
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emitting-tunable Eu(2+/3+)-doped Ca(8−x)La(2+x) (PO4)6−x(SiO4)xO2 apatite phosphor for n-UV WLEDs with high-color-rendering
Yi Weia,
Hui Jiaa,
Hui Xiaoa,
Meng Meng Shangb,
Chun Che Lin*c,
Chaochin Su*c,
Ting-Shan Chand,
Guo Gang Li *a and
Jun Lin*b
*a and
Jun Lin*b
aFaculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, People's Republic of China. E-mail: ggli8312@gmail.com
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: jlin@ciac.ac.cn
cInstitute of Organic and Polymeric Materials, National Taipei University of Technology, Taipei 106, Taiwan. E-mail: cclin0530@gmail.com; f10913@ntut.edu.tw
dNational Synchrotron Radiation Research Center, Hsinchu 300, Taiwan
First published on 12th January 2017
Abstract
Currently, developing single-phased white light phosphors based on a single-doped activator is an efficient approach to blends of bicolor/tricolor phosphors for realizing phosphor-converted white light emitting diodes (pc-WLEDs) with a high color rendering index (CRI) and low correlated color temperature (CCT). Here, we present high CRI (Ra = 93–95) and low CCT (3500–6000 K) white lights by cosubstituting [Ca2+–P5+] for [La3+–Si4+] in the solid solution Ca(8→2)La(2→8)(PO4)6−x(SiO4)xO2:Eu2+/Eu3+ (CLPSO_Eu). The results are attributed to the presence of multi Ca2+ sites due to possible mixing nanophases and the simultaneous occupancy of Ca2+ and La3+ sites by Eu, resulting in the mixing of blue (466 nm) and green emissions (540 nm) for Eu2+, and red emission (613 nm) for Eu3+, which were perfectly confirmed using X-ray Rietveld refinement, photoluminescence spectra and extended X-ray absorption fine structure. These findings not only imply that the as-prepared CLPSO_Eu are promising single-phased white light phosphor for near-UV based WLEDs but also offer a novel avenue to design high CRI white light phosphors based on a tunable Eu2+/Eu3+.
1. Introduction
Near ultraviolet (n-UV) based white light-emitting diodes (WLEDs) that are composed of 380–420 nm LED chips and bicolor/tricolor phosphors are emerging as promising high CRI and low CCT lighting sources.1–5 Unfortunately, this type of WLED easily suffers from a complicated fabrication process and color unbalance. An available alternative is to develop single-phased white light phosphors.6 The common single-phased white light infers the energy transfer among activator ion pairs such as Ce3+/Eu2+ → Mn2+, Ce3+/Eu2+ → Tb3+, Mn2+.7–10 However, the Mn2+ red emission belongs to weak d–d transitions, resulting in the low luminescence efficiency and poor thermal stability.11 To reduce the energy loss in the energy transfer process, single-activator-doped white light phosphors are highly desired, in which the emission spectra of the single activator must cover the entire visible light range.12 Eu2+ is one of the most popular activators and its 4f–5d transition can be tuned from 400 to 700 nm depending on its coordination environment.13–18 Recently, single Eu2+-activated materials were designed as single-phased white light phosphors by changing host component, such as Sr5(PO4)3−x(BO3)xCl:Eu2+.12 However, to obtain enough full width at half maximum (FWHM) of Eu2+ emission is still a challenge. Especially, more red component should be covered for presenting high CRI white light.19 Eu3+-activated materials are potential red-emitting phosphors in pc-WLEDs due to the typical 4f–4f transitions (5D0,1–7FJ, J = 0–6) in the range of 570–700 nm from Eu3+.20–22 Moreover, cation substitution has been confirmed as an effective strategy to adjust luminescent colors and properties of phosphor materials.23–31 In the previous study, we have demonstrated the color-tunable emission from blue or green light to red light based on the valance transformation (3+ to 2+) through cation cosubstitution.32 However, there are few reports to present single-phased white light emission based on simultaneously blue, green, red-emitting Eu2+/Eu3+-activated phosphors. Herein, we realize high CRI white lights with low correlated color temperatures by cosubstituting [La3+–Si4+] for [Ca2+–P5+] in the solid solution Ca(8→2)La(2→8)(PO4)6−x(SiO4)xO2:Eu2+/Eu3+.2. Experimental section
Chemicals and materials
CaCO3 (Aldrich, ≥99.95%), La2O3 (Aldrich, ≥99.999%), SiO2 (Aldrich, ≥99.995%), (NH4)2HPO4 (Aldrich, ≥99.9%), and Eu2O3 (Aldrich, ≥ 99.999%) were purchased from Sigma-Aldrich Corporation. All of the initial chemicals were used without further purification. Aluminum oxide crucibles were used to sinter the phosphor samples.Preparation
