
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green solid-state synthesis and photocatalytic hydrogen production activity of anatase TiO2 nanoplates with super heat-stability†
Jindou Hu,
Yali Cao *,
Kun Wang and
Dianzeng Jia
*,
Kun Wang and
Dianzeng Jia
Key Laboratory of Energy Materials Chemistry, Ministry of Education, Key Laboratory of Advanced Functional Materials, Institute of Applied Chemistry, Xinjiang University, Autonomous Region, Urumqi, Xinjiang 830046, China. E-mail: caoyali523@163.com; Fax: +86-991-8588883; Tel: +86-991-8583083
First published on 16th February 2017
Abstract
A simple and environmentally friendly new method, a low-heating solid-state chemical method (LSCM), has been used to prepare uniform NH4TiOF3 nanoplates with a thickness of about 100 nm in the absence of surfactants. The solid–solid transformation was used to convert NH4TiOF3 nanoplates to TiO2 nanoplates with an exposed (001) facet via a simple sintering process at different temperatures. The anatase TiO2 nanoplates obtained in this work have super heat-stability, and can withstand high temperatures and maintain highly active anatase up to 1000 °C. The obtained compact TiO2 nanoplates have high photocurrent density and photocatalytic hydrogen-production activity, better than as-prepared TiO2 porous nanoplates and reported values in the literature, which is due to the exposed (001) facet and the high degree of crystallinity. This work provides a simple, efficient and environmentally friendly way to prepare TiO2 nanoplates and nanoparticles, and also offers a new approach for preparing anatase TiO2 with super heat-stability and high reactivity, which can be applied to H2-production via photocatalytic water splitting.
1. Introduction
The energy crisis has attracted worldwide attention, due to the extensive use and storage limitations of fossil fuels.1–4 The development and application of new energy sources have become important research areas. Among various new energy sources, and offering the benefits of high energy density and environmental friendliness, hydrogen obtained from the photocatalytic splitting of water is regarded as one of the ideal candidates.5,6 Developing and researching semiconductor photocatalysts for hydrogen production has been recognized as a focus of research in inorganic chemistry, physical chemistry and material chemistry.7To date, among various semiconductor photocatalysts that can produce hydrogen via the photocatalytic splitting of water, TiO2 has the features of superior photocatalytic activity, chemical stability, low-cost and nontoxicity,8–13 so it is regarded as a suitable material for use in photocatalytic water splitting for hydrogen production.14–18 The photocatalytic performance of TiO2 is influenced by many factors, such as the band gap, the active facet, the degree of crystallinity, the size and the morphology. In particular, for anatase TiO2, the high activity of the (001) facet has been verified to influence the photocatalytic activity, through both theory and experiment.19,20 However, the preparation of anatase TiO2 materials with an exposed (001) facet is still a difficult task. Yang et al.19 used HF in a hydrothermal synthetic process to restrain the growth of the (001) facet, and obtained TiO2 with an exposed (001) facet with high photocatalytic activity to produce H2.21,22 Thereafter, a number of methods were used to prepare TiO2 with an exposed (001) facet.19,23–27 These methods usually involved hydrothermal or solvothermal processes at high pressure, and extremely corrosive and toxic HF or NH4F were used as supporting materials for the growing of crystal facets. Consequently, a facile, green and efficient method is needed for the preparation of anatase TiO2 with an exposed (001) facet.
The low-heating solid-state chemical method (LSCM), needing only room temperature or a temperature below 100 °C, is a facile, green, simple and environmentally friendly method to synthesize nanomaterials.28,29 The LSCM has been used to prepare many inorganic nanomaterials, including oxides, sulfides, metals and composites, in our research group.30–36 In this work, the LSCM was applied to the controlled synthesis of crystal surface exposed semiconductors to further develop the simple and effective solid-state method. We designed and prepared fluoro-chemical NH4TiOF3 nanoplates using the LSCM without any surfactant, and then converted the NH4TiOF3 nanoplates to TiO2 nanoplates. In the process of transformation, the presence of fluorine in the NH4TiOF3 nanoplates restrained the growth of the (001) facet. Thus, the anatase TiO2 exposed (001) facet was obtained simply, avoiding the use of toxic substances that have been reported in the literature, such as HF and NH4F. The photocatalytic activity of the as-obtained anatase TiO2 with an exposed (001) facet for H2 production was investigated in detail.
