
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of boron-doped ordered mesoporous carbon by evaporation induced self-assembly under HCl conditions
Yan Zhang *,
Wenwen Dai,
Yujian Liu and
Binge Ma
*,
Wenwen Dai,
Yujian Liu and
Binge Ma
Key Laboratory of Specially Functional Polymeric Materials and Related Technology, Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, MeiLong Road 130, Shanghai 200237, PR China. E-mail: yzhang@ecust.edu.cn; Fax: +86 21 64252659; Tel: +86 21 64252394
First published on 24th January 2017
Abstract
By using hydrochloric acid (HCl) as an acidity regulator, boron-doped ordered mesoporous carbon (B-OMCs) were synthesized via solvent evaporation induced self-assembly (EISA) method. And the self-assembly mechanism of the B-OMCs under HCl conditions was investigated. Not only low-molecular weight boron-modified phenolic resin (BPF), but also triblock copolymer F127 can be protonated by HCl, which results in enhancement of the interaction between precursor and template. Both double-layer hydrogen bonding and electrostatic Coulomb forces act as the driving force for self-assembly of B-OMCs under HCl conditions. In addition, the effect of HCl content on the mesostructure and character of the B-OMCs was studied. With the increase of HCl content, the pore size of B-OMCs decreases, while the surface area, pore order and boron content of B-OMCs increase initially, and then drop off gradually. When pH = 4, the obtained B-OMC has a well-ordered mesoporous structure, highest surface area (690 m2 g−1) and boron content (1.96 wt%). Besides, it possesses excellent electrochemical and capacitance performance (200 F g−1).
1. Introduction
Ordered mesoporous carbons (OMCs) possess excellent physico-chemical properties and potential applications in many fields.1–4 Heteroatoms such as boron, nitrogen, sulfur and phosphorus5–8 have been introduced in the framework of OMCs for higher performance and broader applications ranging from catalysis to hydrogen and energy storage.9–12 Among them, boron-doped OMCs (B-OMCs) have aroused much interest in recent years. Boron bearing three valence electrons can be doped into a carbon lattice as an electron acceptor. And the B–C bonds in the backbone of carbon can also lower the Fermi level of the structure and then tune the properties of oxygen chemisorption and electrochemistry for redox reactions.13–16Compared with the nanocasting process using silica as hard template, which is multistep and costly, the self-assembly of resins and block copolymer route is more efficient. Wang17 synthesized B-OMCs by using resoles and boric acid as precursor, amphiphilic triblock copolymer F127 as a template. The obtained B-OMCs have high boron content (1.8 wt%), but worse-ordered mesoporous structure (472 m2 g−1). B-OMCs with boron contents varying from 0.42 to 2.37 wt% were also prepared by combination of soft-templating and hydrothermal approaches, while its BET surface area is only 300–500 m2 g−1.18 Zhai16 prepared B-OMCs by an evaporation induced self-assembly method using boron-modified phenolic resins as carbon precursor, while the maximum boron content is only 0.33 wt% with BET surface area of 500 m2 g−1.
It seems that B-OMCs with both high boron content and ideal order mesoporous structure are not easy to be obtained. Nevertheless, in most potential utilization, especially in supercapacitors, B-OMCs with a high specific surface area, large pore size and well developed mesoporosity are required.19 The choice of resole as a precursor and triblock copolymer as a template is essential for the successful organization of organic–organic mesostructures because resole with a 3D network structure and plenty of hydroxyl groups (–OH) can interact strongly with the PEO blocks of triblock copolymer templates via hydrogen bonds.20 Therefore, the enhanced hydrogen bond is the key to the preferential arrangement of carbon precursors in the selected domains of PEO–PPO–PEO triblock copolymers by EISA method.21 While for BPFs, it is a great challenge because the reactions of boric acid and hydroxymethyl groups of BPFs will greatly weaken hydrogen bond interaction. And poor solubility of BPF in the solvent like ethanol would result in macrophase separation easily during the EISA process, so well-ordered mesostructures is difficult to be achieved. In addition, the understanding of formation mechanism of B-OMCs with well-developed mesoporosity is still in early stage.
The presence of acids can protonate both boron-modified phenolic resin and F127, so strengthen the hydrogen bonding interactions between soft-template and resins.22 And the solubility of BPF resins can be improved by introducing acid. In addition, phenolic resole resins can be highly reactive when mixed with strong acids and result in severe exothermic reactions. Therefore, acid can promote the curing condensation of phenolic resins. While on the other hand, the reactivity of a catalyzed resin can be so rapid that the processing becomes virtually impossible to control.23
In the study, HCl was selected as acidity and self-assembly regulator. The effect of HCl content on the mesostructure and character of B-OMCs was investigated. And hydrogen bonding interaction between BPFs and F127 was traced by FT-IR. A strategy of both double-layer hydrogen bonding and electrostatic Coulomb forces mechanism for the self-assembly of boron-doped ordered mesoporous carbon under HCl conditions has been deduced.