A series of Ca(8−x)La(2+x)(PO4)6−x(SiO4)xO2:2% Eu (x = 0–6) compounds were prepared by a conventional high-temperature solid state reaction. The doping concentration of Eu was fixed at 2 atom.% of Ca/La. All samples were synthesized in a reductive atmosphere and Ca(8−x)La(2+x)(PO4)6−x(SiO4)xO2:2% Eu (0 ≤ x ≤ 6) samples were correspondingly denoted as CLSPO-x (x = 0–6). Stoichiometric amounts of CaCO3, La2O3, SiO2, (NH4)2HPO4, and Eu2O3 were thoroughly mixed and pestled in an agate mortar for 1 h. Then, the powder mixtures were placed in aluminum oxides crucibles and sintered in a horizontal tube furnace at 1300 °C for 8 h with a reducing atmosphere of H2 (8%) and N2 (92%) atmosphere. After the furnace slowly cooled to room temperature, the sintered products were grinded again, generating the final phosphor powders. The as-prepared Ca(8−x)La(2+x)(PO4)6−x(SiO4)xO2:2% Eu (x = 0, 1, 2, 3, 4, 5, 6) samples were denoted as CLSPO-0, 1, 2, 3, 4, 5, and 6, respectively. Phosphor-converted solid-state lighting devices were fabricated around discrete UV LEDs by using gold wires for electrical operation. The LEDs emit with λmax = 395 nm. Homogeneous mixtures of the as-prepared phosphors and transparent silicone resin were cured on the LED chip. After the packaging was completed, the optical properties of the device were measured in an integrating sphere under forward DC bias conditions.Characterization
Finely ground powders were used in all of the measurements. The phase purity of all samples were analyzed using X-ray diffraction (XRD) obtained in a D8 Focus diffractometer (Bruker, Karlsruhe, Germany) at a scanning rate of 1° min−1 in the 2θ range from 5° to 120°, and the counting time was 5 s per step with Ni-filtered Cu Kα radiation (λ = 0.15406 nm). XRD Rietveld profile refinements of the structural models and texture analysis were performed with the use of General Structure Analysis System (GSAS). The photoluminescence measurements were recorded with a Fluoromax-4P spectrophotometer (Horiba Jobin Yvon, New Jersey, USA) equipped with a 450 W xenon lamp as the excitation source. Both excitation and emission spectra were set up to be 1.0 nm with the width of the monochromator slits adjusted as 0.50 nm. XANES of Eu L3 edge was recorded with a wiggler beamline BL17C (the synchrotron X-ray diffraction wavelength λ = 0.774907 Å) at National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan. The thermal stability of luminescence of phosphor materials were measured by Fluoromax-4P spectrometer connected a heating equipment (TAP-02). The Commission Internationale de l'Eclairage chromaticity color coordinates, color rendering index (Ra), and CCT of WLED devices were measured by Starspec SSP6612.3. Results and discussion
The crystal structure and phase purity of the studied samples were confirmed by powder X-ray diffraction (XRD). Fig. 1a shows the Rietveld XRD refinement of the representative Ca5.88La3.92Eu0.2(PO4)4(SiO4)2O2 (CLSPO-2) sample.33 The refined structure parameters indicate that the studied sample crystallizes in hexagonal phase with space group P63/m (176), a = b = 9.47 Å, c = 6.92 Å, α = β = 90°, γ = 120°, V = 538.13 Å3 and Z = 1. The atom positions, fraction factors, and thermal vibration parameters were refined by convergence and satisfied well the reflection conditions, Rwp = 7.84%, Rp = 4.77%, and χ2 = 6.86. The results imply the formation of single-phased solid solution (CLSPO-2). Fig. 1b shows the XRD patterns for Ca(8−x)La(2+x)(PO4)6−x(SiO4)xO2:2% Eu (0 ≤ x ≤ 6) samples at 30–35°. The XRD patterns of x = 0 and x = 6 samples can be well assigned to the standard Ca8La2(PO4)6O2 (CLPO, JCPDS no. 33-0287) and Ca2La8(SiO4)6O2 (CLSO, JCPDS no. 29-0337) phases, respectively.34,35 A linear shift of XRD diffraction peaks at 30–35° to small-angle direction with the substitution of [Ca2+–P5+] for [La3+–Si4+] (Fig. 1b) is attributed to the smaller ion radius of r[Ca2++P5+] (1.23 Å, CN = 7; 1.35 Å, CN = 9) than that of r[La3+–Si4+] (1.36 Å, CN = 7; 1.476 Å, CN = 9). This phenomenon not only indicates the successful incorporation of [La3+–Si4+] into the lattice by substituting [Ca2+–P5+] but also confirms that the CLSPO-x (x = 0–6) can maintain single phase and form solid solutions in the whole series.32 Interestingly, two (112) crystal planes that respectively belong to CLPO and CLSO hosts simultaneously appear in CLPSO-2 and CLPSO-3 samples. This finding imply a possible mixing nanophases of two crystal structures, which could induce the coexistence of different coordination environments for activator ions.23 A schematic spatial view of the Ca(8−x)La(2+x)(PO4)6−x(SiO4)xO2:2% Eu solid solution unit cell is shown in Fig. 1c. Ca/La/Eu occupy the nine-coordinated 4f sites with C3 point symmetry and the seven-coordinated 6h sites with Cs point symmetry.33–35 Along the c-axis, Ca/LaO9 tetrakaidecahedron chains at 4f sites share faces, while Ca/LaO7 decahedrons at 6h sites share vertexes and edges. The neighbouring tetrakaidecahedron and decahedron are connected through tetrahedral PO4 groups and share edges. With the substitution of Si4+ for P5+, the relative Ca2+/La3+ ratio will change according to the charge compensation principle, accompanying the site change of the Eu activator at Ca2+ and La3+ sites. | ||
| Fig. 1 The Rietveld XRD refinement of (a) Ca5.88La3.92Eu0.2(PO4)4(SiO4)2O2 sample by the GSAS program, and (b) enlarged XRD patterns at 2θ = 30–35° of Ca(8−x)La(2+x)(PO4)6−x(SiO4)xO2:2% Eu (CLPSO-x) (x = 0–6) samples. Observed and calculated XRD patterns, baseline, and their difference are depicted with black crosses, red solid line, green solid line, and blue solid line, respectively. The short vertical lines show the positions of Bragg reflections of the calculated pattern. (c) A schematic crystal structure and cation-cosubstitution of CLPSO-x along c-axis. | ||
Fig. 2a and b depict the normalized photoluminescence excitation (PLE) and emission spectra (PL) of CLPSO-x samples prepared at 1300 °C. In previous report, it was found that the PL properties of Ca7.84La1.96Eu0.2(PO4)6O2 phosphors were related to the synthesis temperature.23,32 The Eu2+ ions in Ca7.84La1.96Eu0.2(PO4)6O2 exhibit blue emission (400–600 nm) and green emission (420–650 nm) when the sintering temperatures are 1250 °C and 1350 °C, respectively.32,33 The current CLSPO-0 sample show a broad absorption band from 250 nm to 500 nm centered at 395 nm due to the typical 5d–4f transitions of Eu2+. Interestingly, under 395 nm UV excitation, it gives a super wide emission band from 420 nm to 700 nm with two emission centers at 467 nm and 556 nm, respectively. Obviously, this super wide emission band also originates from the 5d–4f transitions of Eu2+ ions, and it simultaneously contains the Eu2+ emissions of Ca7.84La1.96Eu0.2(PO4)6O2-1250 °C and Ca7.84La1.96Eu0.2(PO4)6O2-1350 °C. The results reveal that two kinds of Eu2+ lattice environments appear in CLSPO-0 host at an intermediate sintering temperature (1300 °C). According to the previous conclusions in Eu2+-doped (Sr1−xBax)Si2O2N2 system, the super wide Eu2+ emission in Ca7.84La1.96Eu0.2(PO4)6O2-1300 °C single-phased sample should be attributed to the nanophase segregation and mixing Ca7.84La1.96Eu0.2(PO4)6O2-1250 °C and Ca7.84La1.96Eu0.2(PO4)6O2-1350 °C phases, which is consistent with the XRD results.23 It is noted that some previous report also ascribed the same wide Eu2+ emission to replace at two different sites. It maybe the complexes of the two situation. Although the emission spectrum of CLSPO-0 sample covers a wide range from 420 nm to 700 nm, it's CIE color coordinates (0.316, 0.386) locates at yellow-green junction region due to the deficiency of red component. In addition, there is no obvious characteristic emission of Eu3+, revealing that Eu mainly occupy the lattice sites of Ca2+ ions and thus present +2 form. Interestingly, the Eu2+ emission gradually decreases and the characteristic red emissions of Eu3+ that are assigned to 5D0–7F0–3 transitions, i.e., 577 nm (0–0), 588 nm (0–1), 613 nm (0–2), 650 nm (0–3), 700 nm (0–4), appear and gradually rise with increasing [La3+–Si4+] concentration. Moreover, the enhanced relative emission intensity of Eu3+/Eu2+ with increasing x value, implying a gradual transformation from Eu2+ to Eu3+ with the substitution of [La3+–Si4+] for [Ca2+–P5+]. Beyond x ≥ 2, the emission intensity of Eu3+ is obviously higher than that of Eu2+. Finally, at x = 6 the Eu2+ emission vanishes, and only the characteristic emission of Eu3+ can be observed. The result indicates that the Eu mainly enter into the La3+ sites in Ca2La8(SiO4)6O2 parent lattice and show +3 form. When monitoring with the maximum emission at 613 nm (5D0–7F2), the PLE spectra of x = 3–6 samples mainly consist of a O2p → Eu4f charge transfer band (CTB) in 250–335 nm and several 4f–4f transition lines from 350 nm to 500 nm.19Especially, the excitation intensities of 7F0–5L6 (395 nm) and 7F0–5D2 (463 nm) transitions are almost equivalent to or even surpass the O2p → Eu4f CTB absorption intensity, revealing that these samples could be effectively excited by 395 nm n-UV and 463 nm blue light. In general, the transformation of Eu2+ to Eu3+ happens with the substitution of [Ca2+–P5+] for [La3+–Si4+] in the CLSPO-x host, in which the Eu2+/3+ can coexist and be controllably adjusted at appropriate x value. The finding leads to the emission colors of the studied samples from yellow-green region to red light across the white light region, as shown by the luminescence photos in Fig. 2c. The CIE color coordinates of CLSPO-x samples are listed in Table 1 and Fig. 3. Evidently, the color coordinates gradually shift from (0.316, 0.386) to (0.618, 0.383), further confirming the tunable emission colors. Especially, the x = 2–4 are well distributed at different white light region with the different ratio of Eu2+/Eu3+, which could be used as potential single-activator-doped single-phased white light phosphors. The cation-cosubstitution derived.
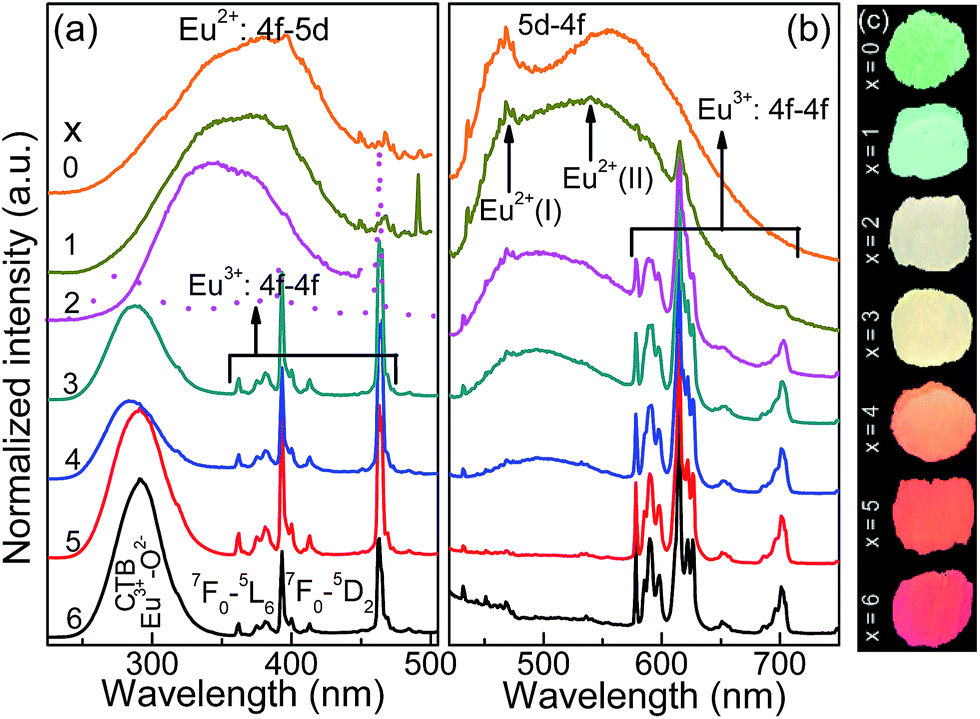 | ||
| Fig. 2 (a) The normalized photoluminescence excitation (PLE, λem = 466, 556, 613 nm) and (b) emission (PL, λex = 395 nm) spectra of CLPSO-x (x = 0–6) samples prepared at 1300 °C. (c) The luminescent photos of CLPSO-x (x = 0–6) samples under 395 nm UV are also shown. Eu2+(I) and Eu2+(II) means the crystal lattice environments of blue and green emission, respectively. | ||
| Sample | X | Y | Peak (nm) |
|---|---|---|---|
| x = 0 | 0.316 | 0.386 | 468 |
| x = 1 | 0.322 | 0.375 | 540 |
| x = 2 | 0.337 | 0.363 | 615 |
| x = 3 | 0.358 | 0.358 | 614 |
| x = 4 | 0.423 | 0.356 | 614 |
| x = 5 | 0.595 | 0.398 | 615 |
| x = 6 | 0.618 | 0.383 | 615 |
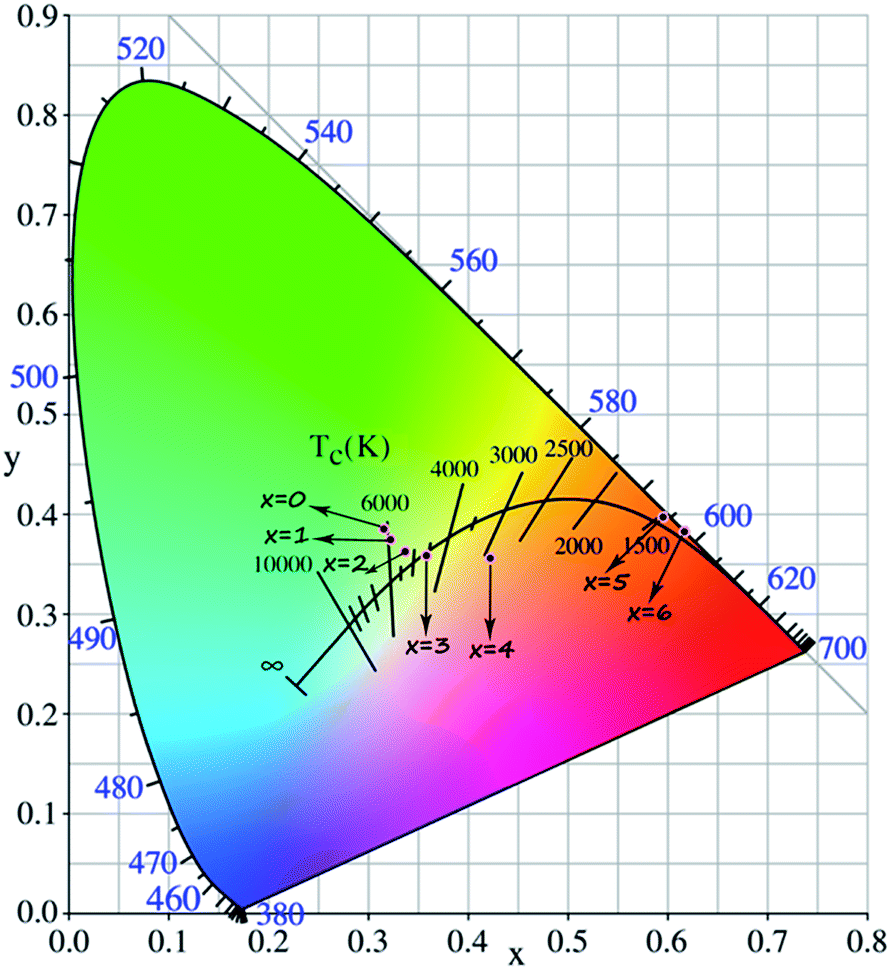 | ||
| Fig. 3 CIE diagram of Ca(8−x)La(2+x)(PO4)6−x(SiO4)xO2:2% Eu (CLPSO-x) (0 ≤ x ≤ 6) samples (λex = 395 nm). | ||
Eu2+/Eu3+-tunable strategy could be employed to design the desired single-phased white light in the apatite system.
To further verify the valence transformation of Eu in CLSPO-x, a powerful evidence, namely, the normalized Eu L3-edge extended X-ray absorption fine structure (XANES) spectra of the studied samples were collected in Fig. 4a.20,32 Clearly, two peaks at 6975 eV and 6985 eV are observed, which are attributed to the electron transitions of 2p3/2 → 5d in Eu2+ and Eu3+, respecitvely.20,32 The reason is the bigger shielding effect of nuclear potential by an additional 4f electron in Eu2+ than that in Eu3+. For the CLSPO-0, Eu almost exists in the form of 2+ except for a small amount of residual Eu3+, and this finding is consistent with the PL results. Moreover, the obviously higher emission of 5D0 → 7F2 at 613 nm with respect to the 5D0 → 7F1 emission at 588 nm indicates that the Eu3+ ions mainly enter the deviating inverse symmetry crystalline sites.32,36 For the substitution of [Ca2+–P5+] for [La3+–Si4+], the absorption intensity of Eu2+ systematically increases, while that of Eu3+ correspondingly decreases, as shown in Fig. 4a. Finally, the Eu almost evolves to the 3+ format x = 6. The results convincingly demonstrate that Eu3+ and Eu2+ could coexist and mutually transform through the cation cosubstitution. A possible transformation mechanism between Eu3+ and Eu2+ is proposed, as shown by Fig. 4b. In the initial Ca8La2(PO4)6O2 lattices, the Eu ions preferentially occupy the Ca2+ (6h) sites, presenting the Eu2+ broad band emission. A moderate sintering temperature induces the mixing nanophases of Ca8La2(PO4)6O2 and Ca2La8(SiO4)6O2 in the domain region, which further generates the multi-emitting sites of Eu2+ ions and produces a yellow-green emission. With the introduction of Si4+, the La3+ ions at 6h sites simultaneously enter the Ca2+/Eu2+ sites to balance charge, and thus Eu2+ ions gradually evolve to be Eu3+ ions. For the starting structure, the La3+ ions occupy all 6h sites and half of 4f sites in the ending Ca2La8(SiO4)6O2 host. Therefore, the Eu2+ ions are finally completely transformed to Eu3+ ions. In general, the cation cosubstitution strategy could drive the coexistence and adjustment of Eu2+/Eu3+ in apatite structures, generating the single-phased white light phosphors for n-UV based WLEDs.
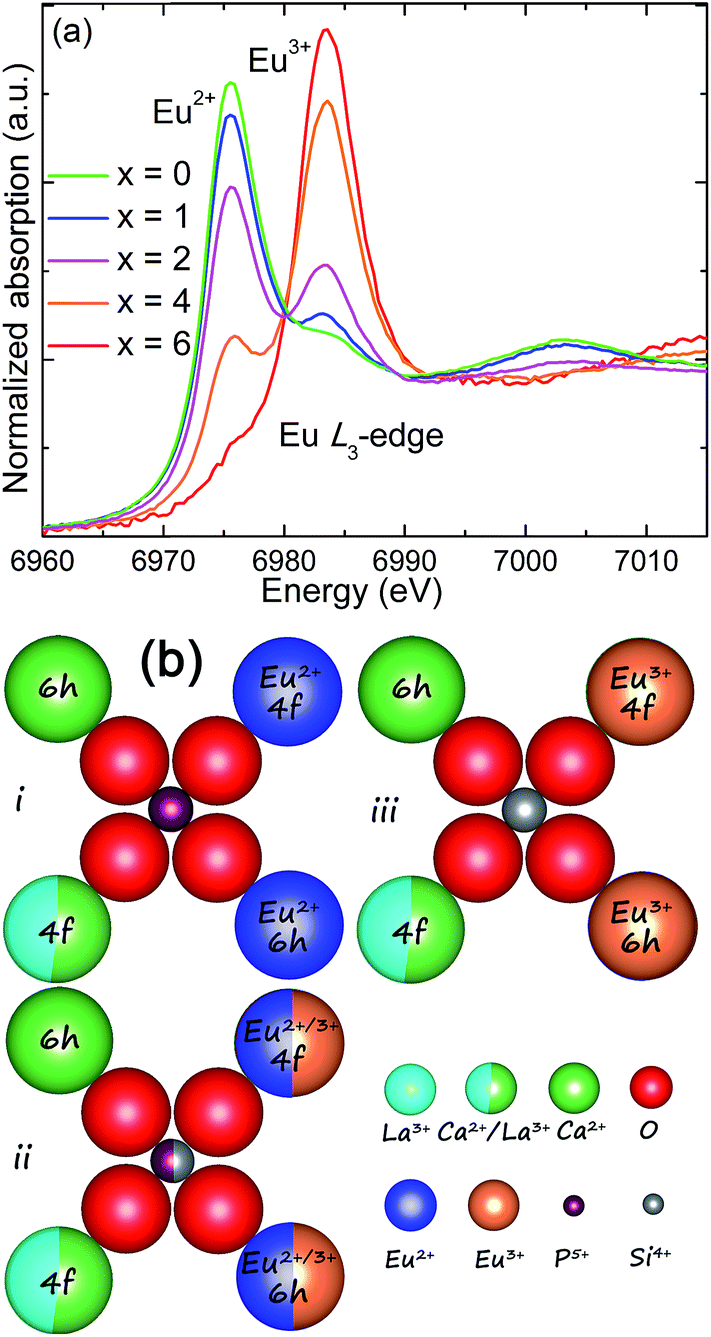 | ||
| Fig. 4 (a) Normalized Eu L3-edge X-ray absorption near edge structure (XANES) spectra of the representative CLPSO-x (x = 0, 1, 2, 4, 6) samples. (b) A schematic mechanism to explain the transformation between Eu2+ and Eu3+ with the cosubstitution of [Ca2+–P5+] for [La3+–Si4+]. | ||
As an important index to evaluate the efficiency and stability of pc-WLEDs devices, the thermal quenching behaviour of CLPSO-x samples from room temperature to 573 K was examined and shown in Fig. 5a.37–42 Given a non-radiative transition under high working temperature, it is reasonable to observe the progressive emission attenuation of the studied samples.43–47 It is noted that the thermal stability of x = 2–6 samples are obviously better than that of x = 0, 1 samples. Especially, a prominent improvement appears at x = 2, which can obtain 56% of the original emission intensity at room temperature. This is mainly because the outside electrons of Eu have a larger shielding effect for the 4f–4f transitions relative to its 5d–4f transitions.32,36 Therefore, a weak electron-photon coupling interaction occurs and leads to a low thermal quenching under high temperature environment.32,36 Finally, the x = 0 and x = 2 samples were fabricated to be single-phased white light-emitting devices combined with the 395 nm n-UV chips. Fig. 5b shows the corresponding electroluminescence (EL) spectra of the packaged WLEDs devices under a 200 mA forward bias current, which are basically in agreement with their PL results. Simultaneously, the packaged WLEDs present high Ra and low CCT, namely, Ra = 95.9, CCT = 5690 K for x = 0 sample and Ra = 93.2, CCT = 3680 K for x = 2 sample, respectively. The CIE color coordinates of x = 0 and 2 samples are calculated to be (0.33, 0.33) and (0.38, 0.34), respectively. Therefore, excellent warm white light with high Ra could be realized in the single Eu-doped apatite phosphor by the cation cosubstitution.
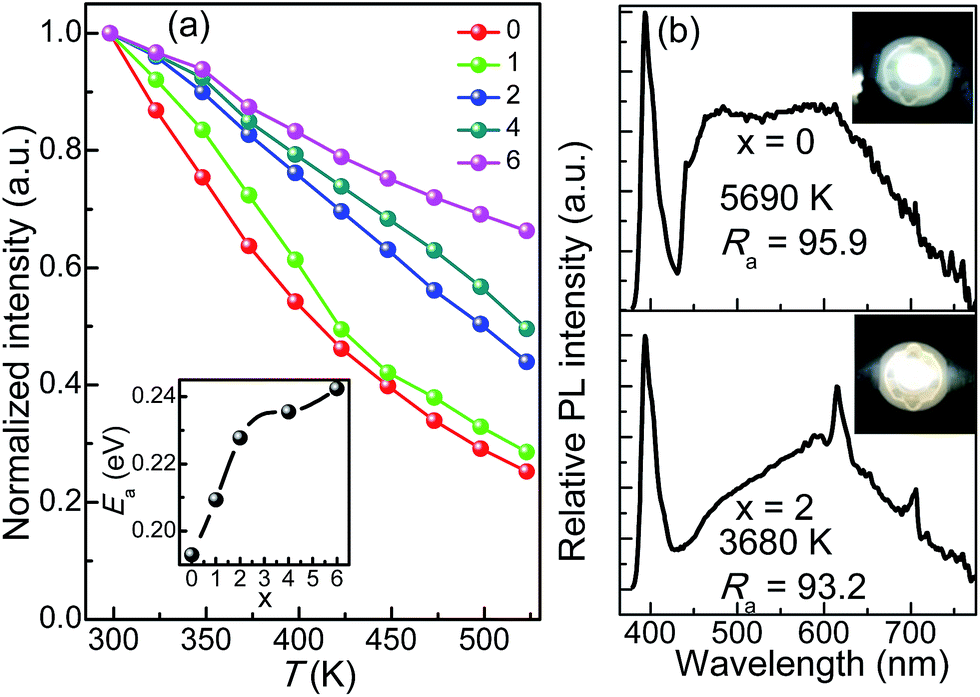 | ||
| Fig. 5 Thermal quenching behaviour of photoluminescence for CLPSO-x (x = 0–6) samples (λex = 395 nm). The insert shows the corresponding activation energy Ea. (b) Electroluminescence spectra of the CLPSO-0 (Top) and CLPSO-2 (Bottom) samples incorporated into an 395 nm-InGaN LED at 200 mA forward bias current. The CCT, Ra and corresponding luminescence photos (the insets) of the white lights are listed. | ||
4. Conclusions
In conclusions, a novel Eu-activated single-phased white light phosphor was developed based on the presence of multi-emitting Eu2+ and the transformation between Eu2+ and Eu3+ by cation cosubstitution of [La3+–Si4+] for [Ca2+–P5+] in apatite analogue. The XRD patterns demonstrate that the formation of Ca8−xLa2+x(PO4)6−x(SiO4)xO2:2% Eu solid solutions. Ultra broad Eu2+ emission from 420 nm to 600 nm imply the mixing nanophases of Ca8La2Eu(PO4)6−x(SiO4)xO2 samples at different sintering temperatures. The PL and XANES spectroscopy of Eu L3 edge convincingly confirm the valence transformation from Eu3+ to Eu2+ by the substitution of [La3+–Si4+] for [Ca2+–P5+]. Because of a whole cover across the blue, green emission from Eu2+ and red emission from Eu3+ in CLSPO phosphors, we obtained high Ra warm white lights (95.9 for x = 0; 93.2 for x = 2), with the corresponding CCT of 5690 K and 3680 K, respectively. The newly developed single Eu-activated white light phosphors exhibit potential applications in single-phase white emitting n-UV pumped LEDs devices.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grants No. NSFC 51672259, 21301162, 51672265, 91433110, U1301242) and the Ministry of Science and Technology of Taiwan (Contract no. MOST 104-2113-M-027-007-MY3).Notes and references
- N. C. George, K. A. Denault and R. Seshadri, Annu. Rev. Mater. Res., 2013, 43, 481–501 CrossRef CAS.
- F. W. Kang, X. B. Yang, M. Y. Peng, L. Wondraczek, Z. J. Ma, Q. Y. Zhang and J. R. Qiu, J. Phys. Chem. C, 2014, 118, 7515–7522 CAS.
- C. H. Huang, T. W. Kuo and T. M. Chen, ACS Appl. Mater. Interfaces, 2010, 2, 1395–1399 CAS.
- P. F. Smet, A. B. Parmentier and D. Poelman, J. Electrochem. Soc., 2011, 158, R37–R54 CrossRef CAS.
- N. Guo, Y. H. Zheng, Y. C. Jia, H. Qiao and H. P. You, J. Phys. Chem. C, 2012, 116, 1329–1334 CAS.
- C. H. Huang, Y. C. Chiu, Y. T. Yeh, T. S. Chan and T. M. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 6661–6667 CAS.
- K. Li, M. M. Shang, H. Z. Lian and J. Lin, J. Mater. Chem. C, 2016, 4, 5507–5530 RSC.
- Z. J. Wang, P. L. Li, Z. P. Yang and Q. L. Guo, J. Lumin., 2014, 151, 170–175 CrossRef CAS.
- P. L. Li, Z. J. Wang, Z. P. Yang and Q. L. Guo, RSC Adv., 2014, 4, 27708–27713 RSC.
- W. R. Liu, C. H. Huang, C. W. Yeh, Y. C. Chiu, Y. T. Yeh and R. S. Liu, RSC Adv., 2013, 3, 9023–9028 RSC.
- X. Ding, G. Zhu, Q. Wang and Y. H. Wang, RSC Adv., 2015, 5, 30001–30004 RSC.
- P. P. Dai, C. Li, X. T. Zhang, J. Xu, X. Chen, X. L. Wang, Y. Jia, X. J. Wang and Y. C. Liu, Light: Sci. Appl., 2016, 5, e16024 CrossRef CAS.
- R. J. Xie and N. Hirosaki, Sci. Technol. Adv. Mater., 2007, 8, 588–600 CrossRef CAS.
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS PubMed.
- X. J. Zhang, J. Wang, L. Huang, F. J. Pan, Y. Chen, B. F. Lei, M. Y. Peng and M. M. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 10044–10054 CAS.
- W. R. Liu, C. W. Yeh, C. H. Huang, C. C. Lin, Y. C. Chiu, Y. T. Yeh and R. S. Liu, J. Mater. Chem., 2011, 21, 3740–3744 RSC.
- D. J. Hou, C. M. Liu, X. M. Ding, X. J. Kuang, H. B. Liang, S. S. Sun, Y. Huang and Y. Tao, J. Mater. Chem. C, 2013, 1, 493–499 RSC.
- Z. Y. Zhao, Z. G. Yang, Y. R. Shi, C. Wang, B. T. Liu, G. Zhu and Y. H. Wang, J. Mater. Chem. C, 2013, 1, 1407–1412 RSC.
- Q. Q. Zhu, L. Wang, N. Hirosaki, L. Y. Hao, X. Xu and R. J. Xie, Chem. Mater., 2016, 28, 4829–4839 CrossRef CAS.
- K. W. Huang, W. T. Chen, C. I. Chu, S. F. Hu, H. S. Sheu, B. M. Cheng, J. M. Chen and R. S. Liu, Chem. Mater., 2012, 24, 2220–2227 CrossRef CAS.
- F. P. Du, Y. Nakai, T. Tsuboi, Y. L. Huang and H. J. Seo, J. Mater. Chem., 2011, 21, 4669–4678 RSC.
- Y. S. Liu, W. Q. Luo, R. F. Li, G. K. Liu, M. R. Antonio and X. Y. Chen, J. Phys. Chem. C, 2009, 113, 5340 CAS.
