
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DFT study of NO adsorption on pristine graphene
Hongwei Gao * and
Zhijie Liu
* and
Zhijie Liu
Key Laboratory of Plant Resources and Chemistry in Arid Regions, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China. E-mail: gaohongw369@ms.xjb.ac.cn; Fax: +86-991-3858319; Tel: +86-991-3858319
First published on 24th February 2017
Abstract
This study reports in detail the adsorption of NO exhaust gas on pristine graphene by a first-principle. Ten kinds of adsorption configurations were built for three different adsorption sites on graphene. Structure optimization was carried out using different approximation methods; we found that the optimal method was the PBEsol approximation under the GGA approximation, and the orbital cutoff was 6 Å. Optimal parameters were used to calculate the adsorption energy, charge Mulliken, electron density difference, LUMO HOMO, and projected density of states (PDOS) for these ten kinds of adsorption configurations. We found that the adsorption of NO molecules on graphene was a physical adsorption process, and electrons were transferred from graphene to a NO molecule. The optimal adsorption site of graphene indicated the B-site, followed by the T-site, and the least negative adsorbed position was the H-site. Meanwhile, adsorption energy analysis showed that O-end graphene was the most stable adsorption configuration.
1. Introduction
Graphene, made up of six-member rings, is a two-dimensional periodic lattice structure. Graphene can be warped into zero-dimensional fullerene, rolled up into one-dimensional carbon nanotubes, and stacked into three-dimensional graphite.1,2 Ideally, graphene is a monolayer plane with a hexagonal type honeycomb lattice structure, in which carbon (C) atoms have a sp2 hybrid orbital, meaning that each C atom has four bonds, and every C atom forms a σ-bond with the nearest neighbor three C atoms. The remaining p electrons, which are perpendicular to the surface of graphene, form π-bonds with the surrounding electrons. Graphene crystallites of high quality were fabricated by Andre G. and Konstantin N., who were awarded the Nobel Prize for Physics in 2010, by using the ‘Scotch-tape’ method, which was invented by Novoselov et al. in 2004.3–5 Enormous amounts of research have been reported concerning the basic characterization of graphene, which used graphene as a carrier for various modifications.4,6–9 Graphene, which has a particular nanostructure and excellent physical and chemical properties, has a wide range of applications in electronics, optics, magnetism, biomedicine, catalysis, energy storage, sensors, and other areas. Applications of graphene, especially in areas of environmental protection, have significant practicability and exploitability.With the enhancement of people's awareness concerning environmental protection, automobile exhaust pollution has become a global issue due to rapid developments in the automobile industry. Automotive exhaust mainly includes hydrocarbons, nitrogen oxide, carbon monoxide, sulfur dioxide, solid suspended particles, etc.10–12 Graphene as an adsorption material to deal with this exhaust has been confirmed by many experiments and research results.13–16 From literature concerning graphene as an adsorption material, we found that most research focused on major emissions, such as CO, NO, and NO2. Liu et al.17 found that CO adsorption energy could be enhanced by an electric field, and Choudhuri et al.18 also demonstrated that B–N doped graphene could heighten adsorption of NO, NO2, CO, and CO2. Based on our previous work, we used Density Functional Theory (DFT) methods to calculate NO, the main pollution gas from exhaust, on pristine graphene surface in this investigation. We finally pinpointed the stable adsorption sites; meanwhile, this paper provides a detailed analysis on electronic structures of the adsorption configurations. The results of this research can provide theoretical guidance for relevant experimental research.
2. Computational details
Adsorption of NO on pristine graphene was investigated by a first principle using the Dmol3 code. Using computer simulation we chose ten kinds of supercells without vacuum layers including 4 × 4 × 1, 5 × 5 × 1, 6 × 6 × 1, 7 × 7 × 1, 8 × 8 × 1, 9 × 9 × 1, 10 × 10 × 1, 11 × 11 × 1, and 12 × 12 × 1, and tried to determine the most stable supercell system. Furthermore, we carried out an optimization test of functions under the LDA and GGA approximates in the process of geometry optimization, and then chose the optimum one. Orbital cutoffs were also tested by the optimum functional, 0.006 smearing and 5 × 5 × 1 k-points.In this computer simulation, the length of N–O bond was set as lN–O = 1.17 Å.19 For this study, a NO molecule was adsorbed on pristine graphene surface, and the adsorption energy Ead was defined as:
| Ead = Etotal − (Egraphene + ENO) | (1) |
3. Results and discussion
3.1 Supercell chosen with calculation parameters at software default values
To obtain detailed comprehension of adsorption structures of NO on the graphene layer, it was necessary to perform a study on selection of a supercell. The C–C bond length of a pristine graphene unit cell was 1.42 Å.20–22In Table 1, the longest C–C bond lengths for pristine graphene supercells in the surface center region are 1.423, 1.422, 1.418, 1.419, 1.417, 1.416, 1.415, 1.415, and 1.414 Å, respectively. We found that the supercell 7 × 7 × 1 was the most appropriate one and the relative deviation was 0.000704. Meanwhile, a short time was spent on calculations for a hexagonal structure that included 96 atoms.
