
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbohydrates as potentially versatile core subcarriers for multivalent immunogens†
Divya Kushwaha ,
Peng Xu and
Pavol Kováč*
,
Peng Xu and
Pavol Kováč*
NIDDK, LBC, National Institutes of Health, Bethesda, MD 20892-0815, USA. E-mail: kpn@helix.nih.gov
First published on 24th January 2017
Abstract
Synthetic multivalent glycoclusters that carry carbohydrate antigen epitopes have been recognized as promising candidates for the development of carbohydrate based vaccines. Here we describe a convergent strategy for the synthesis of conjugation-ready multivalent glycoclusters using sugars as versatile core subcarriers. D-Glucose and gentiobiose were converted into poly-alkyne functionalized cores which were then decorated with an azide bearing model ligand D-glucose using click chemistry, to form structurally well-defined tetra- and heptavalent glycoclusters. Each cluster was conjugated to a model protein bovine serum albumin (BSA) by squaric acid chemistry. Carbohydrate clusters can be prepared in a variety of sizes and spatial arrangements by altering the structure and configuration of the core, depending on the mono-, or oligosaccharides used for their assembly. It is suggested that the use of carbohydrate as core subcarriers provides an opportunity to tailor the size and topology of antigens and modify multivalent presentation of immunogens in a way to optimize cluster effect for stronger immunoreactivity.
Introduction
Binding of carbohydrates to host proteins is a frequent event in biology. Carbohydrate–protein interactions control diverse physiologically important biological processes.1 To these belongs also the recognition of O-specific polysaccharides (O-SP), which are the protective antigens of Gram-negative bacterial pathogens, by the elements of the immune system of vertebrates. The magnitude of the immune response and, in turn, the spectrum and level of antibodies produced upon infection depend on the strength of these interactions. Nature and evolution prepared us to respond to many infections in close to perfect way. Thus, were it not for some, often tolerable level of toxicity and pyrogenicity, vaccines based on killed or attenuated pathogens would be the best and cheapest means of prevention of infectious diseases. Glycoconjugate vaccines, the man-made constructs from detoxified lipopolysaccharides (LPS) and carrier proteins, are often, although still not entirely, free from the undesirable effects bacterial vaccines often have. The ideal vaccine would contain the pure protective antigen as the sole active antigenic ingredient. Lack of a reliable method for preparation of pure O-SP from LPS led to the idea of using synthetic oligosaccharides that mimic structures of O-SPs as antigenic components of conjugate vaccines. For the clinical use, such constructs would be made from pure components, i.e. synthetic oligosaccharide antigens and recombinant proteins. Thus, neoglycoconjugate vaccines, i.e. a vaccine from the fully synthetic carbohydrate antigen, would be well defined, easy-to-characterize and reproduce molecules with long shelf life. The carbohydrate and the recombinant protein fields have progressed to the stage that we can make virtually any structure that would be needed. Thus, making such molecules is not the bottleneck responsible for the lack of progress in the synthetic vaccine development and for the fact that to date only one neoglycoconjugate vaccine is known to be clinically useful.2Making vaccines from synthetic oligosaccharides is a very young field and structural/architectural requirements for a potent vaccine are not known. Optimizing such features is difficult because the formation of a glycoconjugate offers virtually an infinite number of configurational choices, as very ably demonstrated by Stein.3
When monovalent carbohydrate ligands are involved in interactions with receptors, the binding is often weak. To magnify the recognition, biological systems have circumvented this binding limitation through multivalency. The phenomenon of multivalency (cluster effect) was first described in their seminal work by Lee and coworkers.4 They observed that not only the number of ligands in the cluster but also the ligand's spatial arrangement affected the ligand's binding affinity to receptors. These phenomena have not been systematically studied in connection with vaccine development from synthetic oligosaccharides.
We have been interested in developing a conjugate vaccine for cholera from synthetic fragments of O-specific polysaccharides (O-SP) of Vibrio cholerae O1 for a number of years.5 When looking at how some variables affect the immunogenicity of glycoconjugates, we have examined the size of the oligosaccharide,6 the length of the spacer (linker)7 and the carbohydrate–protein ratio.8 The conjugates used in those works were made according to the single-ended conjugation model,9 which ensured that the vaccines were well defined, non cross-linked molecules. For making conjugates containing hapten in the form of clusters10 we designed our own core subcarrier.11 Its attractive feature was that it allowed to vary the number of ligands in the cluster but had the drawback that its use gave rise to cross-linked, high molecular mass constructs which were difficult to reproduce and characterize. Generally, the cross-linking can be avoided only when the fully assembled clusters are conjugated to the carriers according to the single-ended model.9 Thus, the synthetic scheme towards a vaccine where the antigen is present in a form of clusters must include a core subcarrier that allows installation of a reasonable and defined number of antigens and be equipped with the spacer bearing only a single reactive group compatible with the intended chemistry of conjugation to carriers. Towards this goal, here we have devised a generally applicable route for the preparation of conjugation-ready multivalent glycoclusters utilizing sugars as versatile core subcarriers. D-Glucose and gentiobiose have been chosen as model sugars for making the poly-alkyne functionalized cores and 6-azidohexyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside was used as model hapten to be attached to the cores. Cu(I)-catalyzed azide–alkyne “click” cycloaddition (CuAAC)12 was employed for their coupling to form structurally well defined tetra- and heptavalent glycoclusters. Furthermore, we show how the cluster thus formed can be conjugated to the model protein bovine serum albumin (BSA) using squaric chemistry.13,14 When a bacterial antigen or a synthetic fragment thereof would be used as hapten in such conjugation, an experimental immunogen (vaccine) would be formed.
Results and discussion
When searching for the class of substance that was to become the core to carry our antigen in the future cluster-type immunogen (vaccine), we took several factors into consideration. We wanted the core not only to carry a defined number of haptens but to also allow us to vary the size of the cluster and fine-tune the spatial arrangements of the haptens around it. Carbohydrates are among few classes of compounds that can fulfil these requirements. They are inexpensive commodities, many occur naturally, and methods for orthogonal functionalization of this class of compounds are readily available. The rich structural and configurational diversity of carbohydrates allows them to adopt many forms (different ring size and anomeric varieties) or unusual conformations (into which sugars can be forced by deoxygenation at various positions in the pyranose or furanose ring form). When such structures are utilized as cores, a large number of multivalent glycomimetics with diverse spatial arrangements, number of haptens and degree of freedoms can be constructed. Therefore, depending on the nature of the receptor, the use of carbohydrates as core allows optimization of the glycoside-cluster effect by providing the opportunity to tailor the valency and topology of the clusters formed.Using carbohydrates as core molecules for creating clusters as probes for many purposes in the life sciences, also as chiral scaffolds,15 is quite common16–32 but to our knowledge making carbohydrate clusters by attaching fragments of O-SPs to cores by click chemistry and attaching the resulting cluster of antigens to carrier proteins by squaric acid chemistry to make neoglycoconjugate vaccines for bacterial diseases has not been explored. Here, we propose carbohydrates as convenient starting materials to form tailor-made core subcarriers for making clustered immunogens/vaccines. The synthetic strategy and the methods for characterization of the constructs obtained are generally applicable to making clusters from virtually any appropriately functionalized ligand. Clusters can be obtained in many sizes and shapes when the core results from a rational choice of carbohydrate(s). The newly formed constructs can be used as vaccines when the ligand is a mimic of a bacterial carbohydrate antigen, or tailor-made probes in many branches of the life sciences.
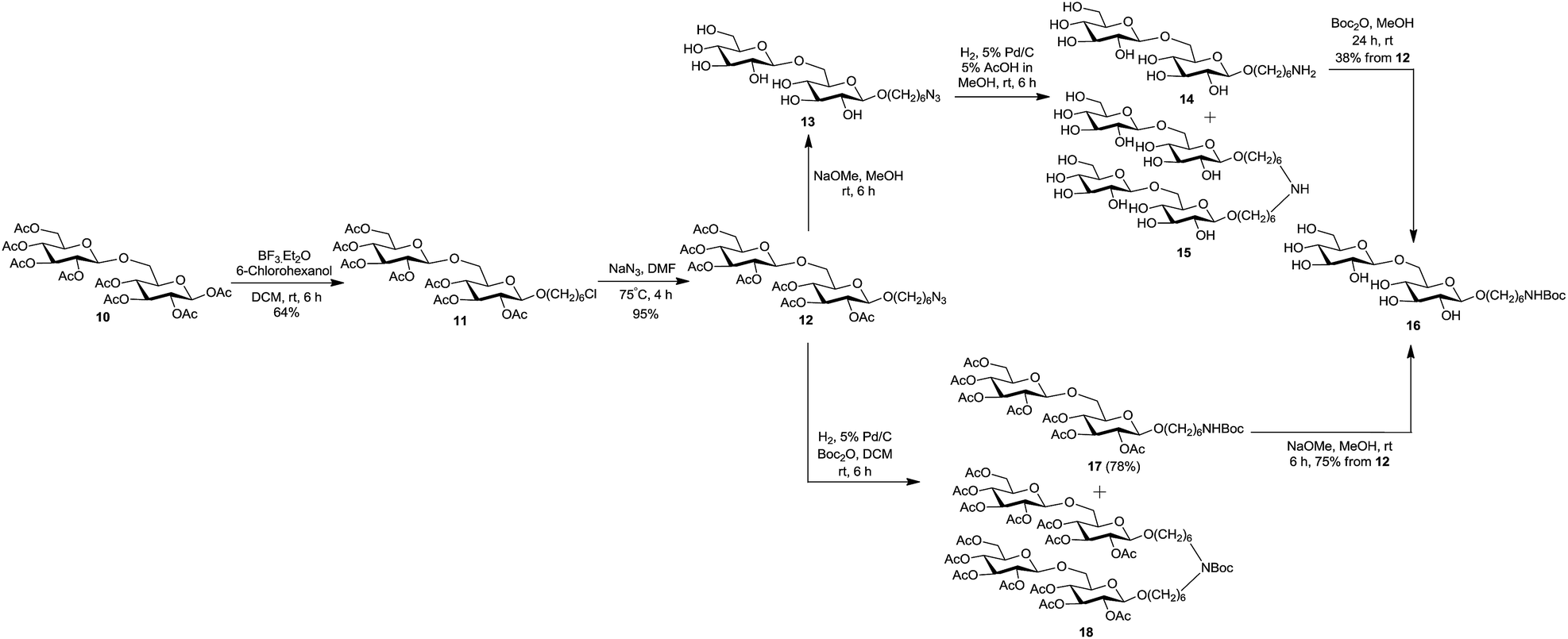
We herein report a straightforward and efficient strategy for the synthesis of two carbohydrate-centered glycoclusters and their conjugation to a model carrier protein BSA by squaric acid chemistry. The latter has become our method of choice for conjugation because of its unsurpassed efficiency and experimental simplicity.33 For the preparation of poly-alkyne functionalized carbohydrate cores 19 and 22, we chose simple monosaccharide per-O-acetylated β-D-glucose 134 and the disaccharide per-O-acetylated β-gentiobiose 10 as starting materials.35 The synthesis began with the introduction of spacer arm to per-O-acetates 1 and 10 by BF3·Et2O catalyzed glycosidation with 6-chlorohexanol, to obtain 2 and 11 in 36% and 64% yield respectively (Scheme 1 and 2). The bromo analog of 2 was previously obtained in the same yield in a similar way by Dubber et al.24 However, the chloro derivatives are preferred intermediates when conversion to the corresponding azides is anticipated because 6-chlorohexanol is a less expensive commodity. The azidation of 2 and 11 was brought about in the subsequent step, by their treatment with NaN3 in DMF at 75 °C to afford corresponding azides 3 and 12 in almost theoretical yields. The starting materials and the products of the aforementioned conversions showed very similar chromatographic mobility in each case. However, the formation of 3 and 12 could be monitored by NMR spectroscopy. It showed confidently that the conversion of chlorides was complete and that these were one-product reactions. In both cases, compared to the 1H spectra of 2 and 11, the 2-proton triplet for H-6′ of 3 and 12 was shifted upfield by ∼0.26 ppm, and the C-6′ signal in 13C NMR spectra was shifted downfield by ∼6.4 ppm.
 | ||
| Scheme 1 Synthesis of the intermediate 7. | ||
 | ||
| Scheme 2 Synthesis of the key intermediate 16. | ||
With the acetylated azide armed glycosides 3 and 12 at hand, we intended to convert them to the corresponding poly-hydroxylated glycosides 7 and 16 carrying a protected amine-functionalized linker. The route employed was de-O-acetylation (Zemplén) of azide 3 to obtain 4, followed by the reduction of 4 under catalytic hydrogenation conditions (H2, 5% Pd/C, experimental, preparation of 7, A). Careful analysis of the reaction mixture of hydrogenation of 4 by TLC revealed the presence of two poorly resolved major products in ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. A challenging chromatographic separation on a small scale led to products amenable to structural analysis (NMR, MS). The compounds isolated were identified as the expected amine 5 and a by-product. Structure of the by-product 6 was determined on the basis of the spectral data (NMR, MS) and the literature precedence.36–38 The same holds for deduction of structures of compounds 9, 15, and 18 described below. Because the proportion of side product 6 was unacceptably high, more favourable conditions for azide reduction were sought. Compound 5 was formed in a significantly improved amount (5/6, ∼10:1) when the reduction of 4 was performed in presence of NH4OH in MeOH (experimental, preparation of 7, B). On preparative scale, the difficult separation of 5 and 6 was omitted and the crude mixture was directly submitted for NH-Boc protection with di-tert-butyl dicarbonate in MeOH. In this way the key intermediate NH-Boc protected amine 7 was isolated in 67% yield (from 3), after chromatography.
