
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinitiators for two-photon polymerisation: effect of branching and viscosity on polymerisation thresholds†
Reece Whitby abdef,
Yael Ben-Talbdef,
Ryan MacMillandef,
Stefaan Janssensa,
Sebastiampillai Raymonda,
Dave Clarkea,
Jianyong Jinbf,
Andrew Kay*ae and
M. Cather Simpson*bcdef
abdef,
Yael Ben-Talbdef,
Ryan MacMillandef,
Stefaan Janssensa,
Sebastiampillai Raymonda,
Dave Clarkea,
Jianyong Jinbf,
Andrew Kay*ae and
M. Cather Simpson*bcdef
aPhotonics Team, Callaghan Innovation, Lower Hutt, New Zealand. E-mail: Andrew.Kay@callaghaninnovation.govt.nz
bSchool of Chemical Sciences, The University of Auckland, Auckland, New Zealand. E-mail: c.simpson@auckland.ac.nz; rwhi899@aucklanduni.ac.nz
cDepartment of Physics, The University of Auckland, Auckland, New Zealand
dThe Photon Factory, The University of Auckland, Auckland, New Zealand
eThe Dodd-Walls Centre for Photonic and Quantum Technologies, New Zealand
fThe MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
First published on 27th February 2017
Abstract
A series of multi-branched two-photon photoinitiators (PIs) based around the well-known triphenylamine donor core were synthesised for use in two-photon polymerisation (TPP) and are designated as compounds 6, 7 and 8. The use of a phenylene-vinylene π-system was used with an ethyl ester acceptor moiety which gives dipolar (6), quadrupolar (7) and octupolar (8) branching. Two-photon absorption cross-sections (δ2PA) of 126 GM, 358 GM and 590 GM were measured at 780 nm for 6, 7 and 8, respectively. The fluorescence quantum yields (ϕF) were determined in both MeOH and the acrylate system employed for TPP, and demonstrate the impact of viscosity upon photophysical properties of multi-branched molecules. Excellent polymerisation thresholds were demonstrated in the μW region: namely 45 μW (6), 61 μW (7) and 27 μW (8) at a writing speed of 50 μm s−1. Finally, an explanation for the disparity of polymerisation thresholds is proposed for these PIs and provides insight into the future development of low threshold PIs for TPP.
1. Introduction
The development of photoinitiators (PIs) for two-photon polymerisation (TPP) has been an area of intense research for nearly two decades.1–5 Many two-photon absorbing (2PA) materials have been developed in recent years to maximise 2PA cross-sections (δ2PA), however often neglected are key factors that make them more useful for TPP.6–10 These include the need for low fluorescence quantum yields (ϕF) and solubility in pre-polymer resins. Consequently, a large δ2PA doesn't necessarily translate into an efficient PI. Many early 2PA materials possessed high δ2PA but either exhibited large ϕF yields or poor monomer solubility.3 Large ϕF yields can lead to poor efficiency during TPP, as relaxation of the excited state via fluorescence and initiation from the triplet state are competing pathways. Therefore, minimising ϕF yields is considered a key factor for maximising a PIs ability to function efficiently.11 The presence of carbonyl groups leads to low-lying n–π* excited states being present and this can result in a lowering of the ϕF yields.12 For aromatic ketones, conjugation reduces the energy gap between singlet and triplet levels, leading to increased rate coefficients for intersystem crossing.13,14 Carbonyl groups play a dual-role, as (1) strong electron acceptors and (2) efficient spin–orbit coupling agents to promote intersystem crossing.15–17 A number of research groups have utilised carbonyl moieties in the creation of 2PA PIs, with high sensitivity and low polymerisation thresholds.2,3,11,17–21 These are often focussed on several well-known functional components, including; α-β-unsaturated ketones,3,16,21 thioxanthones,1,15 keto-coumarins,19,22 1,3-diketones23 and acylo-phosphine oxides.24 Furthermore, many groups have produced dipolar, quadrupolar and octupolar PIs for TPP.3,16,20,25,26 A number of groups have investigated the effects of branching on photophysical properties including, UV-Vis, fluorescence and on δ2PA.27–30 However, to the best of our knowledge no data exist on how branching affects polymerisation thresholds. Many of these chromophores can be considered as twisted intramolecular charge-transfer (TICT) complexes and thus, their fluorescence properties are greatly influenced by local viscosity.31Taking the above into account, we decided to utilise the strong electron donor triphenylamine for its propeller, C3 symmetric shape that can be chemically modified for dipolar, quadrupolar and octupolar branching.10,32,33 By modifying this donor core with ethyl ester functional groups to act as both an electron acceptor and to reduce ϕF yields we believe these could be novel and viable materials. Consequently, we have designed and synthesised a series of branched D–[π–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–R]n chromophores where n = 1, 2, 3 viz. 6–8. Investigation of the photophysical properties of the PIs was conducted using UV-Vis, fluorescence, fluorescence lifetime and, z-scan measurements. Finally, TPP structuring tests conducted using a femtosecond laser system were performed at different intensities and writing speeds (irradiation times) to characterise the efficiency of these PIs for TPP.
O)–R]n chromophores where n = 1, 2, 3 viz. 6–8. Investigation of the photophysical properties of the PIs was conducted using UV-Vis, fluorescence, fluorescence lifetime and, z-scan measurements. Finally, TPP structuring tests conducted using a femtosecond laser system were performed at different intensities and writing speeds (irradiation times) to characterise the efficiency of these PIs for TPP.