- Z. G. Xia, G. K. Liu, J. G. Wen, Z. G. Mei, M. Balasubramanian, M. S. Molokeev, L. C. Peng, L. Gu, D. J. Miller, Q. L. Liu and K. R. Poeppelmeier, J. Am. Chem. Soc., 2016, 138, 1158–1161 CrossRef CAS PubMed.
- Z. G. Xia, C. G. Ma, M. S. Molokeev, Q. L. Liu, K. Rickert and K. R. Poeppelmeier, J. Am. Chem. Soc., 2015, 137, 12494–12497 CrossRef CAS PubMed.
- W. B. Im, N. George, J. Kurzman, S. Brinkley, A. Mikhailovsky, J. Hu, B. F. Chmelka, S. P. DenBaars and R. Seshadri, Adv. Mater., 2011, 23, 2300–2305 CrossRef CAS PubMed.
- M. Seibald, T. Rosenthal, O. Oeckler and W. Schnick, CRC Crit. Rev. Solid State Sci., 2014, 39, 215–229 CrossRef CAS.
- W. B. Park, S. P. Singh and K. S. Sohn, J. Am. Chem. Soc., 2014, 136, 2363–2373 CrossRef CAS PubMed.
- F. W. Kang, H. S. Zhang, L. Wondraczek, X. B. Yang, Y. Zhang, D. Y. Lei and M. Y. Peng, Chem. Mater., 2016, 28, 2692–2703 CrossRef CAS.
- Y. Sato, H. Kato, M. Kobayashi, T. Masaki, D. H. Yoon and M. Kakihana, Angew. Chem., Int. Ed., 2014, 53, 7756–7759 CrossRef CAS PubMed.
- W. T. Chen, H. S. Sheu, R. S. Liu and J. P. Attfield, J. Am. Chem. Soc., 2012, 134, 8022–8025 CrossRef CAS PubMed.
- D. G. Deng, H. Yu, Y. Q. Li, Y. J. Hua, G. H. Jia, S. L. Zhao, H. P. Wang, L. H. Huang, Y. Y. Li, C. X. Li and S. Q. Xu, J. Mater. Chem. C, 2013, 1, 3194–3199 RSC.
- G. G. Li, C. C. Lin, Y. Wei, Z. W. Quan, Y. Tian, Y. Zhao, T. S. Chan and J. Lin, Chem. Commun., 2016, 52, 7376–7379 RSC.
- M. M. Shang, G. G. Li, D. L. Geng, D. M. Yang, X. J. Kang, Y. Zhang, H. Z. Lian and J. Lin, J. Phys. Chem. C, 2012, 116, 10222–10231 CAS.
- Y. F. Xia, J. Chen, Y. G. Liu, M. S. Molokeev, M. Guan, Z. H. Huang and M. H. Fang, Dalton Trans., 2016, 45, 1007–1015 RSC.
- G. S. R. Raju, J. Y. Park, H. C. Jung, B. K. Moon, J. H. Jeong and J. H. Kim, J. Electrochem. Soc., 2011, 158, J20–J26 CrossRef CAS.
- F. W. Kang, Y. Zhang and M. Y. Peng, Inorg. Chem., 2015, 54, 1462–1473 CrossRef CAS PubMed.
- P. Dorenbos, ECS Solid State Lett., 2013, 2, R3001–R3011 CAS.
- W. R. Liu, C. W. Yeh, C. H. Huang, C. C. Lin, Y. C. Chiu, Y. T. Yeh and R. S. Liu, J. Mater. Chem., 2011, 21, 3740–3744 RSC.
- H. Daicho, T. Iwasaki, K. Enomoto, Y. Sasaki, Y. Maeno, Y. Shinomiya, S. Aoyagi, E. Nishibori, M. Sakata, H. Sawa, S. Matsuishi and H. Hosono, Nat. Commun., 2012, 3, 1132 CrossRef PubMed.
- K.-S. Sohn, B. Lee, R.-J. Xie and N. Hirosaki, Opt. Lett., 2009, 34, 3427–3430 CrossRef CAS PubMed.
- X. Piao, K. Machida, T. Horikawa, H. Hanzawa, Y. Shimomura and N. Kijima, Chem. Mater., 2007, 19, 4592–4599 CrossRef CAS.
- W. J. Zhou, F. J. Pan, L. Zhou, D. J. Hou, Y. Huang, Y. Tao and H. B. Liang, Inorg. Chem., 2016, 55, 10415–10424 CrossRef CAS PubMed.
- T. Takeda, N. Hirosaki, S. Funahshi and R. J. Xie, Chem. Mater., 2015, 27, 5892–5898 CrossRef CAS.
- C. W. Yeh, W. T. Chen, R. S. Liu, S. F. Hu, H. S. Sheu, J. M. Chen and H. T. Hintzen, J. Am. Chem. Soc., 2012, 134, 14108–14117 CrossRef CAS PubMed.
- M. Y. Peng, X. W. Yin, P. A. Tanner, M. G. Brik and P. F. Li, Chem. Mater., 2015, 27, 2938–2945 CrossRef CAS.
- Y. Zhang, X. M. Liu, X. J. Li, K. Li, H. Z. Lian, M. M. Shang and J. Lin, Dalton Trans., 2015, 44, 7743–7747 RSC.
- F. W. Kang, M. Y. Peng, D. Y. Lei and Q. Y. Zhang, Chem. Mater., 2016, 28, 7807–7815 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |