
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Three dimensional M × N type aptamer-functionalized solid-phase micro extraction fibers array for selectively sorptive extraction of multiple antibiotic residues in milk†
Haibo Liua,
Ning Gan *a,
Yinji Chenb,
Tianhua Lia and
Yuting Caoa
*a,
Yinji Chenb,
Tianhua Lia and
Yuting Caoa
aFaculty of Materials Science and Chemical Engineering, Ningbo University, Ningbo, 315211, China. E-mail: ganning@nbu.edu.cn; Fax: +86 574 87608347; Tel: +86 574 87608347
bDepartment of Food Science and Engineering/Collaborative Innovation Center for Modern Grain Circulation and Safety, Nanjing University of Finance and Economics, Nanjing, 210046, China
First published on 20th January 2017
Abstract
In this study, a novel three dimensional (3D) M × N type aptamer-functionalized solid-phase micro extraction (SPME) fibers array (M represents the number of targets; N represents the number of samples) was developed for selective enrichment of multiplex antibiotic residues from milk samples, with three chloramphenicols (CAPs) as models. First, gold nanoparticles (AuNPs) were electrodeposited on a gold wire (Φ = 0.2 mm), which was wound around a conductive indium tin oxide (ITO) glassy fiber. Then, the fiber was immersed into thiol-functionalized aptamer solution, which could specifically recognize three CAPs, including chloramphenicol (CAP), thiamphenicol (TAP) and florfenicol (FF). The aptamer can be covalently immobilized on AuNPs through Au–S bonds. Thus, a 3D aptamer-functionalized fiber interface (3D-Apt@AuNPs@Au wire–ITO) was built and employed for specifically sorptive extraction of CAPs from the milk samples with a matrix complex based on the high affinity of the aptamer for the targets. The extraction capacities for CAP, TAP and FF were 887, 840 and 801 ng per fiber, respectively. The enrichment folds were more than 500 times, which is 3.1 folds higher than that of the 2D-Apt@AuNPs–ITO fiber and 6.6 folds higher than that of the 1D-Apt@Au wire–ITO fiber. Afterwards, several similar fibers were assembled together into an array for simultaneous enrichment of three CAPs from 12 samples in one run. Finally, the adsorbed targets were washed away using a pH 8.5 0.1 M Tris–HCl buffer and then detected by high performance liquid chromatography (HPLC)-Diode Array Detector (DAD). The parameters, including extraction temperature, shuttle rates, extraction and desorption pH, and extraction and desorption time, were investigated and discussed. Under the optimized conditions, the limits of detection (LOD) and quantitation (LOQ) were determined as 0.262–0.293 and 0.864–0.967 ng mL−1, respectively, for several CAPs. The fibers array can be applied in replicate batch-extraction for at least 60 extraction cycles with a recovery of over 80%. The SPME fibers assay coupled with HPLC detection possessed advantages of high-throughput, high selectivity and adsorption capacity in one run. Furthermore, the process is environmentally friendly without using organic solutions during the entire extraction process. Thus, the method is a universal platform that can be extended to the selective extraction of other organic pollutant residues if the modified aptamers are changed.
1. Introduction
Antibiotic residues can usually be found in animal foods. For example, chloramphenicols, including chloramphenicol (CAP), thiamphenicol (TAP) and florfenicol (FF), are widely used in veterinary medicine for their disease-resistant properties because of their broad range of activity against bacteria and low cost.1–3 However, their residues in human foodstuffs can provoke serious allergic reactions in some hypersensitive individuals.4,5 Because the antibiotic residues in foods are usually found at very low levels with complex matrices, it is urgent to develop effective sample preparation techniques for selective enrichment of the targets and elimination of the matrix interferences to allow accurate detection.Solid phase micro-extraction (SPME) was introduced in 1990 as a solvent-free sample preparation method for the extraction and enrichment of organic compounds from aqueous matrices.6 Recently, the coupling of SPME has been successfully applied to enrich and determine trace levels of pollutants in food samples, e.g. fruit, pork, and fish.7,8 The coating of SPME fiber plays key roles to effectively extract ultratrace levels of targets in samples with a complex matrix. The common commercial coatings are polydimethylsiloxane (PDMS),9 polyacrylate (PA),10 carboxen/PDMS (CAR/PDMS),10 and PDMS/divinylbenzene (PDMS/DVB).10 Although these coatings are effective for real sample pretreatment, most of them do not have selectivity towards targets, which can lead to non-specific adsorption of other organic pollutants further resulting in serious matrix interference, particularly in animal food stuffs.11–14 Moreover, their adsorption capacities need some improvement in order to enrich the ultratrace level of antibiotics in milk samples. Furthermore, the majority of SPME devices can only pretreat one sample in one run, and the extraction efficiency is limited. It is better to fabricate an SPME assay for simultaneous extraction of several samples in one run. Above all, it is urgent to fabricate new SPME coatings to improve the selectivity and adsorption capacity towards analytes and achieve multi-tasking batch pretreatment.
