DOI:
10.1039/C6RA26855B
(Paper)
RSC Adv., 2017,
7, 5541-5548
Metal organic frameworks with uni-, di-, and tri-nuclear Cd(II) SBU prepared from 1,3-bis(4-pyridyl)propane and different dicarboxylate ligands: syntheses, structures and luminescent properties†
Received
15th November 2016
, Accepted 29th December 2016
First published on 17th January 2017
Abstract
Three new metal organic frameworks (MOFs) derived from the flexible dipyridyl ligand 1,3-bis(4-pyridyl)propane (BPP), Cd(BPP)(OBA)(H2O)·(DMF)(H2O) (1), Cd(BPP)(4-HOIP)·(DMF)0.5(H2O) (2), [Cd6(BPP)(BPDC)7(DMF)2]2−·(H3O+)2(H2O)7 (3), (H2OBA = 4,4′-oxybis(benzoic acid), 4-H3OIP = 4-hydroxyisophthalic acid, H2BPDC = 4,4′-biphenyldicarboxylic acid, DMF = N,N-dimethylformamide) have been synthesized under mild conditions. By changing the co-dicarboxylate ligands, the three structures are based on uni-, di-, and tri-nuclear Cd(II) SBUs (secondary building units). Notably, compound 3 has an unusual 3D pillared framework with 2-fold interpenetrated hex topology. Most interestingly, in compound 3, BPP and BPDC ligands play the roles of two kinds of pillars, and support the same 2D layers alternatively leading to a special anionic 3D framework. To the best of our knowledge, such an arrangement has not been reported to date. Moreover, the three compounds exhibit strong luminescent emission at room temperature in the solid state.
1. Introduction
The rational design and synthesis of MOFs constitutes a current prosperous research field that is attracting intensive attention owing to the scientific interest in the creation of intriguing aesthetic topological structures, as well as their potential applications in gas storage, separation, catalysis, and magnetic and optical devices.1–4 It has been realized that the self-assembly of MOFs is influenced by many subtle factors, such as the pH of the solution, the reaction temperature and time, the nature of the metal ions and counter ions, the coordination modes of the organic ligands, and so on.5–9 Therefore, great efforts have been dedicated to exploring the assembly principle of MOFs. As it is known, different donor groups can hold great promise for incorporation of accessibility, structural diversity and geometrical control of properties into the resulting networks. A lot of research groups have been particularly interested in frameworks with N-donor and O-donor mixed ligands systems, which show extensive functional properties and dynamic features.10–13 However, the influence of the ligands on the resulting networks in flexible N-donor and O-donor mixed ligands systems has not been well investigated. Thereby, we have embarked on a project to investigate the influence of the ligands on the diversity in assembly of the resulting structures from the N-donor and O-donor mixed ligands systems.
During the last two decades, more and more MOFs have been reported with various topologies. Among the diverse structures, the four-connected nets are the most popular according to the reviews by O'Keeffe, Yaghi and co-workers, such as dia, nbo, pts or cds topologies, which have been widely reported.14–18 However, MOFs based on five-connected or eight-connected nets, such as hex-type nets, are comparatively rare.19,20 Therefore, herein we hope to get more new types of structures when employing common organic linkers.
Taking into account all the aforementioned topics, we employed BPP as a common and flexible organic ligand into our synthetic strategy. The BPP ligand can give rise to TT, TG, GG and GG′ conformations (T = trans, G = gauche), thanks to the free twist of the longer intermediate chain (propyl group: –CH2–CH2–CH2–). Thereby it can adopt different conformations to take part in the coordination of the metal, resulting in a large variety of network topologies.21–25 For the mixed O-donor ligands, we employed H2OBA, 4-H3OIP, and H2BPDC. 4-H3OIP and H2OBA exhibit a V-shape architecture with similar angles (around 120°) built between the two carboxyl groups towards the core of the ligand. However, a main difference appears in terms of flexibility since 4-H3OIP is rigid while H2OBA can bend or rotate freely owing to the central O atom (ether group). H2BPDC is commonly considered as a long and rigid linear ligand because it is unable to bend and the angle of the two aromatic carboxyl groups towards the centre of the ligand is 180°. With the systematic variation of the O-donor ligands, three new Cd(II) MOFs have been synthesized under mild conditions. Compound 1 features a 2D four-connected sql network with uni-nuclear Cd(II) SBUs, whereas compound 2 possesses a 3D distorted cds architecture with di-nuclear Cd(II) SBUs and compound 3 has a rare 2-fold interpenetrated 3D pillared hex framework with tri-nuclear Cd(II) SBUs. Their frameworks display outstanding structural diversification owing to the variation of the dicarboxylate ligands. Unexpectedly, we found an unusual phenomenon linked to compound 3 in which there exist two kinds of pillars that are arranged alternately in the same layer to construct an atypical anionic 3D framework. To the best of our knowledge, it is the first MOF reported to date with such a feature. Herein, we describe the three structures and discuss the influence of the nature of the ligands on the remaining topologies. The thermal stability and the luminescent properties are also investigated.
2. Experimental
2.1 Materials and measurements
All chemicals purchased were of reagent grade or better and were used without any further purification. The elemental analyses for C, H, and N and the inductively coupled plasma (ICP) analysis were carried out on a PerkinElmer 240C elemental analyzer and PerkinElmer Optima 3300 DV ICP spectrometer, respectively. The infrared (IR) spectra were recorded (400–4000 cm−1 region) on a Nicolet Impact 410 FTIR spectrometer by using the KBr pellets method. Powder X-ray diffraction (PXRD) data were collected on a Siemens D5005 diffractometer with Cu Kα radiation (λ = 1.5418 Å). TGA (thermogravimetric analyses) were performed under nitrogen with a heating rate of 5 °C min−1 using a PerkinElmer TGA 7 thermogravimetric analyzer. Luminescence spectroscopy data were recorded on an LS55 luminescence spectrometer.
2.2 Synthesis of compounds
Compound 1. Cd(NO3)2·4H2O (0.06 g, 0.2 mmol), BPP (0.04 g, 0.2 mmol), and H2OBA (0.05 g, 0.2 mmol) were dissolved in DMF (10.00 mL)/H2O (1.00 mL) mixture, then a DMF solution (0.50 mL) containing 2-propanolamine (0.05 mL) was slowly diffused into the mixture over 3 day at 85 °C. The resulting colorless, block-shaped single crystals of compound 1 were collected by filtration, washed with DMF and dried in air (yield: 70%, based on cadmium). IR: 3332m, 3062w, 2944w, 2856w, 1668s, 1596s, 1558s, 1500s, 1392s, 1238s, 1159s, 1089m, 1014m, 875m, 790s, 663m, 516m. Elemental analysis and ICP calculated for Cd(BPP)(OBA)(H2O)·(DMF)(H2O) (C30H33N3O8Cd) (676.02 g mol−1) (%): C, 53.30; H, 4.92; N, 6.22; Cd, 16.63; found (%): C, 53.03; H, 5.09; N, 6.57; Cd, 16.35.
Compound 2. The procedure was similar to that used for compound 1, except that H2OBA (0.05 g, 0.2 mmol) was replaced by 4-H3OIP (0.04 g, 0.2 mmol). The resulting colorless, block-shaped single crystals were collected by filtration, washed by DMF and then dried in air (yield: 65%, based on cadmium). IR: 3433m, 3067w, 2928w, 1931w, 1659s, 1593s, 1504s, 1394s, 1267w, 1180m, 1105m, 1063w, 1007m, 860m, 771s, 677m, 526w. Elemental analysis and ICP calculated for Cd(BPP)(4-HOIP)·(DMF)0.5(H2O) (C22.5H27.5N2.5O7.5Cd) (563.39 g mol−1) (%): C, 47.96; H, 4.93; N, 6.22; Cd, 19.95; found (%): C, 48.10; H, 4.79; N, 6.58; Cd, 19.77.
Compound 3. The procedure was similar to that used for compound 1, except that H2OBA (0.05 g, 0.2 mmol) was replaced by H2BPDC (0.04 g, 0.2 mmol). The resulting colorless, rod-shaped single crystals were collected by filtration, washed by DMF and dried in air (yield: 55%, based on cadmium). IR: 3448w, 3064w, 2929w, 1934w, 1660s, 1579s, 1519s, 1394s, 1178m, 1099m, 1063w, 1006m, 854m, 773s, 682m, 523w. Elemental analysis and ICP calculated for [Cd6(BPP)(BPDC)7(DMF)2]2−·(H3O+)2(H2O)7 (C117H104N4O39Cd6) (2864.58 g mol−1) (%): C, 49.06; H, 3.66; N, 1.96; Cd, 23.54; found (%):C, 49.38; H, 3,87; N, 1.79; Cd, 23.69.
2.3 X-ray crystallographic study
Diffraction intensities for compound 1, compound 2 and compound 3 were collected on a computer-controlled Bruker SMART CCD diffractometer equipped with graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation at room temperature by using the ω-scan technique. Raw data for all structures were processed using SAINT-5.0 and absorption corrections were applied using SADABS.26,27 These structures were solved by direct methods. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were fixed at calculated positions and refined by using a riding mode. All calculations were performed using the SHELXTL-97 program.28 The main crystallographic data and structure refinement parameters are summarized in Table 1. CCDC-756103 (1), CCDC-756104 (2) and CCDC-756105 (3) contain the supplementary crystallographic data for this paper.†
Table 1 Crystallographic data and structure refinement for compound 1, compound 2 and compound 3
| |
1 |
2 |
3 |
| R = ∑||Fo| − |Fc||/∑|Fo|. Rw = [∑w(Fo2 − Fc2)/∑w(Fo2)2]1/2. |
| Empirical formula |
C30H29CdN3O7 |
C21H18CdN2O5O5 |
C117H86Cd6N4O31 |
| Fw |
655.96 |
490.78 |
2718.36 |
| T (K) |
293(2) |
293(2) |
293(2) |
| λ (Å) |
0.71073 |
0.71073 |
0.71073 |
| Crystal syst. |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c (no. 14) |
C2/c (no. 15) |
C2/c (no. 15) |
| a (Å) |
15.3271(3) |
16.375(3) |
53.600(11) |
| b (Å) |
17.5154(6) |
12.766(3) |
13.976(3) |
| c (Å) |
11.6216(3) |
25.320(5) |
47.389(9) |
| α (°) |
90.00 |
90.00 |
90.00 |
| β (°) |
101.145(2) |
98.43(3) |
118.27(3) |
| γ (°) |
90.00 |
90.00 |
90.00 |
| V (Å3) |
3061.10(14) |
5235.8(19) |
31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 266(14) 266(14) |
| Z |
4 |
8 |
8 |
| ρcalc. (Mg m−1) |
1.423 |
1.245 |
1.155 |
| μ (mm−1) |
0.762 |
0.861 |
0.859 |
| GOF on F2 |
0.958 |
1.225 |
1.086 |
| Ra [I > 2δ(I)] |
0.0545 |
0.0668 |
0.0873 |
| Rwb |
0.1321 |
0.2401 |
0.2798 |
3. Results and discussion
3.1 Description of the structures
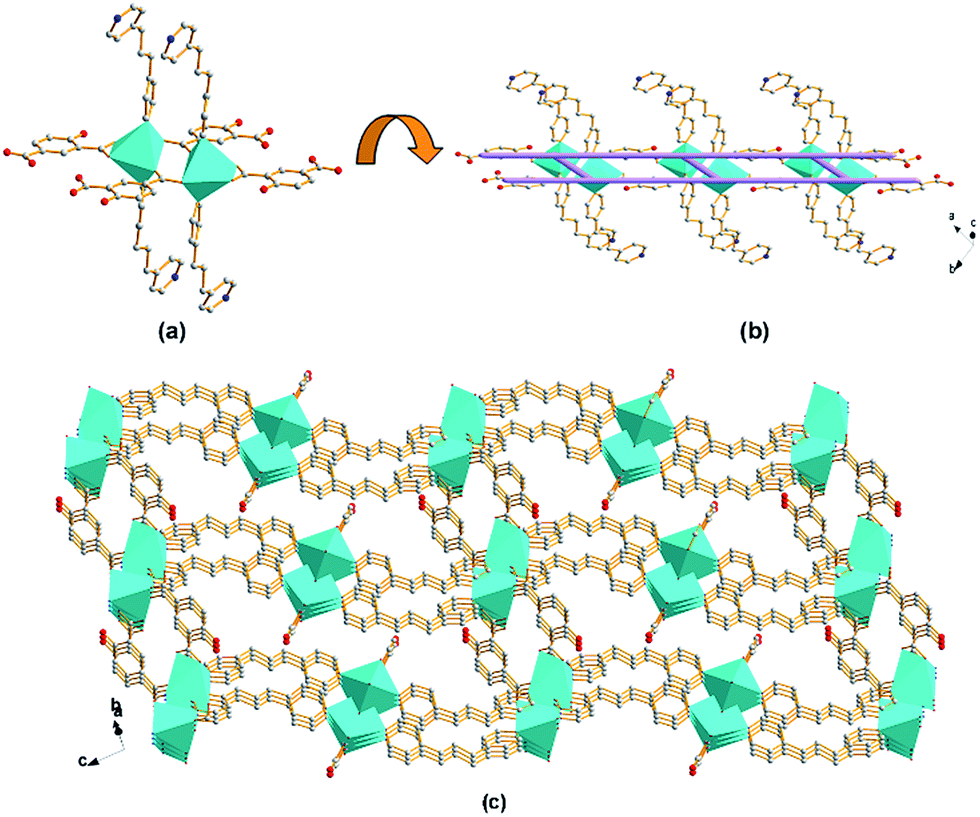
Structure of compound 1. Single crystal X-ray diffraction analysis reveals that compound 1 crystallizes in the monoclinic system, space group P21/c (no. 14). As shown in Fig. 1a, the uni-nuclear Cd(II) SBU consists of a crystallographic hexacoordinated Cd(II). The coordination involves four oxygen from one chelating bidentate and one monodentate carboxyl groups of two OBA (Cd–O of 2.224(3)–2.460(3) Å), and two nitrogen donors from two BPP ligands (Cd–N of 2.289(3) Å and 2.371(4) Å). The uni-nuclear Cd(II) SBUs are connected to each other along the crystallographic a axis by the carboxylate ligands to form 1D zig-zag chains (Fig. 1b). Then the chains are further linked to the adjacent ones by the flexible BPP ligands along the crystallographic c axis, affording a 2D (4, 4) network with square meshes of 6.2 × 10.2 Å2 (dimensions based on van der Waals' radius of carbon) on the ac plane (Fig. 1c). Each undulated layer interacts with successive one to form double 2D nets, which pack in ABAB mode along b axis as shown in Fig. 1d. The DMF guest molecules are statistically disposed inside between two double layers via weak H-bond (d(H⋯A) = 2.54 Å, d D⋯A) = 3.327(10) Å) (Fig. S1†).29,30 Further insight into the structure of compound 1 can be obtained by reducing the 2D structure to simple node-linker geometries and associated nets.31–33 The uni-nuclear SBU can be defined as a 4-connected node, and the ligands only serve as bridging linkers. According to the simplification principle, compound 1 is formed with a uninodal 4-connected 2D net, and its Schläfli symbol is (44), which is a typical sql structure, as shown in Fig. 2.
 |
| | Fig. 1 Schematic illustrations of (a) uni-nuclear Cd(II) SBU as octahedron; (b) a 1D zig-zag chain with the uni-nuclear Cd(II) SBUs connected by OBA along a axis; (c) an infinite 2D puckered (4, 4) network with the square meshes lay on the ac plane; (d) the double 2D layers stacking in ABAB mode viewed along c axis (color code: Cd, cyan; C, grey; O, red; N, blue). | |
 |
| | Fig. 2 Schematic illustration of the sql topology of compound 1. | |
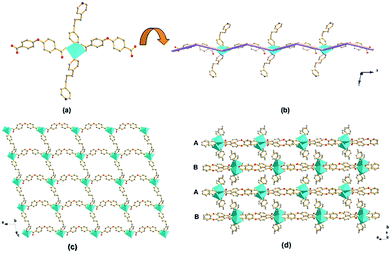
Structure of compound 2. Single crystal X-ray diffraction analysis reveals that compound 2 crystallizes in the monoclinic system, space group C2/c (no. 15). As shown in Fig. 3a, the di-nuclear Cd(II) SBU consists of two hexacoordinated Cd(II) with an identical distorted octahedral coordination environment. Each octahedral coordination mode is completed by four oxygen from one chelating bidentate and two bidentate carboxyl groups borne by three different 4-HOIP (Cd–O of 2.272(6)–2.432(7) Å), and two nitrogen donors (Cd–N of 2.315(7) and 2.305(7) Å). The Cd–Cd separation (center–center distance) is 4.01 Å. As shown in Fig. 3b, the di-nuclear Cd(II) SBUs are linked by 4-HOIP along the a axis to form 1D ladder-like chains. Then two vertical adjacent chains are further connected by the BPP ligands, resulting in a 3D framework with 1D channels along the [110] direction (Fig. 3c). Notably, the channel is separated by two benzene rings of 4-HOIP into three connected parts while all of the hydroxyl groups (–OH) are free in the void. Two of the parts are triangular channels about 1.6 × 5.1 Å2 (calculated based on van der Waals' radius of carbon), while the other one is almost blocked.
 |
| | Fig. 3 Schematic illustrations of (a) di-nuclear Cd(II) SBU; (b) the 1D ladder-like chain with the di-nuclear Cd(II) SBUs connected by 4-HOIP along the a axis; (c) 3D framework of compound 2 formed by vertical adjacent 1D ladder-like chains connected by the BPP ligands while all of the hydroxyl groups (–OH) remain protonated in the void, viewed along the [110] direction (color code: Cd, cyan; C, grey; O, red; N, blue). | |
As previously applied for compound 1, a structural simplification based on the di-nuclear Cd(II) SBU as a four-connected node, di-BPP and di-4-HOIP in four vertical directions as two types of two-connected linkers, a cds 3D network sustained by 4-coordinated planar nodes is obtained, and its Schläfli symbol is (65·8). Noticeably, owing to the different length of the two types of connections related to two kinds of ligands (10.382 Å and 14.029 Å), the distance between two nodes is different. Meanwhile, the torsion of the flexible BPP ligand gives rise to the distortion of the 4-coordinated planar nodes. Consequently, the resulting network of compound 2 is a distorted cds topology (Fig. 4).
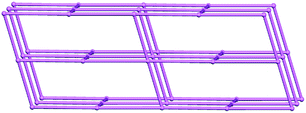 |
| | Fig. 4 Schematic illustration of the distorted cds topology of compound 2. | |
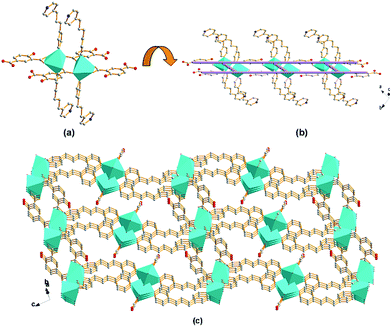
Structure of compound 3. The crystallographic analysis reveals that compound 3 crystallizes in the monoclinic system, space group C2/c (no. 15). As illustrated in Fig. 5, the tri-nuclear Cd(II) SBU consists of three crystallographic Cd(II) centers. The decahedron coordination of heptacoordinated Cd1 is realized by seven O donors, which are two pairs of chelating/bridging-bidentate carboxylic O donors, one bidentate and one pair of chelating bidentate carboxylic O donors separately from four distinct BPDC. Heptacoordinated Cd3 is in the same geometry of Cd1, only with one O donor of the coordinated DMF molecule and one N donor of one BPP ligand taking the place of the couple of chelating carboxylic O donors on the apical position. However, hexacoordianted Cd2 adopts the different coordination mode. The octahedral coordination is achieved by four chelating/bridging-bidentate carboxylic O donors of four distinct BPDC on the equatorial plane and two bidentate carboxylic O donors of two BPDC occupying the two apical coordination sites. The Cd–O distances are between 2.189(5) and 2.809(6) Å, and the Cd–N distances are between 2.281(6) and 2.285(5) Å.
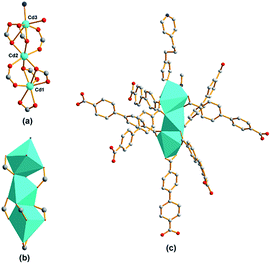 |
| | Fig. 5 Schematic illustrations of tri-nuclear Cd(II) SBU for compound 3 in (a) ball-and-stick mode, (b) polyhedra representation, and (c) SBU coordination mode. | |
The eight vertexes of the tri-nuclear Cd(II) SBU, Cd3(CO2)6ON cluster (Fig. 5a and b), on the equatorial plane are linked by six BPDC to form 2D (3, 6)-connected networks along the [10−1] plane (Fig. 6a). The remaining two opposite apical positions are separately occupied by one BPP ligand and one BPDC through the N donor and a couple of carboxylate O donors leading to a 3D pillared framework. Notably, the adjacent tri-nuclear Cd(II) SBUs display an inverse orientation along the vertical direction of the (3, 6) plane. Therefore the BPDC and BPP ligands occupy the opposite positions compared to the neighboring SBU. Consequently, in compound 3 not only are there two kinds of pillars (BPDC and BPP) but also the distinct pillars alternately support the 2D planes in the same layer, as shown in Fig. 6b. To the best of our knowledge, this fantastic arrangement of two pillars in compound 3 is the first reported to date. So unlike most pillared MOFs, compound 3 has an anionic framework with charge-balancing H3O+ in the channels. As shown in Fig. 6c, two identical networks interpenetrate to generate 1D channels of about 4.8 × 4.8 Å2 (calculated based on van der Waals' radius of carbon) along the [101] direction, with coordinated DMF and guest water molecules residing inside.
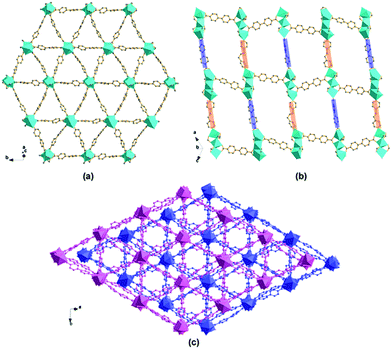 |
| | Fig. 6 Representations of (a) an infinite 2D (6, 3) network constructed from tri-nuclear Cd(II) SBUs connected by BPDC along the [101] plane; (b) 3D framework of compound 3 formed from pillars (BPP and BPDC ligands alternately) supporting the 2D layers, viewed along the [101] direction; (c) 2-fold interpenetrated 3D framework of compound 3 formed by two identical networks interpenetrating, viewed along the [101] direction (color code: Cd, cyan; C, grey; O, red; N, blue). | |
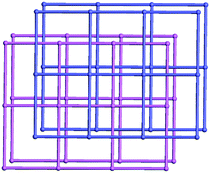
The topology of compound 3 is studied to further understand the structure. By considering the tri-nuclear SBU as an 8-connected node, and the BPP ligand and BPDC as two-connected links, an unusual 2-fold interpenetrating 3D pillared hex type network with 8-coordinate nodes is obtained with a Schläfli symbol of (36·418·53·6), as shown in Fig. 7.
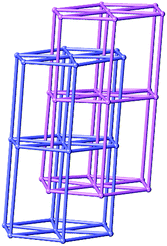 |
| | Fig. 7 Schematic illustration of 2-fold interpenetrating hex topology of compound 3. | |
3.2 Effect of the nature of the ligands on the structures
There is no doubt that the above structure discussion demonstrates the apparent effect of the nature and the configuration of the ligands in the syntheses mixtures on the resulting structures. Although it is impossible to make an exact verdict on the synthesis rule owing to the complexity of the MOFs synthesis system, primitive exploration is still worthwhile for guiding further research. To analyze the structures, a comparison is realized among the different carboxylate ligands and the conformations of the BPP ligand in the three compounds (Tables 2 and 3).