:1 ratio. A challenging chromatographic separation on a small scale led to products amenable to structural analysis (NMR, MS). The compounds isolated were identified as the expected amine 5 and a by-product. Structure of the by-product 6 was determined on the basis of the spectral data (NMR, MS) and the literature precedence.36–38 The same holds for deduction of structures of compounds 9, 15, and 18 described below. Because the proportion of side product 6 was unacceptably high, more favourable conditions for azide reduction were sought. Compound 5 was formed in a significantly improved amount (5/6, ∼10:1) when the reduction of 4 was performed in presence of NH4OH in MeOH (experimental, preparation of 7, B). On preparative scale, the difficult separation of 5 and 6 was omitted and the crude mixture was directly submitted for NH-Boc protection with di-tert-butyl dicarbonate in MeOH. In this way the key intermediate NH-Boc protected amine 7 was isolated in 67% yield (from 3), after chromatography.
Following the optimized reaction conditions for the preparation of 7, the synthesis of its disaccharide analogue 16 was attempted from azide 12 (Scheme 2). De-O-acetylation of 12 (Zemplén)39 gave azide 13 in virtually theoretical yield, but hydrogenation of the latter under the conditions applied to 4 was unsuccessful: more than 95% (TLC) of 13 remained unchanged after several hours. The effort to reduce azide 13 under high pressure (200 psi) also failed. The reduction of 13 could be completed when carried out in 5% AcOH in MeOH as solvent. Sadly, though, the desired amine 14 and the side product 15 (deduced from MS and analogy to formation of 6) were formed in ∼1:1 ratio (TLC). Because the treatment of such mixture with Boc anhydride furnished 16 in only 38% yield (in 3 steps from 12), and neither the yield of 7 was considered quite satisfactory, an alternative, more efficient synthetic route for 7 and 16 was sought.
We have previously described a convenient conversion of a sugar azide to the corresponding N-acetamido derivative by conducting the catalytic hydrogenation (H2, Pd/C) in presence of Ac2O using MeOH as solvent.40 Because in the present situation neither the starting material nor the conversion products contained hydroxyl groups, the use of MeOH as solvent became unimportant. Accordingly, hydrogenation (5% Pd/C) of azide 3 in presence of di-tert-butyl dicarbonate using DCM as solvent effected the reduction to amine and simultaneous NH-Boc protection in one pot (experimental, preparation of 7, C), to give a mixture of desired amine 8 and side product 9 (Scheme 1). A simple chromatographic separation afforded amine 8 (76%), which was de-O-acetylated under Zemplén transesterification conditions, to afford the key intermediate 7 in virtually theoretical yield. Synthesis of disaccharide 16 was achieved from 12 (75%, overall in two steps) in a similar way (Scheme 2).
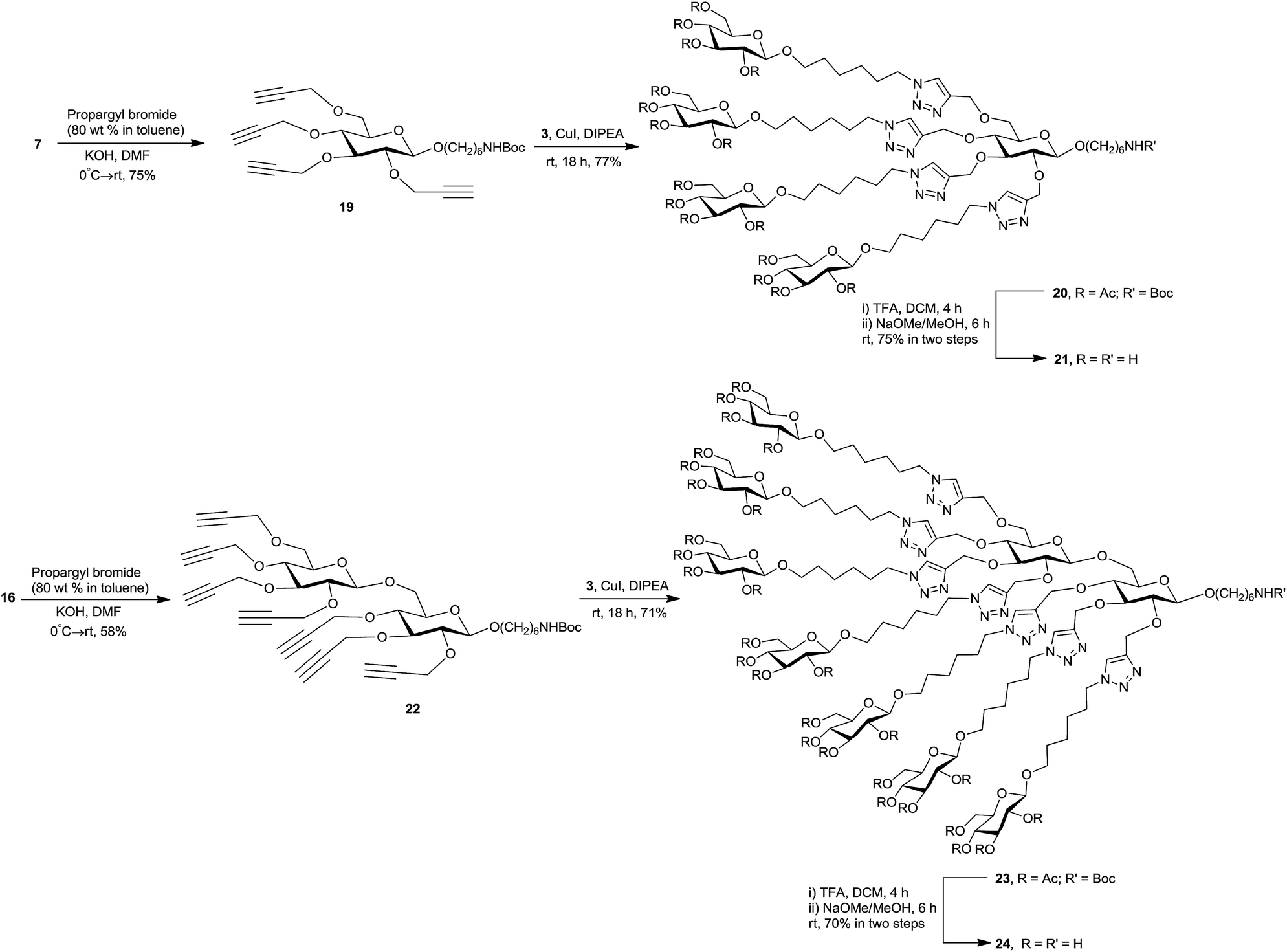
To proceed towards the title cores, glycosides 7 and 16 were treated with propargyl bromide and KOH at 0 °C → rt, to obtain the corresponding alkyne functionalized, clickable intermediates 19 and 22 in 75% and 58% yield, respectively. The structure confirmation for 19 and 22 was provided by spectral analysis (NMR, HRMS). In the 1H NMR spectrum of each of 19 and 22, the characteristic ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H protons appeared as multiplet at δ 2.5 ppm with integral values of four and seven protons respectively. In 13C NMR spectra, the signals for two acetylenic carbons appeared at δ ∼80.0 ppm (CC–H) and ∼74.0 ppm (CC–H). The molecular ion peak [M + Na]+ at m/z 554.2731 for 19 and at 830.3727 for 22 confirmed the complete alkylation in each case.
C–H protons appeared as multiplet at δ 2.5 ppm with integral values of four and seven protons respectively. In 13C NMR spectra, the signals for two acetylenic carbons appeared at δ ∼80.0 ppm (CC–H) and ∼74.0 ppm (CC–H). The molecular ion peak [M + Na]+ at m/z 554.2731 for 19 and at 830.3727 for 22 confirmed the complete alkylation in each case.
To demonstrate the feasibility of our synthetic strategy in glycocluster formation, azidosugar 3 was chosen as click synthon to decorate the carbohydrate-centered building blocks 19 and 22 (Scheme 3). A Cu(I) catalyzed click reaction41–44 was conducted between 19 (1.0 equiv.) and 3 (5.0 equiv.) in presence of CuI and diisopropylethylamine (DIPEA), which afforded 20 (77%). The synthesis of 23 (71%) was achieved in a similar manner through click coupling of 22 (1.0 equiv.) and 3 (8.0 equiv.). The 1H NMR spectra of 20 and 23 showed presence of four (δ 8.16, 7.94, 7.68 ppm in 1:2:1 ratio) and seven newly formed triazolyl proton singlets (δ 8.15, 8.04, 7.94, 7.89, 7.77 ppm in 2:1:2:1:1 ratio), respectively, and the signals for the alkyne C–H protons were absent. This, along with the MS data, confirmed the successful multiple click reactions in both cases. The 13C NMR spectra of 20 and 23 revealed the presence of characteristic carbon signals at δ ∼145–144 and δ ∼124–123 ppm, respectively, for C-4 and C-5 of the 1,4-disubstituted 1,2,3-trizole ring. The complete structural assignment of 20 and 23 was achieved with the aid of 2D NMR experiments (1H COSY, TOCSY and 1H–13C HSQC).
 | ||
| Scheme 3 Synthesis of tetra- and heptavalent glycoclusters 21 and 24 via click chemistry. | ||
The fully protected clusters 20 and 23 were then treated with TFA in DCM to remove the Boc protecting group, and subsequent de-O-acetylation (Zemplén) yielded the conjugation-ready clusters 21 and 24, respectively, in good yields after purification by reverse-phase chromatography. The structures of 21 and 24 were confirmed by NMR (1D and 2D) and HRMS spectral data.
Conjugation of the synthetic glycoclusters to BSA carrier protein was executed by squaric acid chemistry.13,14,45,46 Thus, clusters 21 and 24 were treated with 3,4-dimethoxy-3-cyclobutene-1,2-dione (dimethyl squarate) at pH 7 (phosphate buffer) to give the squaric acid monoester derivatives 25 and 27, respectively, which were readily purified by chromatography (Scheme 4).46 The formation of 25 and 27 was established by HRMS and NMR spectroscopy. In the 1H NMR spectra of each of 25 and 27, the –OCH3 protons attached to the squaric acid moiety resonated at δ 4.36 ppm and their 13C NMR spectra showed –OCH3 carbon as two signals at δ ∼61 ppm. The 13C spectra of monoesters 25 and 27 exhibited typical splitting of some signals due to the double bond nature of the vinylogous amide group, which is characteristic of squaric acid amide esters.14
 | ||
| Scheme 4 Conjugation of the glycoclusters 21 and 24 to BSA by squaric acid chemistry. | ||
Each of the monomethyl ester 25 and 27 was next treated with BSA in 0.5 M borate buffer at pH 9 for 2 days, according to the protocol developed in our lab.45 The conjugations were carried out at a hapten/BSA ratio 6:1 and at a hapten concentration ∼4.0 mM. After freeze drying, the SELDI MS analysis of the purified conjugates showed average molecular mass 73700 Da for the conjugate 26 (molar ratio 25:BSA ∼4.1:1) and 77600 Da for the conjugate 28 (molar ratio 27:BSA ∼3.7:1).
Conclusions
We propose that carbohydrates can serve as versatile subcarriers for clusters of a variety of ligands whose valences and spatial arrangement can be predetermined by judicious choice of natural or chemically modified carbohydrates. Following that rational, two structurally well defined tetra- and heptavalent glycoclusters bearing amino-functionalized linker were synthesized by the Cu(I)-catalyzed click coupling of polyalkyne-functionalized D-glucose and gentiobiose cores and azide-equipped ligand. Squaric acid chemistry was successfully applied for conjugation of the obtained amino group-containing, synthetic, multivalent glycoclusters to the carrier protein BSA. The synthetic materials, many of which were obtained in the analytically pure state, were characterized by physical constants (mp, [α]D) and spectral data (NMR, HRMS).Experimental section
General methods
Melting points were determined with a Kofler hot stage. Optical rotations were measured at ambient temperature for solutions in CHCl3 and MeOH with a Perkin–Elmer automatic polarimeter, Model 341. HPLC grade solvents were used, and reactions requiring anhydrous conditions were carried out under nitrogen or argon. Reactions were monitored by thin-layer chromatography (TLC) on silica gel 60 glass slides. Spots were visualized by charring with H2SO4 in EtOH (5% v/v). Column chromatography was performed with Biotage Isolera chromatograph equipped with a Varian Evaporative Light-Scattering Detector. Nuclear magnetic resonance (NMR) spectra were measured at 600 MHz for 1H, and 150 MHz for 13C for solutions in CDCl3, MeOD and D2O with Bruker Avance spectrometer. Chemical shifts were reported relative to TMS (1H: δ 0.00 ppm), CHCl3 (13C: δ 77.00 ppm), MeOH (1H: δ 3.31 ppm; 13C: δ 49.00 ppm) or H2O (δ 4.79 ppm). Assignments of NMR signals were aided by 1D and 2D experiments (APT, COSY, TOCSY, HSQC, HMQC, and HMBC). When reporting assignment of NMR signals, nuclei associated with the subcarrier spacer (linker) are denoted with a prime and those associated with the ligand spacer are denoted with a double prime. Sugar residues in oligosaccharides are serially numbered, beginning with the one bearing the aglycone, and are identified by a Roman numeral superscript in listings of signal assignments. Liquid Chromatography-Electron Spray-Ionization Mass Spectrometry (ESI-MS) was performed with a Hewlett–Packard 1100 MSD spectrometer. Conjugation of carbohydrates was monitored by the BioRad Protein Chip SELDI system using NP-20 chip arrays. 3,5-Dimethoxy-4-hydroxycinnamic acid (sinapinic acid) was used as matrix. Attempts have been made to obtain correct combustion analysis data for all new compounds. However, some compounds tenaciously retained traces of solvents, despite exhaustive drying, and analytical figures for carbon could not be obtained within ±0.3%. The palladium-on-charcoal catalyst used was purchased from Engelhard Industries (Escat 103, Lot# FC96303). BSA was purchased from Sigma Chemical Company: A. Cat. Number A-0281; B. Fraction V, Sigma Cat. no. A-4503, from which fatty acids were removed by charcoal treatment.47 Buffers used were as follows; A, BuffAR pH 7.0 Reference solution (Macron, Cat. no. 0031-04), concentrated to 1/10 volume; B, 0.5 M borate buffer pH 9, made in house (1 L) from boric acid (30.9 g), KCl (26.1 g), and KOH (8.42 g), and final adjustment to pH 9.0 by addition of solid KOH. During squaric acid monoester formation reaction, pH of the reaction mixture was monitored intermittently with a Corning, Model 440 pH meter equipped with model MI-410 microelectrode (Microelectrodes, Inc. Bedford, NH, USA) and when necessary, pH was maintained at 7 by addition of buffer salts, obtained by freeze-drying Buffer A. Solutions in organic solvents were dried with anhydrous Na2SO4, and concentrated at <40 °C/2 kPa.:1 hexane–acetone), and [α]23D −17.8 (c 0.7, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 5.20 (t, 1H, J 9.5 Hz, H-3), 5.09 (t, 1H, J 9.7 Hz, H-4), 4.98 (dd, 1H, J1,2 8.0 Hz, J2,3 9.6 Hz, H-2), 4.49 (d, 1H, H-1), 4.26 (dd, 1H, J5,6a 4.8 Hz, J6a,6b 12.3 Hz, H-6a), 4.14 (dd, 1H, J5,6b 2.5 Hz, H-6b), 3.87 (dt, 1H, J 6.3, 9.6 Hz, H-1′a), 3.69 (ddd, 1H, J5,4 10.0 Hz, H-5), 3.53 (t, 2H, J 6.7 Hz, H-6′a,b), 3.48 (dt, 1H, J 6.6 Hz, H-1′b), 2.09, 2.04, 2.03, 2.00 (4 s, 12H, 4 COCH3), 1.76 (m, 2H, H-5′a,b), 1.59 (m, 2H, H-2′a,b), 1.44 (m, 2H, H-4′a,b), 1.36 (m, 2H, H-3′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.67, 170.30, 169.34, 169.26 (4 COCH3), 100.80 (C-1), 72.85 (C-3), 71.76 (C-5), 71.34 (C-2), 69.89 (C-1′), 68.47 (C-4), 61.98 (C-6), 44.92 (C-6′), 32.45 (C-5′), 29.21 (C-2′), 26.48 (C-4′), 25.11 (C-3′), 20.72, 20.64, 20.60, 20.58 (4 COCH3) ppm. HRMS (ESI): m/z calcd for C20H35NO10Cl [M + NH4]+: 484.1949; found: 484.1953; anal. calcd for C20H31ClO10: C, 51.45; H, 6.69; found: C, 51.35; H, 6.62.:1 hexane–acetone) and [α]23D −18.9 (c 0.7, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 5.20 (t, 1H, J 9.6 Hz, H-3), 5.09 (t, 1H, J 9.8 Hz, H-4), 4.98 (dd, J1,2 8.0 Hz, J2,3 9.6 Hz, H-2), 4.49 (d, 1H, H-1), 4.26 (dd, 1H, J5,6a 4.8 Hz, J6a,6b 12.3 Hz, H-6a), 4.14 (dd, 1H, J5,6b 2.5 Hz, H-6b), 3.87 (dt, 1H, J 6.3, 9.6 Hz, H-1′a), 3.69 (ddd, 1H, J5,4 10.0 Hz, H-5), 3.48 (dt, 1H, J 6.7 Hz, H-1′b), 3.26 (t, 2H, J 7.0, H-6′a,b), 2.09, 2.04, 2.03, 2.01 (4 s, 12H, 4 COCH3), 1.59 (m, overlapped, 4H, H-2′a,b, 5′a,b), 1.37 (m, overlapped, 4H, H-3′a,b, 4′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.68, 170.31, 169.40, 169.26 (4 COCH3), 100.81 (C-1), 72.85 (C-3), 71.77 (C-5), 71.35 (C-2), 69.89 (C-1′), 68.48 (C-4), 61.99 (C-6), 51.33 (C-6′), 29.24, 28.75, 26.38, 25.40 (C-2′, C-3′, C-4′, C-5′), 20.73, 20.64, 20.61, 20.59 (4 COCH3) ppm. HRMS (ESI): m/z calcd for C20H35N4O10 [M + NH4]+: 491.2353; found: 491.2351; anal. calcd for C20H31N3O10: C, 50.74; H, 6.60; N, 8.87; found: C, 50.63; H, 6.53; N, 8.85.A. To a solution of acetate 3 (500 mg, 1.05 mmol) in MeOH (6 mL), a solution of NaOMe in MeOH was added until strong alkalinity and the mixture was kept at room temperature until the reaction was complete (∼6 h). The mixture was diluted with MeOH, neutralized with Amberlite IR 120H+, filtered and concentrated. Chromatography (4
:3:0.2 DCM–acetone–MeOH) afforded 6-azidohexyl β-D-glucopyranoside 4 (310 mg, 96%) as colorless syrup. Rf 0.25 (4:3:0.2 DCM–acetone–MeOH). 1H NMR (MeOD, 600 MHz): δ 4.24 (d, 1H, J1,2 7.9 Hz, H-1), 3.91 (dt, 1H, J 6.7, 9.6 Hz, H-1′a), 3.86 (dd, 1H, J5,6a 2.2 Hz, J6a,6b 12.0 Hz, H-6a), 3.66 (dd, 1H, J5,6b 5.3 Hz, H-6b), 3.55 (dt, 1H, J 6.7 Hz, H-1′b), 3.34 (t, 1H, J 8.8 Hz, H-3), 3.29–3.24 (m, overlapped, 4H, H-6′a,b, H-4, H-5), 3.17 (dd, 1H, J2,3 9.0 Hz, H-2), 1.62 (m, overlapped, 4H, H-2′a,b, 5′a,b), 1.42 (m, overlapped, 4H, H-3′a,b, 4′a,b) ppm; 13C NMR (MeOD, 150 MHz): δ 104.37 (C-1), 78.14 (C-3), 77.92 (C-5), 75.14 (C-2), 71.69 (C-4), 70.67 (C-1′), 62.79 (C-6), 52.40 (C-6′), 30.63 (C-2′), 29.86 (C-5′), 27.62, 26.64 (C-3′, C-4′) ppm. HRMS (ESI): m/z calcd for C12H23N3O6Na [M + Na]+: 328.1485; found: 328.1479.A stirred solution of azido sugar 4 (310 mg, 1.01 mmol) in MeOH (∼5 mL) was treated with hydrogen in presence of 5% Pd/C (60 mg) at room temperature overnight. TLC (1:2:0.06 DCM–MeOH–25% NH4OH) showed that two major products were formed in an approximate ratio of 1:1 (Rf ∼0.1, partially overlapping). After filtration through Celite pad, the filtrate was concentrated. For identification, a small amount of the mixture was chromatographed to obtain the major products, which were sufficiently pure for spectral identification. Eluted first was material identified as 5 (white solid), 1H NMR (MeOD, 600 MHz): δ 4.24 (d, 1H, J1,2 7.8 Hz, H-1), 3.91 (dt, 1H, J 6.7, 9.6 Hz, H-1′a), 3.86 (m, 1H, 12.0 Hz, H-6a), 3.66 (dd, 1H, J6a,6b 12.0 Hz, J5,6b 5.3 Hz, H-6b), 3.55 (dt, 1H, H-1′b), 3.34 (m, 1H, H-3), 3.29–3.24 (m, overlapped, 2H, H-4, H-5), 3.16 (m, 1H, H-2), 2.67 (t, 2H, J 7.2 Hz, H-6′a,b), 1.64 (m, 2H, CH2 linker), 1.50 (m, 2H, CH2 linker), 1.39 (m, overlapped, 4H, 2 × CH2 linker) ppm; 13C NMR (MeOD, 150 MHz): δ 104.37 (C-1), 78.15 (C-3), 77.93 (C-5), 75.13 (C-2), 71.68 (C-4), 70.73 (C-1′), 62.77 (C-6), 42.19 (C-6′), 32.92, 30.64, 27.66, 26.86 (4 × CH2 linker) ppm. HRMS (ESI): m/z calcd for C12H25NO6Na [M + Na]+: 302.1580; found: 302.1584. Later eluted was the side product 6 (white solid), 1H NMR (MeOD, 600 MHz): δ 4.24 (d, 1H, J1,2 7.8 Hz, H-1), 3.91 (dt, 1H, J 6.7, 9.6 Hz, H-1′a), 3.86 (m, 1H, H-6a), 3.66 (dd, 2H, J6a,6b 11.8 Hz, J5,6b 5.2 Hz, H-6b), 3.55 (dt, 2H, H-1′b), 3.34 (m, 2H, H-3), 3.29–3.24 (m, overlapped, 4H, H-4, H-5), 3.16 (t, 2H, J 8.2 Hz, H-2), 2.61 (t, 4H, J 7.6 Hz, H-6′a,b), 1.64 (m, 4H, 2 × CH2 linker), 1.54 (m, 4H, 2 × CH2 linker), 1.40 (m, overlapped, 8H, 4 × CH2 linker) ppm; 13C NMR (MeOD, 150 MHz): δ 104.36 (C-1), 78.13 (C-3), 77.91 (C-5), 75.12 (C-2), 71.67 (C-4), 70.74 (C-1′), 62.77 (C-6), 50.43 (C-6′), 30.63, 29.96, 28.11, 26.93 (4 × CH2 linker) ppm. HRMS (ESI): m/z calcd for C24H48NO12 [M + H]+: 542.3177; found: 542.3183. Because 5, the intended intermediate to 7, was not the major reaction product, this route to 7 was not pursued further.
B. Compound 3 (500 mg, 1.05 mmol) was deacetylated as described in A and a solution of azido sugar 4 formed was hydrogenated in 1
:20 25% NH4OH–MeOH (∼5 mL) in presence of 5% Pd/C (60 mg) at room temperature overnight. After conventional processing, the crude mixture of 5 and 6 (281 mg) obtained was dissolved in MeOH (5 mL) and treated overnight with di-tert-butyl dicarbonate (330 mg, 1.50 mmol). After concentration, the residue was chromatographed (8:1 CHCl3–MeOH) to yield 7 as white foam (310 mg, 67%, in three steps from 3); Rf 0.5 (6:1 CHCl3–MeOH); [α]23D −30.7 (c 0.8, CHCl3). 1H NMR (MeOD, 600 MHz): δ 4.24 (d, 1H, J1,2 7.8 Hz, H-1), 3.90 (dt, 1H, J 6.8, 9.5 Hz, H-1′a), 3.86 (dd, 1H, J5,6a 2.1 Hz, J6a,6b 11.9 Hz, H-6a), 3.66 (dd, 1H, J5,6b 5.4 Hz, H-6b), 3.54 (dt, 1H, H-1′b), 3.34 (t, 1H, J 8.8 Hz, H-3), 3.28 (t, overlapped, 1H, H-4), 3.25 (m, overlapped, 1H, H-5), 3.16 (dd, 1H, J2,3 9.0 Hz, H-2), 3.02 (t, 2H, J 7.0 Hz, H-6′a,b), 1.63 (m, 2H, H-2′a,b), 1.49–1.38 (m, overlapped, H-5′a,b, H-3′a,b), 1.43 (s, overlapped, NHCOOC(CH3)3), 1.34 (m, 2H, H-4′a,b) ppm; 13C NMR (MeOD, 150 MHz): δ 158.57 (NHCOOC(CH3)3), 104.37 (C-1), 79.78 (NHCOOC(CH3)3), 78.14 (C-3), 77.92 (C-5), 75.13 (C-2), 71.68 (C-4), 70.75 (C-1′), 62.78 (C-6), 41.28 (C-6′), 30.90 (C-5′), 30.69 (C-2′), 28.80 (NHCOOC(CH3)3), 27.65 (C-4′), 26.76 (C-3′) ppm. HRMS (ESI): m/z calcd for C17H33NO8Na [M + Na]+: 402.2104; found: 402.2111; anal. calcd for C17H33NO8: C, 53.81; H, 8.77; N, 3.69; found: C, 53.73; H, 8.85; N, 3.65.
C. A suspension of 5% Pd/C (50 mg) in DCM (2 mL) was added to the solution of azide 3 (250 mg, 0.53 mmol) and di-tert-butyl dicarbonate (230 mg, 1.05 mmol) in DCM (4 mL) and the mixture was stirred at room temperature under H2 atmosphere for 5 h, when TLC showed complete consumption of starting material. After conventional processing, as above, and concentration, the residue was chromatographed (4
:1 hexane–acetone) to give 6-N-(tert-butyloxycarbonyl)hexyl 2,3,4,6-tetra-O-acetyl-β-D-glucopyranoside 8 (218 mg, 76%), Rf 0.4 (2:1 hexane–acetone). 1H NMR (CDCl3, 600 MHz): δ 5.20 (t, 1H, J 9.5 Hz, H-3), 5.07 (t, 1H, J 9.7 Hz, H-4), 4.97 (dd, 1H, J1,2 8.0 Hz, J2,3 9.6 Hz, H-2), 4.57–4.48 (bs, NH), 4.49 (d, 1H, H-1), 4.26 (dd, 1H, J5,6a 4.8 Hz, J6a,6b 12.3 Hz, H-6a), 4.14 (dd, 1H, J5,6b 2.5 Hz, H-6b), 3.85 (dt, 1H, J 6.3, 9.6 Hz, H-1′a), 3.69 (ddd, 1H, J5,4 9.9 Hz, H-5), 3.47 (dt, 1H, J 6.7 Hz, H-1′b), 3.09 (t, 2H, J 6.9 Hz, H-6′a,b), 2.08, 2.03, 2.02, 2.00 (4 s, 12H, 4 COCH3), 1.56 (m, 2H, H-2′a,b), 1.46 (m, overlapped, H-5′a,b), 1.44 (s, overlapped, NHCOOC(CH3)3), 1.32 (m, overlapped, 4H, H-3′a,b, 4′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.56, 170.20, 169.33, 169.19 (4 COCH3), 155.97 (NHCOOC(CH3)3), 100.82 (C-1), 79.03 (NHCOOC(CH3)3), 72.92 (C-3), 71.82 (C-5), 71.44 (C-2), 69.95 (C-1′), 68.61 (C-4), 62.05 (C-6), 40.51 (C-6′), 29.99 (C-5′), 29.29 (C-2′), 28.41 (NHCOOC(CH3)3), 26.41 (C-4′), 25.50 (C-3′), 20.65, 20.58, 20.53, 20.51 (4 COCH3) ppm. HRMS (ESI): m/z calcd for C25H41NO12Na [M + Na]+: 570.2526; found: 570.2526. Eluted later was the side product 9. 1H NMR (CDCl3, 600 MHz): δ 5.20 (t, 2H, J 9.5 Hz, H-3), 5.07 (t, 2H, J 9.7 Hz, H-4), 4.97 (dd, 2H, J1,2 8.0 Hz, J2,3 9.6 Hz, H-2), 4.49 (d, 2H, H-1), 4.26 (dd, 2H, J5,6a 4.8 Hz, J6a,6b 12.2 Hz, H-6a), 4.14 (dd, 2H, J5,6b 2.5 Hz, H-6b), 3.85 (dt, 2H, J 6.5, 9.6 Hz, H-1′a), 3.69 (ddd, 2H, J5,4 10.0 Hz, H-5), 3.47 (dt, 2H, J 6.7 Hz, H-1′b), 3.13 (t, 4H, J 7.1 Hz, H-6′a,b), 2.08, 2.03, 2.02, 2.00 (4 s, 24H, 2 × 4 COCH3), 1.57 (m, 4H, 2 × CH2 linker), 1.48 (m, 4H, 2 × CH2 linker), 1.44 (s, 9H, NHCOOC(CH3)3), 1.39–1.24 (m, overlapped, 8H, 4 × CH2 linker) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.59, 170.24, 169.36, 169.19 (2 × 4 COCH3), 155.58 (NHCOOC(CH3)3), 100.85 (C-1), 78.98 (NHCOOC(CH3)3), 72.95 (C-3), 71.84 (C-5), 71.47 (C-2), 70.02 (C-1′), 68.63 (C-4), 62.08 (C-6), 46.96 (C-6′), 29.42 (CH2 linker), 28.50 (overlapped, NHCOOC(CH3)3, CH2 linker), 26.67 (CH2 linker), 25.63 (CH2 linker), 20.65, 20.60, 20.56, 20.54 (4 COCH3) ppm. HRMS (ESI): m/z calcd for C45H71NO22Na [M + Na]+: 1000.4365; found: 1000.4359.A solution of 8 (218 mg, 0.40 mmol) in MeOH (4 mL), was treated with NaOMe in MeOH as described for the preparation of 4. Chromatography of the residue (8:1 CHCl3–MeOH) afforded 7 (146 mg, 73% in two steps from 3) as white foam, which was in all aspects identical with 7 described in B.
:1 toluene–acetone) of the residue furnished 11 (1.44 g, 64%) as white solid. A portion of 11 was crystallized (EtOH) to give material, mp 148–149 °C; Rf 0.4 (4:1 toluene–acetone); [α]23D −23.5 (c 0.8, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 5.19 (t, overlapped, J 9.5 Hz, H-3I), 5.18 (t, overlapped, J 9.5 Hz, H-3II), 5.07 (t, 1H, J 9.7 Hz, H-4II), 4.99 (dd, 1H, J1II,2II 8.0 Hz, J2II,3II 9.6 Hz, H-2II), 4.93 (dd, 1H, J1I,2I 8.0 Hz, J2I,3I 9.7 Hz, H-2I), 4.88 (t, 1H, J 9.7 Hz, H-4I), 4.60 (d, 1H, H-1II), 4.46 (d, 1H, H-1I), 4.27 (dd, 1H, J5II,6aII 4.8 Hz, J6aII,6bII 12.3 Hz, H-6aII), 4.13 (dd, 1H, J5II,6bII 2.3 Hz, H-6bII), 3.88 (dt, overlapped, J 6.2, 9.6 Hz, H-1′a), 3.85 (dd, overlapped, J5I,6aI 1.6 Hz, J6aI,6bI 11.1 Hz, H-6aI), 3.70–3.65 (m, overlapped, 2H, H-5II, H-5I), 3.62 (m, 1H, 6bI), 3.53 (t, 2H, J 6.7 Hz, H-6′a,b), 3.47 (dt, 1H, J 6.6 Hz, H-1′b), 2.09, 2.04, 2.03, 2.02, 2.00, 1.99 (s, overlapped, 21H, 7 COCH3), 1.77 (m, 2H, H-5′a,b), 1.59 (m, 2H, H-2′a,b), 1.45 (m, 2H, H-4′a,b), 1.37 (m, 2H, H-3′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.59, 170.22, 170.17, 169.60, 169.39, 169.26, 169.18 (7 COCH3), 100.79 (C-1II), 100.55 (C-1I), 73.36 (C-5I), 72.78, 72.74 (C-3II, C-3I), 71.96 (C-5II), 71.37 (C-2I), 71.11 (C-2II), 69.75 (C-1′), 69.17 (C-4I), 68.30 (C-6I), 68.27 (C-4II), 61.81 (C-6II), 44.92 (C-6′), 32.46 (C-5′), 29.21 (C-2′), 26.52 (C-4′), 25.16 (C-3′), 20.71, 20.64, 20.60, 20.59, 20.57 (overlapped, 7 COCH3) ppm. HRMS (ESI): m/z calcd for C32H47ClO18Na [M + Na]+: 777.2349, found: 777.2344; anal. calcd for C32H47ClO18: C, 50.90; H, 6.27, found: C, 50.61; H, 6.18.:1 toluene–acetone) gave 12 (2.44 g, 95%) as white solid. A portion of 12 when recrystallized (EtOH) as described for azide 3 showed mp 129–130 °C; Rf 0.4 (4:1 toluene–acetone); [α]23D −22.3 (c 0.9, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 5.19 (t, overlapped, J 9.5 Hz, H-3I), 5.18 (t, overlapped, J 9.5 Hz, H-3II), 5.07 (t, 1H, J 9.7 Hz, H-4II), 4.99 (dd, 1H, J1II,2II 8.0 Hz, J2II,3II 9.6 Hz, H-2II), 4.93 (dd, 1H, J1I,2I 8.0 Hz, J2I,3I 9.7 Hz, H-2I), 4.88 (t, 1H, J 9.7 Hz, H-4I), 4.59 (d, 1H, H-1II), 4.45 (d, 1H, H-1I), 4.27 (dd, 1H, J5II,6aII 4.9 Hz, J6aII,6bII 12.3 Hz, H-6aII), 4.13 (dd, 1H, J5II,6bII 2.3 Hz, H-6bII), 3.88 (dt, overlapped, J 6.3, 9.6 Hz, H-1′a), 3.85 (dd, overlapped, J5I,6aI 2.0 Hz, J6aI,6bI 11.0 Hz, H-6aI), 3.70–3.66 (m, overlapped, H-5II, H-5I), 3.62 (m, 1H, 6bI), 3.47 (dt, 1H, J 6.6 Hz, H-1′b), 3.27 (t, 2H, J 6.9 Hz, H-6′a,b), 2.09, 2.04, 2.03, 2.02, 2.00, 1.99 (s, overlapped, 7 COCH3), 1.59 (m, overlapped, 4H, H-2′a,b, H-5′a,b), 1.38 (m, overlapped, 4H, H-3′a,b, H-4′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.59, 170.21, 170.17, 169.60, 169.39, 169.26, 169.18 (7 COCH3), 100.79 (C-1II), 100.55 (C-1I), 73.34 (C-5I), 72.78, 72.73 (C-3II, C-3I), 71.96 (C-5II), 71.36 (C-2I), 71.11 (C-2II), 69.75 (C-1′), 69.17 (C-4I), 68.31 (C-6I), 68.27 (C-4II), 61.82 (C-6II), 51.32 (C-6′), 29.23, 28.74 (C-5′,C-2′), 26.40, 25.44 (C-4′, C-3′), 20.71, 20.60, 20.63, 20.60, 20.58, 20.57, 20.56 (overlapped, 7 COCH3) ppm. HRMS (ESI): m/z calcd for C32H47N3O18Na [M + Na]+: 784.2752; found: 784.2765; anal. calcd for C32H47N3O18: C, 50.46; H, 6.22; N, 5.52; found: C, 50.36; H, 6.15; N, 5.48.A. Acetate 12 (0.78 g, 1.02 mmol) in MeOH (5 mL), was treated with a solution of NaOMe in MeOH as described for preparation of 4 to afford 6-azidohexyl β-gentiobioside 13 (0.46 g, 95%) as white foam. Rf 0.46 (1
:1 DCM–MeOH); 1H NMR (MeOD, 600 MHz): δ 4.38 (d, 1H, J1II,2II 7.8 Hz, H-1II), 4.26 (d, 1H, J1I,2I 7.8 Hz, H-1I), 4.14 (dd, 1H, J5I,6aI 2.0 Hz, J6aI,6bI 11.5 Hz, H-6aI), 3.89 (dt, overlapped, J 6.6, 9.6 Hz, H-1′a), 3.87 (dd, overlapped, H-6aII), 3.78 (dd, 1H, J5I,6aI 5.6 Hz, H-6bI), 3.67 (dd, 1H, J5II,6aII 5.5 Hz, J6aII,6bII 11.8 Hz, H-6bII), 3.56 (dt, 1H, J 6.6 Hz, H-1′b), 3.44 (m, 1H, H-5I), 3.37–3.33 (m, overlapped, 6H, H-3I, H-3II, H-4I), 3.30–3.25 (m, overlapped, 4H, H-4II, H-6′a,b, H-5II), 3.22 (dd, 1H, J2II,3II 9.1 Hz, H-2II), 3.18 (m, 1H, H-2I), 1.62 (m, overlapped, 4H, H-2′a,b, H-5′a,b), 1.42 (m, overlapped, 4H, H-3′a,b, H-4′a,b) ppm; 13C NMR (MeOD, 150 MHz): δ 104.82 (C-1II), 104.39 (C-1I), 78.01, 77.97 (C-5I, C-3I, C-3II), 77.01 (C-5I), 75.06 (C-2I, C-2II), 71.57 (C-4II), 71.46 (C-4I), 70.84 (C-1′), 69.76 (C-6I), 62.74 (C-6II), 52.39 (C-6′), 30.64, 29.85, 27.61, 26.64 (C-2′, C-5′, C-4′, C-3′) ppm. HRMS (ESI): m/z calcd for C18H33N3O11Na [M + Na]+: 490.2013, found: 490.2011.A solution of the foregoing azido sugar 13 (0.46 g, 0.98 mmol) in 5% AcOH in MeOH (4 mL) was treated overnight with hydrogen in presence of 5% Pd/C (90 mg). The mixture was filtered through Celite pad, the pad was washed with MeOH, and the filtrate was concentrated. The residue was a mixture of 14 and 15 (MALDI-TOF MS: 14, C18H35NO11Na [M + Na]+: calcd 464.21; found 464.22 and 15, C36H67NO22Na [M + Na]+: calcd 888.40; found 888.39), analogs of 5 and 6.
The foregoing mixture was dissolved in MeOH (3 mL) and treated overnight with di-tert-butyl dicarbonate (0.32 g, 1.47 mmol). After concentration, the residue was chromatographed (5:1 CHCl3–MeOH) to yield 16 as white foam (0.22 g, 38% in three steps from 12). Rf 0.3 (5:1 CHCl3–MeOH); [α]23D −32.9 (c 0.8, MeOH). 1H NMR (MeOD, 600 MHz): δ 4.38 (d, 1H, J1II,2II 7.8 Hz, H-1II), 4.25 (d, 1H, J1I,2I 7.8 Hz, H-1I), 4.14 (dd, 1H, J5I,6aI 2.0 Hz, J6aI,6bI 11.5 Hz, H-6aI), 3.88 (dt, overlapped, H-1′a), 3.87 (dd, overlapped, H-6aII) 3.78 (dd, 1H, J5I,6bI 5.6 Hz, H-6bI), 3.67 (dd, 1H, J5II,6bII 5.4 Hz, J6aII,6bII 11.8 Hz, H-6bII), 3.55 (dt, 1H, J 6.6, 9.6 Hz, H-1′b), 3.44 (m, 1H, H-5I), 3.37–3.33 (m, overlapped, H-3I, H-3II, H-4I), 3.29–3.24 (m, overlapped, H-4II, H-5II), 3.22 (dd, 1H, J2II,3II 9.1 Hz, H-2II), 3.17 (m, 1H, H-2I), 3.03 (t, 2H, J 7.1 Hz, H-6′a,b), 1.63 (m, 2H, H-2′a,b), 1.49–1.38 (m, overlapped, H-3′a,b, H-5′a,b), 1.43 (s, overlapped, NHCOOC(CH3)3), 1.35 (m, 2H, H-4′a,b) ppm; 13C NMR (MeOD, 150 MHz): δ 158.55 (NHCOOC(CH3)3), 104.81 (C-1II), 104.38 (C-1I), 79.79 (NHCOOC(CH3)3), 78.00, 77.96 (C-5I, C-3I, C-3II), 77.02 (C-5I), 75.06 (C-2I, C-2II), 71.57 (C-4II), 71.46 (C-4I), 70.93 (C-1′), 69.74 (C-6I), 62.74 (C-6II), 41.29 (C-6′), 30.90 (C-5′), 30.70 (C-2′), 28.80 (NHCOOC(CH3)3), 27.65 (C-4′), 26.76 (C-3′) ppm. HRMS (ESI): m/z calcd for C23H43NO13Na [M + Na]+: 564.2641; found: 564.2632.
B. A solution of azide 12 (1.0 g, 1.31 mmol) and di-tert-butyl dicarbonate (0.57 g, 2.62 mmol) in DCM (4 mL) was mixed with a suspension of 5% Pd/C (0.20 g) in DCM (3 mL) and the mixture was stirred at room temperature under H2 atmosphere until TLC showed complete consumption of the starting material (∼6 h). After conventional work-up, as described above, chromatography (2
:1 toluene–EtOAc) afforded 6-N-(tert-butyloxycarbonyl)hexyl 2,3,4-tri-O-acetyl-6-O-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-β-D-glucopyranoside [6-N-(tert-butyloxycarbonyl) hexyl 2,3,4,2′,3′,4′,6′-hepta-O-acetyl-β-gentiobioside] 17 (0.867 g, 78%) as white solid. Crystalline 17 showed mp 126–127 °C (EtOH), Rf 0.4 (4:1 toluene–acetone) and [α]23D −16.2 (c 0.7, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 5.18 (t, overlapped, J 9.5 Hz, H-3I), 5.17 (t, overlapped, J 9.5 Hz, H-3II), 5.07 (t, 1H, J 9.6 Hz, H-4II), 4.99 (dd, 1H, J1II,2II 8.0 Hz, J2II,3II 9.6 Hz, H-2II), 4.93 (dd, 1H, J1I,2I 8.0 Hz, J2I,3I 9.6 Hz, H-2I), 4.88 (t, 1H, J 9.6 Hz, H-4I), 4.60 (d, overlapped, H-1II), 4.58 (broad s, overlapped, NH), 4.45 (d, 1H, H-1I), 4.27 (dd, 1H, J5II,6aII 4.9 Hz, J6aII,6bII 12.3 Hz, H-6aII), 4.13 (dd, 1H, J5II,6bII 2.3 Hz, H-6bII), 3.86 (dt, overlapped, H-1′a), 3.85 (dd, overlapped, 6aI), 3.68 (m, overlapped, 2H, H-5II, H-5I), 3.62 (m, overlapped, 6bI), 3.46 (dt, 1H, J 6.6, 9.5 Hz, H-1′b), 3.10 (m, 2H, H-6′a,b), 2.09, 2.04, 2.03, 2.02, 2.00, 1.99 (7 s, overlapped, 21H, 7 COCH3), 1.57 (m, 2H, H-2′a,b), 1.45 (m, overlapped, H-5′a,b), 1.44 (s, overlapped, NHCOOC(CH3)3), 1.33 (m, overlapped, 4H, H-3′a,b, H-4′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.60, 170.21, 170.20, 169.61, 169.39, 169.29, 169.20 (7 COCH3), 155.96 (NHCOOC(CH3)3), 100.78 (C-1II), 100.54 (C-1I), 79.01 (NHCOOC(CH3)3), 73.34 (C-5I), 72.78, 72.75 (C-3II, C-3I), 71.94 (C-5II), 71.36 (C-2I), 71.10 (C-2II), 69.89 (C-1′), 69.18 (C-4I), 68.30 (C-6I), 68.27 (C-4II), 61.81 (C-6II), 40.46 (C-6′), 29.98 (C-5′), 29.27 (C-2′), 28.41 (NHCOOC(CH3)3), 26.48, 25.57 (C-4′, C-3′), 20.71, 20.64, 20.60, 20.58, 20.57 (overlapped, 7 COCH3) ppm. HRMS (ESI): m/z calcd for C32H57NO20Na [M + Na]+: 858.3372; found: 858.3373; anal. calcd for C32H57NO20: C, 53.17; H, 6.87; N, 1.68; found: C, 53.24; H, 6.79; N, 1.72. The side product 18 was subsequently eluted from the column and obtained as white solid. 1H NMR (CDCl3, 600 MHz): δ 5.18 (t, overlapped, J 9.5 Hz, H-3I), 5.17 (t, overlapped, J 9.5 Hz, H-3II), 5.07 (t, 2H, J 9.6 Hz, H-4II), 4.99 (dd, 2H, J1II,2II 8.0 Hz, J2II,3II 9.6 Hz, H-2II), 4.93 (dd, 2H, J1I,2I 8.0 Hz, J2I,3I 9.6 Hz, H-2I), 4.88 (t, 2H, J 9.6 Hz, H-4I), 4.50 (d, 2H, H-1II), 4.45 (d, 2H, H-1I), 4.27 (dd, 2H, J5II,6aII 4.9 Hz, J6aII,6bII 12.3 Hz, H-6aII), 4.13 (dd, 2H, J5II,6bII 2.3 Hz, H-6bII), 3.90–3.85 (m, overlapped, 4H, H-1′a, 6aI), 3.68 (m, overlapped, 4H, H-5II, H-5I), 3.62 (m, 2H, 6bI), 3.45 (dt, 2H, J 6.6, 9.5 Hz, H-1′b), 3.13 (m, 4H, H-6′a,b), 2.09, 2.04, 2.03, 2.02, 2.00, 1.99 (7 s, overlapped, 42H, 7 COCH3), 1.56 (m, 4H, H-2′a,b), 1.48 (m, overlapped, H-5′a,b), 1.44 (s, overlapped, NHCOOC(CH3)3), 1.33 (m, 4H, 2 × CH2 linker), 1.26 (m, 4H, 2 × CH2 linker) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.57, 170.20, 170.15, 169.60, 169.38, 169.23, 169.18 (7 COCH3), 155.51 (NHCOOC(CH3)3), 100.75 (C-1II), 100.53 (C-1I), 78.94 (NHCOOC(CH3)3), 73.22 (C-5I), 72.78, 72.73 (C-3II, C-3I), 71.93 (C-5II), 71.35 (C-2I), 71.07 (C-2II), 69.90 (C-1′), 69.18 (C-4I), 68.32 (C-6I), 68.25 (C-4II), 61.78 (C-6II), 46.86 (C-6′), 29.37 (CH2 linker), 28.46 (overlapped, NHCOOC(CH3)3, CH2 linker), 26.67, 25.67 (each CH2 linker), 20.69, 20.62, 20.61, 20.59, 20.57, 20.55 (overlapped, 7 COCH3) ppm. HRMS (ESI): m/z calcd for C69H103NO38Na [M + Na]+: 1576.6056; found: 1576.6051.Deacetylation of 17 (0.45 g, 0.54 mmol) was performed with NaOMe in MeOH (4 mL) as described above, followed by chromatography (5:1 CHCl3–MeOH) to afford 16 (0.28 g, 96%) as white foam, which was in all aspects identical with the material described above.
:1 toluene–EtOAc) to give 6-N-(tert-butyloxycarbonyl)hexyl 2,3,4,6-tetra-O-propargyl-β-D-glucopyranoside 19 (120 mg, 75%) as colorless syrup, Rf 0.3 (7:1 toluene–EtOAc). 1H NMR (CDCl3, 600 MHz): δ 4.55–4.36 (m, 7H, 3 OCH2CCH, NH), 4.26 (d, overlapped, J1,2 7.9 Hz, H-1), 4.26–4.18 (m, overlapped, OCH2CCH), 3.88 (dt, 1H, J 6.5, 9.5 Hz, H-1′a), 3.84 (dd, 1H, J5,6a 1.9 Hz, J6a,6b 10.8 Hz, H-6a), 3.76 (dd, 1H, J5,6b 4.9 Hz, H-6b), 3.54 (t, 1H, J 8.9 Hz, H-3) 3.47 (m, overlapped, H-1′b), 3.44 (m, overlapped, H-4), 3.38 (ddd, 1H, J5,4 9.8 Hz, H-5), 3.31 (dd, 1H, J1,2 7.9 Hz, J2,3 8.9 Hz, H-2), 3.10 (m, 2H, J 6.0 Hz, H-6′a,b), 2.46 (m, 4H, 4 OCH2CCH), 1.60 (m, 2H, H-2′a,b), 1.49–1.44 (m, overlapped, H-5′a,b), 1.44 (s, overlapped, NHCOOC(CH3)3), 1.37 (m, overlapped, H-3′a,b), 1.33 (m, overlapped, H-4′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 155.92 (NHCOOC(CH3)3), 102.98 (C-1), 83.31 (C-3), 81.33 (C-2), 80.02, 79.93, 79.80, 79.52 (4 OCH2CCH), 78.98 (NHCOOC(CH3)3), 76.14 (C-4), 74.62, 74.33, 74.24, 74.23 (4 OCH2CCH), 74.05 (C-5), 69.90 (C-1′), 68.47 (C-6), 60.20, 59.94, 59.29, 58.61 (4 OCH2CCH), 40.47 (C-6′), 29.95 (C-5′), 29.46 (C-2′), 28.38 (NHCOOC(CH3)3), 26.47 (C-4′), 25.67 (C-3′) ppm. HRMS (ESI): m/z calcd for C29H41NO8Na [M + Na]+: 554.2730; found: 554.2731.To a solution of sugar alkyne 19 (120 mg, 0.23 mmol) and sugar azide 3 (534 mg, 1.13 mmol) in dry DCM (6.0 mL), CuI (43 mg, 0.23 mmol) and DIPEA (39 μL, 0.23 mmol) were added and the mixture was stirred at room temperature under inert atmosphere overnight. The mixture was concentrated and the residue was partitioned between brine and EtOAc. The organic layer was dried with NaSO4, concentrated and chromatography (2:1 CHCl3–acetone) followed by freeze-drying yielded 20 as white solid (426 mg, 77%), Rf 0.2 (2:1 CHCl3–acetone); [α]23D −18.0 (c 0.7, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 8.10 (s, 1H, H-triazole), 7.90 (s, overlapped, 2H, H-triazole), 7.67 (s, 1H, H-triazole), 5.20 (t, 4H, J 9.5 Hz, 4 H-3II), 5.08 (t, 4H, J 9.7 Hz, 4 H-4II), 5.04–4.89 (m, overlapped, OCH2-triazole), 4.97 (t, overlapped, J 8.8 Hz, 4 H-2II), 4.84–4.67 (m, overlapped, OCH2-triazole), 4.69 (bs, overlapped, NH), 4.48 (d, overlapped, 4H, J1II,2II 8.0 Hz, 4 H-1II), 4.35 (m, overlapped, 4 H-6′′a,b), 4.32 (d, overlapped, J1I,2I 7.9 Hz, H-1I), 4.27 (dd, 4H, J5II,6aII 4.7 Hz, J6aII,6bII 12.2 Hz, 4 H-6aII), 4.13 (dd, 4H, J5II,6bII 2.2 Hz, 4 H-6bII), 3.91 (dt, 1H, J 6.6, 9.4 Hz, H-1′a), 3.87–3.82 (m, 4H, 4 H-1′′a), 3.79 (m, 2H, H-6a,bI), 3.69 (ddd, 4H, J4II,5II 9.9 Hz, 4 H-5II), 3.51 (m, overlapped, H-3I, H-4I, H-1′b), 3.49–3.44 (m, 4H, 4 H-1′′b), 3.37 (m, 1H, H-5I), 3.33 (m, 1H, H-2I), 3.10 (m, 2H, H-6′a,b), 2.08 (s, 12H, 4 COCH3), 2.03, 2.02 (s, overlapped, 24H, 8 COCH3), 2.00 (s, 12H, 4 COCH3), 1.91 (m, 8H, 4 H-5′′a,b), 1.65 (m, overlapped, H-2′a,b), 1.56 (m, overlapped, 4 H-2′′a,b), 1.49 (m, overlapped, H-5′a,b), 1.43 (s, overlapped, NHCOOC(CH3)3), 1.42–1.30 (m, 4 H-3′′a,b, 4 H-4′′a,b, H-3′a,b, H-4′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.61, 170.23, 169.37, 169.23, (4 COCH3), 156.01 (NHCOOC(CH3)3), 144.78, 144.72, 144.53, 144.31 (4 triazole C-4), 124.30, 123.73, 123.35, 122.84 (4 triazole C-5), 103.42 (C-1I), 100.77 (C-1II), 83.85 (C-3I), 81.67 (C-2I), 78.91 (NHCOOC(CH3)3), 77.49 (C-4I), 74.45 (C-5I), 72.80 (C-3II), 71.73 (C-5II), 71.30 (C-2II), 69.94 (C-1′), 69.84–69.81 (C-1′′), 69.05 (C-6I), 68.44 (C-4II), 66.28, 65.76, 65.72, 64.91 (4 OCH2-triazole), 61.93 (C-6II), 50.43, 50.30, 50.22, 50.19 (C-6′′), 40.47 (C-6′), 30.13 (C-5′′), 29.94 (C-5′), 29.56 (C-2′), 29.15 (C-2′′), 28.41 (NHCOOC(CH3)3), 26.49 (C-4′), 26.20–26.14 (C-4′′), 25.72 (C-3′), 25.28 (C-3′′), 20.71, 20.66, 20.58, 20.56 (4 COCH3) ppm; HRMS (ESI): m/z calcd for C109H166N13O48 [M + H]+: 2425.0948; found: 2425.0972; anal. calcd for C109H165N13O48: C, 53.98; H, 6.86; N, 7.51; found: C, 53.85; H, 6.87; N, 7.40.
:1 CHCl3–MeOH) showed complete disappearance of 20. After concentration, the residue was deacetylated under conditions described above, and the crude product was subjected to solid-phase extraction (C18 5 g Waters SepPack cartridge, 1:2 → 4:1 MeOH–water) to afford 21 (92 mg, 75%) as white foam, after concentration and freeze drying. Rf 0.3 (C18-silica, 1:1 MeOH–water); 1H NMR (D2O, 600 MHz): δ 8.17, 8.12, 8.04, 8.01 (4 s, 4H, 4 H-triazole), 5.06 (m, 1H, OCH2-triazole), 4.96–4.74 (m, overlapped, 6H, 3 OCH2-triazole), 4.63 (d, overlapped, J1I,2I 7.6 Hz, H-1I), 4.62 (m, overlapped, OCH2-triazole), 4.56–4.47 (m, overlapped, 12H, 4 H-1II, 4 H-6′′a,b), 4.04 (m, overlapped, 4 H-6aII), 4.01 (m, overlapped, H-1′a), 4.00–3.95 (m, overlapped, 4 H-1′′a), 3.87 (m, overlapped, H-6aI), 3.85 (m, overlapped, 4 H-6bII), 3.82 (m, overlapped, H-6bI), 3.77 (m, overlapped, H-1′b), 3.76–3.72 (m, overlapped, 4 H-1′′b), 3.74 (m, overlapped, H-3I), 3.66 (m, overlapped, H-5I), 3.64 (m, overlapped, H-4I), 3.62 (m, overlapped, 4 H-3II), 3.57 (m, 4H, 4 H-5II), 3.51 (m, overlapped, 4 H-4II), 3.48 (m, overlapped, H-2I), 3.39 (m, 4H, 4 H-2II), 3.13 (t, 2H, J 7.6 Hz, H-6′a,b), 1.98 (m, 8H, H-5′′a,b), 1.79 (m, overlapped, H-5′a,b), 1.74 (m, overlapped, H-2′a,b), 1.68 (m, overlapped, 4 H-2′′a,b), 1.53–1.43 (m, overlapped, 12H, H-4′a,b, H-3′a,b, 4 H-3′′a,b), 1.36 (m, 8H, 4 H-4′′a,b) ppm; 13C NMR (D2O, 150 MHz): δ 144.84, 144.34 (4 triazole C-4), 125.73, 125.42, 125.31, 125.28 (4 triazole C-5), 103.07 (C-1I), 102.88 (C-1II), 83.54 (C-3I), 81.70 (C-2I), 77.53 (C-4I), 76.57, 76.55 (overlapped, C-3II, C-5II), 74.04 (C-5I), 73.84 (C-2II), 71.26 (C-1′), 70.92–70.89 (C-1′′), 70.38 (C-4II), 68.39 (C-6I), 66.09, 65.60, 65.32, 63.75 (4 OCH2-triazole), 61.50 (C-6II), 50.98–50.93 (C-6′′), 40.10 (C-6′), 29.94–29.85 (C-5′′), 29.21–29.16 (C-2′′, C-2′), 27.24 (C-5′), 25.96–25.89, 25.35 (C-4′′, C-3′, C-4′) 25.11–25.04 (C-3′′) ppm; HRMS (ESI): m/z calcd for C72H126N13O30 [M + H]+: 1652.8734; found: 1652.8705.:1 toluene–EtOAc) to obtain 6-N-(tert-butyloxycarbonyl)hexyl 2,3,4,2′,3′,4′,6′-hepta-O-propargyl-β-gentiobioside 22 (120 mg, 58%) as colorless syrup; Rf 0.4 (5:1 toluene–EtOAc). 1H NMR (CDCl3, 600 MHz): δ 4.55–4.35 (m, overlapped, NH, 6 OCH2CCH), 4.40 (d, overlapped, H-1II), 4.27 (d, overlapped, J1I,2I 7.9 Hz, H-1I), 4.26–4.18 (m, overlapped, OCH2CCH), 4.23 (m, overlapped, H-6aI), 3.87 (dt, overlapped, 1H, J 6.4, 9.6 Hz, H-1′a), 3.84 (dd, overlapped, J5II,6aII 1.9 Hz, J6aII,6bII 10.8 Hz, H-6aII), 3.76 (dd, 1H, J5II,6bII 4.9 Hz, H-6bII), 3.66 (dd, 1H, J5I,6bI 7.5 Hz, J6aI,6bI 11.4 Hz, H-6bI), 3.54 (t, overlapped, J 9.0 Hz, H-3I), 3.51 (t, overlapped, J 8.8 Hz, H-3II), 3.48–3.42 (m, overlapped, H-4II, H-5I, H-1b′), 3.38 (dd, 1H, J4II,5II 9.8 Hz, H-5II), 3.34 (dd, overlapped, J1II,2II 7.9 Hz, J2II,3II 9.0 Hz, H-2II), 3.32–3.29 (m, overlapped, H-2I, H-4I), 3.10 (m, 2H, H-6′a,b), 2.46 (m, 7H, 7 OCH2CCH), 1.60 (m, 2H, H-2′a,b), 1.46 (m, overlapped, H-5′a,b), 1.44 (s, overlapped, NHCOOC(CH3)3), 1.35 (m, overlapped, 4H, H-3′a,b, H-4′a,b) ppm; 13C NMR (CDCl3, 150 MHz): δ 155.94 (NHCOOC(CH3)3), 103.32 (C-1II), 102.82 (C-1I), 83.42 (C-3I), 83.35 (C-3II), 81.41 (C-2I), 81.23 (C-2II), 80.04, 79.99, 79.96, 79.91, 79.86, 79.80, 79.62 (7 OCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e002.gif) CH), 79.03 (NHCOOC(CH3)3), 76.38 (C-4I), 76.04 (C-4II), 74.77, 74.68, 74.44, 74.37, 74.33, 74.31, 74.27 (7 OCH2CCH), 74.39 (C-5I), 74.13 (C-5II), 69.97 (C-1′), 69.05 (C-6I), 68.44 (C-6II), 60.23, 60.20, 59.99, 59.86, 59.26, 58.68 (7 OCH2CCH), 40.53 (C-6′), 30.02 (C-5′), 29.54 (C-2′), 28.42 (NHCOOC(CH3)3), 26.57, 25.79 (C-4′, C-3′) ppm. HRMS (ESI): m/z calcd for C44H57NO13Na [M + Na]+: 830.3728; found: 830.3727.
CH), 79.03 (NHCOOC(CH3)3), 76.38 (C-4I), 76.04 (C-4II), 74.77, 74.68, 74.44, 74.37, 74.33, 74.31, 74.27 (7 OCH2CCH), 74.39 (C-5I), 74.13 (C-5II), 69.97 (C-1′), 69.05 (C-6I), 68.44 (C-6II), 60.23, 60.20, 59.99, 59.86, 59.26, 58.68 (7 OCH2CCH), 40.53 (C-6′), 30.02 (C-5′), 29.54 (C-2′), 28.42 (NHCOOC(CH3)3), 26.57, 25.79 (C-4′, C-3′) ppm. HRMS (ESI): m/z calcd for C44H57NO13Na [M + Na]+: 830.3728; found: 830.3727.To a solution of the foregoing sugar alkyne 22 (120 mg, 0.14 mmol) and sugar azide 3 (560 mg, 1.19 mmol) in dry DCM, CuI (28 mg, 0.14 mmol) and DIPEA (26 μL, 0.14 mmol) were added and the mixture was stirred at room temperature overnight under inert atmosphere. The mixture was concentrated and the residue was partitioned between brine and DCM. Concentration of the organic phase, chromatography (1:1 CHCl3–acetone) and freeze-drying (benzene) afforded 23 (430 mg, 71%) as white solid; Rf 0.3 (1:1 CHCl3–acetone); [α]23D −18.9 (c 0.8, CHCl3). 1H NMR (CDCl3, 600 MHz): δ 8.15, 8.04, 7.94, 7.89, 7.77 (s, 7H, H-triazole), 5.20 (7 t, overlapped, 7H, J 9.5 Hz, 7 H-3III), 5.08 (7 t, overlapped, 7H, J 9.7 Hz, 7 H-4III), 5.06–4.66 (m, overlapped, 14H, 7 OCH2-triazole), 4.97 (m, overlapped, 7 H-2III), 4.49 (7 d, overlapped, J1III,2III 8.0 Hz, 7 H-1III), 4.47 (m, overlapped, H-1II), 4.38–4.31 (m, overlapped, 7 H-6′′a,b), 4.36 (m, overlapped, H-1I), 4.27 (7 dd, overlapped, 7H, J5III,6aIII 4.5 Hz, J6aIII,6bIII 12.2 Hz, 7 H-6aIII), 4.21 (m, 1H, H-6aI), 4.13 (7 dd, overlapped, 7H, 7 H-6bIII), 3.89–3.82 (m, overlapped, 8H, H-1′a, 7 H-1′′a), 3.80 (m, 2H, H-6a,bII), 3.75 (m, 1H, H-6bI), 3.70 (m, 7H, 7 H-5III), 3.54 (m, overlapped, H-3I), 3.51 (m, overlapped, H-5I), 3.50 (m, overlapped, H-3II, H-4II), 3.46 (m, overlapped, H-1′b, 7 H-1′′b), 3.39 (m, overlapped, H-4I), 3.38 (m, overlapped, H-5II), 3.35 (m, overlapped, H-2II), 3.33 (m, overlapped, H-2I), 3.06 (m, 2H, H-6′a,b), 2.08–2.00 (m, overlapped, 84H, 28 COCH3), 1.95–1.86 (m, 7 H-5′′a,b), 1.61–1.51 (m, overlapped, 7 H-2′′a,b), 1.46–1.27 (m, 7 H-3′′a,b, 7 H-4′′a,b) 1.42 (s, overlapped, NHCOOC(CH3)3) ppm; 13C NMR (CDCl3, 150 MHz): δ 170.63, 170.24, 169.40, 169.25 (28 COCH3), 156.05 (NHCOOC(CH3)3), 144.76, 144.70, 144.56, 144.52, 144.50, 144.31, 144.25 (7 triazole C-4), 124.29, 123.80, 123.72, 123.35, 123.14 (7 triazole C-5), 103.71 (C-1II), 103.23 (C-1I), 100.77 (C-1III), 83.96 (C-3I), 83.90 (C-3II), 81.74 (C-2I), 81.40 (C-2II), 78.83 (NHCOOC(CH3)3), 78.18 (C-4I), 77.36 (C-4II), 74.54 (C-5I), 74.47 (C-5II), 72.81 (7 C-3III), 71.73 (7 C-5III), 71.30 (7 C-2III), 69.86 (7 C-1′′), 68.99 (C-6II), 68.73 (C-6I), 68.44 (7 C-4III), 66.34, 65.92, 65.70, 64.83 (7 OCH2-triazole), 61.94 (7 C-6III), 50.39–50.23 (7 C-6′′), 40.44 (C-6′), 30.20 (7 C-5′′), 29.88, 29.66, 29.50, 29.18 (7 C-2′′), 28.43 (NHCOOC(CH3)3), 26.47, 26.24, 26,21, 26.18, 25.70, 25.29 ppm. HRMS (ESI): m/z calcd for C184H274N22O83 [M]+: 4119.7896; found: 4119.7900; anal. calcd for C184H274N22O83: C, 53.61; H, 6.70; N, 7.48; found: C, 53.84; H, 6.81; N, 7.37.
:1 CHCl3–MeOH) showed complete disappearance of 23. After concentration, the residue (145 mg, 0.036 mmol) was deacetylated (Zemplén condition) and solid-phase extraction (C18 5 g Waters SepPack cartridge, 1:2 → 4:1 MeOH/water) afforded 24 (72 mg, 70%) as white foam, after freeze drying. Rf 0.3 (C18, 1:1 MeOH–water); 1H NMR (D2O, 600 MHz): δ 8.24, 8.21, 8.20, 8.18, 8.15, 8.13, 8.12 (7 s, overlapped, 7H, 7 H-triazole), 5.17 (m, 2H, OCH2-triazole), 5.07–4.75 (m, 12H, 6 OCH2-triazole), 4.76 (m, overlapped, H-1II), 4.69 (d, 1H, J1I,2I 8.0 Hz, H-1I), 4.63 (m, overlapped, 7 H-1III), 4.60 (m, overlapped, 7 H-6′′a,b), 4.33 (m, 1H, H-6aI), 4.13 (m, 7H, 7 H-6aIII), 4.10–4.03 (m, 7H, 7 H-1′′a), 3.99 (m, overlapped, H-6aII), 3.97 (m, overlapped, H-1′a), 3.95 (m, overlapped, 7 H-6bIII), 3.94 (m, overlapped, H-6bI), 3.93 (m, overlapped, H-6bII), 3.89 (m, overlapped, H-3I), 3.85 (m, overlapped, H-3II), 3.83 (m, overlapped, 7 H-1′′b, H-5I), 3.75 (m, overlapped, H-5II, H-4II), 3.74 (m, overlapped, H-1′b), 3.71 (m, 7 H-3III, H-4I), 3.65 (m, overlapped, 7 H-5III), 3.63 (m, overlapped, 7 H-4III), 3.62 (m, overlapped, H-2II), 3.60 (m, overlapped, H-2I), 3.49 (m, 7 H-2III), 3.11 (t, J 7.4 Hz, 2H, H-6′a,b), 2.06 (m, 14H, H-5′′a,b), 1.83–1.72 (m, 16H, H-5′a,b, 7 H-2′′a,b), 1.70 (m, overlapped, H-2′a,b), 1.62–1.39 (m, 32H, H-3′a,b, H-4′a,b, 7 H-3′′a,b, 7 H-4′′a,b) ppm; 13C NMR (D2O, 150 MHz): δ 145.02, 144.96, 144.92, 144.90, 144.57, 144.45 (7 triazole C-4), 125.57, 125.33, 125.29, 125.24 (7 triazole C-5), 103.40 (C-1II), 103.07 (C-1I), 102.95 (C-1III), 83.82 (C-3I), 83.72 (C-3II), 81.84 (C-2I), 81.57 (C-2II), 78.25 (C-4I), 77.55 (C-4II), 76.63, 76.61 (C-3III, C-5III), 74.36, 74.32 (C-5I, C-5II), 73.90 (C-2III), 71.11 (C-1′), 70.88 (C-1′′), 70.46 (C-4III), 69.09 (C-6I), 68.73 (C-6II), 66.22, 66.16, 65.69, 65.65, 65.62, 65.36, 64.11 (7 OCH2-triazole), 61.59 (C-6III), 50.99–50.94 (C-6′′), 40.32 (C-6′), 30.01–29.91 (C-5′′), 29.28–29.24 (C-2′′), 28.19 (C-5′′), 26.10–25.97 (C-4′′) 25.44, 25.14 (C-3′′). HRMS (ESI): m/z calcd for C123H210N22O53 [M]+: 2843.4414; found: 2843.4429.:1 CHCl3–MeOH) to give 25 (17 mg, 53%); Rf 0.13 (2:1 CHCl3–MeOH). 1H NMR (MeOD, 600 MHz): δ 8.04, 8.02, 8.00, 7.97 (s, 4H, 4 H-triazole), 4.98 (m, 1H, OCH2-triazole), 4.89–4.78 (m, overlapped, 4H, OCH2-triazole), 4.68–4.58 (m, overlapped, 4H, OCH2-triazole, NH), 4.41–4.35 (m, overlapped, 4 H-6′′a,b, OCH3), 4.38 (m, overlapped, H-1I), 4.24 (4 d, overlapped, J1II,2II 7.8 Hz, 4H, 4 H–1II), 3.88 (m, overlapped, H-1′a), 3.87 (m, overlapped, 4 H-1′′a), 3.86 (m, overlapped, 4 H-6aII), 3.71 (m, 2H, H-6a,bI), 3.66 (dd, 4H, J5II,6bII 5.2 Hz, J6aII,6bII 11.9 Hz, 4 H-6bII), 3.58 (m, overlapped, H-6′a), 3.55 (m, overlapped, H-1′b), 3.54 (m, overlapped, H-3I), 3.52 (m, overlapped, 4 H-1′′b), 3.46 (m, overlapped, H-4I), 3.40 (m, overlapped, H-5I), 3.39 (m, overlapped, H-6′b), 3.35 (m, overlapped, 4 H-3II), 3.29–3.24 (m, overlapped, 9H, 4 H-4II, 4 H-5II, H-2I), 3.17 (4 t, overlapped, J 8.5 Hz, 4H, 4 H-2II), 1.89 (m, 8H, 4 H-5′′a,b), 1.65–1.55 (m, overlapped, 12H, 4 H-2′′a,b), 1.46–1.37 (m, overlapped, 12H, 4 H-3′′a,b), 1.31 (m, overlapped, 8H, 4 H-4′′a,b) ppm; 13C NMR (MeOD, 150 MHz): δ 190.03 (d, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 184.82 (d, CO), 177.80 (d, NHCCOCH3), 174.49 (d, NHCCOCH3), 146.29, 146.20, 145.87, 145.79 (4 triazole C-4), 125.38, 125.29, 125.22, 125.17 (4 triazole C-5), 102.61 (C-1I), 102.35 (C-1II), 84.88 (C-3I), 82.97 (C-2I), 78.48 (C-4I), 78.12 (C-3II), 77.91 (C-5II), 75.62 (C-5I), 75.13 (C-2II), 71.68 (C-4II), 70.79 (C-1′), 70.57 (C-1′′), 70.07 (C-6I), 67.08, 66.34, 65.32 (overlapped, 4 OCH2-triazole), 62.79 (C-6II), 61.19, 61.07 (two signals, OCH3), 51.30 (C-6′′), 45.58–45.28 (d, C-6′), 31.39, 31.21 (C-5′′), 30.60, 30.50 (C-2′′), 27.22 (C-4′′), 27.08, 26.77, 26.44 (C-3′′) ppm. HRMS (ESI): m/z calcd for C77H128N13O33 [M + H]+: 1762.8738; found: 1762.8719.:1). The mixture was gently stirred at room temperature for 48 h, when SELDI-TOF showed the ratio of hapten–BSA to be 4.1:1 (conjugation efficiency, 68%). The mixture was transferred into an Amicon Ultra (0.5 mL, 30 K cutoff) centrifuge tube and dialyzed (centrifugation at 4 °C, 7 min, 8 times) against 10 mM aqueous (NH4)2CO3. After lyophilization, 19.87 mg (94%, based on BSA) of conjugate 26 was obtained as white solid.:1 EtOAc–MeOH) to yield 27 (13 mg, 50%); Rf 0.2 (1:1 EtOAc–MeOH). 1H NMR (MeOH, 600 MHz): δ 8.07, 8.05, 8.04, 8.03, 7.99 (s, 7H, H-triazole), 5.02–4.79 (m, overlapped, 10H, 5 OCH2-triazole), 4.69–4.61 (m, overlapped, 4H, 2 OCH2-triazole), 4.47 (m, 1H, H-1II), 4.40–4.34 (m, overlapped, 7 H-6′′a,b, OCH3), 4.39 (m, overlapped, H-1I), 4.24 (7 d, overlapped, 7H, 7 H-1III), 4.07 (m, 1H, H-6aI), 3.90–3.84 (m, overlapped, 7 H-6aIII, 7 H-1′′a), 3.81 (m, overlapped, H-1′a), 3.73 (m, 2H, H-6a,bII), 3.66 (7 dd, overlapped, 2H, J5III,6aIII 5.0 Hz, J6aIII,6bIII 12.0 Hz, 7 H-6bIII), 3.63 (m, overlapped, H-6bI), 3.60–3.45 (m, overlapped, H-3I, H-6′a, H-3II, 7 H-1′′b, H-5I, H-1′b, H-4II), 3.45–3.38 (m, overlapped, H-4I, H-5II), 3.37 (m, overlapped, H-6′b), 3.35 (m, overlapped, 7 H-3III), 3.32 (m, overlapped, H-2I), 3.30 (m, overlapped, H-2II), 3.28 (m, overlapped, 7 H-4III), 3.26 (m, overlapped, 7 H-5III), 3.17 (7 t, overlapped, 7 H-2III), 1.89 (m, 14H, 7 H-5′′a,b), 1.63–1.53 (m, 18H, 7 H-2′′a,b), 1.46–1.38 (m, 14H, 7 H-3′′a,b), 1.35–1.27 (m, 18H, 7 H-4′′a,b) ppm; 13C NMR (MeOH, 150 MHz): δ 190.01, 184.87, 178.01, 174.47 (4 d), 146.30, 146.22, 146.19, 146.14, 145.82 (overlapped, 7 triazole C-4), 125.43–125.17 (overlapped, 7 triazole C-5), 104.81 (C-1II), 104.51 (C-1I), 104.35 (C-1III), 85.08 (C-3I, C-3II), 83.05 (C-2I), 82.78 (C-2II), 78.95 (C-4I), 78.45 (C-4II), 78.13 (C-3III), 77.91 (C-5III), 75.72 (C-5II), 75.53 (C-5I), 75.13 (C-2III), 71.68 (C-4III), 70.83 (C-1′), 70.58 (C-1′′), 70.15 (C-6II), 69.69 (C-6I), 67.20, 67.16, 66.45, 66.33, 65.42 (overlapped, 7 OCH2-triazole), 62.80 (C-6III), 61.30, 61.12 (two signals, OCH3), 51.30 (C-6′′), 45.57, 45.28 (d, C-6′), 31.88, 31.40, 31.23 (C-5′′), 30.78, 30.52 (C-2′′), 27.26 (C-4′′), 27.07, 27.04, 26.69, 26.46 (C-3′′) ppm. HRMS (ESI): m/z calcd for C128H212N22O56 [M]+: 2953.4418; found: 2953.4443.:1 (conjugation efficiency, 62%).
O), 184.82 (d, CO), 177.80 (d, NHCCOCH3), 174.49 (d, NHCCOCH3), 146.29, 146.20, 145.87, 145.79 (4 triazole C-4), 125.38, 125.29, 125.22, 125.17 (4 triazole C-5), 102.61 (C-1I), 102.35 (C-1II), 84.88 (C-3I), 82.97 (C-2I), 78.48 (C-4I), 78.12 (C-3II), 77.91 (C-5II), 75.62 (C-5I), 75.13 (C-2II), 71.68 (C-4II), 70.79 (C-1′), 70.57 (C-1′′), 70.07 (C-6I), 67.08, 66.34, 65.32 (overlapped, 4 OCH2-triazole), 62.79 (C-6II), 61.19, 61.07 (two signals, OCH3), 51.30 (C-6′′), 45.58–45.28 (d, C-6′), 31.39, 31.21 (C-5′′), 30.60, 30.50 (C-2′′), 27.22 (C-4′′), 27.08, 26.77, 26.44 (C-3′′) ppm. HRMS (ESI): m/z calcd for C77H128N13O33 [M + H]+: 1762.8738; found: 1762.8719.:1). The mixture was gently stirred at room temperature for 48 h, when SELDI-TOF showed the ratio of hapten–BSA to be 4.1:1 (conjugation efficiency, 68%). The mixture was transferred into an Amicon Ultra (0.5 mL, 30 K cutoff) centrifuge tube and dialyzed (centrifugation at 4 °C, 7 min, 8 times) against 10 mM aqueous (NH4)2CO3. After lyophilization, 19.87 mg (94%, based on BSA) of conjugate 26 was obtained as white solid.:1 EtOAc–MeOH) to yield 27 (13 mg, 50%); Rf 0.2 (1:1 EtOAc–MeOH). 1H NMR (MeOH, 600 MHz): δ 8.07, 8.05, 8.04, 8.03, 7.99 (s, 7H, H-triazole), 5.02–4.79 (m, overlapped, 10H, 5 OCH2-triazole), 4.69–4.61 (m, overlapped, 4H, 2 OCH2-triazole), 4.47 (m, 1H, H-1II), 4.40–4.34 (m, overlapped, 7 H-6′′a,b, OCH3), 4.39 (m, overlapped, H-1I), 4.24 (7 d, overlapped, 7H, 7 H-1III), 4.07 (m, 1H, H-6aI), 3.90–3.84 (m, overlapped, 7 H-6aIII, 7 H-1′′a), 3.81 (m, overlapped, H-1′a), 3.73 (m, 2H, H-6a,bII), 3.66 (7 dd, overlapped, 2H, J5III,6aIII 5.0 Hz, J6aIII,6bIII 12.0 Hz, 7 H-6bIII), 3.63 (m, overlapped, H-6bI), 3.60–3.45 (m, overlapped, H-3I, H-6′a, H-3II, 7 H-1′′b, H-5I, H-1′b, H-4II), 3.45–3.38 (m, overlapped, H-4I, H-5II), 3.37 (m, overlapped, H-6′b), 3.35 (m, overlapped, 7 H-3III), 3.32 (m, overlapped, H-2I), 3.30 (m, overlapped, H-2II), 3.28 (m, overlapped, 7 H-4III), 3.26 (m, overlapped, 7 H-5III), 3.17 (7 t, overlapped, 7 H-2III), 1.89 (m, 14H, 7 H-5′′a,b), 1.63–1.53 (m, 18H, 7 H-2′′a,b), 1.46–1.38 (m, 14H, 7 H-3′′a,b), 1.35–1.27 (m, 18H, 7 H-4′′a,b) ppm; 13C NMR (MeOH, 150 MHz): δ 190.01, 184.87, 178.01, 174.47 (4 d), 146.30, 146.22, 146.19, 146.14, 145.82 (overlapped, 7 triazole C-4), 125.43–125.17 (overlapped, 7 triazole C-5), 104.81 (C-1II), 104.51 (C-1I), 104.35 (C-1III), 85.08 (C-3I, C-3II), 83.05 (C-2I), 82.78 (C-2II), 78.95 (C-4I), 78.45 (C-4II), 78.13 (C-3III), 77.91 (C-5III), 75.72 (C-5II), 75.53 (C-5I), 75.13 (C-2III), 71.68 (C-4III), 70.83 (C-1′), 70.58 (C-1′′), 70.15 (C-6II), 69.69 (C-6I), 67.20, 67.16, 66.45, 66.33, 65.42 (overlapped, 7 OCH2-triazole), 62.80 (C-6III), 61.30, 61.12 (two signals, OCH3), 51.30 (C-6′′), 45.57, 45.28 (d, C-6′), 31.88, 31.40, 31.23 (C-5′′), 30.78, 30.52 (C-2′′), 27.26 (C-4′′), 27.07, 27.04, 26.69, 26.46 (C-3′′) ppm. HRMS (ESI): m/z calcd for C128H212N22O56 [M]+: 2953.4418; found: 2953.4443.:1 (conjugation efficiency, 62%).Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes of Health (NIH) and National Institute of Diabetes and Digestive and Kidney Diseases.References
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS PubMed.
- V. Verez-Bencomo, V. Fernández-Santana, E. Hardy, M. E. Toledo, M. C. Rodrıguez, L. Heynngnezz, A. Rodriguez, A. Baly, L. Herrera, M. Izquierdo, A. Villar, Y. Valdés, K. Cosme, M. L. Deler, M. Montane, E. Garcia, A. Ramos, A. Aguilar, E. Medina, G. Torano, I. Sosa, I. Hernandez, R. Martınez, A. Muzachio, A. Carmenates, L. Costa, F. Cardoso, C. Campa, M. Diaz and R. Roy, Science, 2004, 305, 522–525 CrossRef CAS PubMed.
- K. E. Stein, Int. J. Tech. Assess. Health Care, 1994, 10, 167–176 CrossRef CAS.
- Y. C. Lee, R. R. Townsend, M. R. Hardy, J. Lonngren, J. Arnarp, M. Haraldsson and H. Lonn, J. Biol. Chem., 1983, 258, 199–202 CAS.
- P. Kováč, in Protein-Carbohydrate Interactions in Infectious Diseases, ed. C. Bewley, Royal Society of Chemistry, London, 2006, pp. 175–220 Search PubMed.
- M. D. Meeks, R. Saksena, X. Ma, T. K. Wade, R. K. Taylor, P. Kováč and W. F. Wade, Infect. Immun., 2004, 72, 4090–4101 CrossRef CAS PubMed.
- R. Saksena, X. Ma, T. K. Wade, P. Kováč and W. F. Wade, FEMS Immunol. Med. Microbiol., 2006, 46, 116–128 CrossRef PubMed.
- A. Chernyak, S. Kondo, T. K. Wade, M. D. Meeks, P. M. Alzari, J. M. Fournier, R. K. Taylor, P. Kováč and W. F. Wade, J. Infect. Dis., 2002, 185, 950–962 CrossRef CAS PubMed.
- W. E. Dick Jr and M. Beurret, in Conjugate Vaccines, ed. J. M. Cruse and R. E. Lewis Jr, Krager, Basel, 1989, pp. 48–114 Search PubMed.
- J. Zhang and P. Kováč, Carbohydr. Res., 1999, 321, 157–167 CrossRef CAS PubMed.
- J. Zhang and P. Kováč, Bioorg. Med. Chem. Lett., 1999, 9, 487–490 CrossRef CAS PubMed.
- Click Chemistry in Glycoscience: New developments and Strategies, ed. Z. J. Witczak and R. Bielski, Wiley, 2013 Search PubMed.
- L. F. Tietze, C. Schröter, S. Gabius, U. Brinck, A. Goerlach-Graw and H. J. Gabius, Bioconjugate Chem., 1991, 2, 148–153 CrossRef CAS PubMed.
- L. F. Tietze, M. Arlt, M. Beller, K. H. Glüsenkamp, E. Jähde and M. F. Rajewsky, Chem. Ber., 1991, 124, 1215–1221 CrossRef CAS.
- I. Velter, B. La Ferla and F. Nicotra, J. Carbohydr. Chem., 2006, 25, 97–138 CrossRef CAS.
- M. Dubber and T. K. Lindhorst, J. Carbohydr. Chem., 2001, 20, 755–760 CrossRef CAS.
- B. Gerland, A. Goudot, G. Pourceau, A. Meyer, V. Dugas, S. Cecioni, S. Vidal, E. Souteyrand, J.-J. Vasseur, Y. Chevolot and F. Morvan, Bioconjugate Chem., 2012, 23, 1534–1547 CrossRef CAS PubMed.
- M. Dubber and T. K. Lindhorst, Org. Lett., 2001, 3, 4019–4022 CrossRef CAS PubMed.
- D. J. Lee, S.-H. Yang, G. M. Williams and M. A. Brimble, J. Org. Chem., 2012, 77, 7564–7571 CrossRef CAS PubMed.
- M. L. Uhrig and J. Kovensky, in Click Chemistry in Glycosience: New Developments and Strategies, ed. Z. J. Witczak and R. Bielski, John Wiley & Sons, Inc, 2013, pp. 107–142 Search PubMed.
- M. L. Gening, D. V. Titov, S. Cecioni, A. Audfray, A. G. Gerbst, Y. E. Tsvetkov, V. B. Krylov, A. Imberty, N. E. Nifantiev and S. Vidal, Chem.–Eur. J., 2013, 19, 9272–9285 CrossRef CAS PubMed.
- C. Muller, G. Despras and T. K. Lindhorst, Chem. Soc. Rev., 2016, 45, 3275–3302 RSC.
- C. Kieburg, M. Dubber and T. K. Lindhorst, Synlett, 1997, 1447–1449 CAS.
- M. Dubber and T. K. Lindhorst, J. Org. Chem., 2000, 65, 5275–5281 CrossRef CAS PubMed.
- L. X. Wang, J. Ni and S. Singh, Bioorg. Med. Chem., 2003, 11, 159–166 CrossRef CAS PubMed.
- J. Ni, H. Song, Y. Wang, N. M. Stamatos and L.-X. Wang, Bioconjugate Chem., 2006, 17, 493–500 CrossRef CAS PubMed.
- M. Köhn, J. M. Benito, C. Ortiz Mellet, T. K. Lindhorst and J. M. García Fernández, ChemBioChem, 2004, 5, 771–777 CrossRef PubMed.
- O. Sperling, M. Dubber and T. K. Lindhorst, Carbohydr. Res., 2007, 342, 696–703 CrossRef CAS PubMed.
- Y. Gao, A. Eguchi, K. Kakehi and Y. C. Lee, Bioorg. Med. Chem., 2005, 13, 6151–6157 CrossRef CAS PubMed.
- S. Gouin, F. J. Garcia, M. E. Vanquelef, F.-Y. Dupradeau, E. Salomonsson, H. Leffler, M. Ortega-Munoz, J. Nilsson Ulf and J. Kovensky, ChemBioChem, 2010, 11, 1430–1442 CrossRef CAS PubMed.
- T.-E. Gloe, A. Müller, A. Ciuk, T. M. Wrodnigg and T. K. Lindhorst, Carbohydr. Res., 2016, 425, 1–9 CrossRef CAS PubMed.
- M. Dubber, A. Patel, K. Sadalapure, I. Aumueller and T. K. Lindhorst, Eur. J. Org. Chem., 2006, 5357–5366 CrossRef CAS.
- P. Kováč and P. Xu, in Carbohydrate Chemistry, A Specialist Periodical Report, ed. A. P. Rauter, T. K. Lindhorst and Y. Queneau, RSC, 2017, vol. 42, pp. 83–115 Search PubMed.
- M. L. Wolfrom and A. Thompson, in Methods in Carbohydrate Chemistry, ed. R. L. Whistler and M. L. Wolfrom, Academic Press, New York, 1963, vol. 2, pp. 211–215 Search PubMed.
- D. D. Reynolds and W. L. Evans, J. Am. Chem. Soc., 1938, 60, 2559–2561 CrossRef CAS.
- D. Rygielska-Tokarska, G. Andrei, D. Schools, R. Snoecks and I. E. Glowacka, Monatsh. Chem., 2016, 147, 1081–1090 CrossRef CAS.
- I.-H. An, H.-R. Seong and K. H. Ahn, Bull. Korean Chem. Soc., 2004, 25, 420–422 CrossRef CAS.
- M. Lange, A. L. Pettersen and K. Undheim, Tetrahedron, 1998, 54, 5745–5752 CrossRef CAS.
- G. Zemplén, A. Gerecs and I. Hadácsy, Berichte der Deutschen Chemischen Gesellschaft, 1936, 69, 1827–1829 CrossRef.
- V. Pavliak, P. Kováč and C. P. J. Glaudemans, Carbohydr. Res., 1991, 229, 103–116 CrossRef.
- C. W. Meldal, C. Tornoe and M. Meldal, J. Org. Chem., 2002, 67, 3057–3062 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- D. Kushwaha, P. Dwivedi, S. K. Kuanar and V. K. Tiwari, Curr. Org. Synth., 2013, 10, 90–135 CrossRef CAS.
- D. Kushwaha and V. K. Tiwari, J. Org. Chem., 2013, 78, 8184–8190 CrossRef CAS PubMed.
- S. j. Hou, R. Saksena and P. Kováč, Carbohydr. Res., 2008, 343, 196–210 CrossRef CAS PubMed.
- P. Xu, M. M. Alam, A. Kalsy, R. C. Charles, S. B. Calderwood, F. Qadri, E. T. Ryan and P. Kováč, Bioconjugate Chem., 2011, 21, 2179–2185 CrossRef PubMed.
- R. F. Chen, J. Biol. Chem., 1967, 242, 173–181 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C spectra of all new compounds. SELDI-TOF MS of the BSA conjugates. See DOI: 10.1039/c6ra27181b |
| This journal is © The Royal Society of Chemistry 2017 |