| Supercell | C–C (min.) (Å) | Relative deviation (%) | C–C (max.) (Å) | Relative deviation (%) | Exp. (Å) |
|---|---|---|---|---|---|
| 4 × 4 × 1 | 1.418 | −0.00141 | 1.423 | 0.00211 | |
| 5 × 5 × 1 | 1.412 | −0.00563 | 1.422 | 0.00141 | |
| 6 × 6 × 1 | 1.415 | −0.00352 | 1.418 | −0.00141 | |
| 7 × 7 × 1 | 1.414 | −0.00423 | 1.419 | −0.00070 | |
| 8 × 8 × 1 | 1.413 | −0.00493 | 1.417 | −0.00211 | 1.420 |
| 9 × 9 × 1 | 1.413 | −0.00493 | 1.416 | −0.00282 | |
| 10 × 10 × 1 | 1.413 | −0.00493 | 1.415 | −0.00352 | |
| 11 × 11 × 1 | 1.413 | −0.00493 | 1.415 | −0.00352 | |
| 12 × 12 × 1 | 1.413 | −0.00493 | 1.414 | −0.00423 |
3.2 Select functional and test orbital cutoff
During the process of geometric optimization, LDA and GGA were two kinds of common exchange–correlation energy approximation methods used. The structural optimization of pristine graphene using a 7 × 7 × 1 supercell was conducted by PWC, VWN under LDA, and PW91, BP, PBE, BLYP, BOP, VWN-BP, RPBE, HCTH, and PBEsol under GGA. The computational specific conditions are shown in Table 2.| C–C (Å) | Relative deviation (%) | HOMOa (eV) | LUMOb (eV) | Δc (eV) | ||
|---|---|---|---|---|---|---|
| a HOMO: Highest Occupied Molecular Orbital.b LUMO: Lowest Unoccupied Molecular Orbital.c Δ: HOMO − LUMO. | ||||||
| LDA | PWC | 1.418 | −0.00141 | −5.895 | −5.738 | −0.157 |
| VWN | 1.417 | −0.19230 | −5.896 | −5.739 | −0.157 | |
| GGA | PW91 | 1.424 | 0.00282 | −5.813 | −5.789 | −0.024 |
| BP | 1.425 | 0.00352 | −5.748 | −5.721 | −0.027 | |
| PBE | 1.425 | 0.00352 | −5.786 | −5.761 | −0.025 | |
| BLYP | 1.428 | 0.00563 | −5.596 | −5.575 | −0.021 | |
| BOP | 1.429 | 0.00634 | −5.517 | −5.493 | −0.0.24 | |
| VWN-BP | 1.425 | 0.00352 | −5.749 | −5.722 | −0.027 | |
| RPBE | 1.429 | 0.00634 | −5.784 | −5.755 | −0.029 | |
| HCTH | 1.424 | 0.00282 | −6.145 | −6.118 | −0.027 | |
| PBEsol | 1.423 | 0.00211 | −5.782 | −5.758 | −0.024 | |
By comparing the obtained results with experimental values, we found that LDA approximation methods were more applicable for calculating molecule bond length.23,24 The C–C bond length was 1.418 Å as calculated by PWC, which was closer to the experimental value (1.420 Å). But, the LDA approximation had strong localization about the electron distributions; moreover, it wasn't suitable for analyzing the absolute value of the forbidden band width and the system binding energy. Generally, the semiconductor band gap computed by GGA was closer to the experimental values than when computed by LDA. Through comprehensive consideration of calculated and experimental values of the lattice constant and energy band gap, we selected the GGA approximation PBEsol functional to proceed with further work.
Fig. 1 clearly shows that the convergence test of the orbital cutoff was calculated using a GGA-PBEsol approximation. When orbital cutoff was 4.0 Å, the curve began to appear to converge. When the value of the orbital cutoff was 6.0 Å, then geometry converged very well, so we chose the value of orbital cutoff as 6.0 Å.
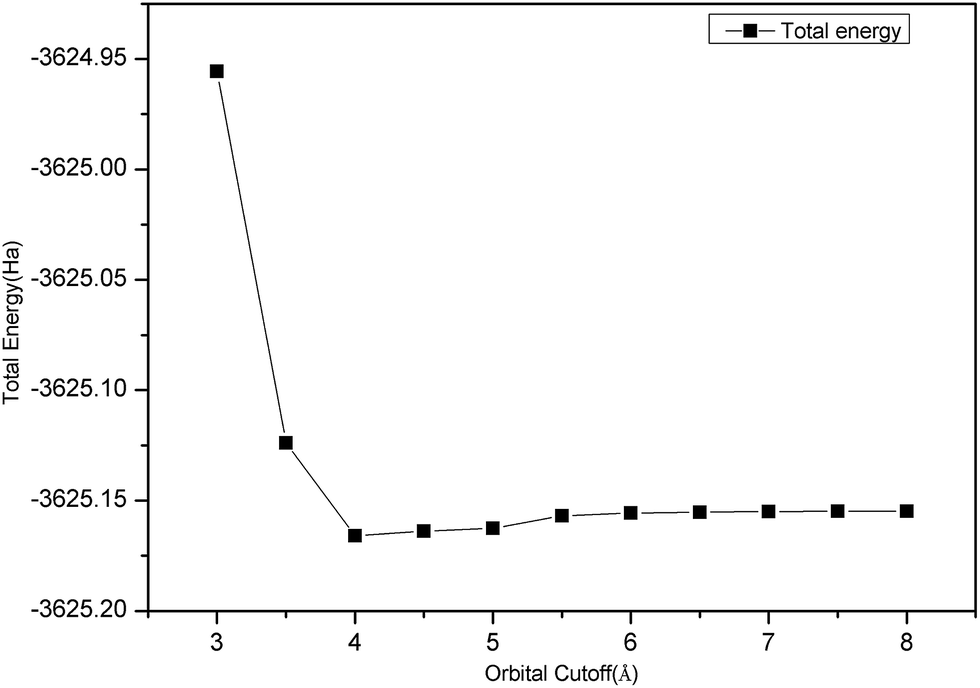 | ||
| Fig. 1 Convergence test image of orbital cutoff. | ||
3.3 NO adsorption on graphene layer
Three sites were considered for NO adsorption on graphene, as shown in Fig. 2(a): bridge site, top site, and hollow site which are abbreviated as B-site, T-site, and H-site, respectively. For the T-site and H-site, three different configurations of NO–graphene were calculated and a N atom, O atom, and N–O bond were placed near adsorption sites, respectively. For the B-site, the N–O bonds placed parallel and perpendicular to the adsorption sites were considered during the adsorption process of the N–O bond. Ten configurations are shown in Fig. 2(b). When NO was adsorbed on three sites of graphene by various adsorption methods, tiny deformations occurred in all graphene layers. The C–C bond length was no longer a fixed value because it changed from 1.242 Å to 1.481 Å. The isolated N–O bond was 1.17 Å and all bond lengths increased after adsorption.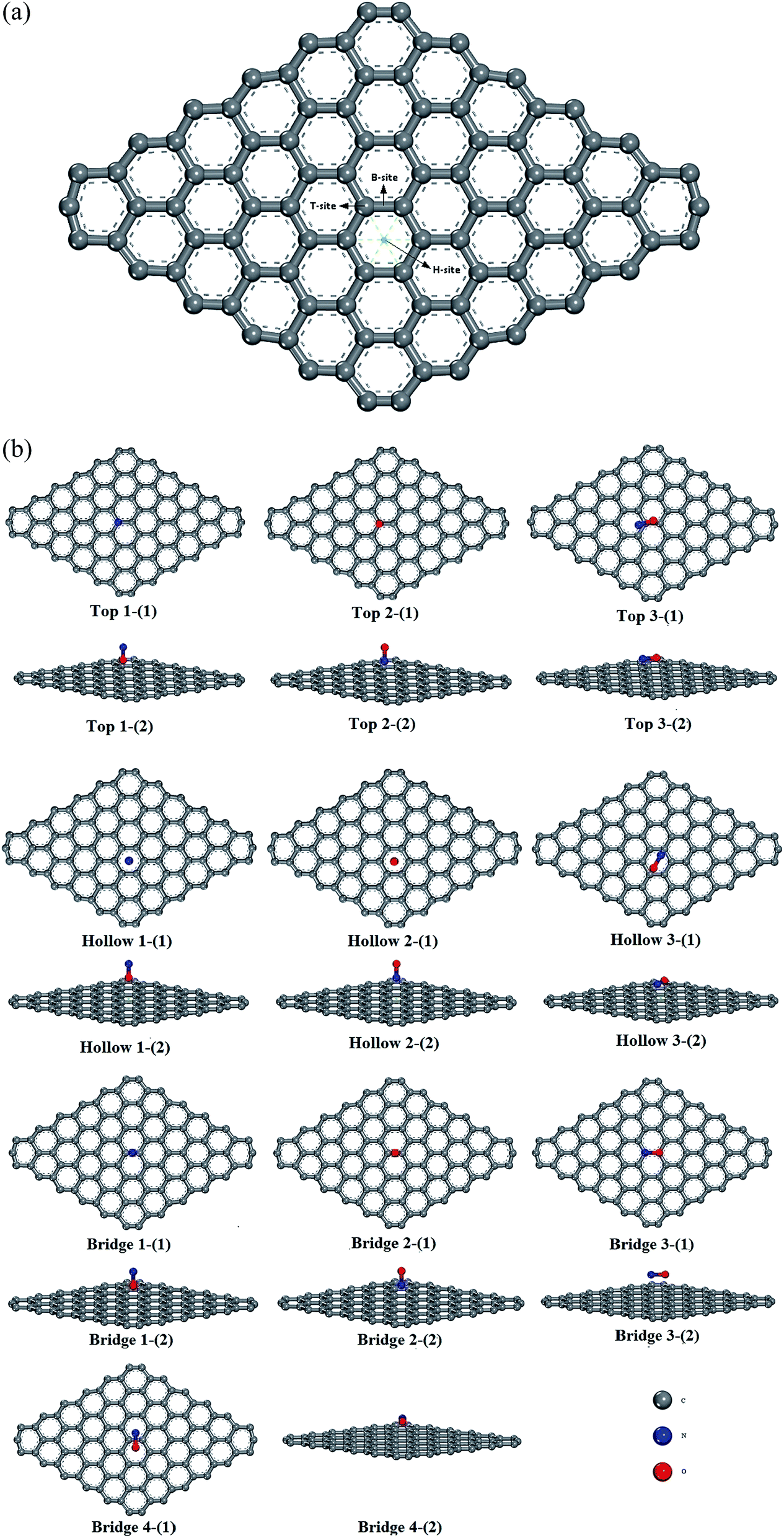 | ||
| Fig. 2 (a): A total of three adsorption sites on pristine graphene surface. (b): Ten different configurations of three sites on pristine graphene surface: (1) top observation; (2) side observation; (1): O terminal approached graphene; (2): N terminal approached graphene; (3, 4): N–O bond parallel or perpendicular approached graphene. | ||
Based on DFT, adsorption energies, distances of NO to graphene surface, and geometry structures of ten configurations are listed in Table 3.
| Configuration | h (Å) | Total energy (eV) | Ead (eV) | C–C (Å) | N–O (Å) | Relative deviation (%) | |
|---|---|---|---|---|---|---|---|
| T-Site | O | 3.144 | −102498.381 | −2.1424 | 1.242–1.480 | 1.201 | 0.02637 |
| N | 3.154 | −102498.347 | −2.1084 | 1.242–1.479 | 1.120 | 0.02517 | |
| N–O | 3.089 | −102498.187 | −1.9489 | 1.243–1.479 | 1.201 | 0.02626 | |
| B-Site | O | 2.849 | −102498.610 | −2.3713 | 1.243–1.480 | 1.201 | 0.02645 |
| N | 3.147 | −102498.440 | −2.2018 | 1.243–1.481 | 1.120 | −0.00147 | |
| N–O(Para.) | 3.086 | −102498.137 | −1.8981 | 1.242–1.481 | 1.202 | 0.02742 | |
| N–O(Perp.) | 2.839 | −102498.565 | −2.3266 | 1.242–1.480 | 1.200 | 0.02604 | |
| H-Site | O | 2.940 | −102498.195 | −1.9569 | 1.243–1.480 | 1.201 | 0.02645 |
| N | 2.845 | −102498.332 | −2.0932 | 1.242–1.481 | 1.194 | 0.02043 | |
| N–O | 3.101 | −102497.984 | −1.7453 | 1.243–1.480 | 1.201 | 0.02678 | |
Comparing the calculated results, we found that the most favorable adsorption position of NO on a graphene surface was the B-site, which is in accordance with the study reported by Leenaerts.25 But the calculated adsorption energy (−2.4 eV) in our paper is larger than that by Leenaerts25 (29 meV, i.e. 0.029 eV). Therefore, we consulted other literature and found that the adsorption energies of NO on a graphene surface were −0.061 eV by using the DMol3 package by Chen et al.,27 −24 kJ mol−1 (about −0.25 eV) using Grimme's D3 dispersion model by Rad et al.,28 −0.801 eV using SIESTA software code by Balangi et al.,29 −0.14 eV using VASP5.2 by Hou et al.,30 −0.30 eV using CASTEP by Zhang et al.,31 and 0.19 eV using DMol3 by Tang,32 respectively. In our previous works, the calculated adsorption energy of NO N-end on Ptn/γ-Al2O3 (ref. 33) using the Dmol3 module in a Materials Studio8.0 package was −1.90 eV. However, the calculated adsorption energy of NO N-end on Pd/γ-Al2O3 (ref. 34) using the CASTEP module in a Materials Studio8.0 package was −0.18 eV. The adsorption energies of NO on different catalysts (Al2O3, GaAlIV, GaAlIII) by Liu et al.35 based on the DMol3 module in a Materials Studio package were −1.08 eV, −1.12 eV, and −1.04 eV, respectively. The adsorption energy of NO on pristine graphene using the Atomistix ToolKit package by Aghaei et al.36 was −2.68 eV, but the distance between NO and the surface was 1.73 Å. We also found that the calculated adsorption energy by Zhang et al.37 has a big difference with one by Aghaei et al.,36 although the distance between NO and the surface was similar. By comparing the adsorption energies in the above literature, and checking our work again, we thought that this inconformity may be due to differences in the application of computational software, computational methods, and calculation parameters. Furthermore, for NO adsorption on a pristine graphene sheet in our paper, the calculated adsorption energy of the B-site was the largest, and those of T-site and H-site were similar. This result was consistent with existing research results in the literature.
