIPAM is constant throughout the gel compositions, the effect of hydrophilicity of monomers and their interactions with solvents at high methanol rich region (xm > 0.40, outside of cononsolvency region) are comparable. So the main role on the swelling ratio variations of the gels depend on the porosity of the gels and the molecular volume of the synthesis media. Hence, with the increase in xm value from 0 to 0.21, the swelling ratio value increased steeply from 16 to 23, as porosity also increased significantly [Fig. 2(a)–(d)]. Further increase of methanol content in synthesis solvent xm, led to an approximately constant swelling ratio between 23 and 24.5. For the conclusive evidence, we have measured the BET isotherms for four of the samples (X0, X0.21, X0.43 and X0.76). The BET results confirm the SEM observations – that is, a significantly higher pore volume and surface area for the gel synthesized in the cononsolvency region (X0.21). The gels synthesized outside of the cononsolvency region have much lower pore volume and surface area. It confirms that the higher swelling ratios of the gels synthesized at methanol rich solvents are mainly due to their lower chain density and not due to porosity. The results are shown in Fig. SI2.† Therefore, this effect cannot be explained by the porosity alone. Hence, its discussion and explanation is moved to the conclusions.
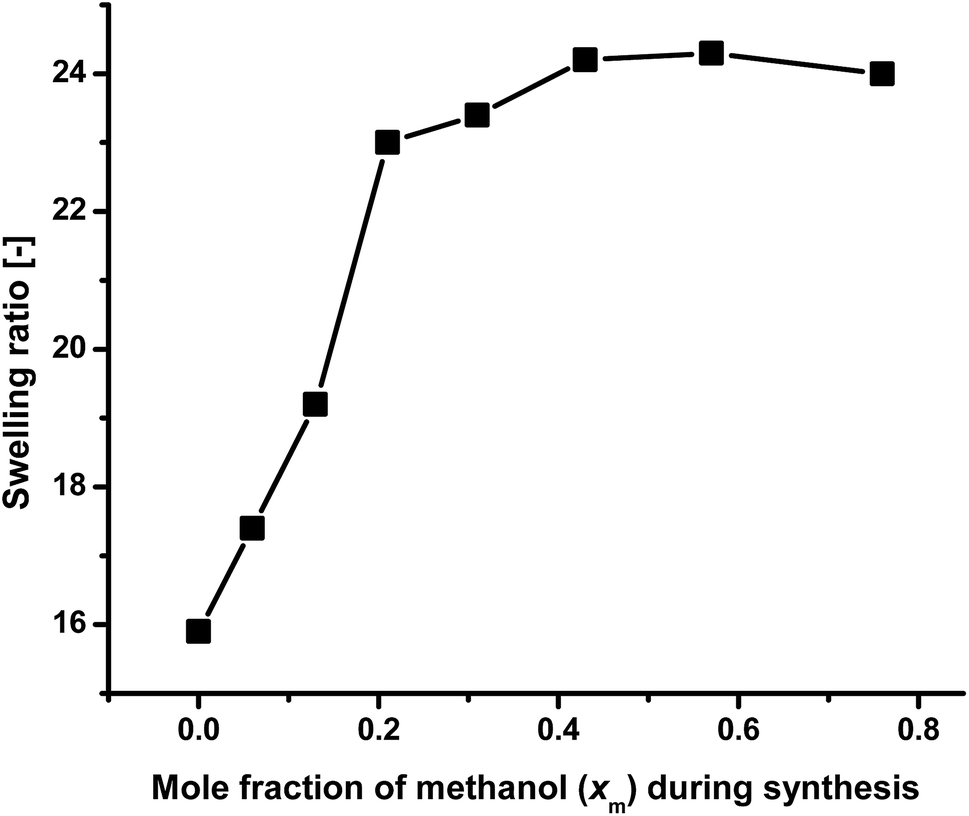 | ||
| Fig. 3 Comparison of swelling ratio of the gels at 20 °C as a function of solvent composition during synthesis when immersed in water. | ||
In these solvents compositions, the gel networks present in more expanded form compared to those synthesized with low xm value. It is interesting to note that these gels are apparently nonporous [Fig. 2(g) and (h)] but the large molecular volume of methanol has played the dominant role for the determination of swelling ratio value than the porosity. Similar kind of results were also observed before.63 This result is in contradictory, with the result we reported earlier with PNIPAM gels where the maximum swelling ratio was observed in cononsolvency zone.
These results need to be interpreted together with the mechanical data, in order to get a coherent molecular picture. Hence, the discussion of these results is moved to the conclusions.
Cononsolvency study
Fig. 4(a) shows cononsolvency phenomenon of all the gels in different methanol–water mixtures. From the figure it is clear that, swelling ratio of all the gels started to decrease with the increase in the methanol content, passing through a minimum and then again increasing at high methanol region.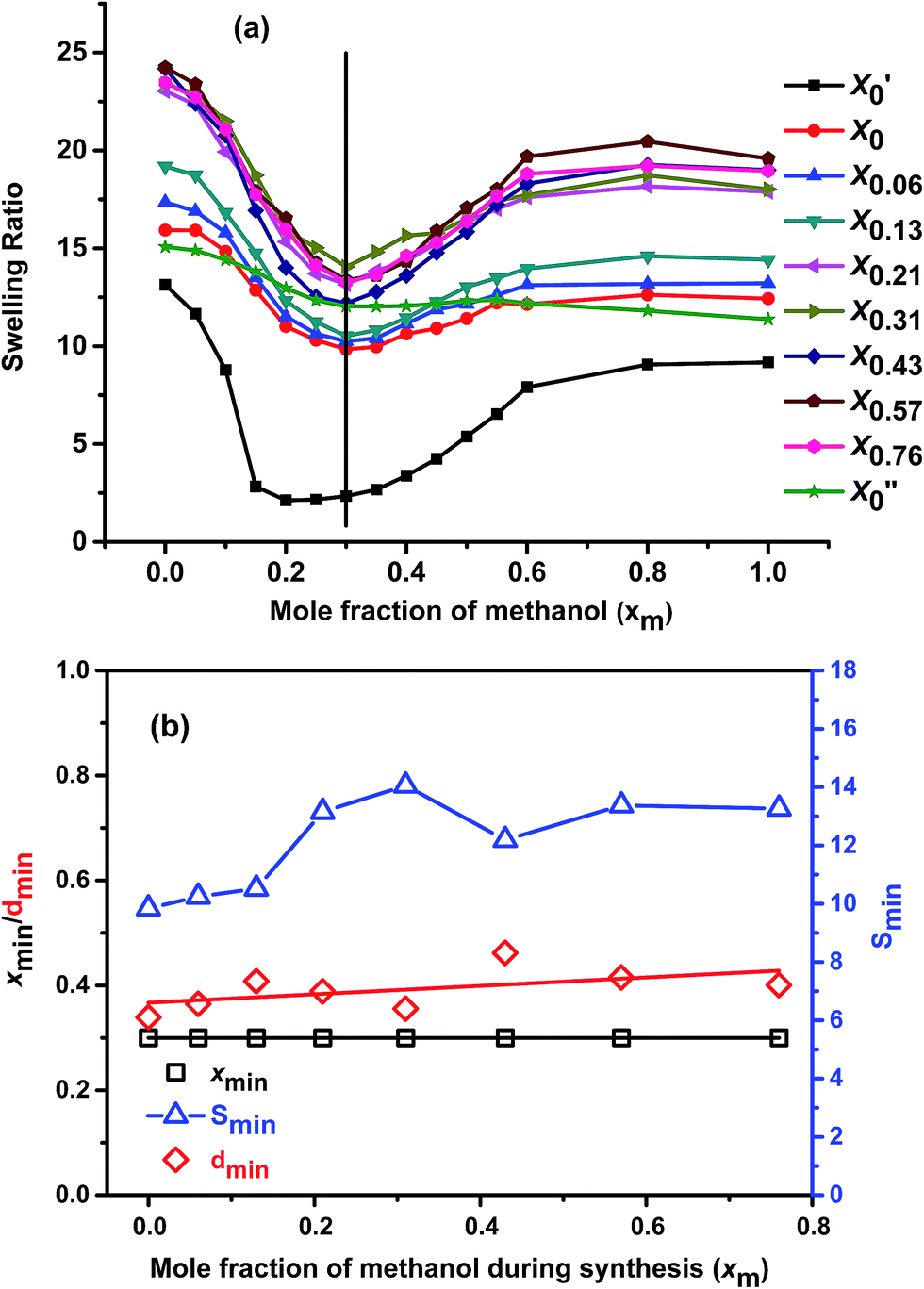 | ||
| Fig. 4 Comparison of (a) cononsolvency of the gels in methanol–water mixture at 20 °C and (b) xmin, dmin and Smin with xm value. | ||
The rate of decrease of swelling ratio is significant after xm = 0.06. This is in agreement with previous reports,8,16 where it showed that at very low methanol concentration (xm ≤ 0.05) methanol is not effective enough to induce cononsolvency. All gels show minimum swelling ratio value at xm = 0.30 (except X′0 which shows its minimum SR at xm = 0.20; and X′′0, which shows negligible cononsolvency at this temperature) and all of them show similar trend in the cononsolvency region. It indicates that the change in the synthesis solvent composition while keeping the molar ratio of monomer fixed does not affect the cononsolvency trend much. Only the depth of the minimum varies with the change in the synthesis solvent. It also proves that, the molar ratio of monomers in the synthesized gels are equal as the xm value of minimum swelling ratio observed is also same. Similar kinds of results also observed before with PNIPAM system in methanol–water mixtures.9
This is easily understood by the fact that the cononsolvency interactions between polymer and solvent do not depend on the exact morphology significantly. However, the morphology determines the overall swelling degree together with the mechanical properties.
For the gel prepared in pure water in presence of NIPAM (X′0) the trend of cononsolvency is quite different from other gels prepared with different 1:1 molar ratio of monomers. It indicates that, the change in the synthesis solvent composition does not affect the cononsolvency much but the change in monomer ratio does.
Fig. 4(b) shows these trends in more detail. The mole fraction at the minimum position (xmin) (left y-axis) remains constant, indicating the abovementioned constant value of hydrophilicity. However, the values of the minimum Smin themselves are somewhat more difficult to grasp, as the swelling of all samples follow a non-trivial pattern. For this reason, it was assumed that the reference for the minimum is chosen by fitting a linear relation through the values for pure water and pure methanol. The expected value Sexp(xmin) for the swelling is calculated from this linear interpolation at the solvent composition of the minimum (xmin). The relative depth of the minimum value dmin is calculated from eqn (4).
| dmin = 1 − Smin/Sexp(xmin) | (4) |
Fig. 4(b) shows that Smin depends slightly on synthesis solvent compositions. Firstly, it shows a slight increase with increase in xm, which can be attributed to the overall lowering of conversion with increasing xm as well as the lower volumetric concentration of monomers with increasing xm due to the lower density of methanol in comparison to water. The former thins out the network due to incomplete reaction of the monomers, the latter further dilutes the network density by lowering the monomer concentration per volume in the synthesis mixture. As a second trend, the samples synthesized in the cononsolvency region, having the strongest porosity show a significantly higher Smin than the rest of the samples, which can be explained by the fact that macroporous network cannot eject the solvent as well as a continuous network when under cononsolvency conditions.
If we analyze the dmin with respect to xm value, we see that, with increasing xm value, the relative depth of the minimum remains almost constant throughout the synthesis solvent compositions with little fluctuation [Fig. 4(b)]. The fluctuations may arise from experimental errors. This means, polymer solvent interactions of the gels synthesized with different xm values are similar at particular solvent compositions, hence the relative depth of the minimum and thus the intensity of the cononsolvency is almost unaffected by solvent composition.
To understand the effect of the temperature on the cononsolvency, we measure the swelling ratio of the gel prepared using 50:50 molar ratios of PNIPAM to PNEAM in 1:1 methanol–water mixture (v/v) (X0.31) were determined at 5, 10, 20, 30, 40, and 50 °C (Fig. 5(a)). It is observed that, the depth of the minimum gradually increases with the increase in temperature and the onset of cononsolvency shifts towards lower xm-value with the increase in the temperature from 5 °C to 50 °C. At 50 °C, the minimum at cononsolvency observed at xm = 0.10, which is very close to the swelling ratio value in water, indicates that, with the increase in the temperature, there is dramatic change in the nature of cononsolvency. This is mainly due to the change in the preferential adsorption where it is observed that early onset of cononsolvency happened with the increase in temperature as minimum gradually shifted towards lesser xm value for PNIPAM system.18 At higher temperatures, the probability of preferential adsorption is also high due to higher competitive hydrogen bonding among solvent and polymer molecules and it is methanol which most preferentially adsorbed as observed before.18 With the increase in the temperature, swelling ratio of the gel in water decreases gradually and after specific temperature (68 °C) it undergoes phase separation. As a result, the onset of cononsolvency is shifted to lower methanol contents as the temperature goes up. This can be comprehended as the consequence of the cononsolvency in combination with the LCST, which shows its onset already significantly below the determined LCST-temperature of ca. 68 °C. The gradually increasing hydrophobicity of the gels affect the solubility at high water content (low methanol content) more than at lower water content. Hence, it is logical that xmin shifts as a function of temperature.
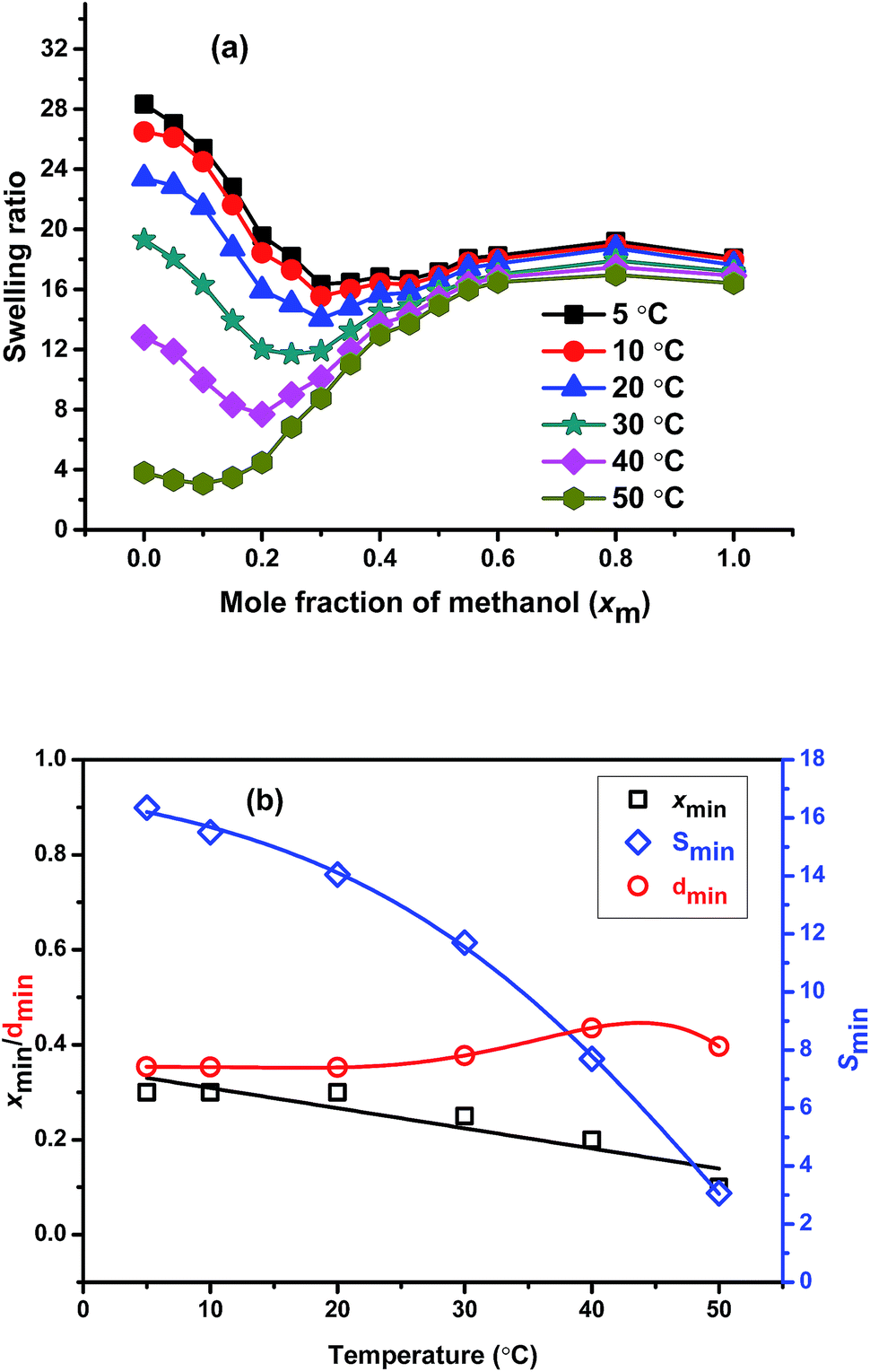 | ||
| Fig. 5 Comparison of (a) swelling ratio of X0.31 as a function of mole fraction of methanol at different temperatures and (b) xmin, dmin, and Smin at different temperatures. | ||
To understand the effect of temperature on cononsolvency clearly, we plotted the xmin, dmin, and Smin value of this sample against temperatures and is shown in the Fig. 5(b). The figure clearly shows that with increase in the temperature, xmin linearly decreases with the increase in temperature, as discussed above. The dmin remains almost constant up to 20 °C and then started to increase with the temperature and shows high value at 40 and 50 °C. It means at higher temperature the preferential adsorption is maximum due to increase in hydrophobicity of the polymer network, consequently, it shows higher dmin value. Smin gradually decreases with the increase in temperature as expected due to the increase in hydrophobicity owing to the predominance of preferential adsorption. At higher temperature the rate of decrement of Smin is more prominent than at lower temperatures. In other words, temperature has a very significant effect on cononsolvency, which can be explained by the influence of the LCST.
Temperature dependence of swelling ratio in water
Swelling ratio measurements of all the gels at different temperatures are shown in Fig. 6, showing that the swelling ratio of all the gels gradually decreases with increasing temperature. This is mainly due to the increasing dominance of hydrophobic interactions among isopropyl groups of PNIPAM, ethyl groups of PNEAM and polymer backbones over hydrophilic interactions between water and amide group. The trend of swelling ratio of all the gels at 20 °C was already explained in the Synthesis section. All the gels show almost in totally collapsed state at around 60 °C (except X′0, and X′′0, which have their LCST at 33 °C and 81 °C respectively), while the LCST determined from rheology study is slightly higher (about 68 °C) than this. It means the gels were still in slightly swollen state after 60 °C and complete collapse happened at around 68 °C. It can be concluded that, LCST does not depend on the synthesis solvent compositions when the monomer compositions are constant, which is due to the fact that the chemical composition and thus the chemical potential does not change with solvent during synthesis. Similar types of results we observed before for the PNIPAM systems.40 But if the monomer composition varied, they would show significant changes as observed here.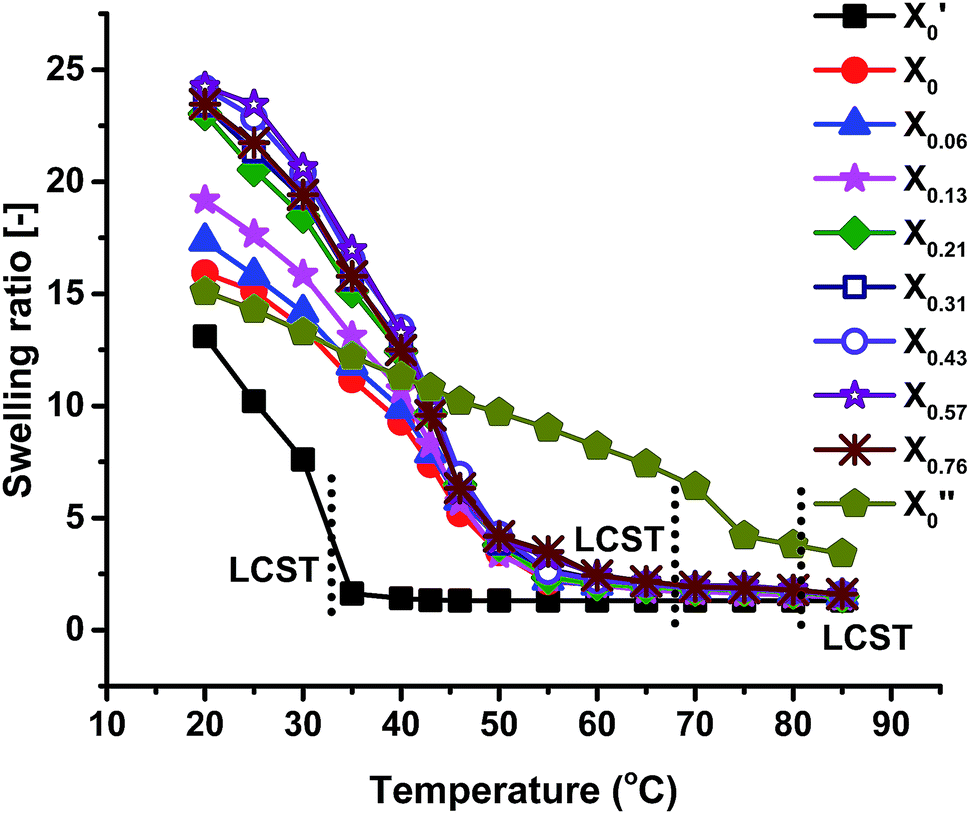 | ||
| Fig. 6 Swelling ratio as a function of temperature. | ||
Temperature dependent deswelling kinetics at 85 °C
Fig. 7 shows the deswelling kinetic study of all the gels at 85 °C. Deswelling rate of the pure NIPAM gel prepared in pure water (X′0) is slow; after 60 min only about 50% of total water released after deswelling. The gel prepared using 50:50 molar ratios of NIPAM and NEAM in water (X0) show higher deswelling rate compared to X′0. This is mainly due to the increase of porosity of the gel compared to X0. The gel synthesized with only PNEAM in water (X′′0) shows lesser deswelling rate compared to X0 (morphology not shown here) as it is less porous and more hydrophilic compared to the later one. Deswelling rate of X0.06 is almost identical to X0. This mainly due to almost similar porosity [Fig. 2(b) and (c)] with similar chemical compositions of the gels. Deswelling rate keeps increasing further with the increase in the xm value from X0.06 to X0.13, due to increase in the porosity [Fig. 2(c) and (d)] owing to the effect of cononsolvency during synthesis. With further increase in the xm value from 0.13 to 0.21, the porosity increases further due to the very significant effect of cononsolvency on the gel [Fig. 2(c) and (d)] and show fastest rate of deswelling among all the gels. The gel prepared at xm = 0.31(X0.31), also shows very rapid deswelling due to high porosity [Fig. 2(e)] owing to the highly cononsolvent medium, but is significantly slower than X0.21 in terms of water release. Deswelling rates keep decreasing thereafter significantly with increasing xm from 0.31 to 0.76 with X0.57 and X0.76 having the slowest deswelling rates observed – even slower than X′0.
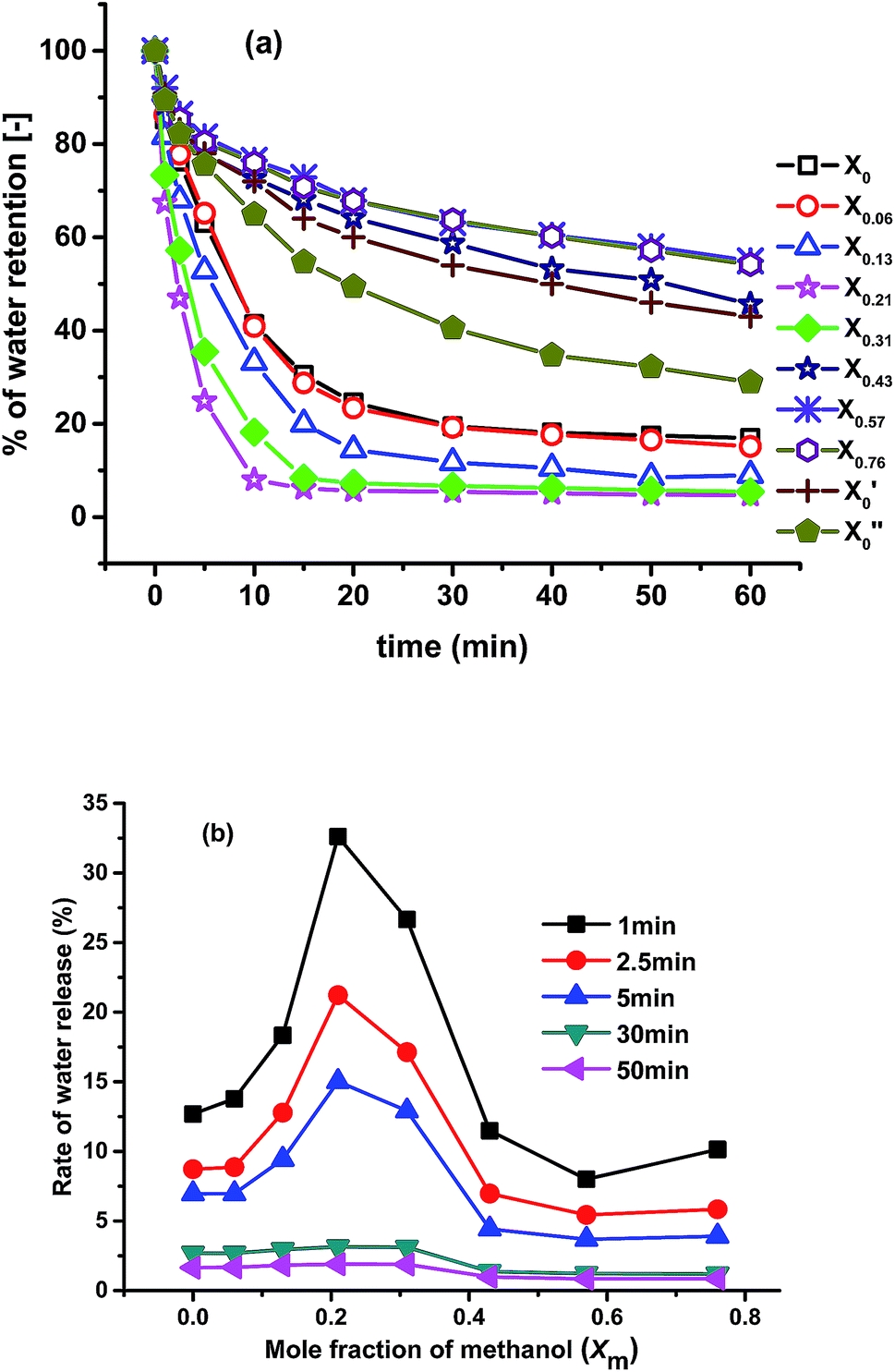 | ||
| Fig. 7 (a) Deswelling rates of the gels in water at 85 °C; (b) comparison of rate of water release of the gels with time. | ||
There are two main reasons behind this observation: firstly, the gels were almost nonporous [Fig. 2(g) and (h)] and secondly the equilibrium swelling ratio of the gels are very high (Fig. 3). As a result, these gel pose very little resistance to swelling, which in turn also means that they cannot eject water effectively. So, by simply tuning the synthesis solvent compositions, we can vary the deswelling rates of the gels at our will.
In Fig. 7(b), the rate of water release of the gels with time is plotted. The rates of release are calculated by using eqn (3). While the absolute values of the water release rate depend on the time, their dependencies on xm are similar for short times (<5 min). For longer times, one can see 2 regimes, in which the water release rates are fairly constant. For xm <0.31 higher release rates are found than for xm >0.31, which again can be explained with the lower porosity of the gels synthesized in methanol dominated solvents as well as with their higher initial swelling, corresponding to a lower release tendency of the water.
Measurements of mechanical properties by AFM
In Fig. 8(a), force (nN) vs. indentation (nm) are given for 3 selected examples. The quantitative analysis using histograms of Young's modulus values in log normal scale with Gaussian distribution fit of three gels synthesized are shown using X0, X0.31, and X0.76 [Fig. 8(b)]. It is evident from the figure that the change in the solvent compositions during the synthesis of hydrogel leads to clear modification in the mechanical properties. A spherical probe, negligible adhesion between probe and sample, sample homogeneity and linearity, allowed using the Hertz-model60 to fit the force–indentation curves [Fig. 8(a)] with good approximation. Gels with low porosity show a more compact and homogeneous structure; hence, more force is required to indent those sample surface [Fig. 8(a)], leading to an increase of Young's modulus distribution values [Fig. 8(b)].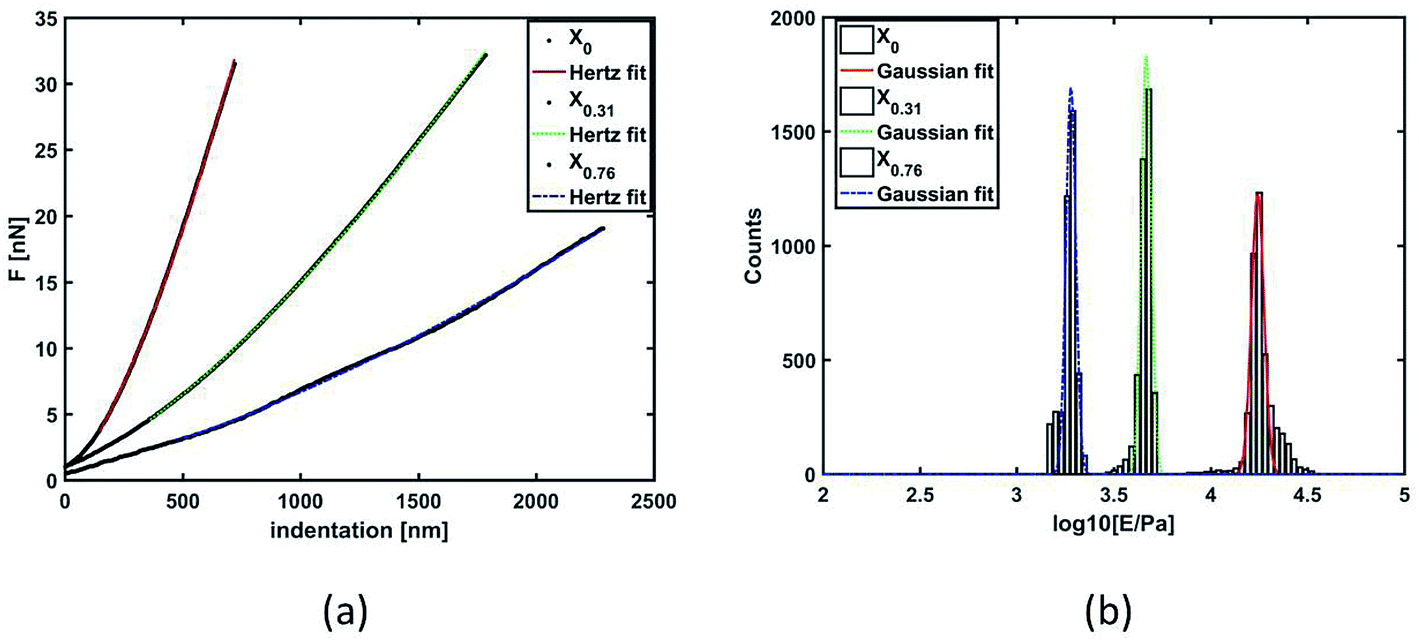 | ||
| Fig. 8 (a) Force (nN) vs. indentation (nm) graph and (b) quantitative analysis using histograms of Young's modulus values in log normal scale with Gaussian distribution fit of the gels X0, X0.31 and X0.76. | ||
A comparison of mechanical properties in terms of Young's modulus with the solvent composition of the gels is shown in Fig. 9. The elastic modulus of the gels prepared in different compositions of methanol–water mixture decreases with the increase in the xm in the gel up to 0.21. This is the consequence of the porosity of the samples due to the cononsolvency of the synthesis solvent as discussed above, being more pronounced for the samples synthesized in cononsolvency region. Although the spherical probe has a great performance for mechanical measurements, the morphology is highly convoluted, due to the micrometric size of the probe, leading to an underestimation of surface roughness. The data for the roughness measurement are not considered. As seen in the SEM-images, the higher porosity was observed for the gels prepared at xm = 0.13, 0.21 and 0.31 respectively, [Fig. 2(d)–(f)]. As a result, the modulus of this gel is also decreasing very rapidly from xm = 0 to 0.21. After that, modulus remains almost constant with further increase in xm value from 0.21 to 0.31. It is understandable that that X0.21 and X0. 31 both are highly porous, so their mechanical properties should be similar. With further increase of xm value from 0.31 to 0.43, the Young's modulus increases slightly as the porosity decreases as expected. With further increase of xm, from 0.43 to 0.76, the Young's modulus decreases significantly.
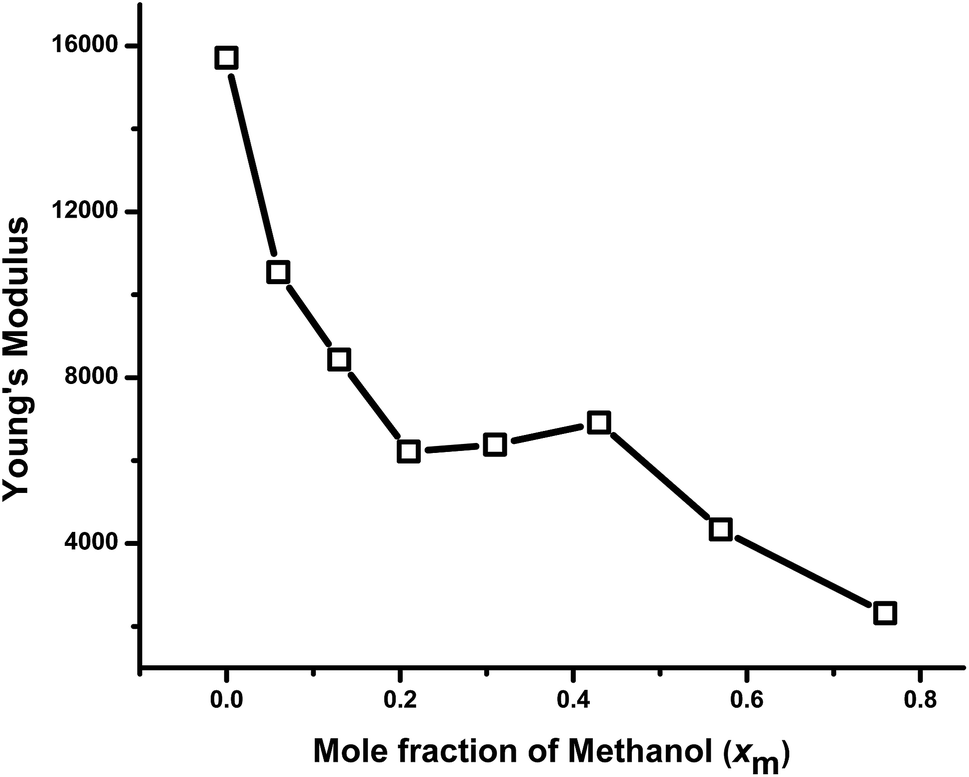 | ||
| Fig. 9 Comparison of Young's modulus as functions of methanol mole fraction. | ||
Rheological properties
In Fig. 10(a), LCST comparison of all the gels are plotted against the synthesis xm value, which shows that LCST is virtually independent of the solvent compositions. This is logical, as the LCST depends on local interactions (in the range of several nm) between solvent molecules and polymer chains.64 Hence, the identical chemical composition determines the LCST, while the different morphology has a negligible influence. Similar kinds of result we observed before with PNIPAM.40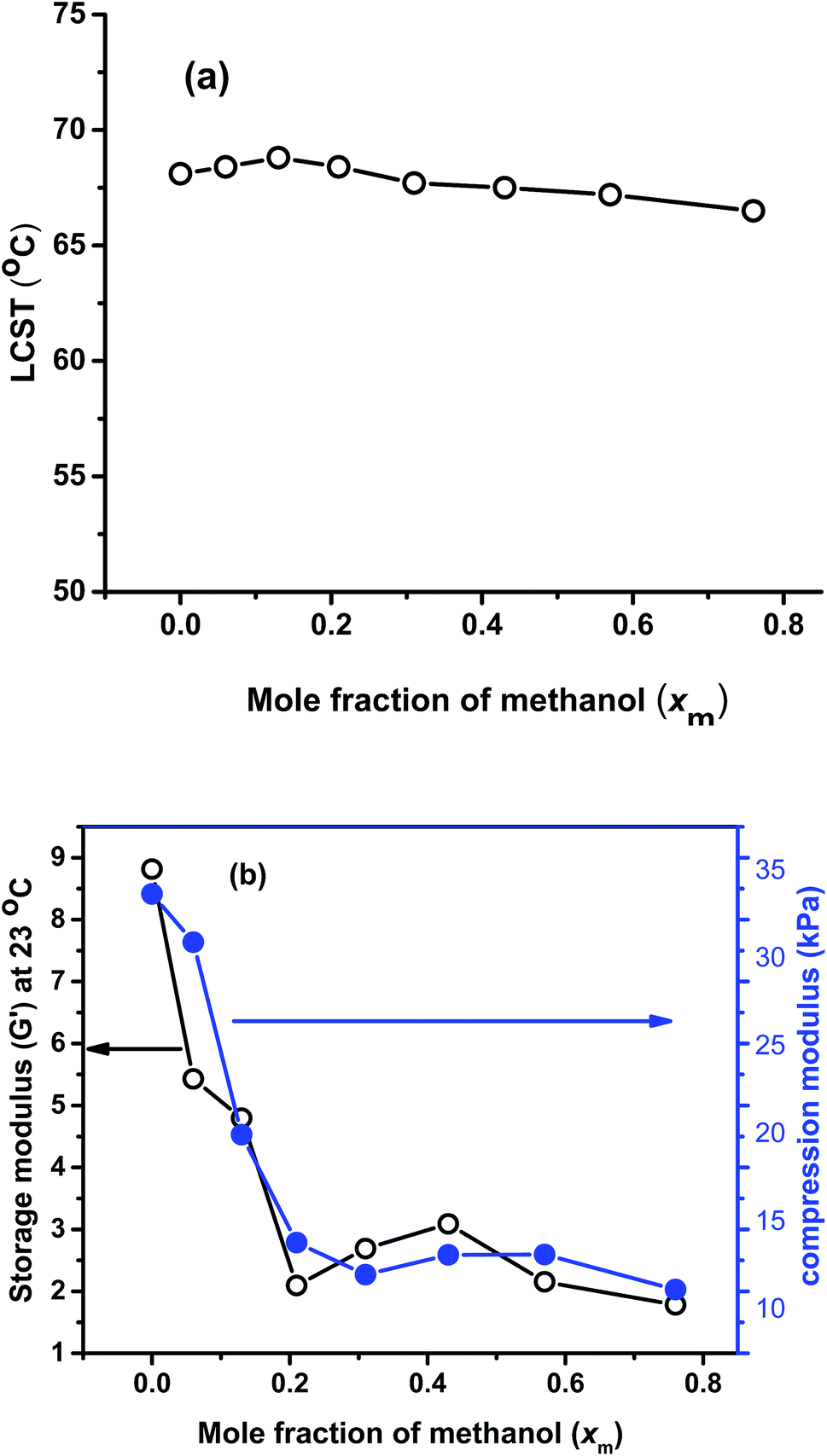 | ||
| Fig. 10 (a) Comparison of LCST with xm value; (b) comparison of G′ with xm value at 23 °C. | ||
In Fig. 10(b), the comparison of storage modulus (G′) at 23 °C and compression modulus are plotted against xm. G′ follows almost similar trend like the G′ obtained from AFM. i.e. it decreasing significantly with the increase in xm value from 0 to 0.21, then slightly increased up to 0.43 and after that it decreased again. Compression modulus also followed almost similar trend like storage modulus, i.e. with the increase in xm value it decreased significantly and at high xm value, it shows much lesser value. This again confirmed that the mechanical properties are varied significantly with the synthesis solvent compositions mainly due to the formation of gels of different porosities and due to the formation of softer gels in presence of methanol owing to its lower density and larger molecular volume.
The compression data show that the compression at break increases slightly from ca. 70% to 75% with increasing methanol content in synthesis solvent xm (Fig. SI3†). This is due to the lower network density. However, as the breaking processes are highly random and the differences between the different samples are not large, the statistical scatter of the results comparable to the size of the effect. Hence, this quantity cannot be regarded as sufficiently reliable. Consequently, also the stress at break cannot be interpreted (Fig. SI4†). This can be seen from the random slope κ of stress with increasing deformation χ [Fig. SI5†].
However, the stress at 65% strain, i.e. before the break of all samples can be evaluated (Fig. SI6†). The resulting trend is almost identical to that of the compression modulus. This is not surprising, as covalent hydrogels in general follow rubber-like behavior almost ideally65 and, hence, their behavior can be described by hyper elastic models, essentially only requiring the elastic modulus to describe the behavior until break.
Conclusions
Random copolymer gels of PNEAM and PNIPAM of 1:1 monomer compositions in different methanol–water mixture are prepared by free radical polymerization. FTIR analysis of the gels show that all the gels have almost identical chemical compositions. Elemental analysis results support these observations. The observed morphologies of the SEM images vary from non-porous to highly porous structure depending on the compositions of the synthesis solvent compositions are supported by BET analysis. Cononsolvency study of the gels at 20 °C shows that, the gels have similar types of cononsolvency behavior. Only relative depth of the minimum varies depending on the synthesis solvent compositions. However, with the variation of temperature, cononsolvency show very interesting change in the behavior. As temperature increases, the minimum in the cononsolvency region becomes deeper and shifts towards lower xm. Swelling ratio value of the gels at 20 °C shows that the gels synthesized at xm, show higher swelling ratio values. Swelling ratio value gradually decreases with increase in the temperature and undergoes phase separation around 60–65 °C. LCST values of the gels are very close to each other irrespective of solvent compositions. Deswelling rates of the gels are high with the gels synthesized in cononsolvent medium and low with the gels synthesized with higher xm value, both being related primarily to porosity. Mechanical properties of the gels vary significantly with synthesis solvent compositions. AFM measurements show that, Young's modulus is very high for the gel synthesized in water (X0) then decreases significantly up to X0.21, after that pass through a plateau up to X0.43 and then decreases again. Quantitative analysis of the gels by AFM using histograms of Young's modulus shows a Gaussian type distribution.
When looking at Fig. 9 and 10(b), it becomes obvious that the data look like an inverted version of Fig. 3. This similarity aids the interpretation of both measurements by utilizing the SEM-images and the understanding of the synthesis conditions.
The first important contribution is the poor solubility of the initiator (APS) in methanol, which can be clearly seen from the poor conversion for the gels synthesized in methanol rich solvent. That alone leads to a significantly lower modulus for obvious reasons. Furthermore, one has to consider that a lower concentration of slower growing polymer chains (the duration of the crosslinking reaction was significantly higher for methanol rich gels) means that the likeliness of forming crosslinks is lower again, thus, leading to a less crosslinked system than what was achieved in water rich solutions. The conversion drop is significantly lower than 100% for X0.31, X0.57, and X0.76 but not for X0.43. The low conversion of X0.31 is due to the high porosity of the gels (leading to a loss of transparency); this gel consists of isolated gel particles in the matrix of loosely connected particles that can be washed out of the gel, ultimately leading to an apparently low conversion, which, however is an artefact of purification. The situation is different for X0.57 and X0.76, as here the gels are macroscopically transparent and virtually non-porous, which leads to the conclusion that washing out gel particles is not a significant contributor to the lower conversion but that simply not enough APS was present in solution to fully polymerize the gel. That in turn leads to a significantly lower concentration of polymer in the original gel.
Such a lower polymer concentration has the consequence that the average distance between 2 crosslinking points is lower and thus the resistance to swelling is lower. At the same time, the driving force for the ejection of water from the gel, when e.g. crossing the LCST, is significantly weaker, which leads to the finding that X0.57 and X0.76 eject water significantly slower than the other samples including X′0, which is also non-porous due to the absence of any visible pores but has a lower LCST and, hence, a higher hydrophobicity.
For this reason, we can conclude that there are two physically different processes increasing the swelling at 20 °C in water, modifying the deswelling kinetics as well as lowering the modulus in AFM, compression as well as in shear rheology.
Furthermore, also the physical properties of methanol could be partially responsible for the trend as follows: the high concentration of methanol used for the synthesis of the gels produces networks with highly expanded form as they have large molecular volume and low density. Their high swelling ratio in comparison with low concentration of methanol (Fig. 3) confirms mechanical results, highlighting a network loosely connected.
Acknowledgements
The authors would like to thank the National Science Foundation of China (21574086), Nanshan District Key Lab for Biopolymers and Safety Evaluation (No. KC2014ZDZJ0001A), Shenzhen Sci & Tech research grant (ZDSYS201507141105130), and Shenzhen City Science and Technology Plan Project (JCYJ20140509172719311) for financial support.References
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl.Chem., 1968, 2, 1441–1455 CrossRef CAS.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossmann, W. S. Pan, M. Garciagaribay and N. J. Turro, Macromolecules, 1993, 26, 4577–4585 CrossRef CAS.
- E. I. Tiktopulo, V. N. Uversky, V. B. Lushchik, S. I. Klenin, V. E. Bychkova and O. B. Ptitsyn, Macromolecules, 1995, 28, 7519–7524 CrossRef CAS.
- H. Yang, X. H. Yan and R. S. Cheng, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 1188–1192 CrossRef.
- K. Kyriakos, M. Philipp, L. Silvi, W. Lohstroh, W. Petry, P. Muller-Buschbaum and C. M. Papadakis, J. Phys. Chem. B, 2016, 120, 4679–4688 CrossRef CAS PubMed.
- F. M. Winnik, M. F. Ottaviani, S. H. Bossmann, M. Garciagaribay and N. J. Turro, Macromolecules, 1992, 25, 6007–6017 CrossRef CAS.
- F. M. Winnik, H. Ringsdorf and J. Venzmer, Macromolecules, 1990, 23, 2415–2416 CrossRef CAS.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, A. K. Mishra, R. Bhimireddi, R. Rai and B. Ray, J. Appl. Polym. Sci., 2012, 125, 2000–2009 CrossRef CAS.
- K. Mukae, M. Sakurai, S. Sawamura, K. Makino, S. W. Kim, I. Ueda and K. Shirahama, J. Phys. Chem., 1993, 97, 737–741 CrossRef CAS.
- P. W. Zhu and D. H. Napper, J. Colloid Interface Sci., 1996, 177, 343–352 CrossRef CAS.
- X. Z. Zhang, Y. Y. Yang and T. S. Chung, Langmuir, 2002, 18, 2538–2542 CrossRef CAS.
- J. K. Hao, H. Cheng, P. Butler, L. Zhang and C. C. Han, J. Chem. Phys., 2010, 132 Search PubMed.
- K. Mukae, M. Sakurai, S. Sawamura, K. Makino, S. W. Kim, I. Ueda and K. Shirahama, Colloid Polym. Sci., 1994, 272, 655–663 CAS.
- N. Osaka and M. Shibayama, Macromolecules, 2012, 45, 2171–2174 CrossRef CAS.
- F. Tanaka, T. Koga and F. M. Winnik, Phys. Rev. Lett., 2008, 101 Search PubMed.
- M. J. A. Hore, B. Hammouda, Y. Y. Li and H. Cheng, Macromolecules, 2013, 46, 7894–7901 CrossRef CAS.
- H. Kojima, F. Tanaka, C. Scherzinger and W. Richtering, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1100–1111 CrossRef CAS.
- C. Scherzinger, A. Schwarz, A. Bardow, K. Leonhard and W. Richtering, Curr. Opin. Colloid Interface Sci., 2014, 19, 84–94 CrossRef CAS.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- C. Yan and T. Mu, Phys. Chem. Chem. Phys., 2015, 17, 3241–3249 RSC.
- C. Yan, Z. Xue, W. Zhao, J. Wang and T. Mu, ChemPhysChem, 2016, 17, 3309–3314 CrossRef CAS PubMed.
- F. O. Obiweluozor, A. GhavamiNejad, S. Hashmi, M. Vatankhah-Varnoosfaderani and F. J. Stadler, Macromol. Chem. Phys., 2014, 215, 1077–1091 CrossRef CAS.
- J. P. Chen and A. S. Hoffman, Biomaterials, 1990, 11, 631–634 CrossRef CAS PubMed.
- L. Wen, X. Tan, Q. Sun and Y. Lv, J. Sep. Sci., 2016, 39, 3267–3273 CrossRef CAS PubMed.
- J. Gregory, J. Cannell, M. Kofron, L. Yeghiazarian and V. Nistor, J. Appl. Polym. Sci., 2015, 132 Search PubMed.
- T. Okano, Y. H. Bae, H. Jacobs and S. W. Kim, J. Controlled Release, 1990, 11, 255–265 CrossRef CAS.
- R.-M. Wang, Q. Liu, Y. Zhang, Z. Hong and H.-F. Wang, RSC Adv., 2016, 6, 50985–50992 RSC.
- Q. Shi, J. Hou, C. Zhao, Z. Xin, J. Jin, C. Li, S. C. Wong and J. Yin, Nanoscale, 2016, 8, 2022–2029 RSC.
- L. Shang, F. Fu, Y. Cheng, Y. Yu, J. Wang, Z. Gu and Y. Zhao, Small, 2016 DOI:10.1002/smll.201600286.
- K. Wu, L. Q. Shi, W. Q. Zhang, Y. L. An and X. X. Zhu, J. Appl. Polym. Sci., 2006, 102, 3144–3148 CrossRef CAS.
- Q. M. Zhang, W. Wang, Y.-Q. Su, E. J. M. Hensen and M. J. Serpe, Chem. Mater., 2016, 28, 259–265 CrossRef CAS.
- C. Wu and S. Q. Zhou, J. Macromol. Sci., Part B: Phys., 1997, 36, 345–355 CrossRef.
- C. Wu and S. Q. Zhou, Macromolecules, 1997, 30, 574–576 CrossRef CAS.
- H. Shirota, N. Kuwabara, K. Ohkawa and K. Horie, J. Phys. Chem. B, 1999, 103, 10400–10408 CrossRef CAS.
- K. Kratz, T. Hellweg and W. Eimer, Polymer, 2001, 42, 6631–6639 CrossRef CAS.
- I. Lynch and K. A. Dawson, Macromol. Chem. Phys., 2003, 204, 443–450 CrossRef CAS.
- Q. F. Liu, P. Zhang, A. X. Qing, Y. X. Lan and M. G. Lu, Polymer, 2006, 47, 2330–2336 CrossRef CAS.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, A. K. Mishra, S. Saha and B. Ray, Langmuir, 2010, 26, 6775–6782 CrossRef CAS PubMed.
- W. F. Lee and S. H. Yen, J. Appl. Polym. Sci., 2000, 78, 1604–1611 CrossRef CAS.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, A. K. Mishra and B. Ray, J. Appl. Polym. Sci., 2011, 121, 2422–2429 CrossRef CAS.
- H. Tokuyama, N. Ishihara and S. Sakohara, Eur. Polym. J., 2007, 43, 4975–4982 CrossRef CAS.
- C. S. Biswas, N. K. Vishwakarma, V. K. Patel, A. K. Mishra, S. Saha and B. Ray, Langmuir, 2012, 28, 7014–7022 CrossRef CAS PubMed.
- T. R. Matzelle, G. Geuskens and N. Kruse, Macromolecules, 2003, 36, 2926–2931 CrossRef CAS.
- D. Goodman, J. N. Kizhakkedathu and D. E. Brooks, Langmuir, 2004, 20, 2333–2340 CrossRef CAS PubMed.
- X. F. Sui, Q. Chen, M. A. Hempenius and G. J. Vancso, Small, 2011, 7, 1440–1447 CrossRef CAS PubMed.
- B. H. Tan, R. H. Pelton and K. C. Tam, Polymer, 2010, 51, 3238–3243 CrossRef CAS.
- S. Hashmi, A. GhavamiNejad, F. O. Obiweluozor, M. Vatankhah-Varnoosfaderani and F. J. Stadler, Macromolecules, 2012, 45, 9804–9815 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Biomacromolecules, 2007, 8, 258–264 CrossRef CAS PubMed.
- G. L. Puleo, F. Zulli, M. Piovanelli, M. Giordano, B. Mazzolai, L. Beccai and L. Andreozzi, React. Funct. Polym., 2013, 73, 1306–1318 CrossRef CAS.
- M. Vatankhah-Varnoosfaderani, S. Hashmi, A. GhavamiNejad and F. J. Stadler, Poly. Chem., 2014, 5, 512–523 RSC.
- W. Xue, M. B. Huglin and T. G. J. Jones, Macromol. Chem. Phys., 2003, 204, 1956–1965 CrossRef CAS.
- J. S. Lowe, B. Z. Chowdhry, J. R. Parsonage and M. J. Snowden, Polymer, 1998, 39, 1207–1212 CrossRef CAS.
- W. Xue, M. B. Huglin and T. G. J. Jones, Eur. Polym. J., 2005, 41, 239–248 CrossRef CAS.
- W. S. Cai and R. B. Gupta, Ind. Eng. Chem. Res., 2001, 40, 3406–3412 CrossRef CAS.
- T. Hirano, A. Ono, H. Yamamoto, T. Mori, Y. Maeda, M. Oshimura and K. Ute, Polymer, 2013, 54, 5601–5608 CrossRef CAS.
- M. T. Savoji, S. Strandman and X. X. Zhu, Macromolecules, 2012, 45, 2001–2006 CrossRef CAS.
- M. T. Savoji, S. Strandman and X. X. Zhu, Soft Matter, 2014, 10, 5886–5893 RSC.
- M. Nichifor and X. X. Zhu, Polymer, 2003, 44, 3053–3060 CrossRef CAS.
- C. S. Biswas and B. Hazer, Colloid Polym. Sci., 2015, 293, 143–152 CAS.
- C. S. Biswas, E. Sulu and B. Hazer, J. Appl. Polym. Sci., 2015, 132 Search PubMed.
- H. J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- W.-F. Lee and S.-H. Yen, J. Appl. Polym. Sci., 2000, 78, 1604–1611 CrossRef CAS.
- B. Ray, Y. Okamoto, M. Kamigaito, M. Sawamoto, K.-i. Seno, S. Kanaoka and S. Aoshima, Polym. J., 2005, 37, 234–237 CrossRef CAS.
- F. J. Stadler, T. Friedrich, K. Kraus, B. Tieke and C. Bailly, Rheol. Acta, 2013, 52, 413–423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27348c |
| This journal is © The Royal Society of Chemistry 2017 |