Fortunately, aptamers, a new class of single stranded DNA/RNA molecules, which has even better affinity towards targets than antibodies, can be selected by the systematic evolution of ligands by exponential enrichment (SELEX).15–17 The aptamers can fold into well-defined three-dimensional shapes to selectively capture specific targets. In contrast to antibodies, aptamers can be easily synthesized in batch via polymerase chain reaction (PCR) at low cost; aptamers also possess higher affinity, specificity, and stability with targets.18,19 Aptamers have been employed for specifically recognizing antibiotics, e.g. chloramphenicol,20,21 oxytetracycline22 and kanamycin,23,24 in food. In addition, Mehta's group reported a DNA aptamer that can specifically recognize chloramphenicols including CAP, TAP and FF.20 Recently, aptamers have been successfully loaded on conducting polymer/gold nanocomposites for solid phase extraction (SPE) of kanamycin.23 The detection method mainly includes colorimetric, electrochemical and fluorescence methods. However, these methods are difficult to use for simultaneous detection. Thus, our group has developed a dSPE and SPME method coupling with GC-MS for simultaneous determination of PCBs in fish samples through the use of aptamers' enrichment.25,26 However, these methods still exhibit some problems. First, the adsorption capacity and enrichment factor are limited due to the limited aptamer labeling amount in the extraction surface. Second, these methods have unsatisfactory efficiency because they can only pretreat one sample in one run. Furthermore, some organic solvents are required to be used for extraction, which are harmful to the environment and personnels. It is urgent for us to develop some effective and environmental aptamer coatings to deal with these problems.
On the other hand, facilitating the immobilization of aptamers on fibers and enhancing their labeling amount is another important factor in the fabrication of a more selective and sensitive SPME assay. Fortunately, the thiol group functionalized organic molecules can be easily self-assembled in a monolayer on gold substrate through Au–S covalent bonds just using the method of soaking in solution.27,28 Moreover, AuNPs have a large specific area and good biocompatibility for labeling biochemical probes, e.g. antibodies, DNA. Moreover, AuNPs can be easily electrodeposited on conductive substrates at a certain potential and time.29–31 Indium tin oxide (ITO) coated glass, which has good conductivity and is robust, can be employed as a good substrate candidate to electrodeposit AuNPs.29 However, the specific surface of the plain ITO is very limited. In order to electrodeposit more AuNPs it is better to increase the roughness of its surface using polishing steps. However, the conductive film can also be corrupted after these steps. In order to protect the conductive film and increase the specific area in ITO, it is a good choice to wind some conductive gold wires on it to build a three dimensional (3D) interface to increase the immobilizing area for aptamers on the bars. After that, we can easily electrodeposit AuNPs on the 3D fiber, and then label the aptamer with thiol groups. Moreover, several fibers can be combined together for multiple sample extraction. Based on this, a solid-phase micro extraction fibers array for selectively sorptive extraction of antibiotics was developed.
In the study, a three dimensional M × N aptamer-functionalized gold nanoparticle modified fiber (3D-Apt@AuNPs@Au wire–ITO) array was developed for selectively sorptive enrichment of multiplex chloramphenicols from milk samples. First, AuNPs were electrodeposited on a thin gold wire being wound around a conductive ITO glass fiber. Then, the fiber was immersed into the solution with thiol-functionalized aptamers, which can recognize three CAPs (CAP, TAP and FF). The aptamer can be covalently immobilized on AuNPs through Au–S bonds. Finally, 12 fibers were assembled together as an array for simultaneous extraction of multiplex CAPs from 12 samples in one run. The device is shown in Fig. S1.† Thus, a type of M × N (M is defined as multiplex antibiotics; N as several samples) solid-phase micro extraction fibers based on the array were fabricated for real sample pretreatment. All the procedures are shown in Fig. 1(A).
 | ||
| Fig. 1 Scheme of the fabrication process and application of the 3D-Apt@AuNPs@Au wire–ITO fibers array. | ||
2. Materials and methods
2.1 Chemicals and instruments
Chloramphenicol (CAP, ≥98%), thiamphenicol (TAP, ≥98%), florfenicol (FF, ≥98%), oxytetracycline (OTC, ≥98%), tetracycline (TC, ≥98%) and kanamycin (KANA, ≥98%) were obtained from Aladdin Chemistry Co., Ltd (Shanghai, China). Stock solutions of the antibiotics' mixtures were prepared at a concentration of 1 mg mL−1 in methyl alcohol and stored at −20 °C. The working solutions were freshly prepared from the stock solutions as required. HPLC-grade acetonitrile was obtained from CNW Technologies (Dusseldorf, Germany). Pure water was obtained from Wahaha foods Co., Ltd. (Hangzhou, China). All the sample solutions used for HPLC analysis were filtered through a 0.22 μm PTFE membrane before injection. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was purchased from Sigma-Aldrich Co., Ltd (St Louis, MO, USA). HAuCl4 was purchased from Aladdin Co., Ltd (Shanghai, China). Gold wires (0.2 mm, 4 N) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). ITO coated glass was obtained from Zhuhai Kaivo Optoelectronic Technology Co., Ltd. (Zhuhai, China). The DNA aptamer20 (Apt) (5′-ACT GAG GGC ACG GAC AGG AGG GGG AGA GAT GGC GTG AGG T-3′) was obtained from Shanghai Sangon Biological Engineering Technology & Services Co., Ltd (Shanghai, China).The commercial SPME fibers with 85 μm polyacrylate (PA), 100 μm polydimethylsiloxane (PDMS), 75 μm carboxen/polydimethylsiloxane (CAR/PDMS) coatings, and 65 μm polydimethyl-siloxane/divinylbenzene (PDMS/DVB), which were purchased from Supelco (Bellefonte, PA, USA), were used for the comparison study.
The chromatographic analysis was performed with a LC-20AT HPLC (Shimadzu, Kyoto, Japan) along with a SPD-M20A detector. The separation column was an Agilent ZORBAX Eclipse XDB-C18 column (5 μm, 250 × 4.6 mm, USA). Microscopy was performed using a TM-3000 scanning electron microscope (SEM) (Hitachi, Tokyo, Japan). The UV-vis absorption spectra were obtained using a UV-1800 UV-vis spectrophotometer (Shimadzu, Kyoto, Japan). Electro-polymerization was performed on a CHI 620D Electrochemical Workstation (Shanghai, China).