2. Experimental
2.1. Synthesis
The various chemicals used in this work were purchased from commercial sources and used without further purification. In a typical experiment, 8 mmol of ammonium hexafluorotitanate was weighed and ground for 10 min in an agate mortar. Subsequently, 6 mmol of boric acid was added. After grinding at room temperature for 0.5 h, the resulting solid samples were heated in a water bath at 60 °C to perform the entire reaction. The as-prepared samples were washed with distilled water and dried under natural conditions for 12 h. The obtained samples were assigned as the precursor and denoted as T1.The final products were fabricated through the calcination of precursor T1. The precursor T1 was annealed at 200 °C, 300 °C, 400 °C, 500 °C, 600 °C, 700 °C, 800 °C, 900 °C or 1000 °C for 2 h under an air atmosphere. The final products were denoted as T2, T3, T4, T5, T6, T7, T8, T9 and T10, respectively, and were collected for further characterization and testing. The detailed description of the obtained samples and the corresponding preparation conditions are provided in Table S1.†
2.2. Characterization
A Bruker D8 Advance diffractometer was used to measure the X-ray diffraction (XRD) patterns of the products, with Cu Kα radiation (k = 1.54184 Å) under operating conditions of 40 kV and 40 mA, over the range of 10–80° at a scanning rate of 2° min−1. The Scherrer formula is used to calculate the average size of the as-obtained products:D = Kλ/B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ |
2.3. Photocatalytic tests
A photocatalytic H2 preparation system (CEL-SPH2NP, CHINA) was used to measure the photocatalytic hydrogen production. Typically, 50 mg of photocatalyst was put into a glass reaction cell containing 74 mL of distilled water, 1.2 mL of H2PtCl6 (1.91 mmol L−1) and 25 mL of methanol. The system was then placed under vacuum for 20 min to remove the O2 in the system prior to irradiation. A 300 W Xe lamp with a different optical reflector or filter (VISREF: 350–780 nm, QD365: 365 nm, QD400: 400 nm, QD600: 600 nm) was used to provide the light source. The reaction temperature was controlled at ca. 6 °C by means of cooling water. The evolved H2 was analyzed every 1 h using an online gas chromatograph with a TCD detector. The following equation was used to calculate the apparent quantum efficiency (QE) value:
3. Results and discussion
3.1. Crystal structures and compositions of the as-obtained samples
Fig. 1 shows the XRD patterns of the as-obtained products from solid-state synthesis. Typical peaks at 2θ = 14.039°, 23.476°, and 27.449° from NH4TiOF3 (PDF#52-1647) are observed in T1 (Fig. 1a), and no peaks from the reactants (NH4)2TiF6 and H3BO3 are observed, indicating that NH4TiOF3 is successfully prepared using the LSCM.37 For the fabrication, environmentally friendly (NH4)2TiF6 and H3BO3 were used as the reactants. The following equation is suggested to account for the reaction:| 4(NH4)2TiF6 + 3H3BO3 = 4NH4TiOF3 + 3NH4BF4 + NH3·H2O + 4H2O |
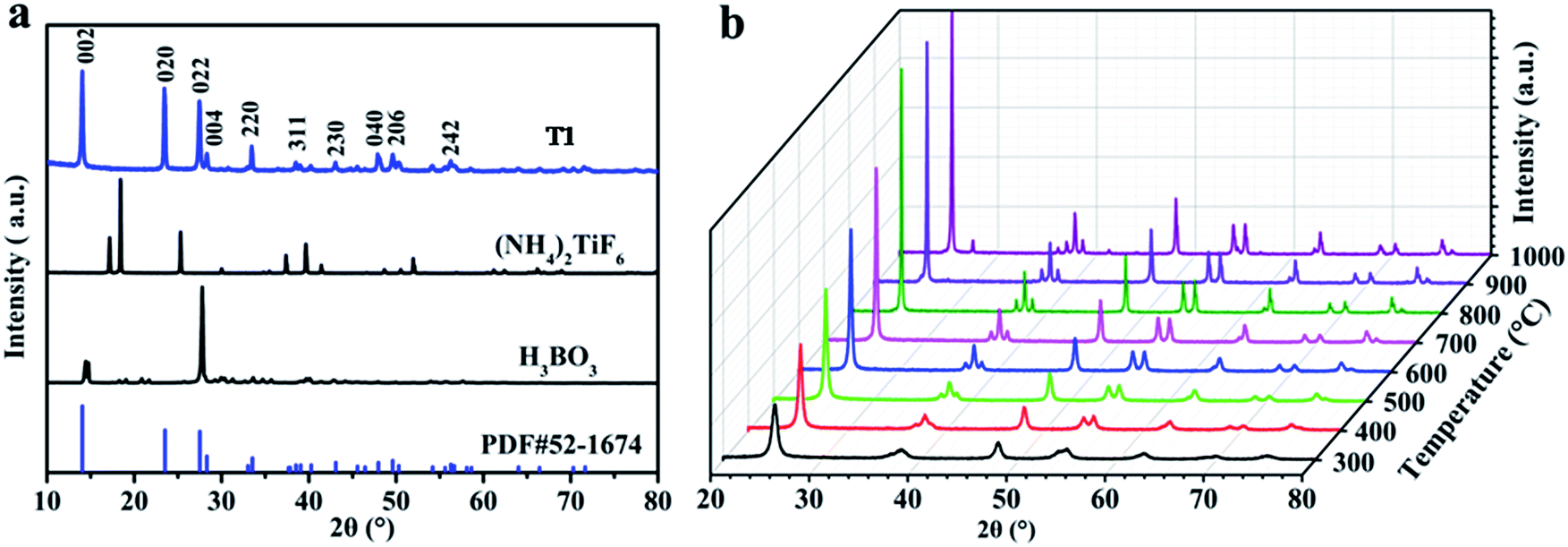 | ||
| Fig. 1 XRD patterns of the samples: (a) raw materials H3BO3, (NH4)2TiF6 and the precursor T1; and (b) samples T3–T10. | ||
In addition, no surfactant was used as an auxiliary material during the process of synthesis, which illustrates the simple, mild, environmentally friendly and low-cost features of the LSCM and indicates the superiority and practical value of green methods in the field of chemical synthesis.