2. Experimental
2.1 Chemicals
Poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) triblock copolymer (EO106PO70EO106, Pluronic F127) with an average molecular weight of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 was provided by Sigma-Aldrich. Phenol, formaldehyde (HCHO, 37 wt%), boric acid, hydrochloric acid (HCl, 35–38 wt%) were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without any further purification. Deionized water was used in all experiments.
600 was provided by Sigma-Aldrich. Phenol, formaldehyde (HCHO, 37 wt%), boric acid, hydrochloric acid (HCl, 35–38 wt%) were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without any further purification. Deionized water was used in all experiments.
2.2 Synthesis of boron-modified phenolic resins
Boron-modified phenolic resins were synthesized following the procedure reported.24 And the schematic illustration is shown in Scheme 1. A low-molecular-weight boron-modified phenolic resin (BPF) was synthesized by mixing phenol, formaldehyde (37 wt%) and sodium hydroxide aqueous solution together in flask, and stirring for 60 min with the molar ratio of phenol and formaldehyde of 1:1.5. Water was removed under vacuum, boric acid solution was added to the mixture solution and kept for 30 min, then water was removed under vacuum again. The final product was dissolved in ethanol for further use. The molecular weight (Mn) of obtained BPF is 1132 g mol−1 according to GPC analysis.
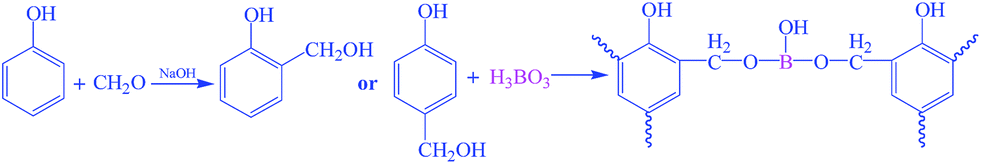 | ||
| Scheme 1 Synthesis of boron modified phenolic resins. | ||
2.3 Synthesis of boron-doped mesoporous carbons (B-OMCs)
B-OMCs were prepared via EISA method25 by using boron-modified phenolic resins obtained above as precursors.26 The poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) triblock copolymer (Pluronic F127) was dissolved in ethanol. BPF resins show low solubility in ethanol, which are not suitable for self-assembly. After adding a small amount of HCl, the hazy BPF solution became clear. Then it was added during stirring to form a homogeneous solution with HCl as the acidity and self-assembly regulator. The mixed solution was poured into the Petri dish and evaporated overnight at room temperature. Then, the films were annealed in an oven at 100 °C for 24 h to crosslink boron-modified phenolic resin. Thermal treatment was performed with flowing nitrogen in the tube furnace at a heating rate of 1 °C min−1 and kept for 2 h at 600 °C. The obtained products were donated as B-OMC-x, where x indicates pH value of the system.2.4 Characterization
Fourier transform infrared (FT-IR) spectra were collected on a Nicolet 5700 spectrometer. X-ray photoelectron spectroscopy (XPS) was conducted on ESCALAB 250 equipped with Al-Kα radiation. The X-ray diffraction (XRD) patterns were achieved on a Rigaku D/max-2550 diffractometer using Cu-Kα radiation. The Raman spectra were collected using a Renishaw inVia Reflex system at an excitation wave length of 514.5 nm. Nitrogen adsorption/desorption isotherms were measured at 77 K on an ASAP 2020 Micromeritics Instrument. The BET surface areas of samples were calculated from the adsorption data using the Brunauer–Emmett–Teller (BET) method. The pore size distributions were derived from the adsorption branches of the isotherms using the BJH (Barrett–Joyner–Halenda) method. TEM images were taken with a JEM-1400 transmission electron microscope operated at 200 keV.2.5 Electrochemical measurements
For preparing a working electrode, a mixture of an active material, acetylene black and poly(tetrafluoroethylene) (PTFE) with a weight ratio of 85:10:5 was homogenized in a mortar and pestle. Then the mixture was rolled into a thin film of uniform thickness (ca. 60 μm), and punched into pellets with a diameter of 1 cm. Each active material was dried overnight at 100 °C, and the electrodes were impregnated with electrolyte under vacuum for several hours prior to the electrochemical tests. The tests were carried out in a three-electrode system (platinum electrode for the auxiliary electrode, saturated calomel electrode for the reference electrode, and B-OMC for the working electrode), which was constructed with two facing carbon electrodes and sandwiched with a separator and a 3 M H2SO4 solution as the electrolyte.
The cyclic voltammetry (CV) tests were conducted using a PARSTAT-2273 (USA) in a potential range of 0–0.9 V. The galvanostatic charge–discharge (GC) measurements were conducted on the PARSTAT-2273 apparatus. The gravimetric capacitance of electrode material was calculated according to the equation Cm = IΔt/(mΔU), where I is the discharge current in ampere, Δt is the discharge time in second, m is the mass of active material on an electrode in gram, and ΔU is the working voltage in volt. In some cases, Cs is presented as F m−2, which is obtained according to the equation Cs = Cm/SBET.