2.2 Preparation of the three-dimensional fiber via electro-deposition
First, ITO glass was cut into small pieces (length × width × height: 50 mm × 5 mm × 1.1 mm) and cleaned with DMF and ethanol in an ultrasonic bath three times. Second, gold wire (250 mm × 0.2 mm) was wound around the conductive ITO glass fiber. Then, 20 mL HAuCl4 (1 wt%) solution was added in a 50 mL beaker, and electrochemical deposition was carried out in a standard three-electrode single-cell setup with a potential of −0.2 V for 300 s to obtain a layer of Au nanoparticle film. The obtained membrane on the gold wire was dried for 24 h at room temperature before it was utilized.2.3 Immobilization of thiol functionalized aptamer on the surface of the three-dimensional fiber
The modification of thiol-functionalized aptamer with 3D-AuNPs@Au wire–ITO fiber was carried out according to a previous report.28 First, 100 μL of 100 μM thiolated Apt-HS was incubated with 1 μL of 10 mM TCEP for 1 h to reduce disulfide bonds, and then diluted to 5.0 μM with TE buffer (pH = 8.0). Subsequently, the 3D-AuNPs@Au wire–ITO fiber was immersed in the aptamer solutions overnight at 4 °C. The prepared aptamer functionalized 3D-AuNPs@Au wire–ITO fiber was taken out and washed several times with PBS buffer (pH = 7.0) and then dipped into the same PBS buffer (pH = 7.0) solution for future use.2.4 Extraction procedures and HPLC analysis
50 mL of sample solution was added into a 100 mL beaker, and then one 3D-Apt@AuNPs@Au wire–ITO fiber was immersed into it. Up and down stirring was then performed for 20 min at the shuttle rate (defined as the times of fiber stirring up and down within one minute, and one cycle was defined as the fiber stirring up and down once) of 120 cycles per min in order to reach equilibrium as soon as possible. Subsequently, the fiber was pulled out and rinsed three times with PBS buffer (5 mL, pH = 7.0) to remove the non-specific adsorption. Then, the fiber was immersed into the 0.1 M Tris–HCl buffer (2 mL, pH = 8.5) for 10 min with a shuttle rate of 120 cycles per min to elute the targets from the fiber. A 12 fiber array can extract 12 samples simultaneously. Afterwards, the desorption solution was dried at ambient temperature with N2 gas and concentrated in 0.1 mL mobile phase (acetonitrile/H2O, 30/70, v/v). Then, the 20 μL solution was filtered and injected into the HPLC for the antibiotics' analysis.The mobile phase consisted of 30% (v/v) of acetonitrile and 70% (v/v) of H2O at 30 °C with a flow rate of 1.0 mL min−1.32 Quantitative analyses of compounds were performed using an SPD detector. The detection wavelength was set at 275 nm for CAP and OTC and 225 nm for TAP and FF.
2.5 Recovery test and determination of antibiotics in a milk sample
To test the applicability of the proposed method for complex food samples, we used the milk sample extraction solutions without CAPs as a matrix interference and determined the corresponding recovery of the spiked CAPs. The milk samples were purchased from Ningbo supermarket (China) and treated according to a previous report.33 40 mL milk samples were spiked with different amounts of CAP, TAP and FF (1.00, 5.00, and 10.0 ng mL−1) into the original sample, equilibrated at room temperature (25 °C) for 1 h and then centrifuged at 5500 rpm for 15 min (Rotor No. 11133, Siama 3K15). The cream layer of the milk sample was carefully removed from the solution and the supernatant was collected. Afterwards, 5 mL acetonitrile was added to promote protein precipitation. The mixture was vortexed for 5 min and then allowed to stand for 10 min at room temperature (25 °C). The contents were centrifuged at 5500 rpm for 15 min (Rotor No. 11133, Siama 3K15) and the supernatant was collected and was subsequently analyzed by the abovementioned SPME procedure.3. Results and discussion
3.1 Characterization of 3D-Apt@AuNPs@Au wire–ITO fiber
To gain a better understanding of the morphological structure of the fiber, all the materials were characterized using SEM. The SEM images of bare gold wire (A), AuNPs@Au wire (B) and (C) are shown in Fig. 2. As can be seen from Fig. 2, the surface of AuNPs@Au wire (B) became rougher than that of bare gold wire (A). We can find some small particles whose size was about 50 nm on the bare gold wire surface (C). Moreover, the hydrosulphonyl-functionalized aptamer was covalently immobilized on the 3D-AuNPs@Au wire–ITO fiber through Au–S bonds. UV-Vis absorption spectrometry was employed to characterize the conjugation of the aptamer. As seen in Fig. 2(D), the coating of gold nanoparticles had characteristic absorption at 520 nm (a). Moreover, the aptamer had characteristic absorption at 253 nm (b), which can be ascribed to a DNA characterized peak. After the aptamer was modified onto the surface of the 3D-AuNPs@Au wire–ITO fiber, an absorption peak at 265 nm was observed (c), and it proved that the aptamer was immobilized on the surface of the 3D-AuNPs@Au wire–ITO fiber. Furthermore, we calculated the aptamer coupling efficiency from the aptamer concentration in sample solutions before and after the immobilizing reaction, which was measured via UV-Vis absorption spectrometry. The results showed that the coupling efficiency was 84.6%. Moreover, in order to stably immobilize the Apt–AuNPs coating on the bare gold wire, AuNPs were electro-deposited on Au wires by adjusting the potential and time. The thickness and degree of consistency of coating could be easily monitored by controlling the deposition time. The voltage (−0.1, −0.2, −0.3, −0.4, −0.5 V) and time (200, 250, 300, 350, 400 s) of the electro-deposition process were optimized. Finally, −0.2 V and 300 s were selected to prepare the AuNPs coating on the bare gold wire because the stable AuNPs and the most amount aptamer coating were obtained at this voltage and deposition time. | ||
| Fig. 2 Characterization of the fiber. (A) SEM of bare gold wire; (B) and (C) AuNPs@Au wire; (D) UV spectra of AuNPs (a), aptamer (b), Apt–Au (c). | ||
Different amounts of aptamer in 2 mL TE buffer solutions (pH = 8.0) were investigated at different concentrations (0.5, 1, 2, 3, 5, 7, 10, and 15 μmol L−1) modified on 3D-AuNPs@Au wire–ITO fiber. Then, the above fiber was used to extract 100 mL 5 ng mL−1 CAP, TAP and FF. As shown in Fig. S2,† the capacity amount was identical when the aptamer concentration was more than 5 μmol L−1. Therefore, we used 5 μmol L−1 aptamer solution to modify 3D-AuNPs@Au wire–ITO fibers in the further experiments. In addition, we investigated the influence of the length of gold wire wound on the ITO fiber on extraction effectiveness. As shown in Fig. S3,† the capacity amount was increased slowly when the length was more than 250 mm. Therefore, 250 mm gold wires were selected for subsequent experiments.
Furthermore, we investigated the reusability of the 3D-Apt@AuNPs@Au wire–ITO fiber, by performing 80 repetitions of the SPME procedure on the wire. The results indicated that the extraction recovery had no evident decrease within 60 repetitions (Fig. S4†). Therefore, it was stable enough to be applied in replicate for at least 60 extraction cycles with a recovery of over 80%.
3.2 Verification of selectivity and capacity
The specificity of the 3D-Apt@AuNPs@Au wire–ITO fiber was evaluated by investigating the recovery of CAP, TAP and FF with the addition of 5 ng mL−1 OTC, TC and KANA as interfering chemicals in PBS buffer. The results shown in Fig. 3(A) indicate that the recovery of CAP, TAP and FF was significantly higher than that of the OTC, TC and KANA. The recovery of OTC, TC and KANA was less than 2%. These results suggest that the prepared 3D-Apt@AuNPs@Au wire–ITO fibers array was able to specifically capture CAP, TAP and FF due to the specific recognition ability of CAP, TAP and FF of its aptamer on the fiber. In addition, we investigated the adsorption of the chaotic sequence (5′-GGCGGGGAACGGTTATGACCCATACAAAATGATCATTATT-3′) for CAP, TAP, FF and OTC. As shown in Fig. S5,† the recoveries of CAP, TAP, FF and OTC were less than 2%. These results also indicate that the prepared 3D-Apt@AuNPs@Au wire–ITO fiber arrays have high selectivity.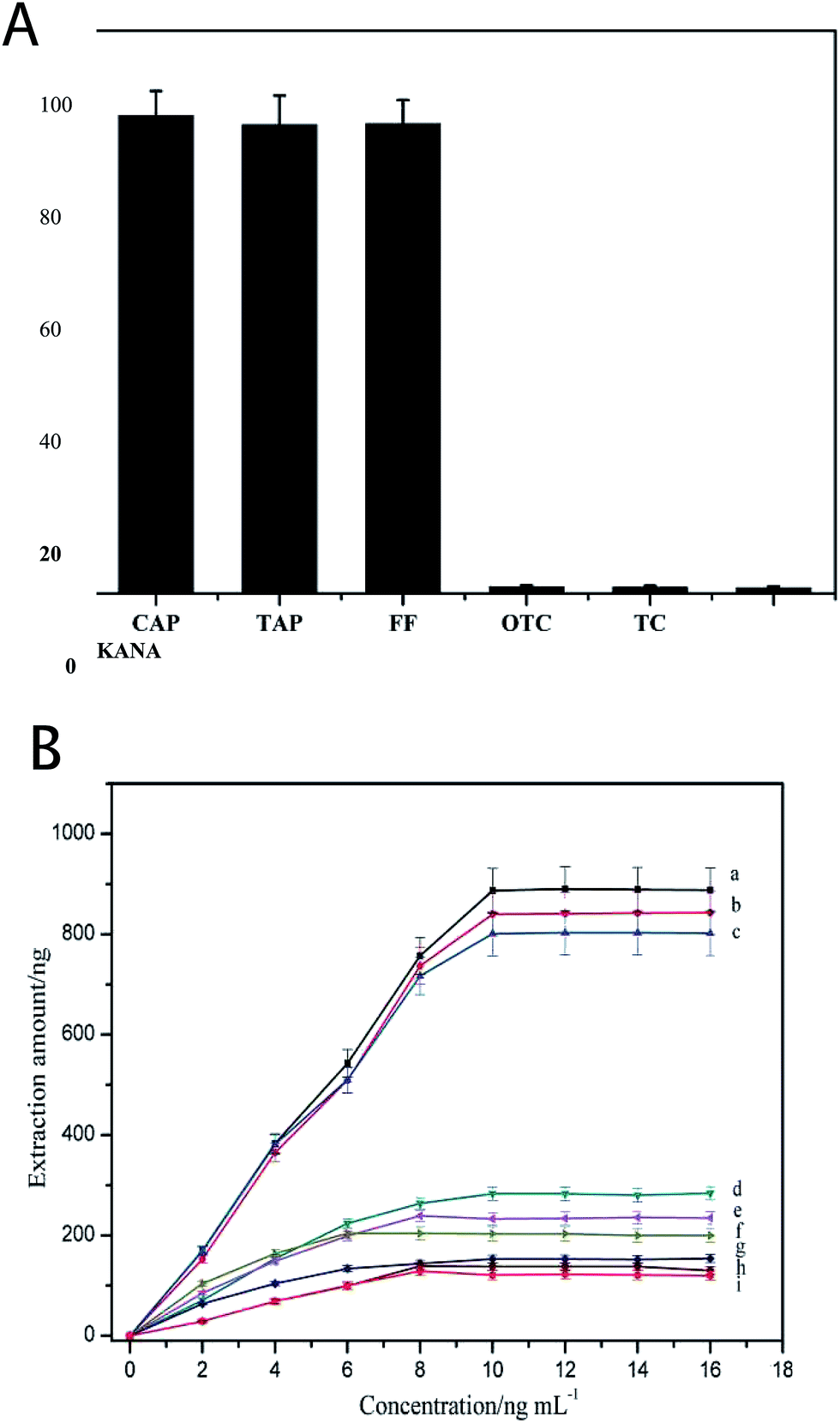 | ||
| Fig. 3 Verification of selectivity and capacity. (A) Selectivity of 3D-Apt@AuNPs@Au wire–ITO fiber; (B) extraction capacity of 3D-Apt@AuNPs@Au wire–ITO fiber (a for CAP, b for TAP, c for FF), 2D-Apt@AuNPs–ITO fiber (d for CAP, e for TAP, f for FF) and 1D-Apt@Au wire–ITO (g for CAP, h for TAP, i for FF). | ||
In order to describe the advantage of three-dimensional gold as a substrate for the immobilizing aptamer, we compared the adsorption capacity of the 1D-Apt@Au wire–ITO fiber, 2D-Apt@AuNPs–ITO fiber and the 3D-Apt@AuNPs@Au wire–ITO fiber. The results are shown in Fig. 3(B). The extraction capacities for CAP, TAP and FF on 3D-Apt@AuNPs@Au wire–ITO were 887, 840 and 801 ng, respectively, which were higher than 283, 234 and 203 ng on 2D-Apt@AuNPs–ITO; they were also higher than 153, 138 and 121 ng on 1D-Apt@Au wire–ITO. This may be ascribed to the fact that the 3D-Apt@AuNPs@Au wire–ITO has the highest labeling amount of aptamer because the deposited AuNPs and the thin gold wire can both be labeled with thiolation aptamers. However, ITO in the 2D-Apt@AuNPs–ITO bar cannot connect with the aptamer.