Fig. 1b shows the XRD patterns of the as-obtained products following the calcination of the precursor T1 at different temperatures. The information from Fig. 1b indicates that NH4TiOF3 can successfully transform to TiO2 (PDF#04-0477) via a simple heating process at 300–1000 °C. Specifically, the precursor starts to convert to anatase TiO2 at 200 °C (see Fig. S1†); pure anatase TiO2 (PDF#04-0477) was obtained after treating the precursor at 300 °C for 2 h. The obtained anatase TiO2 always exists over the annealing temperature range of 300–900 °C. When the temperature reaches 1000 °C, a typical small peak for rutile TiO2 at 2θ = 27.479° is observed.38 This indicates that TiO2 obtained from annealing NH4TiOF3 was able to withstand high temperatures and maintained a stable anatase phase until 1000 °C; similar results are also obtained from the thermal analysis of T1 in Fig. S3,† which indicates that the TiO2 obtained in this work has super heat-stability. With a rise in treatment temperature, the degree of crystallinity of the samples progressively improves. A high degree of crystallinity may be beneficial for H2 production from the splitting of water in sunlight using photocatalysts.39
The elemental compositions and bonding structures were characterized using XPS techniques. The sharp photoelectron peaks appearing at binding energies of 458.9 eV (Ti 2p), 531.1 eV (O 1s) and 564.3 eV (Ti 2s) in Fig. 2a indicate that sample T8 contains the elements Ti and O, and there are no obvious signals from the elements B and F in the sample T8 (Fig. S2†). The high-resolution XPS spectra (HR-XPS) of the Ti 2p peaks are shown in Fig. 2b, and four peaks at 455.18 eV, 458.98 eV, 461.58 eV and 464.68 eV can be observed, which can be assigned to the peaks of Ti3+ 2p3/2, Ti4+ 2p3/2, Ti3+ 2p1/2 and Ti4+ 2p1/2.40,41 Fig. 2c shows the HRXPS spectra of O 1s; the peak at 530.6 eV in Fig. 2c can be assigned to oxygen in the form of Ti–O–Ti, and the peak at 531.8 eV can be ascribed to the Ti–O–H form.42 The hydroxy on the surface of TiO2 may be beneficial for the photocatalytic performance through improving the hydrophilicity of the material. EDS techniques were used to further analyze the elemental compositions. Fig. 2d shows the EDS spectrum of sample T8; the inset is the element content of sample T8. The results from the EDS spectrum and the inset indicate that there is no F element in the samples. The high-resolution XPS spectra of F 1s and B 1s in Fig. S2† also indicate that there is no F or B element in sample T8, because F can easily be removed by heating for 30 min. The results are consisted with those obtained using XPS. The results from XPS and EDS indicate that the simple LSCM has not introduced any impurity elements into the target during the synthesis process, and extra purification after the LSCM is also unnecessary, which indicates the superiority and feasibility of the LSCM in the field of chemical synthesis.
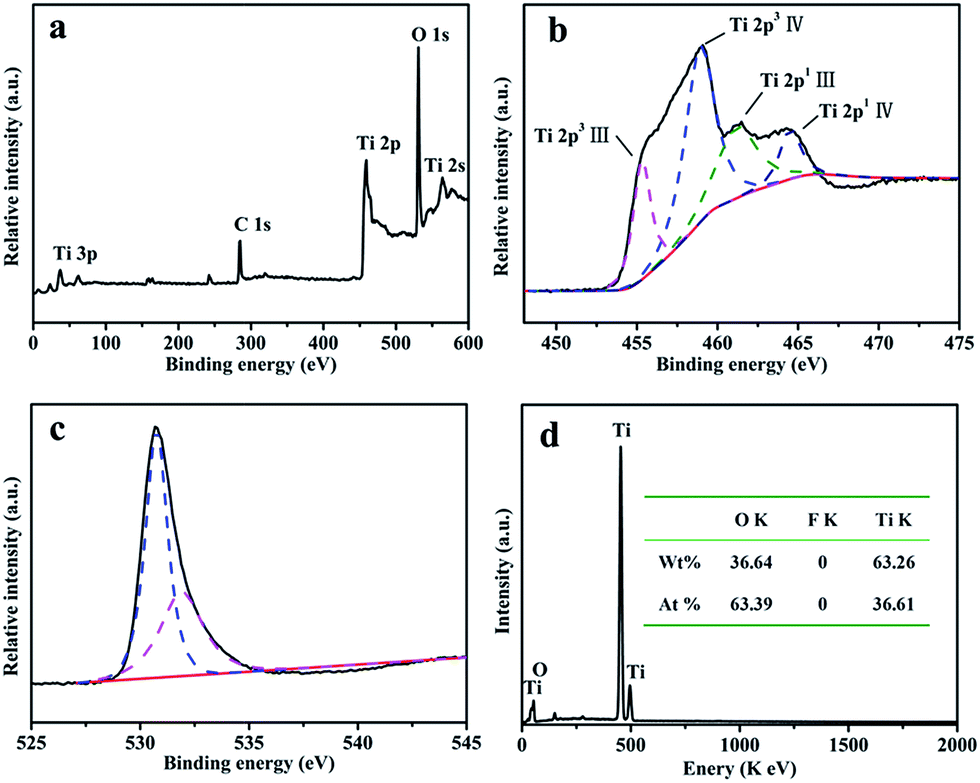 | ||