Table 2 Comparison of the dicarboxylate ligands in compound 1, compound 2 and compound 3
| Compound |
Mixed-dicarboxylate ligands |
| Label |
Shape |
Flexibility |
Coordinated mode |
| 1 |
OBA |
V |
Flexible |
μ2: η1,η0,η1,η1 |
| 2 |
4-HOIP |
V |
Rigid |
μ3: η1,η1,η1,η1 |
| 3 |
BPDC |
Linear |
Rigid |
μ2: η1,η1,η1,η1; μ3: η1,η1,η2,η1; μ3: η1,η1,η1,η1 |
Table 3 Comparison of the configuration of the BPP ligands and frameworks of compound 1, compound 2, and compound 3
| Compound |
BPP ligand |
Frameworks |
| Configuration |
d |
φ |
SBU |
Topology |
Dimension |
| 1 |
TG |
8.41 |
65.09 |
Uni-Cd |
44 |
2D |
| 2 |
TT |
9.99 |
41.28 |
Di-Cd |
65·8 |
3D |
| 3 |
TG |
9.65 |
30.99 |
Tri-Cd |
36·418·53·6 |
3D |
The coordination motifs of the three aromatic dicarboxylate ligands are shown in Scheme 1. The flexibility and the shape of the three ligands as well as the coordination modes of the carboxyl groups are listed in Table 2. Even if OBA and 4-HOIP possess a similar V-shape architecture, the carboxyl groups in the two ligands adopt different coordination modes. The coordination mode of OBA affords μ2: η1,η0,η1,η1 (μn: the bridging mode of the carboxylate ligands; ηn: the coordination mode of oxygen atoms of carboxyl groups), which includes one chelating-bidentate and one monodentate carboxyl group. Owing to the oxygen atom of the ether group in OBA, the free torsion occurs between the two aromatic carboxyl groups, which leads to one of the carboxyl groups adopting a monodentate mode generating the uni-nuclear SBU. The coordination mode of 4-HOIP affords μ3: η1,η1,η1,η1, which includes one chelating-bidentate and one bidentate carboxyl groups. The bidentate carboxyl group of 4-HOIP instead of the corresponding monodentate carboxyl group of OBA leads to di-nuclear SBU. Then the uni- or di-nuclear SBUs are further linked by the carboxylate ligands to form the corresponding zig-zag or ladder-like chains, respectively, in compound 1 and compound 2 (Fig. 1a and b and 3a and b). As a comparison, BPDC can be commonly considered as a linear and rigid ligand. Thus, the relatively long distance, the rigidity and the various coordination modes of the carboxyl groups (μ2: η1,η1,η1,η1; μ3: η1,η1,η2,η1; μ3: η1,η1,η1,η1) facilitate the generation of the tri-nuclear Cd(II) unit, then further form a 2D (3, 6) plane along the [10−1] plane (Fig. 5 and 6a).
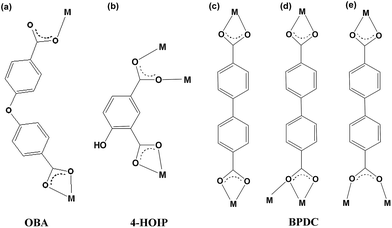 |
| | Scheme 1 The coordination modes of the three dicarboxylate ligands in (a) compound 1, (b) compound 2 and (c–e) compound 3. | |
In addition to the carboxylate ligands, the N-donor ligand also plays a crucial role in the construction of the final framework. Generally, besides the classification of T/G configuration, the conformation of the BPP ligand can be estimated in more details with two factors: (i) the dihedral angle of the two pyridyl rings, named ϕ; and (ii) the N–N distance, defined as d. The T/G configurations of all the bidentate BPP ligands in the three compounds are shown in Scheme 2 and Fig. S4† while ϕ, d values are listed in Table 3. It is not difficult to find that the BPP ligand adopts the more flexible TG mode with ϕ of 65.09° and d of 8.41 Å in compound 1. The twisted conformation of BPP combined with the monodentate carboxyl group of OBA lead to the tendency to form planar 4-connected nodes. It offers an opportunity to the O donor of water to occupy one coordination position furnishing the octahedral coordination mode of Cd(II) in uni-nuclear SBU. In compound 2, two N donors of two parallel BPP ligands occupy the axial position of the di-nuclear Cd(II) SBU. Compared to the flexible conformation of BPP in compound 1, BPP ligands adopt the TT configuration with ϕ of 41.26° and d of 9.99 Å, which may be more in favor of inter-paralleling. Simultaneously, just due to TT configuration and the torsions of the two pyridyls, the two ladder-like chains joined by the same BPP are almost vertical, resulting in a distorted cds topological structure. Although the BPP ligands in compound 3 adopt the TG configuration as in compound 1, the stretch of the propyl group (–CH2–CH2–CH2–) makes the BPP ligands longer (d = 9.65 Å), which is approximately equal to the space of the two carboxyl groups of BPDC. Therefore, this makes it possible to generate the two kinds of pillars alternately supporting the (3, 6) planes in the same layer to form an 8-connected hexagonal primitive framework.
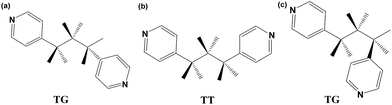 |
| | Scheme 2 The T/G conformations of the BPP ligands in (a) compound 1, (b) compound 2, and (c) compound 3. | |
The three structures in this work clearly reveal that the shape and the flexible abilities of carboxylate ligands, the position and the coordination modes of the carboxyl groups, as well as the configuration of the N-donor ligand, all have a great influence on the target structures. The multi-coordination modes are in favor of the formation of multinuclear SBUs, which are the nodes of the network that profoundly affect the whole topology. Multinuclear SBUs consequently facilitate construct multidimensional frameworks.
3.3 Thermogravimetric analysis
The TGA curve of compound 1 exhibits the first weight loss of 14.0% from 35 to 90 °C which is attributed to the releasing of free DMF and H2O molecules (calc. 13.5%). The second weight loss of 2.9% from 90 to 260 °C corresponds to the dissociation of coordinated H2O (calc. 2.6%). When the temperature is above 260 °C, compound 1 decomposes and the remaining weight of 19.1% corresponds to the percentage of Cd and O components, CdO (calc. 19.0%).
For compound 2, the TGA curve indicates that the first weight loss of 12.9% from 35 to 135 °C corresponds to DMF and H2O free molecules (calc. 12.9%). Then, the structure is stable to 270 °C. When the temperature is above 270 °C, compound 2 decomposes and the remaining weight of 23.2% corresponds to the percentage of Cd and O components, CdO (calc. 22.9%).
For compound 3, the TGA curve indicates that the first weight loss of 5.7% from 35 to 140 °C is attributed to decomposition of all free H2O molecules (calc. 5.7%). The second weight loss of 10.1% from 140 to 260 °C corresponds to the dissociation of one coordinated DMF molecule and BPP (calc. 9.5%). The weight loss of the third step of 11.5% from 260 to 400 °C corresponds to the removal of another coordinated DMF molecule and one BPDC (calc. 11.3%). Above 400 °C, compound 3 decomposes completely, and the remaining weight of 26.9% is in agreement with the percentage of Cd and O components, CdO (calc. 26.9%).
3.4 Luminescent properties
MOFs that contain d10 metal centers are frequently reported to exhibit dramatic luminescent properties, thereby to be promising candidates for photoactive materials with potential applications in chemical sensors, photochemistry, and electroluminescent displays.34–36 The solid-state photoluminescence spectra of the compounds have been investigated at room temperature. It can be observed that the intense emission occurred at 396 nm, 404 nm and 369 nm for compound 1, compound 2 and compound 3 distinctively when the compounds are excited at 330 nm, 318 nm and 308 nm (Fig. 8b–d). Since Cd(II) is difficult to oxidize or reduce due to its d10 configuration, the emissions of the compounds are essentially ligand-based, neither metal-to-ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT).40–42 To analyze the fluorescence properties of the compounds, the solid-state emission spectra of H2OBA, 4-H3OIP, H2BPDC and the pure BPP ligand were measured. Although the free carboxylate ligands, H2OBA, 4-H3OIP and H2BPDC can also exhibit fluorescence at room temperature (Fig. S5†), the emission resulting from the π* → n transition is relatively weak compared to that from the π* → π transition of BPP. With the addition of fluorescence quenching due to the strong electron withdrawing carboxyl group, the carboxylate ligands almost have no contribution to the fluorescent emission of the compounds.37–39 Therefore, the emission band of the compounds may be attributed to the intra-ligand or inter-ligand charge transfer of the BPP ligand. The emission spectra for the compounds exhibit different blue shifts with respect to the emission peak of the free BPP ligand (455 nm excited at 345 nm, as shown in Fig. 8a), which may be ascribed to the entirely distinctive coordination environment of the BPP ligand.39,43 Furthermore, an obvious blue shift of compound 3 relative to compound 1 and compound 2 may be due to the degree of π-overlap of the organic chromophore. The BPP ligands are well separated and have minimal cofacial distances in low-density 3D compound 3 compared to the other two, leading to minimal inter-chromophore interactions, and then a consequence of the obvious blue shift.44 Though more detailed theoretical and spectroscopic studies should be performed in order to better understand the luminescent mechanism, the strong fluorescence emissions of those compounds suggest that they may be candidates for potential photoactive applications.
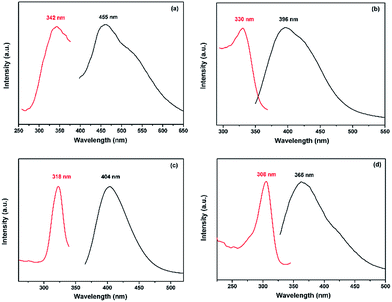 |
| | Fig. 8 Solid-state photoluminescence spectra of (a) free BPP, (b) compound 1, (c) compound 2, and (d) compound 3 at room temperature (excitation spectrum, red/left; emission spectrum, black/right). | |
4. Conclusions
In summary, we have successfully synthesized three new Cd(II) MOFs prepared from the flexible ligand BPP in the presence of three distinct aromatic dicarboxylate ligands. The systematic variation of the carboxylate ligands leads to various structures based on uni-, di-, and tri-nuclear SBUs, and from 2D to 3D frameworks. The diverse crystal structures indicate the crucial influence of the organic ligands on the synthesis of the target frameworks. The successful preparation of the three different MOFs provides a meaningful approach for the construction of other coordination polymers with diverse structures under different synthesis conditions. Simultaneously, this work proves again that common organic linkers can be employed to get new types of structures. The successful achievement of compound 3 indicates for the first time that not only can two different kinds of pillars be involved in the same framework, but they can also alternately arrange in the same layer to support the 2D planes, which constructed a special anionic 3D framework. Finally, the strong fluorescence properties of those compounds indicate that they may be suitable as candidates for potential photoactive materials.
Acknowledgements
The project was supported by the National Natural Science Foundation of China (51502021) and the Special Fund for Basic Scientific Research of Central Colleges, China (2014GB11091).
Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- D. M. Chen, J. Y. Tian, M. Chen, C. S. Liu and M. Du, ACS Appl. Mater. Interfaces, 2016, 8, 18043 CAS.
- M. Du, C. P. Li, M. Chen, Z. W. Ge, X. Wang, L. Wang and C. S. Liu, J. Am. Chem. Soc., 2014, 136, 10906 CrossRef CAS PubMed.
- D. M. Chen, J. Y. Tian, C. S. Liu and M. Du, Chem. Commun., 2016, 52, 8413 RSC.
- B. S. Zheng, J. F. Bai, J. G. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- Q. Chen, J. B. Lin, W. Xue, M. H. Zeng and X. M. Chen, Inorg. Chem., 2011, 50, 2321 CrossRef CAS PubMed.
- S. Shimomura, N. Yanai, R. Matsuda and S. Kitagawa, Inorg. Chem., 2011, 50, 172 CrossRef CAS PubMed.
- Y. Zhang, T. Wu, R. Liu, T. Dou, X. H. Bu and P. Y. Feng, Cryst. Growth Des., 2010, 10, 2047 CAS.
- Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- J. Zhang, W. B. Yang, X. Y. Wu, L. Zhang and C. Z. Lu, Cryst. Growth Des., 2016, 16, 475 CAS.
- M. O'Keeffe, M. Eddaoudi, H. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 1 CrossRef.
- N. Ockwig, O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS PubMed.
- M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343 CrossRef CAS PubMed.
- C. Wang and W. B. Lin, J. Am. Chem. Soc., 2011, 133, 4232 CrossRef CAS PubMed.
- J. Q. Liu, Y. Y. Wang and Y. S. Huang, CrystEngComm, 2011, 13, 3733 RSC.
- M. Xue, Z. J. Zhang, S. C. Xiang, Z. Jin, C. D. Liang, G. S. Zhu, S. L. Qiu and B. L. Chen, J. Mater. Chem., 2010, 20, 3984 RSC.
- C. Wang and W. B. Lin, J. Am. Chem. Soc., 2011, 133, 4232 CrossRef CAS PubMed.
- H. Q. Hao, J. Wang, W. T. Liu and M. L. Tong, CrystEngComm, 2008, 10, 1454 RSC.
- J. Zhang, E. Chew, S. M. Chen, J. T. H. Pham and X. H. Bu, Inorg. Chem., 2008, 47, 3495 CrossRef CAS PubMed.
- A. J. Hernández-Maldonado, R. R. Arrieta-Pérez, J. N. Primera-Pedrozo and J. Exley, Cryst. Growth Des., 2015, 15, 4123 Search PubMed.
- G. G. Luo, H. B. Xiong and J. C. Dai, Cryst. Growth Des., 2011, 11, 507 CAS.
- L. F. Ma, L. Y. Wang, D. H. Lu, S. R. Batten and J. G. Wang, Cryst. Growth Des., 2009, 9, 1741 CAS.
- SHELXTL, Version 5.1, Siemens Industrial Automation, Inc., 1997 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Coordination for Area Detector Data, University of Gottingen, Gottingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL Version 5.1 Software Reference Manual, Bruker AXS. Inc., Madison, WI, 1997 Search PubMed.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Son, Chichester, 2000 Search PubMed.
- A. F. Wells, Further studies of three-dimensional nets, American Crystallographic Association (distributed by Polycrystal Book Service, Pittsburgh, PA), New York, 1979 Search PubMed.
- A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117 CrossRef CAS.
- O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 22 CrossRef.
- Q. Wu, M. Esteghamatian, N. X. Hu, Z. Popovic, G. Enright, Y. Tao, M. D'Iorio and S. Wang, Chem. Mater., 2000, 12, 79 CrossRef CAS.
- J. E. McGarrah, Y. J. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510 CrossRef CAS PubMed.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- L. L. Wen, D. B. Dang, C. Y. Duan, Y. Z. Li, Z. F. Tian and Q. J. Meng, Inorg. Chem., 2005, 44, 7161 CrossRef CAS PubMed.
- J. G. Lin, S. Q. Zang, Z. F. Tian, Y. Z. Li, Y. Y. Xu, H. Z. Zhu and Q. J. Meng, CrystEngComm, 2007, 9, 915 RSC.
- C. Ping, Z. Chen, D. L. Gao, B. Zhao, W. Shi and P. Cheng, Cryst. Growth Des., 2010, 10, 4370 Search PubMed.
- X. Shi, G. S. Zhu, Q. R. Fang, G. Wu, G. Tian, R. W. Wang, D. L. Zhang, M. Xue and S. L. Qiu, Eur. J. Inorg. Chem., 2004, 1, 185 CrossRef.
- X. Shi, G. S. Zhu, X. H. Wang, G. H. Li, Q. R. Fang, X. J. Zhao, G. Wu, G. Tian, M. Xue, R. W. Wang and S. L. Qiu, Cryst. Growth Des., 2005, 5, 341 CAS.
- H. Y. Bai, J. F. Ma, J. Yang, L. P. Zhang, J. C. Ma and Y. Y. Liu, Cryst. Growth Des., 2010, 10, 1946 CAS.
- X. Y. Yu, L. Ye, X. Zhang, X. B. Cui, J. P. Zhang, J. Q. Xu, Q. Hou and T. G. Wang, Dalton Trans., 2010, 39, 10617 RSC.
- C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information files (CIF), PXRD patterns, TGA curves, the selected bond lengths and angles as well as the extensive figures. CCDC 756103–756105. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26855b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Dongdong Chigan,
Luke Yan and
Huaxin Chen
*,
Dongdong Chigan,
Luke Yan and
Huaxin Chen