Moreover, in order to test if the substrate could non-specifically extract the targets (CAP, TAP and FF), the ITO fiber was employed as an internal quality control to extract CAPs. Fig. S6† showed that the extraction amount of CAP, TAP and FF on the control ITO fiber was only 10, 8.0 and 9.0 ng, respectively. Furthermore, the 3D-AuNPs@Au wire–ITO fiber (without aptamer functionalization) was also investigated and the extraction amount increased to 18, 15 and 13 ng, respectively. However, these are still significantly lower than 887, 840 and 801 ng respectively obtained using 3D-Apt@AuNPs@Au wire–ITO fibers. All this means that AuNPs and ITO fibers have very weak non-specific adsorption for CAPs, and therefore they will not disturb the detection of targets.
3.3 Optimization of extraction and desorption conditions
The extraction conditions, including extraction temperature, shuttle rates, extraction pH, desorption pH, extraction time and desorption time, were also optimized. Heating the solution can promote molecular mass transfer and adsorption equilibrium. The effect of extraction temperature was studied from 30 °C to 80 °C (Fig. 4(A)). The peak areas of the antibiotics increased steadily as the extraction temperature increased from 25 °C to 40 °C. Further increasing the temperature resulted in the recovery being reduced. On one hand, a high temperature can promote molecular mass transfer. On the other hand, too high a temperature is unfavorable for the exothermic adsorption of the analytes onto the SPME fiber. Therefore, an extraction temperature of 40 °C was chosen for further experiments. | ||
| Fig. 4 Optimization of extraction and desorption conditions. (A) Extraction temperature; (B) shuttle rates; (C) extraction time; (D) desorption time. | ||
In order to obtain the highest recovery and shorten the pretreatment time, the fiber shuttle rates (60, 90, 120, 150 and 180 cycles per min) were investigated. We found (Fig. 4(B)) that the recovery obtained with 120 cycles per min was higher than that obtained with 90 cycles per min and similar to those obtained with 150 cycles per min. Thus, 120 cycles per min was the parameter selected for subsequent experiments.
Extraction pH can influence the binding between aptamer and targets; the effect of extraction pH was studied from 4 to 11 (Fig. S7†). The results showed that we can obtain higher extraction efficiency when the pH value is 7. A low or high pH can destroy the aptamer (as a single DNA strand), which makes the fiber unusable. Thus, the PBS buffer of pH = 7.0 was selected as the optimal value.
In this study, in order to completely elute the antibiotics captured by the aptamer, we adjusted the pH value of the buffer to change the conformation of the aptamer and release the antibiotics further. Different desorption pH values from 5.0 to 9.5 were employed in this study. The results are presented in Fig. S8.† It was found that a satisfactory recovery was acquired with pH = 8.5. Considering that low pH may destroy the aptamer structure and lead to poor reproducibility for the fiber, pH = 8.5 was selected as the desorption pH for subsequent experiments.