| Mulliken | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | C5 | C6 | N | O | Electron transfer | ||
| Graphene | 0.000 | 0.009 | 0.000 | 0.000 | 0.009 | 0.001 | — | — | — | |
| NO | — | — | — | — | — | — | 0.066 | 0.066 | 0.000 | |
| T-Site | O | 0.001 | 0.012 | 0.001 | −0.002 | 0.010 | 0.003 | 0.071 | −0.075 | −0.004 |
| N | 0.000 | 0.011 | 0.001 | −0.003 | 0.009 | 0.002 | 0.072 | −0.074 | −0.002 | |
| N–O | −0.020 | 0.009 | 0.001 | −0.003 | 0.011 | −0.002 | 0.071 | −0.074 | −0.003 | |
| B-Site | O | 0.002 | 0.012 | −0.002 | −0.001 | 0.012 | 0.002 | 0.070 | −0.075 | −0.005 |
| N | 0.001 | 0.012 | −0.001 | −0.001 | 0.012 | 0.001 | 0.073 | −0.074 | −0.001 | |
| N–O(Para.) | −0.019 | 0.012 | 0.000 | −0.002 | 0.012 | −0.009 | 0.070 | −0.077 | −0.007 | |
| N–O(Perp.) | −0.013 | 0.009 | −0.001 | −0.001 | 0.009 | −0.013 | 0.071 | −0.072 | −0.001 | |
| H-Site | O | 0.001 | 0.011 | 0.001 | 0.001 | 0.010 | 0.002 | 0.073 | −0.078 | −0.005 |
| N | −0.002 | 0.007 | 0.000 | −0.001 | 0.007 | −0.003 | 0.087 | −0.045 | 0.042 | |
| N–O | −0.007 | 0.004 | −0.002 | −0.004 | 0.000 | −0.009 | 0.071 | −0.075 | −0.004 | |
 | ||
| Fig. 3 Six C atoms on the hexagon of the three adsorption sites. | ||
Taken together in Table 4, electrons were transferred from a NO molecule to the graphene surface. When a NO molecule is vertical and its O atom is close to the graphene surface, the number order of electrons transferred on different adsorption sites was B-site = H-site > T-site. When a NO molecule is vertical and its N atom is close to the graphene surface, then the number order of electrons transferred on different adsorption sites was H-site > T-site > B-site. When a NO molecule is parallel to the graphene surface, the number order of electrons transferred on different adsorption sites was B-site > H-site > T-site.
Table 4 also shows the effect of the direction for NO adsorption on the graphene surface. The number of electrons transferred is not the same for different directions. The number (0.007e) of electrons transferred for a parallel NO molecule is much larger than that (0.001e) for a perpendicular NO molecule. The H-site has the greatest change attributed to the direction. Therefore, the charge transfer is not only affected by adsorption sites, but also by molecular orientation. We found that the data in Table 5 of Leenaerts et al.25 have the similar rule, but they focused on the role of orientations.
| T-Site | B-Site | H-Site | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| O | N | N–O | O | N | N–O(Para.) | N–O(Perp.) | O | N | N–O | |
| HOMO (eV) | −5.437 | −5.415 | −5.444 | −5.394 | −5.448 | −5.429 | −5.469 | −5.543 | −5.446 | −5.447 |
| LUMO (eV) | −5.331 | −5.314 | −5.201 | −5.343 | −5.335 | −5.276 | −5.428 | −5.315 | −5.443 | −5.408 |
| HOMO–LUMO (eV) | −0.106 | −1.101 | −0.243 | −0.051 | −0.113 | −0.153 | −0.041 | −0.115 | −0.013 | −0.039 |
| Δρ = ρNO–graphene − ρgraphene − ρNO | (2) |
 | ||
| Fig. 4 The electron density difference of the ten adsorption configurations. (para.): N–O bond parallel approached graphene, (perp.): N–O bond perpendicular approached graphene. | ||
In the ten sectional views, there were no overlaps of the electron density between a NO molecule and C atom. This also showed that the ten kinds of adsorption configurations in the three sites were physical adsorption, which is consistent with the conclusion of the adsorption part of this paper. The red part represents the gained electrons and the blue part represents the lost electrons in different charge density maps. From Fig. 4, we found that graphene acted as an acceptor, which meant the charge was transferred from a NO molecule to the monolayer graphene, and the gained charge in all systems was mainly derived from the NO molecule that was closest to the C atom. By comparing the size of the red area and blue area, we found that most of the electrons were obtained by the O-end to the graphene B-site, the second was on the T-site, and the least was on the H-site. When N-end to a NO molecule was near three adsorption sites, then the graphene B-site lost the most electronic area, the second was located at the T-site, and the least was at the H-site. When the N–O bond was parallel or perpendicular to the graphene layer, we found that two configurations of the B-site had obvious symmetry, and that most electrons were transferred from the NO molecule to graphene. The electron values that NO lost and gained on the H-site were similar to the T-site. Through the comprehensive comparison of ten configurations gaining and losing of electrons, we found that a NO molecule got its number of electrons from the graphene layer following the order: B-site > T-site > H-site. This conclusion was consistent with our previous conclusion of the adsorption energy discussion.
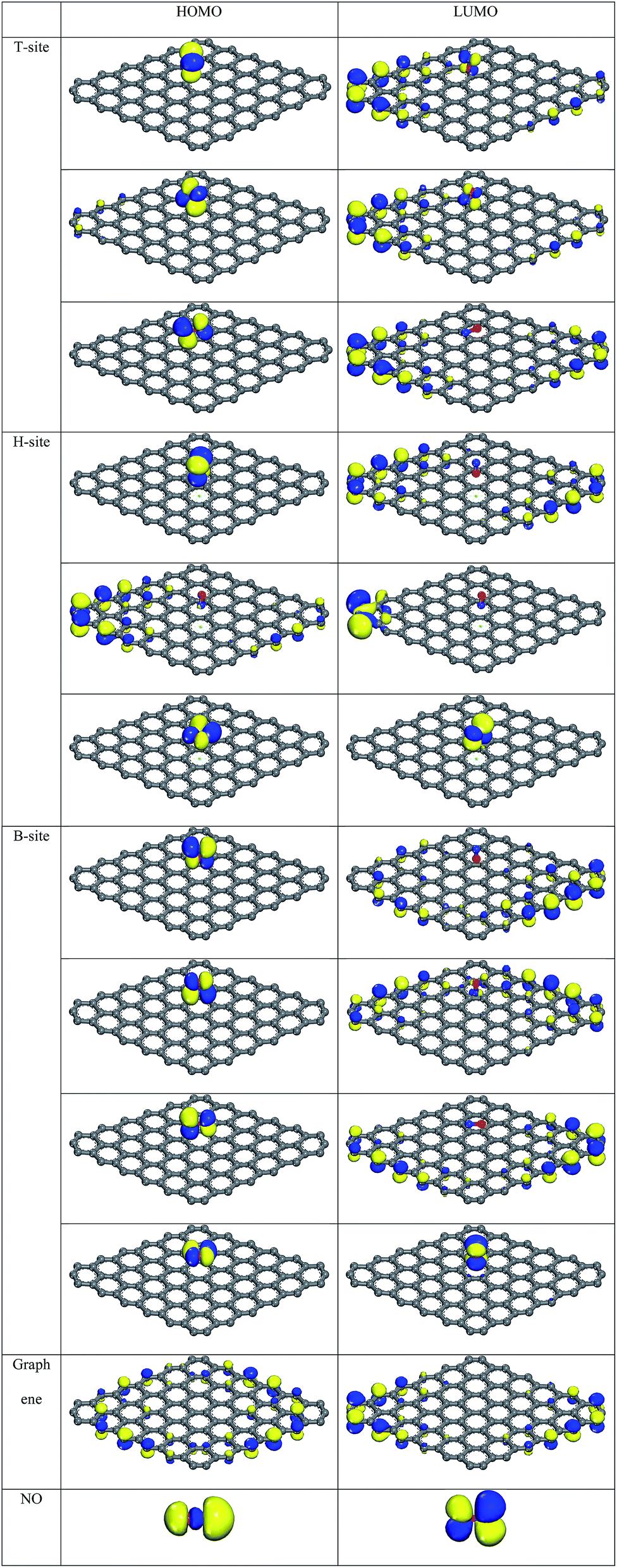 | ||
| Fig. 5 HOMO and LUMO of the ten adsorption configurations, graphene, and NO molecule. | ||
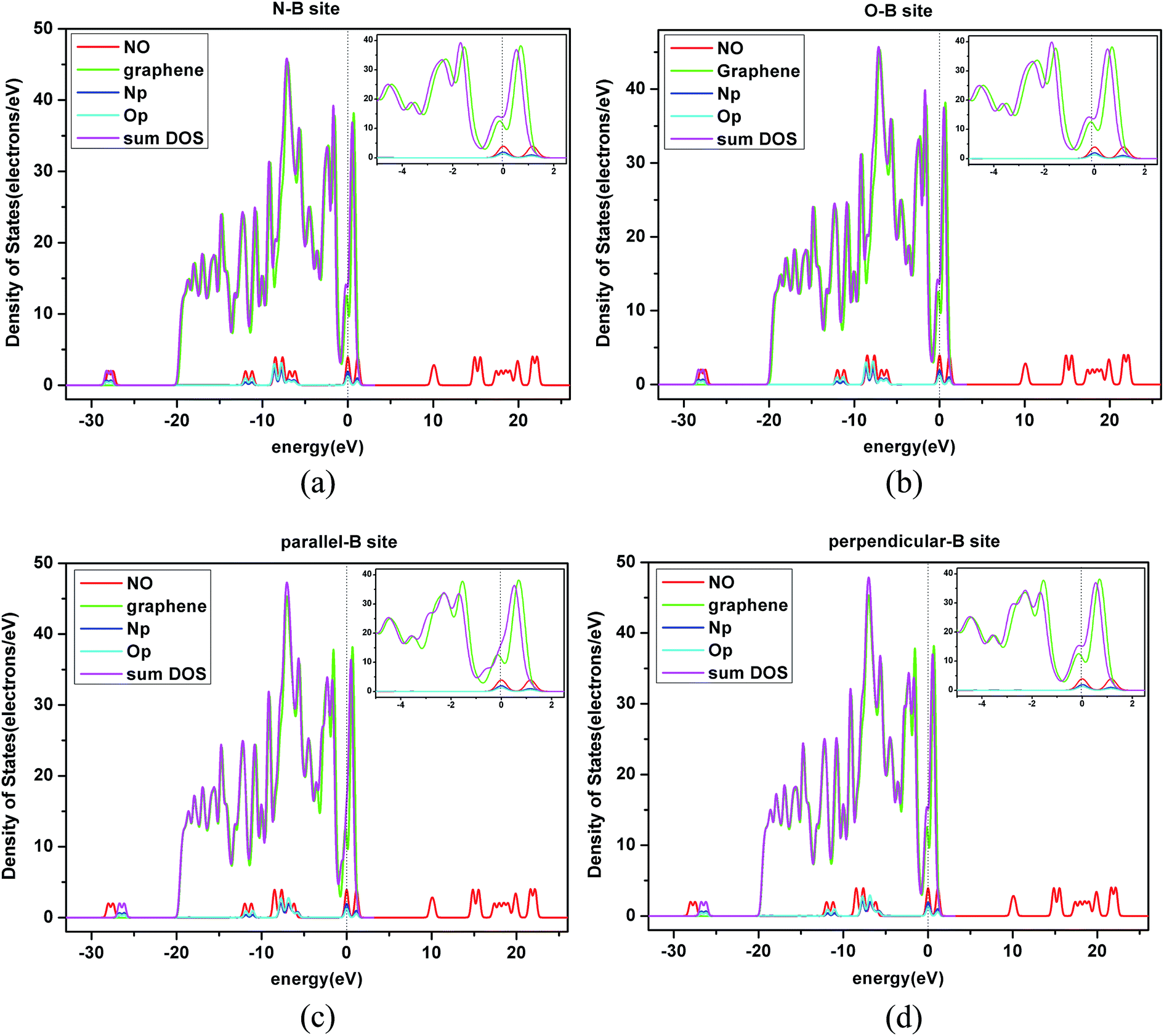 | ||
| Fig. 6 The density of states (DOS) of a NO molecule and graphene sheet. The total density of states (TDOS) of NO adsorbed on the B-site of a pristine graphene layer with the projected density of states (PDOS) for the p state of N and O atoms in four orientations. (a) N-B site: N terminal approaching the B-site; (b) O-B site: O terminal approaching the B-site; (c) parallel-B-site: N–O bond parallel approaching the B-site; (d) perpendicular-B-site: N–O bond perpendicular approaching the B-site. | ||
4. Conclusions
Along with more and more people paying attention to environmental pollution problems, exhaust gas processing has become a key problem for research focus. In this paper, based on previous studies, we studied the adsorption of NO gas on pristine graphene, which is a basic guide for future experiments and the treatment of NO gas under real life situations.Using DFT computer simulation, we investigated the adsorption of NO on a pristine graphene surface. Through an optimizing test, we selected a PBEsol approximation and GGA approximation to carry out further work. We studied the graphene surface of three adsorption sites, and calculated the adsorption energy, Mulliken charge, electron density difference, and the HOMO, LUMO, and PDOS of these three kinds of adsorption sites. We found that the adsorption process was physical adsorption and the adsorption energies were −2.37 to −1.74 eV. The number of transferred electrons from molecular NO to graphene was −0.007 to −0.001e. Calculated results showed that the largest adsorption energy of NO on the graphene surface was the B-site, followed by the T-site, and finally the H-site. Meanwhile, these results were verified by the analysis of Mulliken charge, electron density difference, HOMO, and LUMO.
Results showed that the graphene played a role in adsorbing NO exhaust gas. When a B-site acted as an adsorption site, the adsorption effect was better. Research results have a certain theoretical support and guidance for future NO exhaust gas treatment.
Acknowledgements
This work was supported by Natural Science Foundation of Xinjiang, China, Grant No. 2016D01A073. This work was also financially supported by Recruitment Program of Global Experts, and the Director Foundation of XTIPC, CAS, Grant No. 2015RC011.References
- Z. M. Ao and F. M. Peeters, J. Phys. Chem. C, 2010, 114, 14503–14509 CAS.
- A. Geim and K. Novoselov, R. Swed. Acad. Sci., 2011, 181, 1283 Search PubMed.
- K. S. Novoselov, Int. J. Mod. Phys. B, 2011, 25, 4081–4106 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- W. Wu, Q. Yu, P. Peng, Z. Liu, J. Bao and S.-S. Pei, Nanotechnology, 2012, 23, 035603 CrossRef PubMed.
- T. Gao, S. Xie, Y. Gao, M. Liu, Y. Chen, Y. Zhang and Z. Liu, ACS Nano, 2011, 5, 9194–9201 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- M. Liu, Y. Zhang, Y. Chen, Y. Gao, T. Gao, D. Ma, Q. Ji, Y. Zhang, C. Li and Z. Liu, ACS Nano, 2012, 6, 10581–10589 CrossRef CAS PubMed.
- P. S. Arun, B. P. Ranjith and S. M. A. Shibli, Environ. Sci. Technol., 2013, 47, 2746–2753 CrossRef CAS PubMed.
- F. Ahmed, M. K. Alam, A. Suzuki, M. Koyama, H. Tsuboi, N. Hatakeyama, A. Endou, H. Takaba, C. A. Del Carpio, M. Kubo and A. Miyamoto, J. Phys. Chem. C, 2009, 113, 15676–15683 CAS.
- N. Takahashi and H. Imagawa, J. Jpn. Pet. Inst., 2009, 52, 90–101 CrossRef CAS.
- K. J. Berean, J. Z. Ou, M. Nour, M. R. Field, M. M. Y. A. Alsaif, Y. Wang, R. Ramanathan, V. Bansal, S. Kentish, C. M. Doherty, A. J. Hill, C. McSweeney, R. B. Kaner and K. Kalantar-zadeh, J. Phys. Chem. C, 2015, 119, 13700–13712 CAS.
- E. C. Mattson, K. Pande, M. Unger, S. Cui, G. Lu, M. Gajdardziska-Josifovska, M. Weinert, J. Chen and C. J. Hirschmugl, J. Phys. Chem. C, 2013, 117, 10698–10707 CAS.
- S. C. Xu, S. Irle, D. G. Musaev and M. C. Lin, J. Phys. Chem. B, 2006, 110, 21135–21144 CrossRef CAS PubMed.
- S. Wannakao, T. Nongnual, P. Khongpracha, T. Maihom and J. Limtrakul, J. Phys. Chem. C, 2012, 116, 16992–16998 CAS.
- H. Liu and J. Y. Lee, J. Phys. Chem. C, 2012, 116, 3034–3041 CAS.
- I. Choudhuri, N. Patra, A. Mahata, R. Ahuja and B. Pathak, J. Phys. Chem. C, 2015, 119, 24827–24836 CAS.
- M. P. Jigato, K. Somasundram, V. Termath, N. C. Handy and D. A. King, Surf. Sci., 1997, 380, 83–90 CrossRef.
- R. Heyrovska, ArXiv08044086 Phys., 2008.
- E. V. Castro, K. S. Novoselov, S. V. Morozov, N. M. R. Peres, J. M. B. L. dos Santos, J. Nilsson, F. Guinea, A. K. Geim and A. H. C. Neto, J. Phys.: Condens. Matter, 2010, 22, 175503 CrossRef PubMed.
- P. R. Wallace, Phys. Rev., 1947, 71, 622–634 CrossRef CAS.
- D. Bensaid, J. Mod. Phys., 2011, 2, 642–650 CrossRef CAS.
- P. Haas, F. Tran, P. Blaha, K. Schwarz and R. Laskowski, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 195109 CrossRef.
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125416 CrossRef.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef CAS PubMed.
- Y. Chen, B. Gao, J.-X. Zhao, Q.-H. Cai and H.-G. Fu, J. Mol. Model., 2012, 18, 2043–2054 CrossRef CAS PubMed.
- A. S. Rad and E. Abedini, Appl. Surf. Sci., 2016, 360, 1041–1046 CrossRef CAS.
- H. R. Balangi and A. A. Shokri, Phys. E, 2015, 74, 515–526 CrossRef CAS.
- M. Hou, W. Cen, H. Zhang, J. Liu, H. Yin and F. Wei, Appl. Surf. Sci., 2015, 339, 55–61 CrossRef CAS.
- Y.-H. Zhang, Y.-B. Chen, K.-G. Zhou, C.-H. Liu, J. Zeng, H.-L. Zhang and Y. Peng, Nanotechnology, 2009, 20, 185504 CrossRef PubMed.
- S. Tang and Z. Cao, J. Chem. Phys., 2011, 134, 044710 CrossRef PubMed.
- H. Gao, RSC Adv., 2016, 6, 102914–102923 RSC.
- H. Gao, Appl. Catal., A, 2017, 529, 156–166 CrossRef CAS.
- Z. Liu, L. Ma and A. S. M. Junaid, J. Phys. Chem. C, 2010, 114, 4445–4450 CAS.
- S. M. Aghaei, M. M. Monshi and I. Calizo, RSC Adv., 2016, 6, 94417–94428 RSC.
- N. C. Zhang, J. Ren and X. J. Peng, Fullerenes, Nanotubes, Carbon Nanostruct., 2016, 24, 298–304 CrossRef CAS.
- S. Tang and Z. Cao, J. Chem. Phys., 2011, 134, 044710 CrossRef PubMed.
- Y. Lin and K. A. Fichthorn, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 165303 CrossRef.
- Y. Dong, D. Tang and C. Li, Sci. China: Technol. Sci., 2014, 57, 2153–2160 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |