
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid membrane biomaterials from self-assembly in polysaccharide and peptide amphiphile mixtures: controllable structural and mechanical properties and antimicrobial activity†
V. Castelletto a,
A. Kaura,
I. W. Hamley*a,
R. H. Barnesa,
K.-A. Karatzasa,
D. Hermida-Merinob,
S. Swiokloc,
C. J. Connonc,
J. Stasiakd,
M. Rezae and
J. Ruokolainene
a,
A. Kaura,
I. W. Hamley*a,
R. H. Barnesa,
K.-A. Karatzasa,
D. Hermida-Merinob,
S. Swiokloc,
C. J. Connonc,
J. Stasiakd,
M. Rezae and
J. Ruokolainene
aSchool of Chemistry, Pharmacy and Food Biosciences, University of Reading, Whiteknights, Reading RG6 6AD, UK. E-mail: I.W.Hamley@reading.ac.uk
bEuropean Synchrotron Radiation Facility, ESRF, 71 avenue des Martyrs, 38000 Grenoble, France
cInstitute of Genetic Medicine, Newcastle University, International Centre for Life, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK
dDepartment of Chemical Engineering and Biotechnology, Pembroke Street, Cambridge CB2 3RA, UK
eDepartment of Applied Physics, Aalto University School of Science, P.O. Box 15100, FI-00076 Aalto, Finland
First published on 26th January 2017
Abstract
Macroscopic capsules, with tunable properties based on hierarchical self-assembly on multiple lengthscales, are prepared from the co-operative self-assembly of polysaccharide and peptide amphiphiles. Different formulations can be used to create flexible membrane sacs in solution, soft capsules or rigid free-standing capsules. Samples are prepared by injecting a solution containing sodium alginate, with or without graphene oxide (GO), into a matrix consisting of a solution containing the peptide amphiphile PA C16-KKFF (K: lysine, F: phenylalanine), with or without CaCl2. Graphene oxide is added to the hybrid materials to modulate the mechanical properties of the capsules. Injection of sodium alginate solution into a pure PA matrix provides a flexible membrane sac in solution, while injection of NaAlg/GO solution into a PA matrix gives a soft capsule. Alternatively, a rigid free-standing capsule is made by injecting a NaAlg/GO solution into a PA + CaCl2 matrix solution. A comprehensive insight into the hierarchical order within the capsules is provided through analysis of X-ray scattering data. A novel “Langmuir–Blodgett” mechanism is proposed to account for the formation of the sacs and capsules as the alginate solution is injected at the interface of the PA solution. The capsules show a unique antibacterial effect specific for the Gram positive bacterium Listeria monocytogenes, which is an important human pathogen. The hybrid nanostructured capsules thus have remarkable bioactivity and due to their tunable structural and functional properties are likely to have a diversity of other future applications.
1. Introduction
Peptide amphiphiles (PAs) are a class of remarkable self-assembling molecules that incorporate the diverse functionality of amino acid residues. An important class of PA is that of lipopeptides in which a hydrophilic peptide sequence is attached to one or more lipid chains. Such molecules are produced in living systems1 (for example as part of the bacterial host defense system) and are designed and synthesized by researchers interested in their self-assembly properties and biological activity.1–11 In many examples, it has been demonstrated that PAs have enhanced stability and bioactivity compared to the constituent peptide7,12–16 because self-assembly into fibrillar structures presents the functional peptide motif at high density, and because the lipid confers compatibility with biological membranes.The possibility to prepare novel hybrid materials by combining these properties of PAs and peptides with those of biopolymers leads to the potential to produce novel hybrid biomaterials with remarkable and unique structural and functional properties. Hybrid systems examined in early pioneering studies include PAs with polysaccharides,17–20 PAs with recombinant structural proteins,21 and amyloid peptides22 or polysaccharides such as sodium alginate (NaAlg)23–26 with graphene oxide (GO). Introduction of GO is of interest in the development of hybrid materials with enhanced electrical conductivity, mechanical and/or barrier properties23–26 among others. In a pioneering study, membrane sac formation was observed at the interface of aqueous solutions of high molecular weight polysaccharide (hyaluronic acid) and a cationic designer PA.17 It was proposed that the membrane structure results from the formation of a PA fibril network diffusion barrier, leading to polysaccharide nanofibril bundle growth perpendicular to the interface.17 Stem cell growth and slow release of encapsulated proteins is supported by sacs enclosing PA gel. In subsequent work, the mechanical properties and membrane permeability were examined in more detail.18 Membranes formed by PAs bearing the KLAKLAK peptide (which has anti-cancer activity) mixed with hyaluronic acid were also fabricated and their cytotoxicity towards breast cancer cells was examined.19 A change in PA aggregate structure from cylindrical fibrils to spheres was also noted for this system.19 Membranes formed by complexation of a PA bearing a heparin-binding sequence and several polysaccharides have been prepared and their structure has been probed by SAXS and electron microscopy.20 Remarkably, in some cases, evidence for cubic phase structure within the membrane instead of lamellar of hexagonal ordering was observed.20 Mata and coworkers have recently shown that complexation of cationic PAs with elastin-like proteins produces robust membranes with remarkable mechanical properties such as exemplified by the ability to draw out tubes from membranes, and to extend tubules from the tube walls.21 These materials could be used to create substrates for stem cell growth, showing their great potential in tissue engineering applications.21
We have previously investigated the self-assembly of the PA C16-KKFF (C16: palmitoyl chain, K: lysine, F: phenylalanine) in water and showed that it forms small (5–6 nm diameter) spherical micelles.27 This PA was studied as part of a work on the enzymatic degradation of C16-KKFFVLK (L: leucine, V: valine)27 which itself self-assembles into helically twisted ribbons and nanotubes at sufficiently high concentration.28 The peptide sequence in C16-KKFFVLK is based on a core aggregating sequence from the amyloid β (Aβ) peptide,29 KLVFF, the self-assembly of which we previously examined.30 In C16-KKFFVLK, the KLVFF sequence is attached right-to-left from the C terminus with an additional KKF sequence. As a result, KK and FF sequences are expected to improve the water solubility and promote β-sheet formation through aromatic stacking (π–π) interactions31 respectively, for both C16-KKFFVLK and C16-KKFF PAs.
Here we investigate the formation of macroscopic objects including membrane sacs and soft or rigid capsules. These objects are self-assembled by injecting NaAlg sol with or without GO into a matrix consisting of a C16-KKFF solution with or without CaCl2. We examine the structural and mechanical properties of the sac and capsules using several scattering, microscopy and rheology instruments. In addition, we perform biological assays to examine the cytocompatibility and antimicrobial activity of the membrane sac.
We find evidence for unique hierarchical ordering in these novel self-assembled macroscopic objects and a significant influence of GO and CaCl2 on their mechanical properties. In particular, membrane sacs have excellent intrinsic antimicrobial properties against Listeria monocytogenes and other Gram positive bacteria.
2. Experimental
Materials
Peptide amphiphiles C16-KKFF and C16-G3RGD were synthesized by CS Bio (USA) and supplied as a TFA salts. The molecular weight was found to be 807.31 Da (expected 807.11 Da) and 755.47 Da (expected 755.91 Da) for C16-KKFF and C16-G3RGD respectively, as revealed by electrospray ionization mass spectrometry. Purity of the samples was 99.88% and 98.4%, for C16-KKFF and C16-G3RGD respectively, by analytical HPLC in a 0.1% TFA water/acetonitrile gradient.Sodium alginate (NaAlg) and graphene oxide (GO) were purchased from Sigma Aldrich (UK). The molecular weight of NaAlg was determined to be very polydisperse, in the range 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–40000 g mol−1, according to viscosity experiments. GO was bought as a 4 mg ml−1 dispersion in water.
000–40000 g mol−1, according to viscosity experiments. GO was bought as a 4 mg ml−1 dispersion in water.
GO has a negative electrostatic charge within the pH range 2–7.32 C16-KKFF and C16-G3RGD are positively charged for pH 1–3 (+2 and +1 for C16-KKFF and C16-G3RGD respectively),33 while NaAlg is negatively charged at pH 5–7. This was empirically confirmed by the measured ζ-potential and pH values of the solutions listed in Table 1. Solutions containing only PA were mixed using an ultrasound bath for 20 minutes. Pure NaAlg and GO/NaAlg solutions were mixed by alternating ultrasound bath and vortex mixing for 20 minutes. All solutions were allowed to equilibrate for at least three hours prior to use.
| PA solution | Alginate solution | ||||
|---|---|---|---|---|---|
| 0.03 wt% NaAlg (M), pH 6.22, ζ = −77.6 ± 4.7 mV | 0.2 wt% NaAlg (I), pH 6.72, ζ = −65.1 ± 5.0 mV | 2 wt% NaAlg (I), pH 6.16, ζ = −118 ± 6.9 mV | 2 wt% NaAlg/0.02% GO (I), pH 5.48, ζ = −89.7 ± 6.2 mV | 2 wt% NaAlg/0.34% GO (I), pH 4.41 | |
| a Sample conditions. M: mixed, I: injected. | |||||
| 1 wt% C16-KKFF | Sol I | Sol II | |||
| pH 2.45 | |||||
| ζ = 32.7 ± 2.2 mV | |||||
| 2 wt% C16-KKFF | Membrane sac | ||||
| pH 2.51 | |||||
| ζ = 37.3 ± 2.9 mV | |||||
| 5 wt% C16-KKFF | Cap I | ||||
| pH 2.23 | |||||
| ζ = 28.4 ± 1.3 mV | |||||
| 2 wt% C16-KKFF (0.144 wt% CaCl2) | Cap II | ||||
| pH 2.77 | |||||
| ζ = 27.3 ± 1.9 mV | |||||
Zeta potential measurements
The zeta potential was measured using a Zetasizer Nano 2s from Malvern Instruments. An aliquot 1 ml of sample was placed inside a disposable folded capillary cell. The sample was left to equilibrate for 120 s before measuring the zeta potential, using an applied voltage of 50.0 V. The results presented are the average over three measurements. The zeta potential of 2 wt% NaAlg with 0.34% GO (Table 1) was not measured due to the dark colour of the sample.Scanning electron microscopy (SEM)
After sac/capsule production, the excess peptide solution was exchanged by water. The water was subsequently exchanged by a fixative solution containing 3% paraformaldehyde, 1.5% glutaraldehyde and 2.5% sucrose. This was followed by gradual dehydration from 10 to 100% ethanol (10, 30, 50, 70, 90, 100% ethanol), waiting 30 seconds between dehydration steps. The sample was then extracted from the 100% ethanol solution and subjected to critical point drying. An incision was made in the dried sac/capsule, to allow for observation of internal and external surfaces. The dried sample was placed on a stub covered with a carbon tab (Agar Scientific, UK), and then coated with gold. A FEI Quanta FEG 600 environmental scanning electron microscope (SEM) was used to study and record SEM images of the dried sacs.Laser scanning confocal microscopy (LSCM)
Experiments were performed on a Leica TCS SP2 confocal system mounted on a Leica DM-IRE2 upright microscope, using an objective ×20 with 0.4 NA numerical aperture. After 12 h membrane sac production, the peptide solution excess was removed and replaced by water. The water was then replaced by a solution containing 3 × 10−4 wt% rhodamine B (RhoB) fluorescent dye. The sample was left to rest for 90 minutes before LSCM examination. The excitation wavelength generated by an argon laser was 514 nm, while the emission detection was in the range 558–617 nm. Samples were put between a glass slide and a coverslip.Cryogenic transmission electron microscopy (cryo-TEM)
Experiments were carried out using a field emission cryo-electron microscope (JEOL JEM-3200FSC) operating at 300 kV. Images were taken using bright-field mode and zero loss energy filtering (omega type) with a slit with 20 eV. Micrographs were recorded using a Gatan Ultrascan 4000 CCD camera. The specimen temperature was maintained at −187 °C during the imaging. Vitrified specimens were prepared using an automated FEI Vitrobot device using Quantifoil 3.5/1 holey carbon copper grids with 3.5 μm hole sizes. Grids were cleaned using a Gatan Solarus 9500 plasma cleaner just prior to use and then transferred into an environmental chamber of FEI Vitrobot at room temperature and 100% humidity. Thereafter, 3 μl of sample solution was applied on the grid, blotted once for 1 second and then vitrified in a 1/1 mixture of liquid ethane and propane at −180 °C. Grids with vitrified sample solutions were maintained in a liquid nitrogen atmosphere and then cryo-transferred into the microscope.X-ray diffraction (XRD)
XRD was performed on a peptide stalk prepared by drying a sample between the ends of wax-coated capillaries, resulting in a stalk left on the end of one capillary. The sample was mounted (vertically) onto the goniometer of an Oxford Instruments Gemini X-ray diffractometer, equipped with a Sapphire 3 CCD detector. The sample to detector distance was 45 mm. CrystalClear software was used to reduce the 2D-data to a one-dimensional intensity profile.Small angle X-ray scattering (SAXS)
Synchrotron SAXS experiments on solutions were performed on beamline B21 at Diamond (Didcot, UK) and on beamline BM29 at the ESRF (Grenoble, France), using a BioSAXS robot. Synchrotron SAXS experiments on the membrane sac and capsules were performed on beamline BM26B (DUBBLE) at the ESRF.At B21 and BM29, solutions were loaded into the 96 well plate of an EMBL BioSAXS robot, and then injected via an automated sample exchanger into a quartz capillary (1.8 mm internal diameter) in the X-ray beam. The quartz capillary was enclosed in a vacuum chamber, in order to avoid parasitic scattering. After the sample was injected in the capillary and reached the X-ray beam, the flow was stopped during the SAXS data acquisition. B21 operated with a fixed camera length (3.9 m) and fixed energy (12.4 keV) while BM29 operated with λ = 1.03 Å (12 keV). The images were captured using a PILATUS 2M and 1M detector at B21 and BM29 respectively. Data processing was performed using dedicated beamline software Scatter (B21) or ISPYB (BM29).
On BM26B, capsules or membrane sacs were placed in DSC pans modified with mica windows to enable transmission of the X-ray beam. The sample to SAXS detector distance was 3.16 m using a wavelength of 1.033 Å. A Dectris-Pilatus 1M detector with a resolution of 981 × 1043 pixels and a pixel size of 172 × 172 μm was used to acquire the 2D SAXS scattering patterns. Standard corrections for sample absorption and background subtraction were performed. The data were normalized to the intensity of the incident beam (in order to correct for primary beam intensity fluctuations) and were corrected for absorption, background scattering. Diffraction from silver behenate was used to calibrate the wavevector scale of the scattering curve.
Texture analysis
Experiments were performed using a XT-Plus Texture Analyser from Stable Micro Systems. One capsule, placed on the surface of a 5 kg load cell, was compressed against the cell surface using a circular flat tip tool of 4 mm diameter. The tool compressed the sample at a 0.1 mm s−1 speed. Compression started after the tool measured a 0.1 g resistant force from the sample.Rheology
Rheological properties were determined using a controlled stress TA Instruments AR-2000 rheometer (TA Instruments). The experiments were performed using a cone-and-plate geometry (cone radius = 20 cm; cone angle = 1°). The surface to be compressed by the cone was coated with membrane sacs or capsules. The linear regime was first determined performing oscillatory stress experiments at a constant angular frequency of 6.28 rad s−1. Frequency sweep experiments were performed a constant oscillatory stress within the linear regime, and angular frequencies between between 0.1 and 627 rad s−1.Examination of cytocompatibility
Human adipose-derived stem cells (hASCs) obtained from the subcutaneous fat of healthy donors (Invitrogen, UK), were used for biocompatibility experiments. Following recovery from cryostorage, cells were seeded at 800 cells per cm2 and maintained in reduced-serum (RS) growth medium [MesenPRO™ RS medium containing 2 mM GlutaMAX™ and 1% (v/v) antibiotic–antimycotic (all from Life Technologies, UK)] in a humidified incubator at 37 °C and 5% CO2 with medium changes every 3–4 days until approximately 80% confluence was reached. Cells were harvested using TrypLE™ Express enzyme (Life Technologies, UK) and seeded at a density of 20000 cells per cm2 in ultra-low attachment plates (Corning, UK) containing films on glass coverslips coated with dried membrane sacs or soft capsules. hASCs were seeded onto tissue culture plastic, calcium alginate films, and sodium alginate films using the same methodology. Cells were maintained in growth medium for 24 hours before washing with PBS and staining cells with 1 μM calcein-AM (eBioscience, UK) and 2 μM ethidium homodimer-1 (Sigma Aldrich, UK) for 15 minutes at 37 °C. Live and dead cells were then examined using an Axiovert 200M inverted microscope (Zeiss, UK).
Bacterial strains and growth conditions
Experiments were performed in five different microorganisms namely, Staphylococcus aureus, Listeria monocytogenes, Enterococcus faecalis, Pseudomonas aeruginosa and Escherichia coli. The strains used were as follows: a wild type Staphylococcus aureus previously isolated from ham and used in experiments,34 L. monocytogenes LO28 that has also widely been used in various publications35,36 E. coli K-12 is one of the most widely used strains of this bacterium, E. faecalis NCTC 775 has been utilised in previous antimicrobial experiments and P. aeruginosa NCTC 10299 was isolated from human faeces. Stock cultures were stored at −80 °C in 7% (vol/vol) DMSO (Sigma-Aldrich, Dorset, UK). Prior to experiments, stock cultures of S. aureus, L. monocytogenes E. faecalis and P. aeruginosa were streaked onto brain heart infusion (BHI) agar (LAB M, Lancashire, UK) while those of E. coli were streaked onto Lysogeny Broth (LB) agar (Oxoid, UK) and incubated overnight at 37 °C. Three colonies from these cultures were then transferred to 3 ml sterile broth. BHI broth (LAB M, Lancashire, UK) was used for L. monocytogenes, S. aureus, E. faecalis and P. aeruginosa while LB broth (LAB M, Lancashire, UK) was used for E. coli at 37 °C under agitation for 24 hours (overnight). Subsequently, 100 μl of these overnight cultures was used for the microbial peptide challenge.Microbial peptide challenge
Prior to experiments, the initial cell concentration of the overnight cultures was estimated via sampling and preparation of decimal dilutions in maximum recovery diluent (MRD; Oxoid, Hampshire, UK). More specifically, MRD used for S. aureus alone was supplemented with 5% Tween 80 (ICI Surfactants, Wilmington, DE) to alleviate the clumping of cells, as shown previously.34 Subsequently, 10 μl of each dilution were plated onto BHI agar (LAB M, Lancashire, UK) for L. monocytogenes and S. aureus or LB agar (Oxoid, UK) for E. coli. All samples were plated in duplicate and plates were incubated at 37 °C for 24 hours. Subsequently, colonies were counted to assess the bacterial count of each target organism in the initial cultures.Thermo Scientific Nunc MicroWell 96-Well round-bottomed Microplates (Thermo scientific, Denmark) were used for the microbial peptide challenge experiments. Wells were treated by coating with either a 2 wt% NaAlg solution or a membrane sac, and allowed to dry for 24 hours. Following the treatment of the wells, 100 μl of the same overnight culture from each of the microorganisms was placed in equal numbers of wells coated with membrane sacs or NaAlg solution (control). Subsequently, plates were placed in a fume hood for 5 days or outside the fume hood and wrapped in plastic to prevent any evaporation of the medium present in the culture. This was done to assess the antimicrobial activity under possible applications on a dry surface or in a liquid medium. It was noticed that in the fume hood the 100 μl of all culture samples were evaporated completely within 24 h. Following the placement of the cultures in the wells, samples were taken to assess cell viability every 24 h for a period of 5 days (dried wells) or 2 days (liquid in wells). Specifically, samples from the dry wells in plates placed in the fume hood, were taken by the addition of 200 μl of sterile MRD in each well and using the pipette to mix 20 times to resuspend the cells. Subsequently, samples were serially diluted in decimal dilutions and plated onto the corresponding solid media, as described above. It should be mentioned that the first dilution was prepared by transferring the 200 μl from the well into 800 μl of MRD while all other dilutions were prepared by using 100 μl of the previous dilution into 900 μl of fresh MRD. Following incubation of the plates at 37 °C for 24 h, colony counts were taken to assess viability. All measurements of the viable counts at each time points were performed in triplicate.
3. Results
We first investigated the influence of the mixing procedure on the self-assembly in mixtures of the PA C16-KKFF bearing two cationic lysine residues and NaAlg, the Alg bearing a negative charge. A profound and unexpected difference in morphology was observed comparing a sample made by dissolving PA powder into a NaAlg solution (sol I, Table 1) to one prepared by injecting NaAlg solution into PA solution (sol II). Stupp's group used the latter technique to prepare sacs in hybrid polysaccharide/PA mixtures.17 The concentrations of the PA and NaAlg are listed in Table 1. Fig. 1 presents images from cryo-TEM along with SAXS data, highlighting the completely distinct self-assembled nanostructures. The cryo-TEM image for sol I shows spherical micelles coexisting with micellar clusters. The spherical micelle structure is very similar to that for C16-KKFF previously reported,27 as confirmed by SAXS (to be discussed shortly). However, micelle clusters were not observed for a pure C16-KKFF solution27 and the clusters observable in Fig. 1b are therefore ascribed to attractive interactions between C16-KKFF and the NaAlg. In complete contrast to the morphology shown in Fig. 1a and b, the presence of planar membranes is evident in the cryo-TEM images shown in Fig. 1c for sol II. | ||
| Fig. 1 Transition from micelles to nanosheets upon injection of sodium alginate into a PA solution. (a and b) Cryo-TEM images for sol I, showing micelles (examples highlighted with white arrows) and micelle clusters (highlighted with black arrows) in (b), (c) cryo-TEM image for sol II, (d) SAXS from sol II. | ||
The SAXS data for sol II shown in Fig. 1c provides further insight including quantitative information on nanostructure dimensions. In particular, there is a weak Bragg peak in the data, corresponding to a period d = 42.8 Å (Fig. 1c). This weak Bragg peak is ascribed to the formation of a layered assembly associated with the membrane structure revealed by cryo-TEM. Since the SAXS profile for sol II also contains features associated with a spherical micelle form factor, it was fitted37 to a co-existence of core–shell spherical micelle and Gaussian lipid bilayer structures, the fitting parameters being listed in Table S1.† SAXS fitting provided an outer micelle radius R1 = 24.2 Å, in good agreement with the previously reported value27 and the fitting of the SAXS data for 2 wt% C16-KKFF (Fig. S1,†) indicates a total bilayer thickness l = (36 ± 5) Å, similar to the period d, obtained from the Bragg peak position.
The Bragg peak for sol II is ascribed to a bilayer of C16-KKFF with estimated length l of a hexadecyl chain (18 Å) plus the tetrapeptide in a β-sheet (4 × 3.4 Å = 13.6 Å). Since the measured d and l values are more than one molecular length but less than twice this value, it suggests interdigitation of the molecules within the bilayer, as commonly observed for PA assemblies.38–41 SEM data (to be discussed shortly) suggests that the PA bilayer is preferentially sequestered at the surface of a membrane wall with the NaAlg forming the bulk of the wall. SAXS shows that there is a significant population of spherical micelles present in sol II as well as the planar membranes evident in Fig. 1c. The addition of NaAlg into the PA solution leads to a complex structuring process, a new model for which is discussed below in the context of analysis of SEM images.
Having established that injection of NaAlg into the PA solution leads to membrane structures based on bilayers of the PA with NaAlg, we next investigated the production of membrane sacs and soft capsules in PA/NaAlg mixtures and novel rigid capsules by incorporation of GO and/or CaCl2. Fig. 2 shows images of such structures. For example, by increasing the concentration of NaAlg (and PA) used for sol II, it was possible to prepare a membrane sac as illustrated in Fig. 2a (Table 1). The membrane sac is highly flexible and takes up the cationic dye Rho B (an image from laser scanning confocal microscopy is provided in Fig. S2a†), as expected given that it comprises a significant fraction of anionic NaAlg polysaccharide.
 | ||
| Fig. 2 Images of sacs/capsules and SEM images. Photographs of (a) membrane sac in solution in a vial, (b) soft cap I, (c) self-supporting cap II. SEM images of (d) membrane sac, (e) cap I and (f) cap II. | ||
We further show that it is possible to rationally increase the stiffness of the PA/NaAlg membrane sac such that self-supporting capsules are formed, by addition of GO and/or CaCl2. Cap I, obtained by adding GO and increasing the PA concentration in the membrane sac formulation, corresponds to a soft capsule that can be retrieved from water (Table 1 and Fig. 2b). Cap I was further modified by adding CaCl2 and increasing the GO concentration, to produce cap II which is a rigid self-standing structure when retrieved from water (Table 1 and Fig. 2c).
The second row in Fig. 2 shows SEM images. The walls of sacs produced from the membrane sac present three layers with different thickness and textures (Fig. 2d). The outer layers of the membrane sac walls on each side are several tens of nanometres thick, whereas the interior is several tens of microns thick (33.8 ± 7.6 μm thick in the case of the image in Fig. 2d), with a porous hydrogel-like structure sandwiched between the external and internal layers of the membrane. Fig. S2b† includes an enlarged SEM image showing the structure of the interior part. We ascribe the outer layers of membrane sac wall to PA bilayers because the estimated outer layer thickness is very close to the period obtained from the SAXS profile to be discussed shortly. It has to be considered that the sample is prepared by injection of the NaAlg solution into the PA solution. We suggest that this occurs in the form of a “Langmuir–Blodgett deposition” type event as the NaAlg membrane picks up a monolayer of PA (at the air–water interface) as a coating (Fig. 3). This leads to a monolayer of PA at the surfaces of the membrane sac wall. The exposed cationic residues from the peptide may be neutralized preferentially on the surface facing the solution by the Na counterions in solution enabling the (asymmetric) deposition of further PA layers on one sac wall (ultimately the exterior sac wall). The novel proposed “Langmuir–Blodgett” mechanism shown in Fig. 3 for the structure of our membrane sac differs from the diffusional model proposed by Stupp et al. to account for observed perpendicular PA fibril growth at the surface of polysaccharide sacs with their fibril-forming PA system.17 Their PA was designed to incorporate a β-sheet domain in the peptide to drive fibril formation. In contrast, C16-KKFF forms micelles in bulk but a population of layered structures in the presence of NaAlg. Cap II formed in the presence of CaCl2 as a cross-linker of NaAlg also has an ordered morphology in the capsule wall, as confirmed directly by the SAXS profiles below. For cap II, mediation of PA interferes in the crosslinking of the injected NaAlg by the Ca2+ ions in solution.
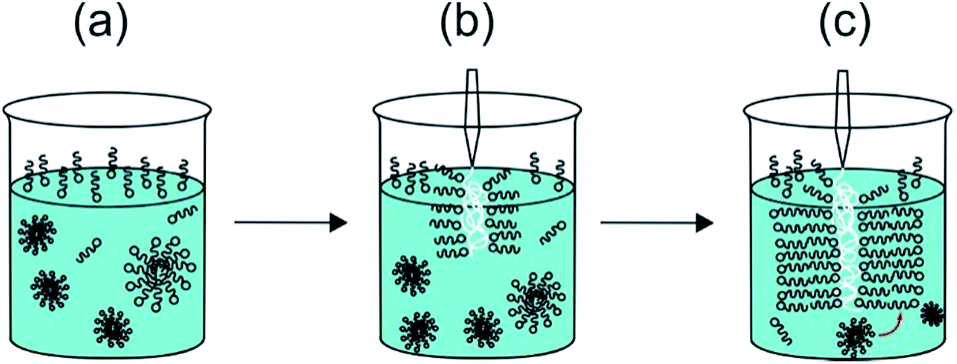 | ||
| Fig. 3 Schematic of “Langmuir–Blodgett” deposition of PA layers (shown as schematic surfactant-like molecules) onto injected NaAlg (polymer shown as white coil). The size of the NaAlg polymer is not shown in scale with that of the PA. The PA bilayer is 42 Å thick, the NaAlg layer in the sacs formed by the membrane is tens of microns thick. (a) The PA solution comprises a monolayer of molecules at the air–water interface with micelles in equilibrium with a few unassociated molecules in solution, (b) the surface monolayer is adsorbed onto the injected NaAlg in a Langmuir–Blodgett type process via electrostatic interaction between PA molecule headgroups and anionic carboxyl groups in the NaAlg, (c) further PA molecules adsorb from micelles and monomers in bulk, forming a multi-bilayer structure coating the NaAlg. | ||
The Langmuir–Blodgett mechanism shown in Fig. 3 applies to the case of injected NaAlg and will not apply in the case of direct dissolution of PA into NaAlg solution as in the case of sol I, where self-assembly occurs via non-interfacial interaction between PA and NaAlg molecules leading to the formation of bulk spherical micelles.
The SEM images for cap I and cap II (Fig. 2e and f) show collapsed capsule-type structures consistent with the photographic images shown in Fig. 2b and c respectively. A cut across the capsule wall shown in Fig. 2e and f exposes the capsule internal area. The capsule diameter is typically several hundred μm. An enlarged SEM image showing the wall of cap I along with an image of the interior structure of cap II are shown in Fig. S3.†
SAXS and WAXS data for the membrane sac, cap I and cap II are shown in Fig. 4. The SAXS data (Fig. 4a) shows a strong Bragg reflection which can be fitted to a layered structure (Table S3†), with a similar spacing to that of the weak, broad Bragg peak for sol II discussed above. The Bragg peak for the membrane sac is much stronger and sharper than for sol II, indicating the presence of ∼tens of repeats of the layer period.
 | ||
| Fig. 4 (a) SAXS and (b) WAXS data from the membrane sac, cap I and cap II as indicated. The full line in (a) represents the fitting to the SAXS data. Parameters extracted from the SAXS fitting in (a) are listed in Table S3.† XRD spacings in (b) are listed in Table S4.† | ||
This same peak is weaker for cap I and cap II, with a weaker slope at low q arising from the bilayer form factor. The SAXS peak position in Fig. 4a differs from that measured for a NaAlg control sample (Fig. S4 and Table S2†).
The SAXS features in Fig. 4a are also different from those measured for PA solutions with and without CaCl2, used to prepare the samples in Fig. 4, which were fitted using a spherical shell form factor (Fig. S1 and Table S2†). The WAXS data for the membrane sac and cap I show a peak at 4.4 Å, tentatively assigned to the β-strand spacing in a β-sheet structure (Table S4†), although the value is somewhat lower than typically observed (4.6–4.8 Å). The membrane sac, cap I and cap II retain features associated with NaAlg or/and GO, as shown by the control WAXS experiments measured for the NaAlg and GO on their own (Fig. S5 and Table S4†).
Together, the SAXS and WAXS data show that the structures of membranes and capsules are both based on PA bilayers. In addition, SAXS shows that the bilayer structure is intrinsic to the membrane sac and capsules, as it is not found in the PA and NaAlg sols used to produce the membrane sac, cap I or cap II. In particular, in two separate experiments, cap I was stained with solutions of the anionic dye Congo red or the cationic dye Rho B. Similarly to the membrane sac shown in Fig. S2a,† cap I absorbed Rho B (Fig. S6†), but did not absorb Congo red, denoting a negatively charged capsule surface dominated by the anionic NaAlg.
The frequency dependence of the storage (G′) and loss (G′′) moduli measured for a membrane sac, cap I and cap II (Fig. S7†), reveals a solid-like behaviour. During frequency sweep experiments, the capsules are compressed by the shear tool against the rheometer plate releasing the liquid entrapped inside them; this liquid is then mixed by the shear tool with the capsule walls, leading to a gel formation. As a consequence, G′ values measured for cap I and cap II are lower than those measured for the membrane sac. However, G′ is higher for cap II than for cap I, in agreement with the texture analysis results shown in Fig. S8.† The observed values of the shear modulus match those found in different types of tissue.42
The cytocompatibility of the membrane sac was studied by examining cell attachment and survival following seeding. Human adipose-derived stem cells at high density were allowed to attach to microscope coverslips coated with a membrane sac. After 24 hours, a proportion of cells had attached to the membrane (approximately 50%) and were relatively dispersed across the surface. Cells maintained a rounded appearance and exhibited a high incidence of cell death, with live/dead (calcein-AM/ethidium homodimer-1) staining demonstrating 31 ± 1% viability (Fig. 5, top row). As an alternative, the cap I formulation was modified to improve cytocompatibility by using a PA matrix consisting of 1.5 wt% C16-KKFF + 0.5 wt% C16-G3RGD in 0.026 wt% CaCl2, instead of a pure PA matrix (Table 1). This formulation provided a soft capsule that was used to coat a microscope coverslip. Whilst the number of cells attached was similar to the membrane sac coating experiment, after 24 hours cells exhibited a less dispersed, more clustered appearance. Live/dead cell staining revealed a better level of cell viability (72 ± 5%) demonstrating good cytocompatibility (Fig. 5, bottom row). Live cells did persist after 1 week of culture but they are only seen off the sample with a line of red (dead) cells between the glass and the gel (Fig. S9†) demonstrating toxicity over longer terms in culture. Results from control experiments are shown in Fig. S10.† Either calcium alginate or sodium alginate films were prepared from the same alginate sample. Cells persist on both calcium alginate and sodium alginate after 24 hours but exhibit a clumped/rounded appearance similar to that seen on the capsule. They do however maintain a high level of viability suggesting that the presence of RGD is not necessary for maintaining viability but that the reduction of content of the PA C16-KKFF is the important factor.
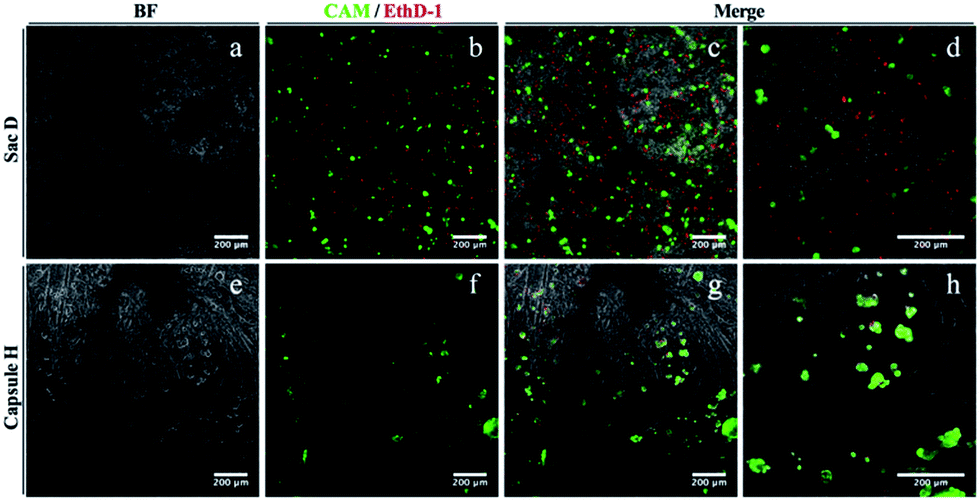 | ||
| Fig. 5 Biocompatibility testing after 24 hours of (a–d) membrane sacs or (e–h) modified cap I films. 24 hours after seeding, hASCs were stained with calcein-AM (live indicator; green) and ethidium homodimer-1 (dead indicator; red) before capturing images by fluorescent microscopy. (a and e) Bright field, (b and f) fluorescence and (c–d, g–h) merged images; (d) and (h) are higher magnifications of (c) and (g) respectively. | ||
Antimicrobial assays were performed against S. aureus, L. monocytogenes, E. faecalis, P. aeruginosa and E. coli. The antimicrobial activity of the peptide was similar under both dry and liquid conditions. Out of the five organisms tested, no effect of the peptide treatment was found against E. coli or P. aeruginosa which both are Gram negative organisms (data not shown). The logarithmic reduction in the number of colony-forming units was similar both the presence and absence of the peptide, during the course of the experiment. In contrast, an antimicrobial effect of the peptide was found against the Gram positive microorganisms S. aureus (Fig. 6) and L. monocytogenes (Fig. 6) and E. faecalis (Fig. S10†). A dramatic effect was seen in the case of L. monocytogenes, a significant log reduction of more than 6.30 orders of magnitude (counts were below the detection limit on the 4th day) was measured in the presence of the peptide, while in its absence a 1.20 log reduction occurred. Similarly to S. aureus, the majority of the antimicrobial effect seemed to occur during the first day.
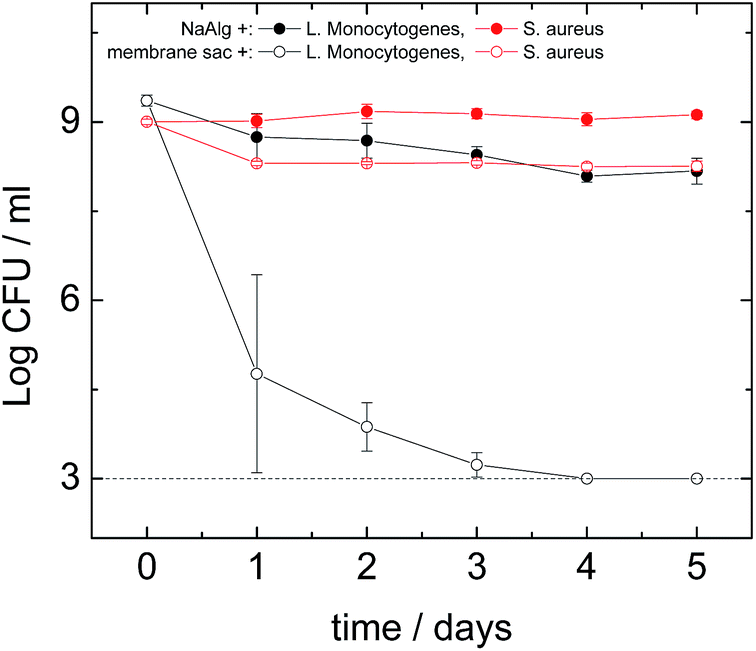 | ||
| Fig. 6 Survival of S. aureus (red lines) and L. monocytogenes (black lines) after exposure to the presence (membrane sac+) or absence (NaAlg+) of the peptide under dry conditions. Estimations of the cell numbers (CFU: colony-forming units) at each time point were performed in triplicate (3 biological replicates) while each dilution was plated in duplicate (2 technical replicates). Markers represent an average of the measurements performed in triplicate, and error bars represent the standard deviation. The dotted line represents the detection limit of the method. | ||
To study the antimicrobial activity of the peptide we selected both Gram negative and Gram positive microorganisms. One of the Gram negative bacteria was E. coli, which is one of the most studied microorganisms with a variety of pathogenic strains causing conditions ranging from foodborne illness to urinary infections.43 The other Gram negative bacterium was P. aeruginosa that is the most common cause of nosocomial infections affecting the respiratory system (pneumonia) and the urinary tract.44 Three Gram positive bacteria were also used of which one was S. aureus, a highly important human pathogen responsible for many nosocomial infections and deaths.34 Another bacterium tested was E. faecalis that is responsible for many nosocomial infections45 (while the last one was L. monocytogenes, currently the most deadly foodborne pathogen46 which can also affect transplantation patients).47 The membrane sac did not have any effect on the Gram negative bacteria E. coli and P. aeruginosa (data not shown), however it had a moderate antibacterial effect on S. aureus (Fig. 6) and E. faecalis (Fig. S10†) and a significant effect on L. monocytogenes (Fig. 6). These results clearly give an indication that the antibacterial effect might be specific to Gram positive bacteria. In the case of L. monocytogenes we found a major antibacterial effect with the membrane sac eliminating completely all measurable cells within 4 days (Fig. 6). This antimicrobial activity pattern is comparable to that of the amphiphilic peptide nisin48 that is active mainly against Gram positive bacteria such as L. monocytogenes and S. aureus.49 Further work is required to identify the antimicrobial activity of the peptide against other organisms and establish the mode(s) of action. However, It is well-known that the activity of antimicrobial peptides against Gram-positive bacteria is much larger than for Gram-negative bacteria.50 It has been suggested that high hydrophobicity may prevent peptide translocation through the outer membrane of Gram negative bacteria. Therefore, it is not surprising that we found an effect against L. monocytogenes, E. faecalis and S. aureus but no effect against E. coli or P. aeruginosa suggesting that the antimicrobial effects are specifically against Gram positive organisms but not Gram negative ones.
4. Conclusions
We have created remarkable hybrid biomaterials based on the co-operative self-assembly of the polysaccharide sodium alginate with peptide amphiphiles. The mechanical properties of sacs and capsules can be controlled through the injection process (e.g. by addition of CaCl2 to modulate electrostatic interactions) or via addition of graphene oxide, as a model ultrathin planar inorganic nanostructure which is able to modulate the rigidity of the capsules. These capsules represent new hybrid organic/inorganic nanocomposites. A novel “Langmuir–Blodgett” mechanism for the interfacial formation of the sac/capsule wall structures is presented, based on the aggregation of the cationic PA bilayers by electrostatic interaction with the injected anionic alginate, starting from the PA solution interface.The capsules are cytocompatible and show highly selective antimicrobial activity against an important human pathogen, L. monocytogenes and to a lesser extend against S. aureus and E. faecalis. The membranes have elasticity comparable to that of human tissue (several kPa range) which may be useful in the development of materials for stem cell differentiation although the cytocompatibility may need to be improved, by incorporation of more of cell adhesion motif (e.g. RGD) lipopeptides. In contrast to established Listeria antibacterial agents such as essential oils,51 our capsules contain bioactive lipopeptides which are embedded in the membrane-like polymeric material which might be processed into a film or coating. These hybrid materials therefore have great potential in the development of selective and highly active antibacterial materials, addressing a major global healthcare challenge. Another demonstrated application is in the selective uptake of cationic molecules (here – dyes), pointing towards potential applications in water treatment. Many other novel uses of these materials are envisaged exploiting their unique structural and functional properties. For example, by reduction of graphene oxide to graphene it may be possible to create materials with interesting electronic properties, and the encapsulation and barrier properties of the capsules are also of great interest for future research.
Statement of contributions
VC performed measurements except cryo-TEM, LSCM, cytotoxicity and antimicrobial activity experiments. AK performed SEM, assisted by VC. RB performed the antimicrobial activity measurements under the guidance of KAA. Cytotoxicity was assayed by SS under the supervision of CJC, JS performed the texture analysis measurements in collaboration with VC, and MR working in the group of JR performed the cryo-TEM imaging. DHM performed the SAXS measurements on ESRF beamline BM26B. IWH and VC prepared the figures and wrote the manuscript.Acknowledgements
This work was supported by EPSRC grant EP/L020599/1. IWH is the recipient of a Royal Society-Wolfson Research Merit Award. We thank K. Inoue (B21 beamline; project SM10077-1) and G. Giachin (BM29 beamline; project MX1769) for support with SAXS experiments. We acknowledge the CAF Laboratory (University of Reading) for access to XRD and SEM instruments.Notes and references
- I. W. Hamley, Chem. Commun., 2015, 51, 8574–8583 RSC.
- Y. C. Yu, P. Berndt, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1996, 118, 12515–12520 CrossRef CAS.
- D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- H. G. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- F. Versluis, H. R. Marsden and A. Kros, Chem. Soc. Rev., 2010, 39, 3434–3444 RSC.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed.
- A. Trent, R. Marullo, B. Lin, M. Black and M. Tirrell, Soft Matter, 2011, 7, 9572–9582 RSC.
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- E. Arslan, I. C. Garip, G. Gulseren, A. B. Tekinay and M. O. Guler, Adv. Healthcare Mater., 2014, 3, 1357–1376 CrossRef CAS PubMed.
- A. Dehsorkhi, V. Castelletto and I. W. Hamley, J. Pept. Sci., 2014, 20, 453–467 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed. Engl., 2013, 53, 6866–6881 CrossRef PubMed.
- J. T. Meijer, M. Roeters, V. Viola, D. Lowik, G. Vriend and J. C. M. van Hest, Langmuir, 2007, 23, 2058–2063 CrossRef CAS PubMed.
- D. Missirlis, H. Khant and M. Tirrell, Biochemistry, 2009, 48, 3304–3314 CrossRef CAS PubMed.
- M. J. Webber, J. Tongers, C. J. Newcomb, K. T. Marquardt, J. Bauersachs, D. W. Losordo and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13438–13443 CrossRef CAS PubMed.
- M. J. Webber, J. Tongers, C. J. Newcomb, K. T. Marquardt, J. Bauersachs, D. W. Losordo and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9220 CAS.
- J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed.
- R. M. Capito, H. S. Azevedo, Y. S. Velichco, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS PubMed.
- D. Carvajal, R. Bitton, J. R. Mantei, Y. S. Velichko, S. I. Stupp and K. R. Shull, Soft Matter, 2010, 6, 1816–1823 RSC.
- R. H. Zha, S. Sur and S. I. Stupp, Adv. Mater., 2013, 2, 126–133 CAS.
- R. Bitton, L. W. Chow, R. H. Zha, Y. S. Velichko, E. T. Pashuck and S. I. Stupp, Small, 2014, 10, 500–505 CrossRef CAS PubMed.
- K. E. Inostroza-Brito, E. Collin, O. Siton-Mendelson, K. H. Smith, A. Monge-Marcet, D. S. Ferreira, R. P. Rodriguez, M. Alonso, J. C. Rodriguez-Cabello, R. L. Reis, F. Sagues, L. Botto, R. Bitton, H. S. Azevedo and A. Mata, Nat. Chem., 2015, 7, 897–904 CrossRef CAS PubMed.
- C. Li, J. Adamcik and R. Mezzenga, Nat. Nanotechnol., 2012, 7, 421–427 CrossRef CAS PubMed.
- K. Vilcinskas, B. Norder, K. Goubitz, F. M. Mulder, G. J. M. Koper and S. J. Picken, Macromolecules, 2015, 48, 8323–8330 CrossRef CAS.
- M. Ionita, M. A. Pandele and H. Iovu, Carbohydr. Polym., 2013, 94, 339–344 CrossRef CAS PubMed.
- K. T. Cao, Z. Y. Jiang, J. Zhao, C. H. Zhao, C. Y. Gao, F. S. Pan, B. Y. Wang, X. Z. Cao and J. Yang, J. Membr. Sci., 2014, 469, 272–283 CrossRef CAS.
- L. Valentini, N. Rescignano, D. Puglia, M. Cardinali and J. Kenny, Eur. J. Inorg. Chem., 2015, 1192–1197 CrossRef CAS.
- A. Dehsorkhi, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 6665–6672 CrossRef CAS PubMed.
- I. W. Hamley, A. Dehsorkhi, V. Castelletto, S. Furzeland, D. Atkins, J. Seitsonen and J. Ruokolainen, Soft Matter, 2013, 9, 9290–9293 RSC.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- M. J. Krysmann, V. Castelletto, A. Kelarakis, I. W. Hamley, R. A. Hule and D. J. Pochan, Biochemistry, 2008, 47, 4597–4605 CrossRef CAS PubMed.
- E. Gazit, FASEB J., 2002, 16, 77–83 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- http://www.pepcalc.com, Innovagen's Peptide Property Calculator.
- K. A. G. Karatzas, A. Zervos, C. C. Tassou, C. G. Mallidis and T. J. Humphrey, Appl. Environ. Microbiol., 2007, 73, 1873–1881 CrossRef CAS PubMed.
- P. D. Cotter, K. O'Reilly and C. Hill, J. Food Prot., 2001, 64, 1362–1368 CrossRef CAS PubMed.
- M. I. Begley, C. G. M. Gahan and C. Hill, Appl. Environ. Microbiol., 2002, 68, 6005–6012 CrossRef CAS PubMed.
- I. Breßler, J. Kohlbrecher and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 1587–1598 Search PubMed.
- V. Castelletto, I. W. Hamley, J. Perez, L. Abezgauz and D. Danino, Chem. Commun., 2010, 46, 9185–9187 RSC.
- I. W. Hamley, A. Dehsorkhi and V. Castelletto, Langmuir, 2013, 29, 5050–5059 CrossRef CAS PubMed.
- V. Castelletto, R. Gouveia, C. J. Connon and I. W. Hamley, Faraday Discuss., 2013, 166, 381–397 RSC.
- I. W. Hamley, A. Dehsorkhi, V. Castelletto, M. N. M. Walter, C. J. Connon, M. Reza and J. Ruokolainen, Langmuir, 2015, 31, 4490–4495 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- A. R. Manges, Clin. Microbiol. Infect., 2016, 22, 122–129 CrossRef CAS PubMed.
- https://www.cdc.gov/hai/organisms/pseudomonas.html.
- A. M. Guzman Prieto, W. van Schaik, M. R. Rogers, T. M. Coque, F. Baquero, J. Corander and R. J. Willems, Front Microbiol, 2016, 7, 788, DOI:10.3389/fmicb.2016.00788.
- R. Paudyal and K. A. G. Karatzas, in Food Hygiene and Toxicology in Ready to Eat Foods, Academic Press, San Diego, 2016 Search PubMed.
- S. Wiesmayr, W. Tabarelli, I. Stelzmueller, D. Nachbaur, C. Boesmueller, H. Wykypiel, B. Pfausler, R. Margreiter, F. Allerberger and H. Bonatti, Wien. Klin. Wochenschr., 2005, 117, 229–233 CrossRef PubMed.
- I. Wiedemann, R. Benz and H.-G. Sahl, J. Bacteriol., 2004, 186, 3259–3261 CrossRef CAS PubMed.
- A. Campion, P. G. Casey, D. Field, P. D. Cotter, C. Hill and R. P. Ross, BMC Microbiol., 2013, 13, 1–8 CrossRef PubMed.
- N. Malanovic and K. Lohner, Pharmaceuticals, 2016, 9, 59 CrossRef PubMed.
- S. Burt, Int. J. Food Microbiol., 2004, 94, 223–253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SAXS data fitting parameters, additional SAXS data, LSCM image of membrane sac, SEM images, WAXS data, dye stain experiment images, rheology and texture analysis data. See DOI: 10.1039/c6ra27244d |
| This journal is © The Royal Society of Chemistry 2017 |