3. Results and discussion
3.1 Chemical state of B-OMCs
The FT-IR spectra of BPF and B-OMC are displayed in Fig. 1. The skeleton vibration absorption of carbon–carbon double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) of benzene rings appears in the regions of 1500–1650 cm−1. And the peaks at 881 cm−1, 830 cm−1, 755 cm−1 are assigned to the C–H flexure of benzene rings. The absorption band at 1396 cm−1 corresponds to B–O bond.27 After carbonization, the bands at 1100 cm−1 (CH2OH), 1450 cm−1 (–CH2–) and 2900 cm−1 (–CH2) almost disappear, indicating resole decomposition and small molecular volatilization. However, the B–O absorption band is still observed in B-OMCs, suggesting that boron has been introduced into the backbone of the phenolic resin and well preserved after calcination at 600 °C.
C) of benzene rings appears in the regions of 1500–1650 cm−1. And the peaks at 881 cm−1, 830 cm−1, 755 cm−1 are assigned to the C–H flexure of benzene rings. The absorption band at 1396 cm−1 corresponds to B–O bond.27 After carbonization, the bands at 1100 cm−1 (CH2OH), 1450 cm−1 (–CH2–) and 2900 cm−1 (–CH2) almost disappear, indicating resole decomposition and small molecular volatilization. However, the B–O absorption band is still observed in B-OMCs, suggesting that boron has been introduced into the backbone of the phenolic resin and well preserved after calcination at 600 °C.
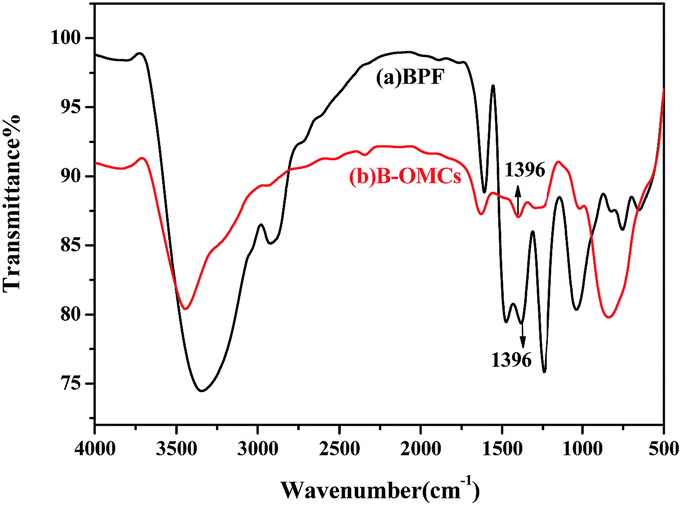 | ||
| Fig. 1 Spectra of BPF resin (a) and B-OMCs (b). | ||
The precise formation of B-OMCs was further analyzed by X-ray photoelectron spectroscopy (XPS), and the results are shown in Fig. 2 and Table 1. XPS survey spectrum confirms that the sample contains boron, carbon and oxygen, and B atoms have been successfully doped into the framework of OMCs. The B 1s spectrum for B-OMC can be fitted into three peaks located at 188.5, 191.6 and 192.4 eV, as shown in Fig. 2b. The B 1s with a binding energy of 188.5 eV suggests that the boron species are incorporated into the carbon structure.28 And the boron with B 1s at 191.6 eV can be assigned to the mixed B–C and B–O bonding, respectively.28,29 Moreover, the binding energy of B 1s centered at 192.4 eV is attributed to the bonding B–O–C,30 which consists with the structure (–O–B–O–C–) obtained from the three-polymerization reaction of boric acid, phenol and formaldehyde. Boron is introduced into the backbone of the phenolic resins by boric acid reacting with hydroxymethyl.