Extraction time can affect the extraction efficiency. In this study, the effect of different extraction times (5, 10, 15, 20, 25, 30 min) on the extraction of the target analytes was studied. Experimental results are shown in Fig. 4(C). Extraction equilibrium for target antibiotics can almost reach a plateau when the time is greater than 20 min. Therefore, 20 min of extraction time was selected for subsequent experiments.
Different desorption times (2, 4, 6, 8, 10, 12, 14, 16 min) were employed in this study. The results are shown in Fig. 4(D). It was found that a satisfactory recovery was acquired for the 10 min desorption time. Therefore, 10 min was selected for subsequent experiments.
3.4 Analytical method validation
The proposed method was applied to the analysis of CAPs in spiked milk samples under optimized conditions. The linear range, reproducibility (RSD), limits of detection (LODs) and limits of quantification (LOQs) were calculated, and the results are summarized in Table 1. The LOD and LOQ were obtained from the diluted samples' signal-to-noise (S/N) ratio. The calculated LOD was 3 and LOQ was 10. As can be observed, the linear ranges for the three CAPs were 1.00–1.00 × 103 ng mL−1. The low LOD values indicated that the 3D-Apt@AuNPs@Au wire–ITO-SPME method was suitable for the extraction of CAP, TAP and FF from the milk samples. Moreover, the accuracy and reproducibility of the proposed method were evaluated by calculating the recoveries of the spiked milk samples at different levels (1.00, 5.00, and 10.0 ng mL−1) based on five independent experiments. The recovery of CAP, TAP and FF in the spiked milk samples ranged from 75.7% to 94.5%.| TAP | FF | CAP | |||
|---|---|---|---|---|---|
| Linear range (ng mL−1) | 1.00–1.00 × 103 | 1.00–1.00 × 103 | 1.00–1.00 × 103 | ||
| Linear equation | Y = 1967.0 + 12400X | Y = 634.1 + 4710X | Y = 355.0 + 4091X | ||
| R2 | 0.9908 | 0.9979 | 0.9982 | ||
| LODs (ng mL−1) | 0.262 | 0.216 | 0.293 | ||
| LOQs (ng mL−1) | 0.864 | 0.713 | 0.967 | ||
| Intra-day | 1.00 ng mL−1 | Recovery | 79.5 | 94.5 | 80.2 |
| RSD (%, n = 5) | 9.10 | 4.62 | 6.23 | ||
| 5.00 ng mL−1 | Recovery | 92.4 | 75.7 | 84.3 | |
| RSD (%, n = 5) | 6.94 | 7.32 | 7.65 | ||
| 10.0 ng mL−1 | Recovery | 76.5 | 89.6 | 91.5 | |
| RSD (%, n = 5) | 8.11 | 9.34 | 5.73 | ||
| Inter-day | 1.00 ng mL−1 | Recovery | 81.6 | 82.3 | 90.1 |
| RSD (%, n = 5) | 3.66 | 3.82 | 7.65 | ||
| 5.00 ng mL−1 | Recovery | 91.9 | 90.4 | 80.2 | |
| RSD (%, n = 5) | 4.93 | 7.05 | 4.36 | ||
| 10.0 ng mL−1 | Recovery | 82.3 | 91.6 | 87.7 | |
| RSD (%, n = 5) | 7.38 | 3.69 | 8.52 |
3.5 Application in milk samples
To validate the practical performance of 3D-Apt@AuNPs@Au wire–ITO fibers in real samples with a complex matrix, milk samples were selected for spiking analysis at three levels of 1.00, 5.00, and 10.0 ng mL−1 with CAP, TAP and FF as analytes. The chromatograms of 5.00 ng mL−1 spiked milk sample solutions and their extracts with 3D-Apt@AuNPs@Au wire–ITO, 2D-Apt@AuNPs–ITO, and commercial PA, PDMS, CAR/PDMS and PDMS/DVB coatings are presented in Fig. 5. The results indicated that direct HPLC analysis with a traditional SPD detector could not monitor CAP, TAP and FF with direct injection of the extract solution before SPME (curve b). In addition, CAPs were not quantified reliably, even after extraction through commercial coatings including PA, PDMS, CAR/PDMS and PDMS/DVB, due to the inefficient enrichment and influence by the presence of other analytes in the sample (curves f, g, h and i). However, the chromatogram obtained after use of the 3D-Apt@AuNPs@Au wire–ITO fiber shows prominent peaks of CAP, TAP and FF with greater intensity as compared with chromatograms obtained through commercial fiber extraction, which reveals highly specific adsorption of CAPs (curve e) with 3D-Apt@AuNPs@Au wire–ITO fiber. Furthermore, it explains that the presence of interfering species could not affect the efficiency and specificity of the aptamer. The results also showed that the recovery of 3D-Apt@AuNPs@Au wire–ITO was preferable to that of the 2D-Apt@AuNPs–ITO (curve d). It was indicated that the 3D-Apt@AuNPs@Au wire–ITO fiber could be used for selective separation and enrichment of trace CAPs in complicated samples. Moreover, CAP was discovered with a content of 1.60 ng mL−1.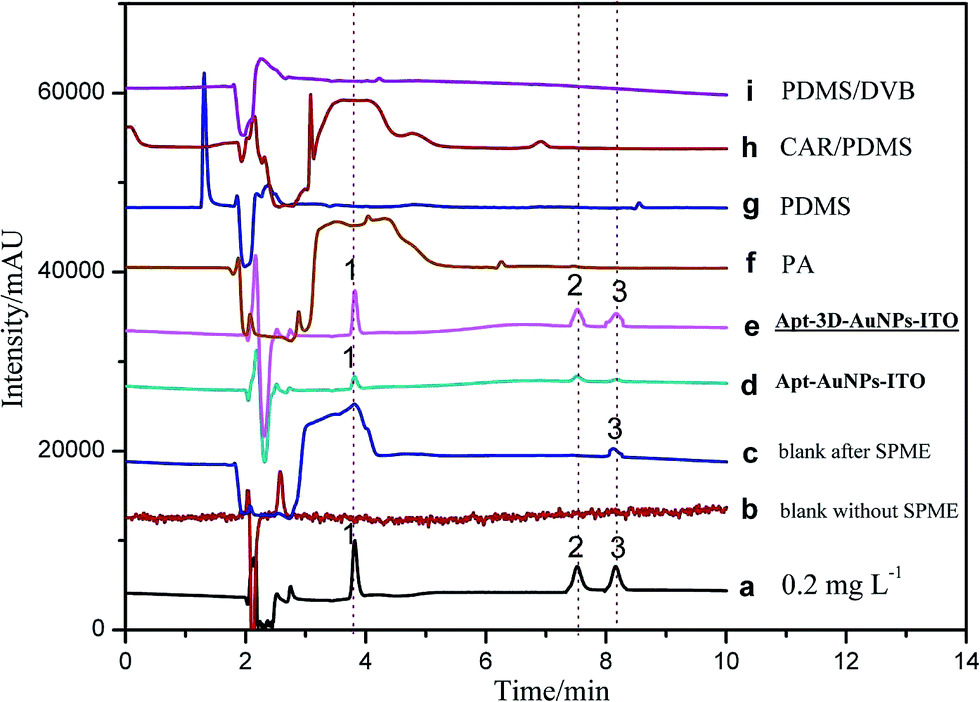 | ||
| Fig. 5 Chromatograms of 0.2 mg L−1 CAP, TAP and FF mixed standard solution (a), direct injection of the blank milk sample extract solution before SPME (b), blank milk sample solutions after extraction with 3D-Apt@AuNPs@Au wire–ITO (c), and 5.00 ng mL−1 spiked milk sample solutions after extraction with 2D-Apt@AuNPs–ITO (d), 3D-Apt@AuNPs@Au wire–ITO (e), commercial PA (f), PDMS (g) CAR/PDMS (h) and PDMS/DVB (i) coatings, respectively. (1) CAP, (2) TAP, (3) FF, injection volume for standard solution and spiked sample solutions: 20 μL. | ||
In addition, a comparison study of different methods for the determination of CAPs in different samples was performed, and the results are shown in Table 2. It can be seen that a shorter pretreatment time was used for the method developed in this study. In our experiments, the extraction time is 20 min and the desorption time is 10 min for three chloramphenicols. The total extraction time is about 30 min. However, the HPLC analysis for three chloramphenicols is about 15 min. Therefore, extraction procedures are the rate determining step of the method and not HPLC. Thus, the device is even more time-saving when several samples are pretreated simultaneously. The LOD and LOQ found for this method were less than those of most previous methods. Moreover, our method has higher selectivity due to the usage of an aptamer, which is very specific toward targets.
| Matrix | Extraction method | Analytical method | LOD | Selectivity | Timea | References |
|---|---|---|---|---|---|---|
| a The time pretreatment of the sample was calculated by summing the sample preparation, preconditioning, extraction and desorption time.b Dispersive liquid–liquid microextraction (DLLME), solid phase extraction (SPE), liquid–liquid extraction (LLE), subcritical water extraction (SWE). | ||||||
| Honey | DLLME | HPLC-VWD | 0.1–0.6 μg g−1 | No | 0.5 h | 34 |
| Wastewater | PS-DVB-SPE | HPLC-DAD | 0.03 μg L−1 | No | 1.5 h | 35 |
| Milk | HLB-SPE | HPLC-DAD | 20 ng mL−1 | No | 1.5 h | 36 |
| Chicken | C18-SPE | HPLC-UV | 2.5 ng g−1 | No | 1.5 h | 37 |
| Meat | LLE | HPLC-UV | 1–10 μg kg−1 | No | 1.0 h | 38 |
| Animal feeds | SWE | HPLC-UV | 0.011 mg L−1 | No | 0.8 h | 32 |
| Milk | Aptamer fiber array | HPLC-UV | 0.262–0.293 ng mL−1 | Yes | 1.0 h (12 samples) | This work |
4. Conclusion
In this study, a novel 3D aptamer-functionalized fiber (3D-Apt@AuNPs@Au wire–ITO) array was developed for selectively sorptive batch extraction of antibiotic residues from multiple food samples simultaneously. We named it the M × N array (where M represents the species of antibiotic and N represents the number of samples). Due to the high labeling amount of the aptamer for the 3D-AuNPs@Au wire and high selective recognition of the aptamer towards target analytes, the prepared 3D-Apt@AuNPs@Au wire–ITO fiber showed high selectivity and pre-concentration ability for extracting trace level CAPs from milk samples with a complex matrix. The extraction capacities for CAP, TAP and FF per fiber were higher than that for the 2D fiber Apt@AuNPs ITO and 1D fiber Apt@Au wire ITO. Interestingly, we also prepared a type of M × N fiber extract array, and we can simultaneously extract multiple amide alcohol antibiotics from 12 samples in one run. Using the novel pretreatment method, it was easy to extract large amounts of samples and the sample pretreatment procedures were accurately simplified. Moreover, the method can be extended to selective extraction of other antibiotic residues if the corresponding modified aptamer is changed in the bars.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51403110), the Natural Science Foundation of Zhejiang (LY15B050002, LY16B050004, LY17C200007), the Natural Science Foundation of Ningbo (2016A610084), and the K. C. Wong Magna Fund in Ningbo University.References
- F. Barreto, C. Ribeiro, R. Barcellos Hoff and T. Dalla Costa, J. Chromatogr. A, 2016, 1449, 48–53 CrossRef CAS PubMed.
- N. N. Naing, S. F. Yau Li and H. K. Lee, J. Chromatogr. A, 2016, 1440, 23–30 CrossRef CAS PubMed.
- M. F. Festing, W. P. Diamanti and J. A. Turton, Food Chem. Toxicol., 2001, 39, 375–383 CrossRef CAS PubMed.
- Z. Xiao, R. Song, Z. Rao, S. Wei, Z. Jia, D. Suo and X. Fan, J. Chromatogr. A, 2015, 1418, 29–35 CrossRef CAS PubMed.
- X. D. Pan, P. G. Wu, W. Jiang and B. j. Ma, Food Control, 2015, 52, 34–38 CrossRef CAS.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- C. Hu, M. He, B. Chen and B. Hu, J. Agric. Food Chem., 2012, 60, 10494–10500 CrossRef CAS PubMed.
- N. Campillo, P. Vinas, N. Aguinaga, G. Ferez and M. Hernandez-Cordoba, J. Chromatogr. A, 2010, 1217, 4529–4534 CrossRef CAS PubMed.
- N. R. Neng, M. L. Pinto, J. Pires, P. M. Marcos and J. M. Nogueira, J. Chromatogr. A, 2007, 1171, 8–14 CrossRef CAS PubMed.
- C. Bicchi, C. Iori and P. Rubiolo, J. Agric. Food Chem., 2002, 50, 449–459 CrossRef CAS PubMed.
- Z. Bai, A. Pilote, P. K. Sarker, G. Vandenberg and J. Pawliszyn, Anal. Chem., 2013, 85, 2328–2332 CrossRef CAS PubMed.
- S. Lin, N. Gan, L. Qiao, J. Zhang, Y. Cao and Y. Chen, Talanta, 2015, 144, 1139–1145 CrossRef CAS PubMed.
- E. Canellas, P. Vera and C. Nerin, Food Chem., 2016, 197, 24–29 CrossRef CAS PubMed.
- Y. Xu, J. Ding and H. Chen, Food Chem., 2013, 140, 83–90 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- L. C. Bock, L. C. Griffin and J. A. Latham, Nature, 1992, 355, 564–566 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- V. Pavlov, B. Shlyahovsky and I. Willner, JACS, 2005, 127, 6522–6523 CrossRef CAS PubMed.
- Y. G. Liu and R. F. Whittier, Genomics, 1995, 25, 674–681 CrossRef CAS PubMed.
- J. Mehta, B. Van Dorst, E. Rouah-Martin, W. Herrebout, M. L. Scippo, R. Blust and J. Robbens, J. Biotechnol., 2011, 155, 361–369 CrossRef CAS PubMed.
- S. Wu, H. Zhang, Z. Shi, N. Duan, C. Fang, S. Dai and Z. Wang, Food Control, 2015, 50, 597–604 CrossRef CAS.
- J. H. Niazi, S. J. Lee, Y. S. Kim and M. B. Gu, Med. Chem., 2008, 16, 1254–1261 CAS.
- Y. Zhu, P. Chandra, K. M. Song, C. Ban and Y. B. Shim, Biosens. Bioelectron., 2012, 36, 29–34 CrossRef CAS PubMed.
- K. M. Song, M. Cho, H. Jo, K. Min, S. H. Jeon, T. Kim, M. S. Han, J. K. Ku and C. Ban, Anal. Biochem., 2011, 415, 175–181 CrossRef CAS PubMed.
- S. Lin, N. Gan, Y. Cao, Y. Chen and Q. Jiang, J. Chromatogr. A, 2016, 1446, 34–40 CrossRef CAS PubMed.
- S. Lin, N. Gan, J. Zhang, L. Qiao, Y. Chen and Y. Cao, Talanta, 2016, 149, 266–274 CrossRef CAS PubMed.
- T. M. Herne and M. J. Tarlov, JACS, 1997, 119, 8916–8920 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- M. C. Tsai and P. Y. Chen, Talanta, 2008, 76, 533–539 CrossRef CAS PubMed.
- O. Shulga and J. R. Kirchhoff, Electrochem. Commun., 2007, 9, 935–940 CrossRef CAS.
- X. Dai and R. G. Compton, Anal. Sci., 2006, 22, 567–570 CrossRef CAS PubMed.
- L. Wang, H. Yang, C. Zhang, Y. Mo and X. Lu, Anal. Chim. Acta, 2008, 619, 54–58 CrossRef CAS PubMed.
- V. Samanidou, M. Kehagia and A. Kabir, Anal. Chim. Acta, 2016, 914, 62–74 CrossRef CAS PubMed.
- H. Chen, H. Chen, J. Ying, J. Huang and L. Liao, Anal. Chim. Acta, 2009, 632, 80–85 CrossRef CAS PubMed.
- M. Vosough and H. Mashhadiabbas Esfahani, Talanta, 2013, 113, 68–75 CrossRef CAS PubMed.
- M. C. Vargas Mamani, F. G. Reyes Reyes and S. Rath, Food Chem., 2009, 117, 545–552 CrossRef.
- H. Tajik, H. Malekinejad, S. M. Razavi-Rouhani, M. R. Pajouhi, R. Mahmoudi and A. Haghnazari, Food Chem. Toxicol., 2010, 48, 2464–2468 CrossRef CAS PubMed.
- H. Y. Shen and H. L. Jiang, Anal. Chim. Acta, 2005, 535, 33–41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27005k |
| This journal is © The Royal Society of Chemistry 2017 |