| Fig. 2 (a) An XPS spectrum of sample T8; the high-resolution XPS spectra of (b) Ti 2p and (c) O 1s in sample T8; and (d) the EDS spectra and element content of sample T8. | ||
3.2. Morphologies of the products
Fig. 3a shows that the morphology of the as-prepared precursor T1 consists of uniform platelets with a thickness of about 100 nm. The uniform morphology and the simple synthesis process indicate the simple features of the LSCM. The morphology of sample T2 is the same as for the precursor T1 (Fig. 3b). But the morphologies of samples T4 (Fig. 3c) and T6 (Fig. 3d) consist of nanoplates composed of abundant nanoparticles, and the thickness of the single-layer nanoplates is about 50 nm. When the temperature is at or below 800 °C, the morphology of the obtained sample T8 consists of smooth nanoplates with a thickness of about 50 nm (Fig. 3e). But when the temperature is above 800 °C, the samples quickly grow to bigger particles and reunification phenomena occur (Fig. 3f). The average size of the obtained samples and the increment in average size during the calcination process at different temperatures were further calculated using the Scherrer formula and are shown in Table S2.† | ||
| Fig. 3 SEM micrographs of the obtained samples: (a) T1; (b) T2; (c) T4; (d) T6; (e) T8; and (f) T10. | ||
In order to explore the specific formative processes of the TiO2 nanoplates under high temperatures, the progressive changes in the TiO2 nanoplates during the heating process were observed and a simulated diagram is presented in Fig. 4a. Typically, F− and NH4+ will disappear from the NH4TiOF3 nanoplates during the heating process, and the precursor will start to convert to TiO2 during calcination above 200 °C. During the heating process, the particles in the middle of the nanoplates may migrate to the surface of the nanoplates. Thus, the middle of the nanoplates will become hollow; when the treatment temperature is up to 300 °C, the morphology of the as-prepared TiO2 will involve double-layer nanoplates consisting of particles. With an increase in the treatment temperature, the double-layer nanoplates will be decomposed to single-layer nanoplates or even nanoparticles with a size of about 50 nm. At 800 °C, the nanoparticles will become nanoplates with a smooth surface, with orientated fusion between nanocrystals.43 This orientated fusion phenomenon is affirmed from the SEM images (Fig. 4b–d). With prolongation of the annealing time at 800 °C, the morphology of TiO2 will change from porous nanoplates (Fig. 4c) to compact nanoplates (Fig. 4d). Finally, after 2 h at 800 °C, the nanoplates composed of nanoparticles will generate smooth nanoplates. If the temperature keeps increasing above 800 °C, the samples will quickly grow to become large particles (Fig. 3f), which is in accordance with the regularity seen in Table S2,† and the reunification phenomenon was also observed when the temperature was higher than 900 °C. During the process of growing the products, no auxiliary materials were used to control the formation of the nanoplates, which also indicates the simple and low-cost features of the solid-state method.
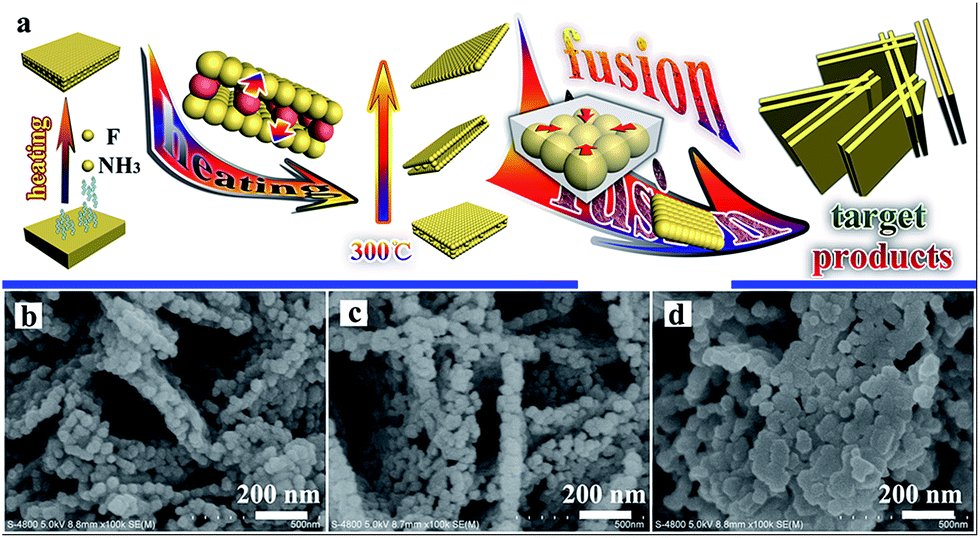 | ||
| Fig. 4 A simulated diagram of the progressive change in the morphology of TiO2 during the heating process (a), and SEM images of TiO2 sintered at 800 °C for different times: 30 min (b), 60 min (c), and 90 min (d). | ||
Fig. 5 shows micrographs of sample T8. The abundant nanoplates are displayed in the low-resolution SEM (Fig. 5a) and high-resolution SEM (Fig. 5b and c) images. The TEM image of T8 (Fig. 5d) also indicates that the morphology of sample T8 is nanoplates with a thickness of about 50 nm. The lattice rules from the SAED in Fig. 5e indicate that the as-prepared TiO2 is monocrystalline. The interplanar spacing in Fig. 5f and g indicates that the as-prepared samples are anatase TiO2 with an exposed (001) facet.44–46 Zhou et al. also reported that heating at 700–900 °C will lead to TiO2 with an exposed (001) facet.43 The surface energy and reactivity of the (001) facet of TiO2 is relative higher than for the other ones.23 Therefore, the exposed (001) facet may be beneficial for the photocatalytic performance. The results from electron microscope observation indicate that TiO2 with a high-activity (001) facet can also be synthesized using the simple solid-state method without any additional surfactant to control the process of growing the products.
 | ||
| Fig. 5 Micrographs of sample T8: (a) SEM; (b and c) high-resolution SEM images; (d) TEM; (e) SAED; and (f and g) HRTEM. | ||
3.3. UV-vis absorption spectra
The absorbance properties of the as-obtained products are displayed in Fig. 6a. The absorption intensities of the samples obtained at different temperatures all start to increase at 400 nm (Fig. 6a), which is similar to the intrinsic band gap of anatase TiO2 (about 3.2 eV (ref. 47)). A similar band gap of about 3.2 eV for all the samples is also shown in Fig. 6b. This indicates that the absorbance properties and band gaps of the samples have nothing to do with the sintering temperature. There are no absorption peaks caused by N or B elements observed in Fig. 6a, which also indicates that pure TiO2 can be obtained using the solid-state method.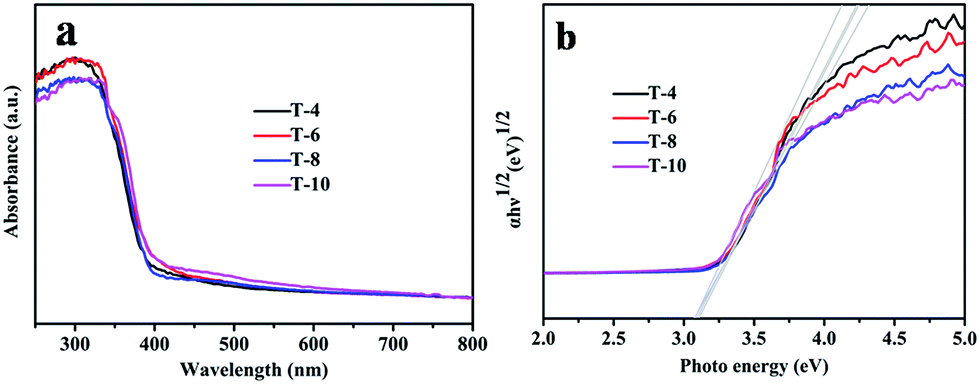 | ||
| Fig. 6 (a) UV-vis absorption spectra of samples T4, T6, T8 and T10; and (b) Tauc plots of samples T4, T6, T8 and T10. | ||
3.4. Photocatalytic activity and photocurrent density
Photocatalytic H2 production from water splitting was used to evaluate the photocatalytic activities of all samples, and the results are shown in Fig. 7. The information in Fig. 7a indicates that the rate of H2 generation firstly increases, and then decreases, with a rise in the sintering temperature, and the rate using sample T8 is 13 mmol h−1 g−1, which is the highest rate from all the as-prepared photocatalysts in this work and better than reported values in the literature.48,49 But the rate of H2 generation decreases when the sintering temperature is above 800 °C. In comparison, the amount of H2 production after 5 h is shown in Fig. 7b; it can clearly be seen that the amount of H2 generation using sample T8 is 4 times that of sample T4, and 9.5 times that of sample T10. The corresponding quantum efficiency of sample T8 under 365 nm, 400 nm and 600 nm light is shown in Fig. 8. The reason behind the high photocatalytic activity may be the comprehensive impact of the size of the photocatalyst (see Table S2†), the degree of crystallisation, the specific surface area (see Table S3†) and the exposed (001) facet. The amount of photocatalytic hydrogen production relative to the specific surface area is shown in Fig. 7c. This demonstrates that the photocatalytic performance of sample T8 is also excellent without the effect of specific surface area. Thus, the high photocatalytic activity of T8 may be ascribed to the exposed (001) facet and the high degree of crystallinity.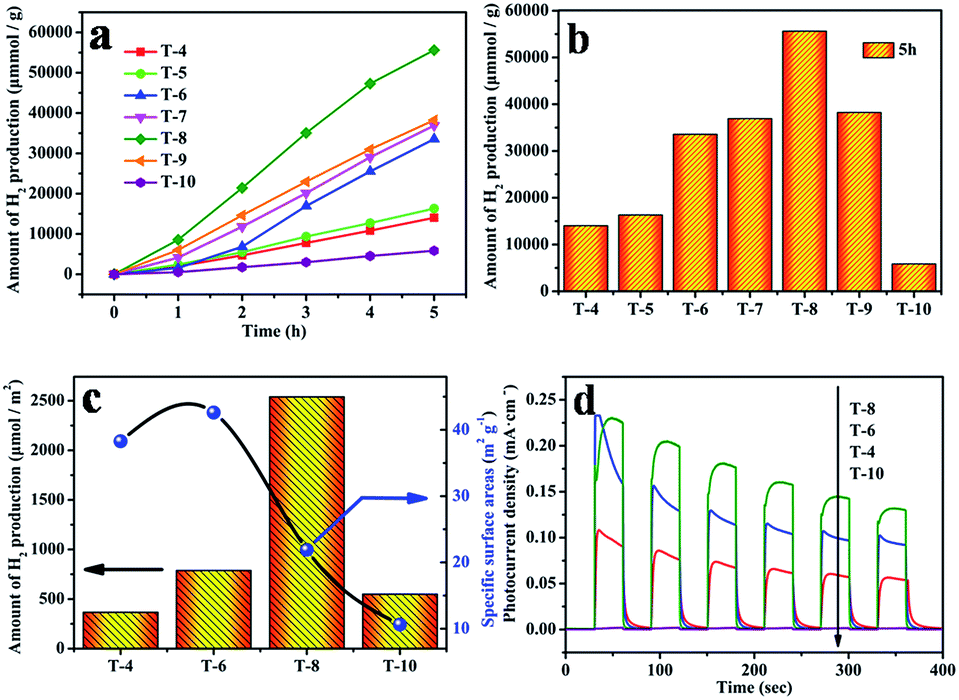 | ||
| Fig. 7 (a) Photocatalytic hydrogen evolution using the obtained TiO2; (b) the amount of photocatalytic hydrogen production after 5 h of irradiation; (c) the amount of photocatalytic hydrogen production relative to the specific surface area – the histogram is the amount of photocatalytic hydrogen production for each specific surface area after 5 h of irradiation and the line is the specific surface area of the samples T4, T6, T8, and T10; and (d) the photocurrent density for samples T4, T6, T8, and T10 under UV irradiation at 0.36 V vs. Ag/AgCl. | ||
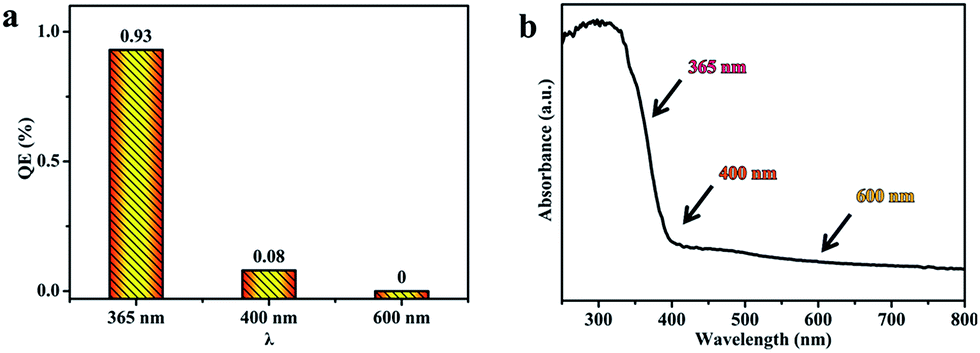 | ||
| Fig. 8 (a) The quantum efficiency of sample T8 under 365, 400 and 600 nm light; and (b) the UV-vis absorption spectrum of sample T8. | ||
The semiconductor's ability to generate and transfer photo-generated charge carriers will be indirectly reflected by the value of the photocurrent under irradiation.50 In order to confirm the active photocatalytic performance of the photocatalysts obtained using the simple solid-state method, the as-obtained photocatalysts were coated on FTO electrodes to evaluate the photocurrent, and the results are displayed in Fig. 7d. Prompt generation of photocurrent was observed when the electrodes were illuminated. With an increase in the annealing temperature, the photocurrent density of the as-prepared TiO2 became higher. In particular, sample T8 shows the highest photocurrent density. It reached a value as high as 0.22 mA cm−2, which is twice that of sample T4, and 20 times that of sample T10. The photocurrent density decreases quickly when the sintering temperature is above 800 °C. Note that sample T10 shows a low photocurrent density, which may be caused by a larger size of crystal and a rapid recombination of the photo-generated electrons and holes in TiO2. The highest photocurrent density from sample T8 may be a consequence of the exposed (001) facet and the more perfect crystalline nature of the sample. Zhao et al. also reported that with an exposed (001) facet the slow recombination rate of photo-induced charges will lead to more favorable photocatalytic activity for TiO2.23,51 The more perfect crystalline nature would reduce obstacles to electronic transmission and allow more electrons to reach the surface of the photocatalyst to assist the catalytic reactions.
The QE values for the photocatalyst T8 were also calculated, using the amount of H2 production under light of different wavelengths (365 nm, 400 nm and 600 nm). The QE is 0.93%, 0.08% and 0% under 365 nm, 400 nm and 600 nm, respectively. The results in Fig. 8a show that the QE decreases with an increase in the wavelength, which is consistent with the absorption intensity in the UV-visible absorption spectra (Fig. 8b).
4. Conclusions
In summary, we have successfully prepared uniform NH4TiOF3 nanoplates with a thickness of about 100 nm using a green, solvent-free LSCM in the absence of a template. The simple solid–solid transformation was used to convert NH4TiOF3 to TiO2 nanoplates with an exposed (001) facet, with a single layer thickness of about 50 nm. The TiO2 obtained in this work has good heat-stability, and is able to withstand high temperatures and maintain a stable anatase phase up to 1000 °C. The anatase TiO2 nanoplates exhibit a high rate of H2 generation, due to the exposed (001) facet and the high crystallinity. This work provides an efficient and environmentally friendly method to prepare TiO2 nanoplates with good heat-stability and high photocatalytic activity for hydrogen production.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21361024, 21471127 and U1503392) and the Natural Science Foundation of Xinjiang Province (No. 2014711004 and 2014211A013).Notes and references
- S. Landsmann, Y. Surace, M. Trottmann, S. Dilger, A. Weidenkaff and S. Pokrant, ACS Appl. Mater. Interfaces, 2016, 8, 12149–12157 CAS.
- W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed.
- T. Montini, V. Gombac, L. Sordelli, J. J. Delgado, X. Chen, G. Adami and P. Fornasiero, ChemCatChem, 2011, 3, 574–577 CrossRef CAS.
- X. An, Y. Wen, A. Almujil, D. Cheng, J. Li, X. Jia, J. Zou and Y. Ni, RSC Adv., 2016, 6, 89457–89466 RSC.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- H. X. Han and C. Li, Natl. Sci. Rev., 2015, 2, 143–145 CrossRef.
- D. Lang, T. Shen and Q. Xiang, ChemCatChem, 2015, 7, 943–951 CrossRef CAS.
- J. Yu, T. Ma and S. Liu, Phys. Chem. Chem. Phys., 2011, 13, 3491–3501 RSC.
- B. Kenens, M. Chamtouri, R. Aubert, K. Miyakawa, Y. Hayasaka, H. Naiki, H. Watanabe, T. Inose, Y. Fujita, G. Lu, A. Masuhara and H. Uji-i, RSC Adv., 2016, 6, 97464–97468 RSC.
- S. A. Bakar and C. Ribeiro, RSC Adv., 2016, 6, 89274–89287 RSC.
- U. Mandi, N. Salam, S. K. Kundu, A. Bhaumik and S. M. Islam, RSC Adv., 2016, 6, 73440–73449 RSC.
- G. B. Soares, R. A. P. Ribeiro, S. R. de Lazaro and C. Ribeiro, RSC Adv., 2016, 6, 89687–89698 RSC.
- H. Zhang, Q. Lin, S. Ning, Y. Zhou, H. Lin, J. Long, Z. Zhang and X. Wang, RSC Adv., 2016, 6, 96809–96815 RSC.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- F. Cao, J. Xiong, F. Wu, Q. Liu, Z. Shi, Y. Yu, X. Wang and L. Li, ACS Appl. Mater. Interfaces, 2016, 8, 12239–12245 CAS.
- J. M. Macak, M. Zlamal, J. Krysa and P. Schmuki, Small, 2007, 3, 300–304 CrossRef CAS PubMed.
- I. Paramasivam, H. Jha, N. Liu and P. Schmuki, Small, 2012, 8, 3073–3103 CrossRef CAS PubMed.
- Y. Tang, R. Hao, Y. Fu, Y. Jiang, X. Zhang, Q. Pan and B. Jiang, RSC Adv., 2016, 6, 96803–96808 RSC.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- M. Maisano, M. V. Dozzi, M. Coduri, L. Artiglia, G. Granozzi and E. Selli, ACS Appl. Mater. Interfaces, 2016, 8, 9745–9754 CAS.
- J. G. Yu, L. F. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CAS.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, J. Pan, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2009, 131, 12868–12869 CrossRef CAS PubMed.
- Y. Zhao, Q. Zhao, X. Li, Y. Hou, X. Zou, J. Wang, T. Jiang and T. Xie, Mater. Lett., 2012, 66, 308–310 CrossRef CAS.
- Q. Xiang and J. Yu, Chin. J. Catal., 2011, 32, 525–531 CrossRef CAS.
- C. X. Lei, X. L. Jiang, X. Huang, X. Liu, D. Q. Zeng, Y. T. Ma, L. S. Wang and D. L. Peng, Appl. Surf. Sci., 2015, 359, 860–867 CrossRef CAS.
- H. Li, T. Li, H. Liu, B. Huang and Q. Zhang, J. Alloys Compd., 2016, 657, 1–7 CrossRef CAS.
- S. Liu, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS PubMed.
- H. Yu, Q. F. Xu, Z. R. Sun, S. J. Ji, J. X. Chen, Q. Liu, J. P. Lang and K. Tatsumi, Chem. Commun., 2001, 2614–2615 RSC.
- C. Zhang, Y. Song, X. Wang, F. E. Kühn, Y. Wang, Y. Xu and X. Xin, J. Mater. Chem., 2003, 13, 571–579 RSC.
- L. Wang, Y. D. Huang, R. R. Jiang and D. Z. Jia, J. Electrochem. Soc., 2007, 154, 1015–1019 CrossRef.
- J. X. Guo, L. Liu, G. F. Liu, D. Z. Jia and X. L. Xie, Org. Lett., 2007, 9, 3989–3992 CrossRef CAS PubMed.
- Y. L. Cao, P. F. Hu, W. Y. Pan, Y. D. Huang and D. Z. Jia, Sens. Actuators, B, 2008, 134, 462–466 CrossRef CAS.
- Y. L. Cao, W. Y. Pan, Y. Zong and D. Z. Jia, Sens. Actuators, B, 2009, 138, 480–484 CrossRef CAS.
- Y. L. Qi, Y. D. Huang, D. Z. Jia, S. J. Bao and Z. P. Guo, Electrochim. Acta, 2009, 54, 4772–4776 CrossRef CAS.
- R. Y. Wang, D. Z. Jia, L. Zhang, L. Liu, Z. P. Guo, B. Q. Li and J. X. Wang, Adv. Funct. Mater., 2006, 16, 687–692 CrossRef CAS.
- Y. Z. Li, Y. L. Cao and D. Z. Jia, J. Mater. Chem. A, 2014, 2, 3761–3765 CAS.
- M. Hojamberdiev, G. Zhu, P. Sujaridworakun, S. Jinawath, P. Liu and J. P. Zhou, Powder Technol., 2012, 218, 140–148 CrossRef CAS.
- B. Liu, X. Li, Q. Zhao, J. Ke, M. Tadé and S. Liu, Appl. Catal., B, 2016, 185, 1–10 CrossRef CAS.
- L. Yan, X. He, Y. Wang, J. Li and D. Wang, J. Mater. Sci.: Mater. Electron., 2016, 27, 4068–4073 CrossRef CAS.
- X. Liu, S. Gao, H. Xu, Z. Lou, W. Wang, B. Huang and Y. Dai, Nanoscale, 2013, 5, 1870–1875 RSC.
- D. Kumar, M. S. Chen and D. W. Goodman, Thin Solid Films, 2006, 515, 1475–1479 CrossRef CAS.
- W. Zhao, Z. Ai, J. Dai and M. Zhang, PLoS One, 2014, 9, 103671–103680 Search PubMed.
- L. Zhou, J. Chen, C. Ji, L. Zhou and P. O'Brien, CrystEngComm, 2013, 15, 5012–5015 RSC.
- J. Q. Yan, H. Wu, H. Chen, Y. X. Zhang, F. X. Zhang and S. Z. F. Liu, Appl. Catal., B, 2016, 191, 130–137 CrossRef CAS.
- S. Yu, Y. Q. Zhong, B. Q. Yu, S. Y. Cai, L. Z. Wu and Y. Zhou, Phys. Chem. Chem. Phys., 2016, 18, 20338–20344 RSC.
- W. Lin, H. Zheng, P. Zhang and T. Xu, Appl. Catal., A, 2016, 521, 75–82 CrossRef CAS.
- M. Miyauchi, A. Ikezawa, H. Tobimatsu, H. Irie and K. Hashimoto, Phys. Chem. Chem. Phys., 2004, 6, 865–870 RSC.
- L. Li, L. Yu, Z. Lin and G. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 8536–8545 CAS.
- G. L. Chiarello, E. Selli and L. Forni, Appl. Catal., B, 2008, 84, 332–339 CrossRef CAS.
- Z. A. Huang, Q. Sun, K. L. Lv, Z. H. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420–427 CrossRef CAS.
- K. Takanabe and K. Domen, ChemCatChem, 2012, 4, 1485–1497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figure (Fig. S1–S3) and tables (Tables S1–S3). See DOI: 10.1039/c6ra27160j |
| This journal is © The Royal Society of Chemistry 2017 |