2. Experimental
2.1 Materials and characterisation
Commercially available reagents were purchased from Sigma-Aldrich New Zealand, Pure Science Ltd, and SiliCycle Canada and were used without further purification. For our TPP tests we purchased commercially available monomers SR415 and SR315, these were purchased directly from Sartomer and used without further purification. Reagent and analytical grade solvents were used without further purification unless otherwise stated. Flash column chromatography was carried out using Silicycle SiliaFlash F60 silica (230–400 mesh) with the stated solvent systems. Analytical thin-layer chromatography (TLC) analyses were performed on pre-coated plates (Merck aluminium sheets, silica gel 60F 254, 0.2 mm). Visualisation of chromophores was achieved by illumination under ultraviolet light (254 and 365 nm). NMR were performed on a BRUKER-500 MHz – FT-NMR spectrometer. The chemical shifts (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet) are stated in ppm using TMS as the internal standard. NMR solvents used were of ≥99.5% purity. High-resolution mass spectrometry was performed using a Waters Q-TOF Premier™ tandem mass spectrometer. Melting points were recorded with an EZ-Melt automated melting point apparatus and are uncorrected.2.2 Measurements
The UV-Vis absorption spectra were recorded on PerkinElmer Lambda 1050 WB UV/VIS/NIR spectrometer at a concentration of 1 × 10−5 mol L−1. Fluorescence spectra were recorded on a Cary Eclipse fluorescence spectrometer at a concentration of 2 × 10−6 M. The fluorescence quantum yields were measured using quinine bisulphate (QBS) in (0.05 M) sulphuric acid (ϕF = 0.52) as a reference standard.34 Two-photon absorption measurements were performed using the z-scan method, using a Toptica femtosecond fibre laser with a pulse duration of 100 fs, a repetition rate of 80 MHz and an operating wavelength of 780 nm. The resulting optical intensity at the focal point was 4.8 GW cm−2, further experimental details are provided in the ESI.† Measurements were performed using a concentration of 2 × 10−2 mol L−1 in CHCl3. Rhodamine 6G (0.5 M) in MeOH was used as a reference standard, a measured value of 33 GM was obtained and this is consistent with previous literature reports.35 The fluorescence decay profiles were acquired using the time correlated single photon counting method exciting the fluorescence with 375 nm pulsed photodiode with a pulse duration of ∼100 ps. Fluorescence decay times were determined by analysis of the decay profiles by iterative reconvolution method.2.3 Synthesis and characterisation
The intermediates 1–5 were prepared using known literature procedures and details can be found in the ESI, Fig. S1 and S2.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :petroleum spirit 60–80 °C) to give pure (E)-ethyl 4-(4-(diphenylamino)styryl)benzoate (6) 300 mg, 15% yield.36 (E)-ethyl 4-(4-(diphenylamino)styryl)benzoate (6): yellow solid, Rf 0.75 (50% DCM:petroleum spirit 60–80 °C), mp 128–130 °C (lit. 120–122 °C).36 1H NMR (CDCl3): δ 8.00 (d, J = 8.25 Hz, Ar-CO2Et, 2H), 7.52 (d, J = 8.25 Hz, Ar-CO2Et, 2H), 7.44–7.41 (m, Ph2N-Ar, 2H), 7.28–7.24 (m, Ph2-NAr, 4H), 7.15 (d, J = 16.51 Hz, –NPh–CHCH–, 1H), 7.13–7.10 (m, Ph2-NAr, 4H), 7.06–7.03 (m, Ph-N-Ar–CHCH, 4H), 7.00 (d, J = 16.51 Hz, –NPh–CHCH–, 1H), 4.38 (q, J = 7.13 Hz, 2H), 1.40 (t, J = 7.13 Hz, 3H). 13C NMR (CDCl3): 166.46, 147.97, 147.43, 142.11, 130.71, 129.98, 129.34, 128.90, 127.68, 126.01, 125.79, 124.73, 123.29, 123.18, 60.86, 14.37. HREI-MS: calcd for C29H25NO2 419.1885, found m/z [M] 419.1886 (100%); Δ = 0.2 ppm.:petroleum spirit 60–80 °C) to give pure diethyl 4,4′-((1E,1′E)-((phenylazanediyl)bis(4,1-phenylene))bis(ethene-2,1-diyl))dibenzoate (7) 100 mg, 5% yield. Diethyl 4,4′-((1E,1′E)-((phenylazanediyl)bis(4,1-phenylene))bis(ethene-2,1-diyl))dibenzoate (7): yellow solid, Rf 0.50 (50% DCM:petroleum spirit 60–80 °C), mp 109–111 °C. 1H NMR (CDCl3): δ 8.01 (d, J = 8.28 Hz, Ar-CO2Et, 4H), 7.54 (d, J = 8.28 Hz, Ar-CO2Et, 4H), 7.44–7.41 (m, J = 8.37 Hz, Ph2N-Ar, 4H), 7.30 (t, Ph-NAr2, 2H), 7.18–7.14 (m, Ph-N-Ar–CHCH, 4H), 7.11–7.07 (m, Ph-N-Ar–CHCH, 5H), 7.03 (d, Ar–CHCH, 2H), 4.38 (q, J = 7.13 Hz, 4H), 1.40 (t, J = 7.13 Hz, 6H). 13C NMR (CDCl3): 166.44, 147.46, 142.00, 131.35, 130.53, 130.00, 129.47, 129.00, 127.76, 126.16, 125.13, 123.84, 60.87, 14.36. HREI-MS: calcd for C40H35NO4 593.2561, found m/z [M] 593.2566 (90%); Δ = −0.8 ppm.:petroleum spirit 60–80 °C) to yield pure triethyl 4,4′,4′′-((1E,1′E,1′′E)-(nitrilotris(benzene-4,1-diyl))tris(ethene-2,1-diyl))tribenzoate (8) 400 mg, 46% yield. Triethyl 4,4′,4′′-((1E,1′E,1′′E)-(nitrilotris(benzene-4,1-diyl))tris(ethene-2,1-diyl))tribenzoate (8): yellow solid, Rf 0.50 (50% DCM:petroleum spirit 60–80 °C), mp 182–185 °C. 1H NMR (CDCl3): δ 8.02 (d, J = 8.24 Hz, Ar-CO2Et, 6H), 7.54 (d, J = 8.24 Hz, Ar-CO2Et, 6H), 7.45 (d, J = 8.66 Hz, Ar2N-Ar, 6H), 7.18 (d, J = 16.07 Hz, -NAr-CHCH–, 3H), 7.13 (d, J = 8.66 Hz, Ar2N-Ar, 6H), 7.05 (d, J = 16.07 Hz, -NAr–CHCH–, 3H), 4.38 (q, J = 7.10 Hz, 6H), 1.41 (t, J = 7.10 Hz, 9H). 13C NMR (CDCl3): 166.42, 147.02, 141.90, 131.89, 130.41, 130.01, 129.10, 127.85, 126.47, 126.10, 124.34, 60.89, 14.37. HREI-MS: calcd for C51H45NO6 767.3347, found m/z [M] 767.3248; Δ = 0.7 ppm.
:petroleum spirit 60–80 °C) to give pure (E)-ethyl 4-(4-(diphenylamino)styryl)benzoate (6) 300 mg, 15% yield.36 (E)-ethyl 4-(4-(diphenylamino)styryl)benzoate (6): yellow solid, Rf 0.75 (50% DCM:petroleum spirit 60–80 °C), mp 128–130 °C (lit. 120–122 °C).36 1H NMR (CDCl3): δ 8.00 (d, J = 8.25 Hz, Ar-CO2Et, 2H), 7.52 (d, J = 8.25 Hz, Ar-CO2Et, 2H), 7.44–7.41 (m, Ph2N-Ar, 2H), 7.28–7.24 (m, Ph2-NAr, 4H), 7.15 (d, J = 16.51 Hz, –NPh–CHCH–, 1H), 7.13–7.10 (m, Ph2-NAr, 4H), 7.06–7.03 (m, Ph-N-Ar–CHCH, 4H), 7.00 (d, J = 16.51 Hz, –NPh–CHCH–, 1H), 4.38 (q, J = 7.13 Hz, 2H), 1.40 (t, J = 7.13 Hz, 3H). 13C NMR (CDCl3): 166.46, 147.97, 147.43, 142.11, 130.71, 129.98, 129.34, 128.90, 127.68, 126.01, 125.79, 124.73, 123.29, 123.18, 60.86, 14.37. HREI-MS: calcd for C29H25NO2 419.1885, found m/z [M] 419.1886 (100%); Δ = 0.2 ppm.:petroleum spirit 60–80 °C) to give pure diethyl 4,4′-((1E,1′E)-((phenylazanediyl)bis(4,1-phenylene))bis(ethene-2,1-diyl))dibenzoate (7) 100 mg, 5% yield. Diethyl 4,4′-((1E,1′E)-((phenylazanediyl)bis(4,1-phenylene))bis(ethene-2,1-diyl))dibenzoate (7): yellow solid, Rf 0.50 (50% DCM:petroleum spirit 60–80 °C), mp 109–111 °C. 1H NMR (CDCl3): δ 8.01 (d, J = 8.28 Hz, Ar-CO2Et, 4H), 7.54 (d, J = 8.28 Hz, Ar-CO2Et, 4H), 7.44–7.41 (m, J = 8.37 Hz, Ph2N-Ar, 4H), 7.30 (t, Ph-NAr2, 2H), 7.18–7.14 (m, Ph-N-Ar–CHCH, 4H), 7.11–7.07 (m, Ph-N-Ar–CHCH, 5H), 7.03 (d, Ar–CHCH, 2H), 4.38 (q, J = 7.13 Hz, 4H), 1.40 (t, J = 7.13 Hz, 6H). 13C NMR (CDCl3): 166.44, 147.46, 142.00, 131.35, 130.53, 130.00, 129.47, 129.00, 127.76, 126.16, 125.13, 123.84, 60.87, 14.36. HREI-MS: calcd for C40H35NO4 593.2561, found m/z [M] 593.2566 (90%); Δ = −0.8 ppm.:petroleum spirit 60–80 °C) to yield pure triethyl 4,4′,4′′-((1E,1′E,1′′E)-(nitrilotris(benzene-4,1-diyl))tris(ethene-2,1-diyl))tribenzoate (8) 400 mg, 46% yield. Triethyl 4,4′,4′′-((1E,1′E,1′′E)-(nitrilotris(benzene-4,1-diyl))tris(ethene-2,1-diyl))tribenzoate (8): yellow solid, Rf 0.50 (50% DCM:petroleum spirit 60–80 °C), mp 182–185 °C. 1H NMR (CDCl3): δ 8.02 (d, J = 8.24 Hz, Ar-CO2Et, 6H), 7.54 (d, J = 8.24 Hz, Ar-CO2Et, 6H), 7.45 (d, J = 8.66 Hz, Ar2N-Ar, 6H), 7.18 (d, J = 16.07 Hz, -NAr-CHCH–, 3H), 7.13 (d, J = 8.66 Hz, Ar2N-Ar, 6H), 7.05 (d, J = 16.07 Hz, -NAr–CHCH–, 3H), 4.38 (q, J = 7.10 Hz, 6H), 1.41 (t, J = 7.10 Hz, 9H). 13C NMR (CDCl3): 166.42, 147.02, 141.90, 131.89, 130.41, 130.01, 129.10, 127.85, 126.47, 126.10, 124.34, 60.89, 14.37. HREI-MS: calcd for C51H45NO6 767.3347, found m/z [M] 767.3248; Δ = 0.7 ppm.2.4 Two-photon polymerisation
:50 wt% mixture of the commercial acrylates ethoxylated trimethylopropane triacrylate (SR415) and trimethylolpropane triacrylate (SR351) with a photoinitiator concentration of 6.3 × 10−6 M. The same fabrication process was implemented for all structures: the optical material was drop-cast onto a glass substrate and flattened to give a consistent thickness (2 mm). The laser beam was focussed inside the material and the focus was scanned across the material to create the desired pattern, a 150 × 150 cross-hatched square. The stage was then lowered 5 μm in the z direction to raise the focus within the material and the pattern was then written again. This process was repeated 10 times to ultimately give a 3D structure 50 μm tall. Final structures were developed by washing with methanol and drying. Scanning electron microscopy (SEM) was employed to image the structures. The structures were sputter coated prior to SEM imaging (∼56 nm).3. Results and discussion
3.1 Synthesis of triphenylamine photoinitiators 6–8
The target compounds, 6–8, were synthesised via Vilsmeier–Haack formylation and Horner–Wadsworth–Emmons coupling as shown in Scheme 1.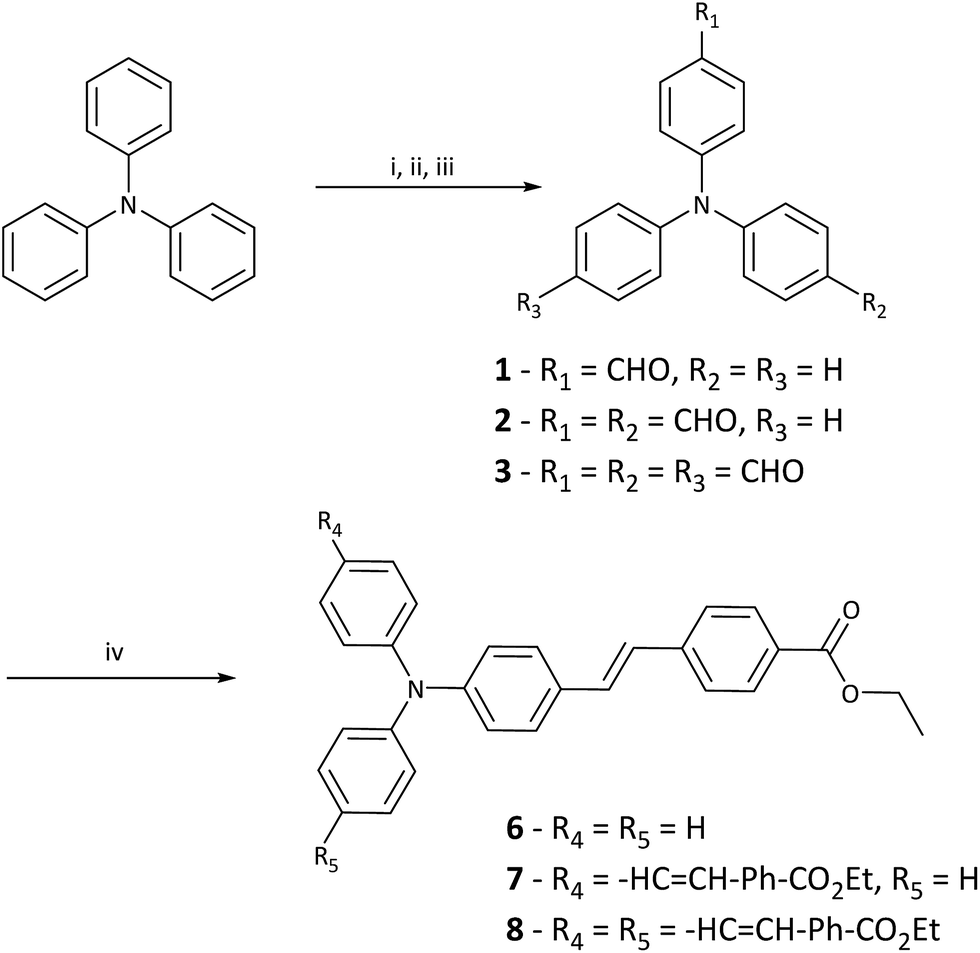 | ||
| Scheme 1 Synthetic route for 1–3 and final PIs 6–8. Reagents and conditions: (i) DMF, POCl3 (1.05 eq.), 95 °C, overnight. (ii) DMF, POCl3 (10.5 eq.), 95 °C, overnight. (iii) 1. DMF (23 eq.), POCl3 (25 eq.), 95 °C, overnight, aqueous workup and then 2. DMF (23 eq.), POCl3 (25 eq.), 95 °C, overnight, aqueous workup. (iv). t-BuOK, THF, 0 °C > r.t., overnight. | ||
4-Formyltriphenylamine (1) and 4,4′-diformyltriphenylamine (2) were prepared from triphenylamine via a Vilsmeier–Haack formylation according to the literature method.37 4,4′,4′′-trisformyltriphenylamine (3) was prepared in two steps, also using a known procedure.38
Ethyl 4-methylbenzoate was converted into ethyl 4-(bromomethyl)benzoate (4) by radical bromination followed by treatment of 4 with triethyl phosphite to yield ethyl 4-((diethoxyphosphoryl)methyl)benzoate (5).39 The dipolar (E)-ethyl 4-(4-(diphenylamino)styryl)benzoate (6) was prepared by Horner–Wadsworth–Emmons coupling of 4-formyltriphenylamine (1) and ethyl 4-((diethoxyphosphoryl)methyl)benzoate (5) under standard conditions. The synthesis of 7 and 8 was conducted in the same manner as described for 6.
3.2 Linear optical properties
The normalised one-photon absorption and fluorescence spectra of 6–8 in MeOH are shown in Fig. 1 and results outlined in Table 1. Full experimental findings are detailed in the ESI.† The absorptions around 400 nm are assigned to localised π–π* transitions of the phenylene moieties, whereas the absorption at longer wavelengths (around 400 nm) are attributed to intramolecular charge-transfer (ICT) absorption bands.40,41 The absorption maxima appear at 384 nm for 6, 404 nm for 7 and 410 nm for 8, respectively. The moderate red-shift with increased branching indicates relatively weak coupling between successive branches.42 A 20 nm red-shift in λmax between 6 and 7 is indicative of a enlargement of the ICT.43 However, as there is only a 6 nm shift between 7 and 8 this suggests that the addition of a third branch doesn't improve ICT as significantly.20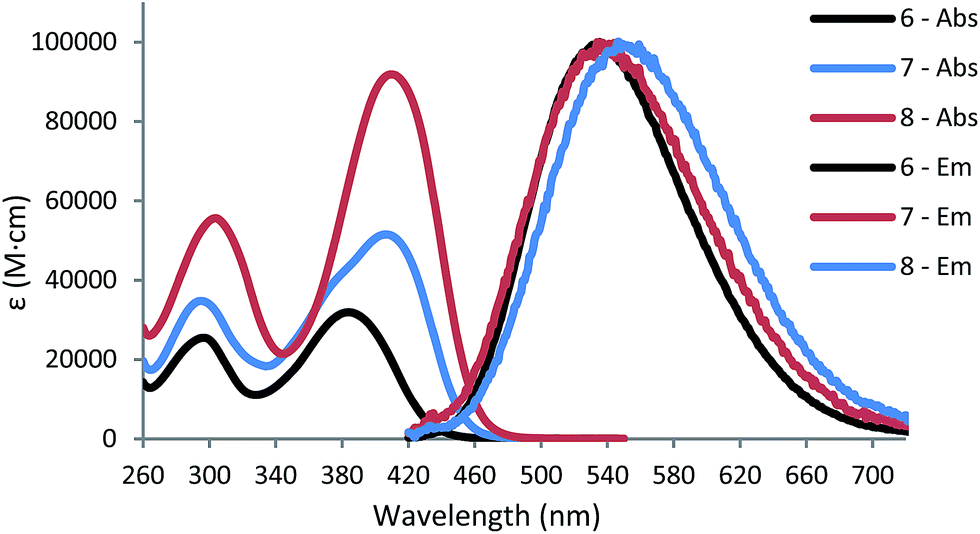 | ||
| Fig. 1 Normalised UV/Vis absorption spectrum of 6, 7 and 8 in MeOH, concentration of 1 × 10−5 M. Normalised fluorescence emission spectrum of 6, 7 and 8 in MeOH, concentration of 2 × 10−6 M and excitation at 385 nm. | ||
| Solvent | λmax | λEm | ε | ST | ϕF | δ2PA | |
|---|---|---|---|---|---|---|---|
| a λmax is the one-photon absorption maximum. λEm is the fluorescence emission maximum, excitation at 385 nm. ε = molar extinction coefficient (104 M−1 cm−1) calculated at λmax. ST = stokes shift; ST = (λEm − λAbs). ϕF is the fluorescence quantum yield. δ2PA = two-photon absorption as measured at 780 nm in CHCl3; concentration 2 × 10−2 M. 1 GM = 1 × 10−50 cm4 s per photons per molecule. | |||||||
| 6 | MeOH | 384 | 536 | 3.49 | 152 | 0.198 | 126 |
| Acrylate | — | 487 | — | 0.216 | |||
| 7 | MeOH | 404 | 542 | 5.46 | 138 | 0.021 | 358 |
| Acrylate | — | 495 | — | 0.182 | |||
| 8 | MeOH | 410 | 546 | 9.48 | 126 | 0.019 | 590 |
| Acrylate | — | 506 | — | 0.183 | |||
The molar extinction coefficients (ε) at the absorption maxima increase with the number of branches and values of 3.49, 5.46 and 9.48 × 104 M−1 cm−1 in MeOH are found for 6, 7 and 8, respectively. The fact that the magnitude of the extinction coefficient is approximately proportional to the number of branches suggests relatively independent behaviour of the branches in these PIs.27,42,44 However, the ε for 7 is slightly lower than expected and is likely due to the weak coupling of the two branches. In the absorption spectra for 7 (Fig. 1), a shoulder exists on the high-energy side of the peak resulting from coupling between the branches which splits the excited state into two bands, and are positioned symmetrically with respect to the dipolar species.29,41,45 This splitting is explained by the Frenkel exciton model, which is also applied to H- and J-aggregates and assumes electrostatic interaction of the molecules.41,46,47 The observed bandwidth broadening of 7, relative to the absorption of 6 and 8 further indicates the presence of two split one-photon excited states.27,29 This splitting is also predicted to occur in 8 with two equivalent excited states being one-photon allowed and the third being two-photon allowed only. The fluorescence emission spectra in MeOH show a bathochromic shift of the emission band (Fig. 1) and thus suggests the excited state is more polar and therefore, more stabilised by increasing solvent polarity.48
This trend can be seen with increasing solvent polarity and more details can be found in the ESI, Fig. S6–S8.†
Alongside this bathochromic shift, band broadening was observed with increased solvent polarity and importantly, the emission spectra for 6, 7 and 8 are similar; this indicates that excited state emission occurs from a single dipolar branch in all PIs.42,46 This is consistent with previous reports of triphenylamine containing chromophores, where photoexcitation produces an emissive charge-transfer (CT) state that is localised to a single branch.27,29,49–51 The large Stokes shifts (ST) that are observed indicate that significant electronic reorganisation takes place after excitation and prior to emission.52 The observed ST decreases from 6–8 in MeOH and this trend is also seen with lower polarity solvents, implying overall excited state polarity decreases with increased branching. The excited state of the dipolar 6 species possesses higher CT character than the respective quadrupolar 7 and octupolar 8 species.45,53
Fluorescence quantum yields (ϕF) were measured in MeOH using a known standard (QBS) (Table 1). The ϕF of the PIs in MeOH was 19.8%, 2.1% and 1.9% for 6, 7 and 8, respectively. Increased π-conjugation and carbonyl acceptor groups are known to reduce overall ϕF.12 Furthermore, from a molecular structure point of view multi-branched species have more tendency to adopt twisted configurations and these consume more energy, thus reducing the overall molecular ϕF.54 This can be rationalised by the effect of phenyl rotation on the CT state. The rotation of the phenyl-N bond on the branches compete to become part of the dipolar CT state and this in turn, reduces the ϕF.28 Moreover, viscosity can play an important role in this process by limiting phenyl-N rotation and its ability to adversely affect fluorescence emission and ϕF.31,51,55 To assess this we measured fluorescence emission and ϕF in increasingly viscous solvent mediums; including MeOH, ethylene glycol (EG), diethylene glycol (DEG) and the acrylate system we employed for our TPP tests, which consists of a 50:50 wt% mixture of SR415 and SR351 (Table S3, ESI†). SR415 and SR351 are commercially available acrylate monomers and have been previously utilised for TPP tests.3,18 The fluorescence emission of 7 and 8 in MeOH are much smaller than that of 6 in the same solvent.
When acrylate is the solvent, however, large increases in fluorescence emission are observed for all three PIs compared to their levels in MeOH (Fig. 2). The increased local viscosity of the acrylates hinders rotation about the phenyl-N bonds in the branches, and reduces the effectiveness of this conformational dynamics decay pathway relative to the formation of the CT state. Thus the emission spectra of 6–8 in acrylates largely mimic that of the dipolar 6 in MeOH, insofar as the emission is limited to the dipolar branch in all PIs.
 | ||
| Fig. 2 Fluorescence emission of 6–8 in acrylate mixture (SR415:SR351, 50:50 wt%) with excitation at 385 nm. | ||
To further investigate the excited state behaviour of our compounds, fluorescence lifetime measurements were performed in MeOH and in our acrylate system (Table 2 and Fig. 3). We observed a bi-exponential decay and calculated weighted average lifetimes for our PIs.56 The radiative decay constant (kf) and can be calculated using kf = ϕF/τ for 6, 7 and 8. The nonradiative decay constants (knr) can be calculated from kf and ϕF according to the following equation:57
 | (1) |
| Solvent | τ (ns) | kf | knr | |
|---|---|---|---|---|
| a τ is the fluorescence lifetime in ns as measured at 375 nm. kf = radiative decay constant (s−1) (kf = ϕF/τ). knr = non-radiative decay constant (s−1) calculated from eqn (1). | ||||
| 6 | MeOH | 1.57 | 1.26 × 108 | 5.11 × 108 |
| Acrylate | 2.08 | 1.04 × 108 | 3.77 × 108 | |
| 7 | MeOH | 0.55 | 3.82 × 107 | 1.78 × 109 |
| Acrylate | 2.17 | 8.39 × 107 | 3.77 × 108 | |
| 8 | MeOH | 0.33 | 5.76 × 107 | 2.97 × 109 |
| Acrylate | 2.26 | 8.10 × 107 | 3.62 × 108 | |
 | ||
| Fig. 3 Fluorescence lifetime decay curves of 6–8 in MeOH and acrylates with excitation pulse at 375 nm. | ||
For 6, 7 and 8 in MeOH, lifetimes of 1.57 ns, 0.55 ns and 0.33 ns were measured respectively. The low ϕF and shorter lifetimes for 7 and 8 can be attributed to the dominant nonradiative decay mechanisms through internal conversion.58 In acrylates, the fluorescence lifetimes were 2.08 ns, 2.17 ns and 2.26 ns for 6, 7 and 8, respectively. The kf and knr values across the series become close to those displayed for 6, suggesting that radiative pathways become the dominant process (Table 2). The acrylate viscosity reduces the ability of the branches in 7 and 8 to rotate and disrupt the dipolar CT state evidenced by the similar knr values (Table 2). Further evidence of emission being limited to a single dipolar branch, like that observed for 6 can be seen in the similar lifetime decay curves of 6–8 in acrylates (Fig. 3).
3.3 Two-photon absorption
The 2PA values were measured using the z-scan technique at 780 nm in CHCl3 and a sample concentration of 2 × 10−2 M. The δ2PA is calculated by open aperture z-scan which can be used to directly obtain the nonlinear absorption coefficient (β). When third order nonlinear losses are small, the normalised change in transmittance due to 2PA can be approximated.59 Our results are shown in Table 1 and the normalised data is shown in Fig. 4. | ||
| Fig. 4 Normalised z-scan data of 6–8 in CHCl3 as measured at 780 nm. | ||
δ2PA values of 126 GM, 358 GM and 590 GM were found for 6, 7 and 8 respectively; a linear relationship between δ2PA and the number of branches is seen, with an increase of 232 GM per branch. The branching effect and cooperative enhancement seen in multi-branched triphenylamine core molecules is well reported in the literature.7,8,60 With increasing π-conjugation and the number of acceptors both maximise ICT and thus, δ2PA.7 The measured one-photon λmax in CHCl3 were 388 nm, 411 nm and 415 nm for 6, 7 and 8, respectively, meaning that the measured GM values are likely to be close to their maximum. However, to obtain a full assessment of the 2PA activity of these molecules, measurements over a broader wavelength range would be necessary as 2PA enhancement is strongly wavelength dependant.27 Furthermore, due to the splitting of the excited states predicted by the Frenkel exciton model a broad wavelength would give insights into the 2PA of these higher excited states. Previous studies have shown that for octupolar branches the high-excited state can be blue-shifted with respect to the one-photon maxima.10,27,61
3.4 Two-photon polymerisation
There exist several methods for evaluating polymerisation efficiency of PIs used in TPP, including laser direct writing of single-lines.62 More recently, complicated 3D shapes have been fabricated by laser irradiation under a variety of conditions.3,18 Here, we evaluated PI efficiency by fabricating a 3D structure consisting of a cross-hatched square (dimensions: 150 × 150 × 50 μm, Fig. 5) using 800 nm, 110 femtosecond laser pulses with a variety of pulse energies and write-speeds (total irradiation). A 1:1 mixture of SR415 and SR351 (see ESI†) with a constant PI concentration of 6.3 × 10−6 mol g−1 was used, as good results have been obtained using such a formulation previously.3,18 We employed the previously studied PI E,E-1,4-bis[4′-(N,N-di-n-butylamino)styryl]benzene (R1B) as a reference for our TPP system. It is known to have a δ2PA of 995 GM at 730 nm and polymerisation threshold of 3.0 mW.5,63 Another study found R1B to polymerise high-quality structures at 12–15 μW at 800 nm and was the basis for our reference system and PI concentration.3,18
 | ||
| Fig. 5 3D structures showing the generic structure produced by TPP. The structures were fabricated at the minimum ‘good quality’ polymerisation threshold going from left to right, 6, 7 and 8 were used. | ||
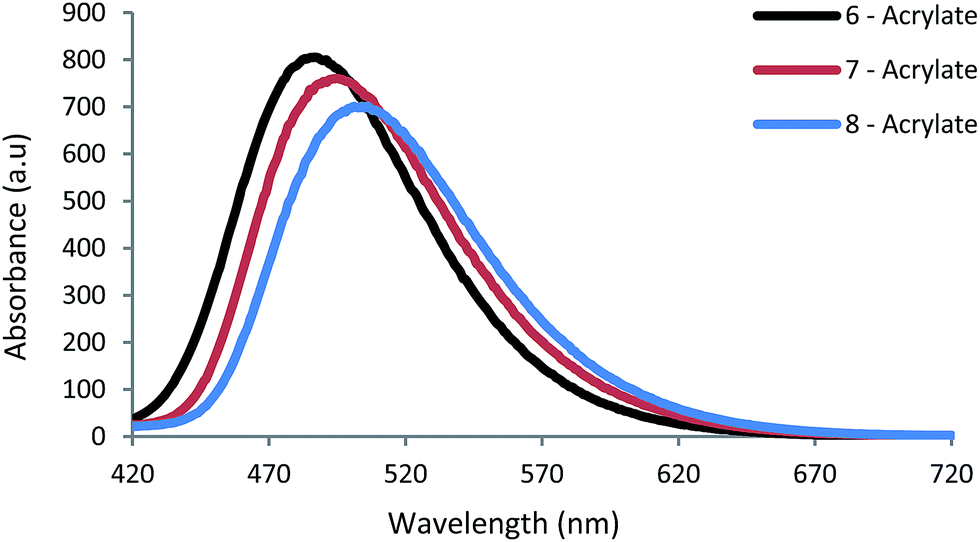
Our structuring tests found high quality polymerisation thresholds with an average power (500 Hz) of 36 μW with our TPP system, using a concentration of 6.3 × 10−6 mol g−1 and writing speed of 50 μm s−1. The average laser power employed ranged from 10 to 400 μW and writing speeds of 50 μm s−1, 100 μm s−1, 150 μm s−1 and 200 μm s−1 were explored. A colour code system as seen in Fig. S13† was used to determine polymerisation thresholds of our PIs at varying writing speeds. Full details of the processing windows, structure quality assignments and polymerisation setup are outlined in the ESI.†
In our measurements, we obtained high-quality structures in the low average μW power range for all PIs, at an order of magnitude smaller than many commercial and literature PIs.2,26,64–67 Additionally, we observe large high-quality processing windows of 50–100 μW for our PIs (Fig. 6). The PIs 6 and 7 displayed excellent solubility in our chosen monomer system even over a period of >6 months. However, over this period 8 began to precipitate out of solution and required stirring and gentle heating to redissolve. Fig. 5 shows the TPP structuring test results at a writing speed of 50 μm s−1 and shows the power range required to create high-quality structures.
 | ||
| Fig. 6 TPP structuring tests of 6, 7 and 8 with writing speed of 50 μm s−1. The data above 200 μW are omitted here. A full description of our TPP tests at varying writing speeds are detailed fully in the ESI.† | ||
The colour of each bar represents the qualitative assessment of structure quality of our fabricated structures. For efficient PIs, broad processing windows and low polymerisation thresholds are desired for high throughput manufacturing.67 Fabricated structures of the highest quality are in green and possess well defined and smooth walls (Fig. 5). Average quality (orange) contain small imperfections including, holes or over-polymerised filaments originating from the structures. Poor quality (yellow) contain major imperfections like collapsed walls and over or under-polymerised features. Finally, very poor quality (red) show very little polymerisation at low power or ablation at high power and the structures barely resemble the desired ones.
At writing speeds of 50 μm s−1 we observed good polymerisation thresholds at 27 μW, 45 μW and 61 μW for 8, 6 and 7, respectively. For 6, the powers of 45–60 μW were seen to polymerise high-quality structures across all the writing speeds and conversely, powers between 80–170 μW for the upper limit of the excellent processing window. For 7, a larger minimum power is required for high-quality structures (61–64 μW) and a maximum of 122–170 μW. The octupolar 8, displayed the lowest polymerisation threshold (27–36 μW) and upper limits of 76–108 μW across the writing speeds used.
From our photophysical measurements and δ2PA values, the expected results would be 8 > 7 > 6. However, our TPP results show this isn't the case and in fact, the order is 8 > 6 > 7 for the highest quality structures. The difference in polymerisation thresholds for this series is relatively narrow. As discussed previously, the localised viscosity of the acrylate system adversely affects the overall properties of these PIs, in particular, the ϕF yields due to limited bond rotation. Consequently, this indicates that while photophysical measurements can suggest the preferred materials for TPP, this does not always translate to the actual structuring tests. These results have important implications for PI design, due to the dipolar nature of the excited state and thus, the origin of initiation. This suggests that synthesising multi-branched PIs to maximise TPP sensitivity may be of limited use.
4. Conclusions
We have synthesised a series of multi-branched D–π–An (n = 1, 2 or 3) PIs for use in TPP. We measured large δ2PA up to 590 GM for 8 at 780 nm and excellent polymerisation thresholds were displayed across the series down to 27 μW. We showed that due to the dipolar nature of active species in these molecules, viscosity plays a key role in determining their photophysical properties like ϕF yields. This is particularly important for large multi-branched PIs like 7 and 8 as branching effects become mitigated. To the best of our knowledge, this is the first report of such effects on TPP thresholds. These results indicate the limited necessity of synthesising large, multi-branched species as a way of maximising polymerisation thresholds for TPP. Consequently, future studies should focus on improvements to structures based on the relatively simple compound 6, and could include further extension of conjugation and the incorporation of stronger acceptors to increase the dipolar nature of the molecules.Acknowledgements
This work was supported by New Zealand Ministry of Business, Innovation and Employment Grant UOAX1202. Further thanks are also given to those who have contributed to this work including; Reece Oosterbeek, Robert Breukers, the Photon Factory team and members of the Photonics team, Callaghan Innovation.Notes and references
- R. Nazir, E. Balčiūnas, D. Buczyńska, F. Bourquard, D. Kowalska, D. Gray, S. Maćkowski, M. Farsari and D. T. Gryko, Macromolecules, 2015, 48, 2466–2472 CrossRef CAS.
- Z. Li, M. Siklos, N. Pucher, K. Cicha, A. Ajami, W. Husinsky, A. Rosspeintner, E. Vauthey, G. Gescheidt, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3688–3699 CrossRef CAS.
- Z. Li, N. Pucher, K. Cicha, J. Torgersen, S. C. Ligon, A. Ajami, W. Husinsky, A. Rosspeintner, E. Vauthey, S. Naumov, T. Scherzer, J. Stampfl and R. Liska, Macromolecules, 2013, 46, 352–361 CrossRef CAS.
- R. Nazir, F. Bourquard, E. Balčiūnas, S. Smoleń, D. Gray, N. V. Tkachenko, M. Farsari and D. T. Gryko, ChemPhysChem, 2015, 16, 682–690 CrossRef CAS PubMed.
- Y. Lu, F. Hasegawa, T. Goto, S. Ohkuma, S. Fukuhara, Y. Kawazu, K. Totani, T. Yamashita and T. Watanabe, J. Lumin., 2004, 110, 1–10 CrossRef CAS.
- B. A. Reinhardt, L. L. Brott, S. J. Clarson, A. G. Dillard, J. C. Bhatt, R. Kannan, L. Yuan, G. S. He and P. N. Prasad, Chem. Mater., 1998, 10, 1863–1874 CrossRef CAS.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641–4678 CrossRef CAS.
- Z. Wang, X. Wang, J. Zhao, W. Jiang, P. Yang, X. Fang, M. Zhou and M. Cheng, Dyes Pigm., 2008, 79, 145–152 CrossRef CAS.
- R. Lartia, C. Allain, G. Bordeau, F. Schmidt, C. Fiorini-Debuisschert, F. Charra and M.-P. Teulade-Fichou, J. Org. Chem., 2008, 73, 1732–1744 CrossRef CAS PubMed.
- Z. Li, J. Torgersen, A. Ajami, S. Mühleder, X. Qin, W. Husinsky, W. Holnthoner, A. Ovsianikov, J. Stampfl and R. Liska, RSC Adv., 2013, 3, 15939–15946 RSC.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS.
- J. Perez-Prieto, R. Galian and M. Miranda, Mini-Rev. Org. Chem., 2006, 3, 117–135 CrossRef CAS.
- G. Angulo, J. Grilj, E. Vauthey, L. Serrano-Andrés, Ò. Rubio-Pons and P. Jacques, ChemPhysChem, 2010, 11, 480–488 CrossRef CAS PubMed.
- J.-P. Malval, M. Jin, F. Morlet-Savary, H. Chaumeil, A. Defoin, O. Soppera, T. Scheul, M. Bouriau and P. L. Baldeck, Chem. Mater., 2011, 23, 3411–3420 CrossRef CAS.
- J. Wu, M. Shi, Y. Zhao and F. Wu, Dyes Pigm., 2008, 76, 690–695 CrossRef CAS.
- G. Lemercier, C. Martineau, J.-C. Mulatier, I. Wang, O. Stephan, P. Baldeck and C. Andraud, New J. Chem., 2006, 30, 1606–1613 RSC.
- N. Pucher, A. Rosspeintner, V. Satzinger, V. Schmidt, G. Gescheidt, J. Stampfl and R. Liska, Macromolecules, 2009, 42, 6519–6528 CrossRef CAS.
- J. Xue, Y. Zhao, F. Wu and D.-C. Fang, J. Phys. Chem. A, 2010, 114, 5171–5179 CrossRef CAS PubMed.
- J. Wu, Y. Zhao, X. Li, M. Shi, F. Wu and X. Fang, New J. Chem., 2006, 30, 1098–1103 RSC.
- R. Nazir, T. T. Meiling, P. J. Cywiński and D. T. Gryko, Asian J. Org. Chem., 2015, 4, 929–935 CrossRef CAS.
- R. Nazir, P. Danilevicius, A. I. Ciuciu, M. Chatzinikolaidou, D. Gray, L. Flamigni, M. Farsari and D. T. Gryko, Chem. Mater., 2014, 26, 3175–3184 CrossRef CAS.
- R. Nazir, B. Thorsted, E. Balciunas, L. Mazur, I. Deperasinska, M. Samoc, J. Brewer, M. Farsari and D. T. Gryko, J. Mater. Chem. C, 2016, 4, 167–177 RSC.
- R. Nazir, P. Danilevicius, D. Gray, M. Farsari and D. T. Gryko, Macromolecules, 2013, 46, 7239–7244 CrossRef CAS.
- X. Wang, F. Jin, W. Zhang, X. Tao, X.-M. Duan and M. Jiang, Dyes Pigm., 2011, 88, 57–64 CrossRef CAS.
- X. Wang, F. Jin, Z. Chen, S. Liu, X. Wang, X. Duan, X. Tao and M. Jiang, J. Phys. Chem. C, 2011, 115, 776–784 CAS.
- C. Katan, F. Terenziani, O. Mongin, M. H. V. Werts, L. Porrès, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024–3037 CrossRef CAS PubMed.
- S. Easwaramoorthi, P. Thamaraiselvi, K. Duraimurugan, A. J. Beneto, A. Siva and B. U. Nair, Chem. Commun., 2014, 50, 6902–6905 RSC.
- D. Cvejn, E. Michail, K. Seintis, M. Klikar, O. Pytela, T. Mikysek, N. Almonasy, M. Ludwig, V. Giannetas, M. Fakis and F. Bures, RSC Adv., 2016, 6, 12819–12828 RSC.
- B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed.
- M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669–1678 CAS.
- K.-S. Lee, K. Ran Hee, Y. Dong-Yol and P. Sang Hu, Prog. Polym. Sci., 2008, 33, 50 CrossRef.
- A. Sobolev, V. Belsky, I. Romm, N. Y. Chernikova and E. Guryanova, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 967–971 CrossRef.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS PubMed.
- P. J. Holliman, M. Mohsen, A. Connell, M. L. Davies, K. Al-Salihi, M. B. Pitak, G. J. Tizzard, S. J. Coles, R. W. Harrington, W. Clegg, C. Serpa, O. H. Fontes, C. Charbonneau and M. J. Carnie, J. Mater. Chem., 2012, 22, 13318–13327 RSC.
- X. R. B. Gaifa Lai, J. Santos and E. A. Mintz, Synlett, 1997, 1275–1276 Search PubMed.
- T. Mallegol, S. Gmouh, M. A. A. Meziane, M. Blanchard-Desce and O. Mongin, Synthesis, 2005, 2005, 1771–1774 CrossRef.
- D. Ding and G. Yan, Chin. J. Chem., 2012, 30, 1906–1908 CrossRef CAS.
- D. Beljonne, J. L. Brédas, M. Cha, W. E. Torruellas, G. I. Stegeman, J. W. Hofstraat, W. H. G. Horsthuis and G. R. Möhlmann, J. Chem. Phys., 1995, 103, 7834–7843 CrossRef CAS.
- L. Yan, X. Chen, Q. He, Y. Wang, X. Wang, Q. Guo, F. Bai, A. Xia, D. Aumiler, S. Vdović and S. Lin, J. Phys. Chem. A, 2012, 116, 8693–8705 CrossRef CAS PubMed.
- F. Terenziani, C. Le Droumaguet, C. Katan, O. Mongin and M. Blanchard-Desce, ChemPhysChem, 2007, 8, 723–734 CrossRef CAS PubMed.
- J. Brunel, O. Mongin, A. Jutand, I. Ledoux, J. Zyss and M. Blanchard-Desce, Chem. Mater., 2003, 15, 4139–4148 CrossRef CAS.
- C. Liu, K.-C. Tang, H. Zhang, H.-A. Pan, J. Hua, B. Li and P.-T. Chou, J. Phys. Chem. A, 2012, 116, 12339–12348 CrossRef CAS PubMed.
- C. Katan, F. Terenziani, C. L. Droumaguet, O. Mongin, M. H. V. Werts, S. Tretiak and M. Blanchard-Desce, Proc. SPIE-Int. Soc. Opt. Eng., 2005, 5935, 593503 CrossRef.
- C. Katan, S. Tretiak, M. H. V. Werts, A. J. Bain, R. J. Marsh, N. Leonczek, N. Nicolaou, E. Badaeva, O. Mongin and M. Blanchard-Desce, J. Phys. Chem. B, 2007, 111, 9468–9483 CrossRef CAS PubMed.
- F. Terenziani, C. Sissa and A. Painelli, J. Phys. Chem. B, 2008, 112, 5079–5087 CrossRef CAS PubMed.
- Y. Zhang, M. Jiang, G.-C. Han, K. Zhao, B. Z. Tang and K. S. Wong, J. Phys. Chem. C, 2015, 119, 27630–27638 CAS.
- J. E. Rogers, J. E. Slagle, D. G. McLean, R. L. Sutherland, B. Sankaran, R. Kannan, L.-S. Tan and P. A. Fleitz, J. Phys. Chem. A, 2004, 108, 5514–5520 CrossRef CAS.
- B. Li, R. Tong, R. Zhu, F. Meng, H. Tian and S. Qian, J. Phys. Chem. B, 2005, 109, 10705–10710 CrossRef CAS PubMed.
- B. Carlotti, E. Benassi, C. G. Fortuna, V. Barone, A. Spalletti and F. Elisei, ChemPhysChem, 2016, 17, 136–146 CrossRef CAS PubMed.
- L. Porrès, C. Katan, O. Mongin, T. Pons, J. Mertz and M. Blanchard-Desce, J. Mol. Struct., 2004, 704, 17–24 CrossRef.
- G. Ramakrishna and T. Goodson, J. Phys. Chem. A, 2007, 111, 993–1000 CrossRef CAS PubMed.
- X. Wang, P. Yang, G. Xu, W. Jiang and T. Yang, Synth. Met., 2005, 155, 464–473 CrossRef CAS.
- T. Förster and G. Hoffmann, Z. Phys. Chem., 1971, 75, 63–76 CrossRef.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- C. A. Zoto and R. E. Connors, J. Mol. Struct., 2010, 982, 121–126 CrossRef CAS.
- C. Toro, L. De Boni, S. Yao, K. D. Belfield and F. E. Hernández, J. Phys. Chem. B, 2008, 112, 12185–12190 CrossRef CAS PubMed.
- I. Asselberghs, J. Pérez-Moreno and K. Clays, in Non-Linear Optical Properties of Matter: From Molecules to Condensed Phases, ed. M. G. Papadopoulos, A. J. Sadlej and J. Leszczynski, Springer, Netherlands, Dordrecht, 2006, vol. 14, pp. 419–459, DOI:10.1007/1-4020-4850-5.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- M. A. Albota, C. Xu and W. W. Webb, Appl. Opt., 1998, 37, 7352–7356 CrossRef CAS PubMed.
- Y. Ren, X.-Q. Yu, D.-J. Zhang, D. Wang, M.-L. Zhang, G.-B. Xu, X. Zhao, Y.-P. Tian, Z.-S. Shao and M.-H. Jiang, J. Mater. Chem., 2002, 12, 3431–3437 RSC.
- M. Rumi, J. E. Ehrlich, A. A. Heikal, J. W. Perry, S. Barlow, Z. Hu, D. McCord-Maughon, T. C. Parker, H. Röckel, S. Thayumanavan, S. R. Marder, D. Beljonne and J.-L. Brédas, J. Am. Chem. Soc., 2000, 122, 9500–9510 CrossRef CAS.
- K. D. Belfield, K. J. Schafer, Y. Liu, J. Liu, X. Ren and E. W. V. Stryland, J. Phys. Org. Chem., 2000, 13, 837–849 CrossRef CAS.
- H.-B. Sun and S. Kawata, in NMR 3D Analysis Photopolymerization, Springer, 2004, pp. 169–273 Search PubMed.
- L. Li and J. T. Fourkas, Mater. Today, 2007, 10, 30–37 CrossRef CAS.
- C. N. LaFratta, J. T. Fourkas, T. Baldacchini and R. A. Farrer, Angew. Chem., Int. Ed., 2007, 46, 6238–6258 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further details of starting material synthesis (1–5), photophysical measurements, TPP and NMR data for final PIs 6–8. See DOI: 10.1039/c6ra27176f |
| This journal is © The Royal Society of Chemistry 2017 |