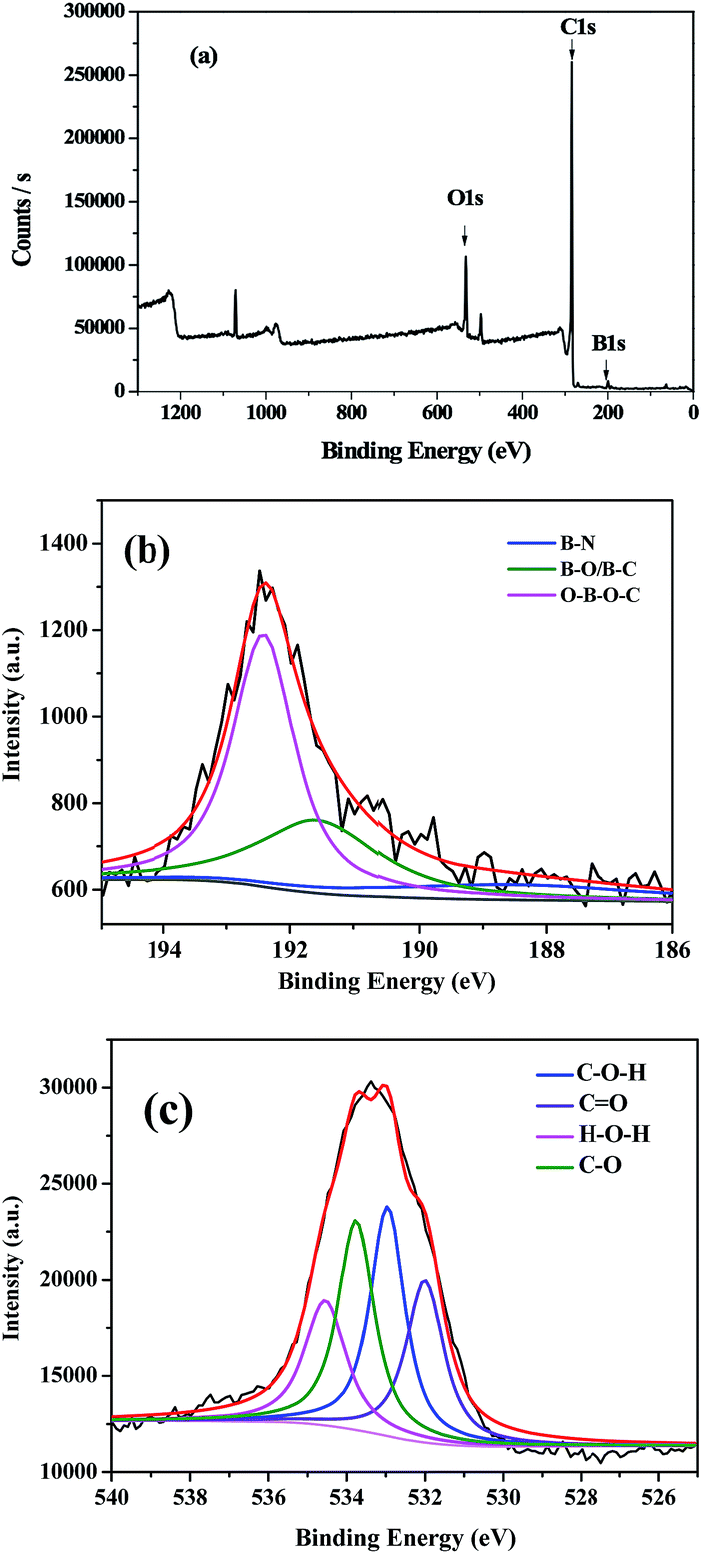 | ||
| Fig. 2 XPS spectra of B-OMC (a), B 1s (b), O 1s (c). | ||
| Element | B.E (eV) | Content (wt%) |
|---|---|---|
| O | 532.0 (CO) |
2.74 |
| 533.0 (C–O) | 3.36 | |
| 533.8 (C–OH) | 2.71 | |
| 534.5 (H2O) | 2.60 | |
| Total content | — | 10.40 |
| B | 188.5 (B–N) | 0.29 |
| 191.6 (B–C, B–O) | 0.59 | |
| 192.4 (B–O–C) | 1.08 | |
| Total content | — | 1.96 |
In addition, the O 1s spectrum for B-OMC, as shown in Fig. 2c, can be grouped into four peaks attributed to the bond of CO (532.0 eV), C–O (533.0 eV), C–OH (533.8 eV) and H2O (534.5 eV) respectively.31 Total oxygen group of B-OMCs reaches 10.40 wt%, much higher than that of undoped OMCs (4.3 wt%).27 The enriched oxygen groups on the carbon surface will be beneficial to the capacitance retention at a high scan rate.32
3.2 Textural and structural characterization
The porous structure of obtained B-OMCs with different HCl addition is further revealed by small-angle XRD (SAXRD) patterns, as displayed in Fig. 3. For samples of B-OMC-2, B-OMC-3, B-OMC-4, the distinct diffraction peaks at around 2θ = 1° imply the uniform and even ordered mesoporous structures. Moreover, with the pH value decreasing, the (100) peak of samples slightly shifts to a higher angle (from 0.86° to 0.93°), indicating the shrinkage of the mesoporous channels. However, for B-OMC-5, the XRD patterns disappear, suggesting the loss of long range order in the mesostructure.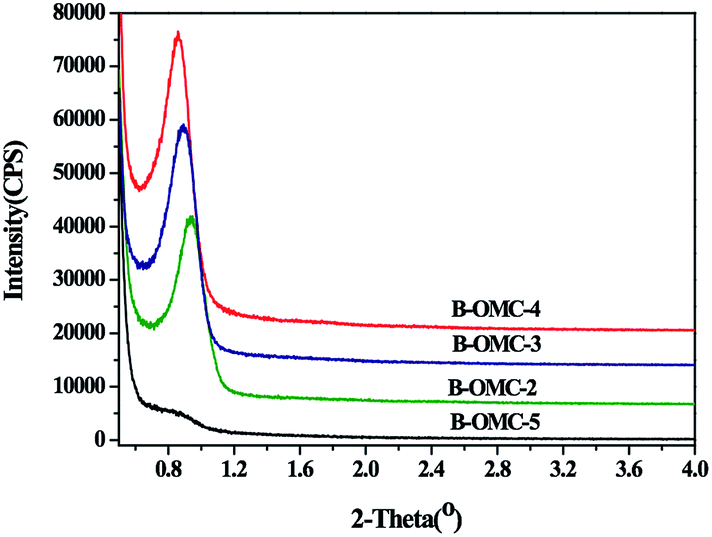 | ||
| Fig. 3 Small-angel XRD patterns of B-OMCs prepared by using F127 as a template via the EISA method under the different pH and calcined at 600 °C under N2. | ||
Wide-angle XRD and Raman spectra of B-OMC-4 are shown in Fig. 4. Two broad maxima, corresponding to (002) and (101) reflections of a turbostratic carbon structure, are observed at 2θ angles about 22.5° and 43.5° (Fig. 4a). In addition, there exist two main Raman bands (Fig. 4b) at about 1358 cm−1 (D band) and 1598 cm−1 (G band), respectively in the plot. The former, corresponding to the E2g mode of graphite, is assigned to the “in-plane” displacement of the carbons strongly coupled in the hexagonal sheets. The latter, corresponding to the defect-induced Raman band, are ascribed to the defects and hexagonal graphene plane.33 The intensity ratio of the D to G bands (ID/IG) is 2.41, which is proportional to the degree of structural order with respect to the graphite structure. The results of WXRD and Raman spectra indicate a low graphitic crystallinity of B-OMCs, because the temperature of 600 °C is still too low for phenolic resin graphitization. However, this value is much lower than that of carbonized products of the unmodified phenolic resins (ID/IG = 2.85).34 It demonstrates an increase of graphite structure and a decrease of disordered structure after introducing boron into the backbone of the resin.
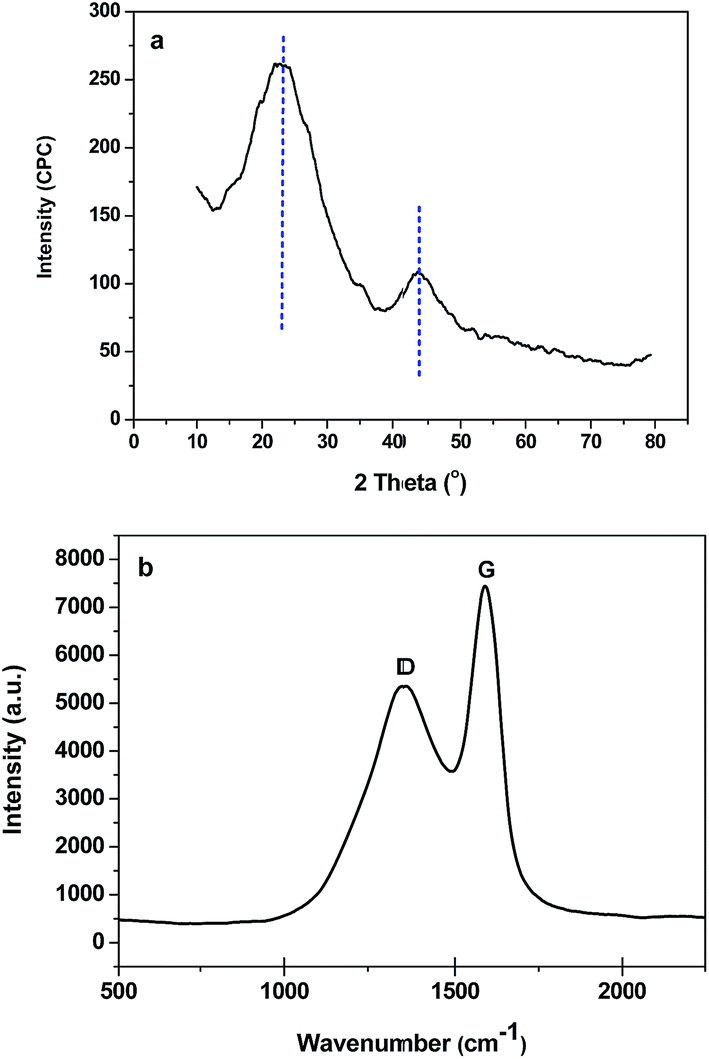 | ||
| Fig. 4 Wide-angle XRD pattern (a), and Raman spectrum (b) of the B-OMC-4 prepared by using F127 as a template via the EISA method under pH = 4 and calcined at 600 °C under N2. | ||
N2 adsorption–desorption isotherms of all samples are presented in Fig. 5. The structural properties and B content of obtained B-OMCs are listed in Table 2. All samples except B-OMC-5 display typical-IV isotherms with well-defined H1 hysteresis loops and clear capillary condensation steps, implying a uniform mesoporous structure. The B-OMC-5 also has typical type IV isotherms. Nevertheless, the distinct capillary condensation steps occur at relative pressures of 0.3–0.8, indicating that the pore size distribution is not very concentrated. Moreover, with the increment of HCl, BET surface area and microporous volume of B-OMCs increase first, then decrease gradually, and the pore diameter reduces from 5.6 nm to 4.5 nm, which consists with the results of XRD. It demonstrated that a little addition of HCl tends to help resins form ordered micelles, while too much acid may make resole resins more reactive and result in severe exothermic reactions, which would interfere self-assembly process. So the BET surface area and pore size of B-OMCs decrease and the order of B-OMCs gets worse under very strong acid conditions.
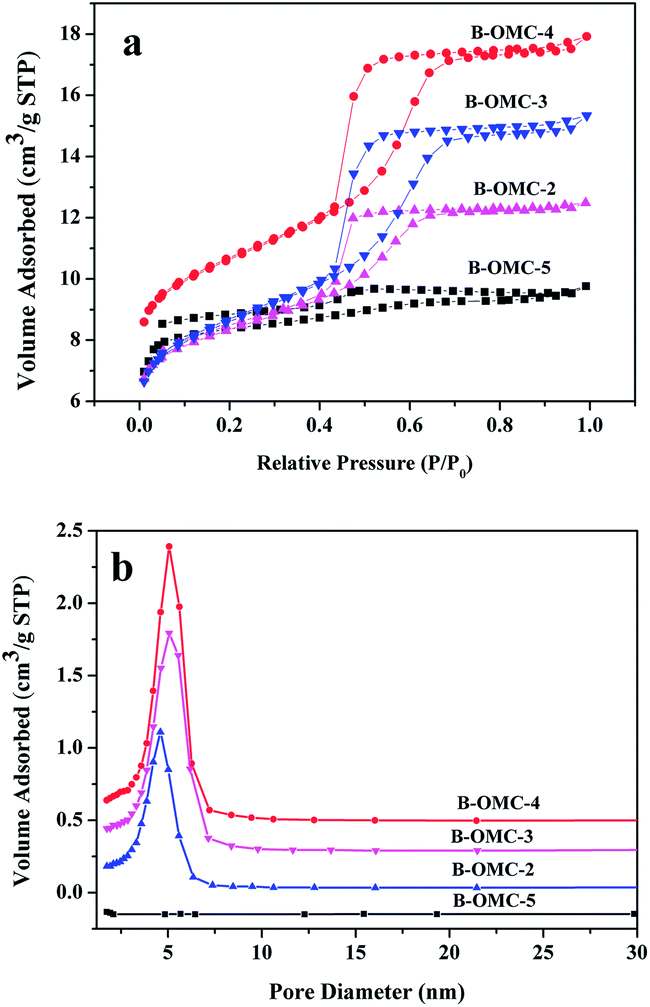 | ||
| Fig. 5 Adsorption–desorption isotherms (a) and pore size distributions (b) of B-OMCs prepared by using F127 as a template via the EISA method under the different pH and calcined at 600 °C under N2. | ||
| Sample | SBET (m2 g−1) | Smic (m2 g−1) | Vpb (m3 g−1) | Vmic (m3 g−1) | Dpa (nm) | Bconb (wt%) |
|---|---|---|---|---|---|---|
| a Pore diameter, calculated from the adsorption branch of the nitrogen isotherm by the Barrett–Joyner–Halenda (BJH) method and the corresponding N2 adsorption and desorption isotherms of B-OMCs.b Determined by XPS. | ||||||
| B-OMC-5 | 122 | 116 | 0.06 | 0.06 | 5.6 | 1.69 |
| B-OMC-4 | 690 | 338 | 0.49 | 0.18 | 5.0 | 1.96 |
| B-OMC-3 | 678 | 345 | 0.38 | 0.18 | 4.6 | 1.86 |
| B-OMC-2 | 589 | 327 | 0.34 | 0.17 | 4.5 | 1.86 |
The addition of HCl can also improve the solubility of resoles in ethanol, so more boron modified phenolic resins will take part in the self-assembly process. Therefore, the boron content of OMCs increases with the addition of hydrochloric acid. By further adding HCl, owing to fast condensation rate, the viscosity of system grow quickly and a very small part of resins with too high molecular weight may precipitate from the solvent, which is not suitable for self-assembly any more. Therefore, the boron content of B-OMCs dropped a little when the pH value reaches 3.
TEM images of B-OMCs are illustrated in Fig. 6. B-OMC-2, B-OMC-3 and B-OMC-4 show well-ordered hexagonal arrangement of mesopores with long straight channels along both the [001] and [110] directions, consistent with a hexagonal mesostructured. The order degree of B-OMC-3 and B-OMC-2 seem to be inferior to that of B-OMC-4. Besides, B-OMC-5 exhibits a wormy structure and its spatial distribution regularity is not as good as that of other samples.
 | ||
| Fig. 6 TEM images of B-OMC-5 (a), B-OMC-4 (b), B-OMC-3 (c) and B-OMC-2 (d) prepared by using F127 as a template via the EISA method under the different pH and calcined at 600 °C under N2. | ||
3.3 Hydrogen bond interaction
Hydrogen bonding between the amphiphilic surfactants and organic polymer frameworks is a very important driving force for the formation of ordered mesostructure. While for boron modified phenolic resins, this interaction is greatly weakened because the reactions of boric acid and hydroxymethyl (–CH2OH) groups of BPFs will lower hydroxyl (–OH) density. It would further result in the reduction of miscibility between organic frameworks and surfactants after polymerization and consequent macroscopic phase separation.35 Both low-molecular-weight boron-modified phenolic resin (BPF) and F127 can be protonated by acid. In order to strengthen interaction force, hydrochloric acid was selected as acidity regulator and the influence of HCl on hydrogen bonding interaction between BPF and F127 was studied by using FTIR spectroscopy.FT-IR spectra belong to a high band in regions from 3600 to 3000 cm−1, where the hydroxyl stretching vibration can be observed. Two unresolved bands (Fig. 7a) correspond to free OH groups at 3527 cm−1 and hydrogen-bonded OH groups (self-association) at 3329 cm−1, respectively.36 And bands at 1113 and 2872 cm−1 can be assigned to the C–O and C–H stretching of copolymer F127. When BPF resin was blended with F127, the intensity of free OH groups decreased. And with the increasing of HCl, the intensity dropped down gradually. At the same time, the absorption at 3329 cm−1 became even wider and shifted to lower wavenumber (3253 cm−1). The band at 1113 cm−1 in Fig. 7b, corresponding to the C–O–C absorptions of free PPO and PEO block37 also shifted to lower wavenumber (1106 cm−1) slightly. It indicates that the hydrogen bonding interaction between OH groups of the phenolic resins and the ether groups of PEO blocks increases with the addition of HCl.
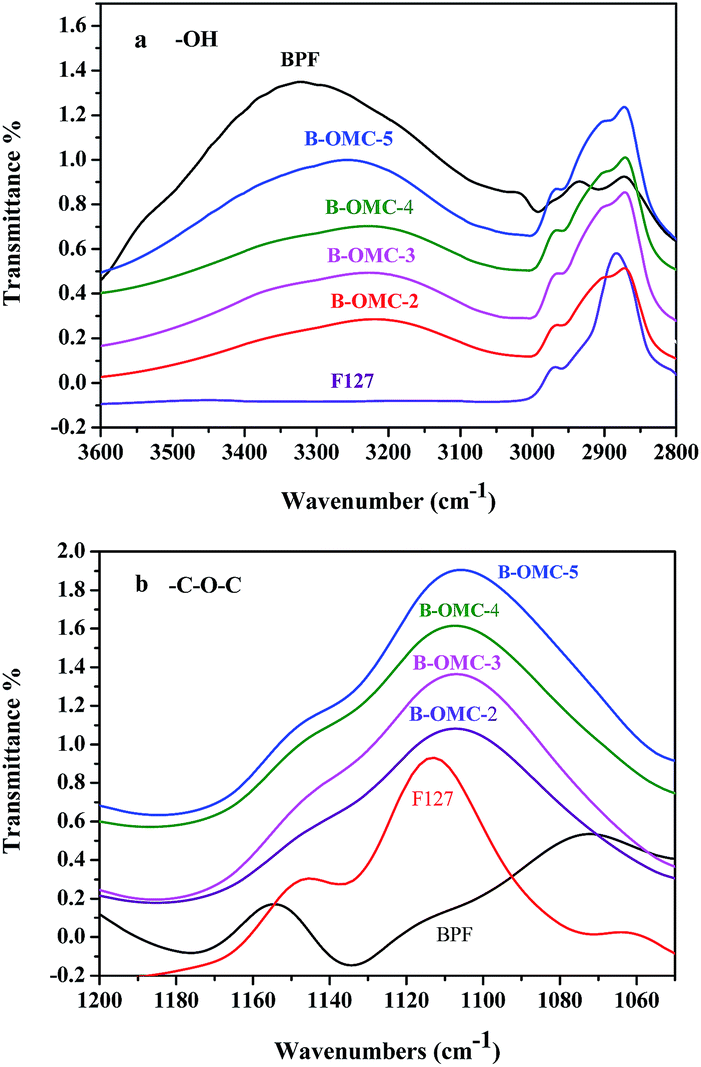 | ||
| Fig. 7 FT-IR spectra of BPF resin/F127 mixed system under different pH conditions (pH = 5, pH = 4, pH = 3, pH = 2), (a) OH stretching, (b) CO stretching. | ||
3.4 Self-assembly mechanism
Both boron-modified phenolic resin and EO blocks of F127 will be protonated by the strong acid, HCl.38 The oxygen in C–O–C bond of F127 is protonic by H+, which produces strong hydrogen bond with –OH of boron-modified phenolic resins. Meanwhile, same as the synthesis of mesoporous silica under acid media conditions, the EO moieties of the surfactant in strong acid media will associate with hydronium ions.39 These charge-associated EO unities and cationic phenolic resins are assembled together by a combination of electrostatic, enhanced hydrogen bonding, and van der Waals interactions, which can be designated as (S0H+)(X−I+).40 Hence, HCl is a very effective self-assembly regulator, which is used to help mesoporous carbons obtain order structure in some researches.41–45 The self-assembly process of BPF and F127 under high HCl concentrations is illustrated in Scheme 2. Therefore, BPF resins, with much less hydromethyl groups than ordinary PF resins can also form order-well mesoporous structure by both coulomb force and hydrogen bonding interaction under hydrogen chloride acid regulator. In addition, the strong hydrogen bond and the Coulomb force interaction will lead to forming small micelle during the self-assembly process. So the pore size of B-OMCs decreases with the increasing of HCl.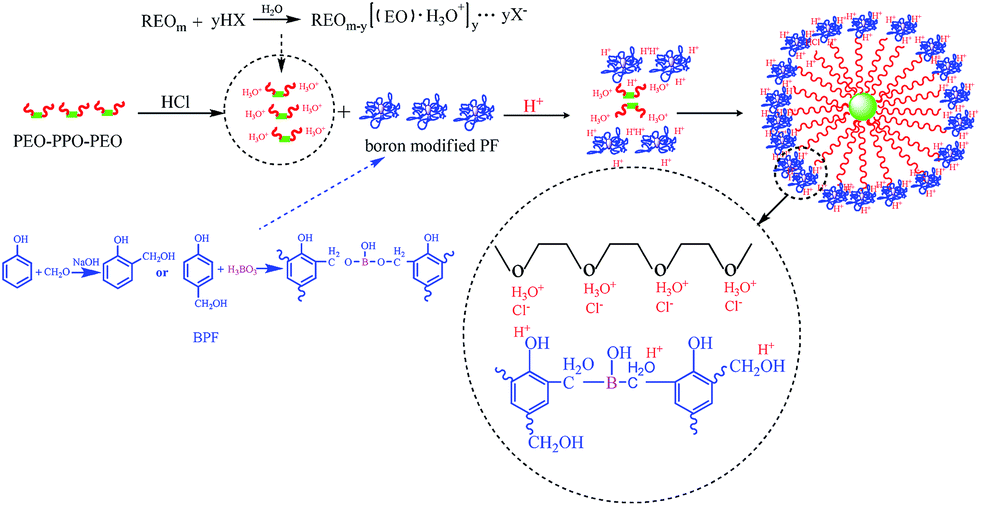 | ||
| Scheme 2 Self-assembly mechanism of BPF and F127 under HCl conditions. | ||
3.5 Electrochemical properties
The B-OMCs synthesized under different pH were subsequently evaluated as the electrodes of super capacitors. Fig. 8 shows the cyclic voltammetry (CV) curves of the B-OMC-x at a scan rate of 5 mV s−1 and the galvanostatic charge–discharge (GC) profile at a current density of 0.2 A g−1. According to Fig. 8a, all samples present well-rectangular CV curves, indicating good electric double layer capacitor (EDLC). With the reduction of pH (from 5 to 2), the current density of B-OMCs changed from 0.44, 1.02, 0.97 to 0.60 A g−1, respectively. When the pH value is 4, the system has the maximum current density, 1.02 A g−1. In addition, it is very important for the electrode materials of the super capacitor to provide high power density. The specific capacitance of the electrode according to GC test at a current of 0.2A g−1 is calculated. It is found that doping boron will lead to an increase in specific capacitance, from 112 F g−1 (OMC) to 200 F g−1 (B-OMC-4), which is also much higher than most of the previous reports of B-OMCs (122–159 F g−1).46–48 It is noted that, the CV curves of B-OMCs show an obvious redox peak around 0.4 V, indicating the increase of capacitance arising from both electric double layer capacitance (EDLC) and pseudocapacitance. Boron atom with three valence electrons can introduce a hole charge carrier by replacing a carbon atom in the grapheme lattice, which can raise the charge density and the density of state, so the double layer capacitance would be enhanced.30 At the same time, the increased oxygen group by boron substitution can also help OMCs obtain a higher redox capacitance.16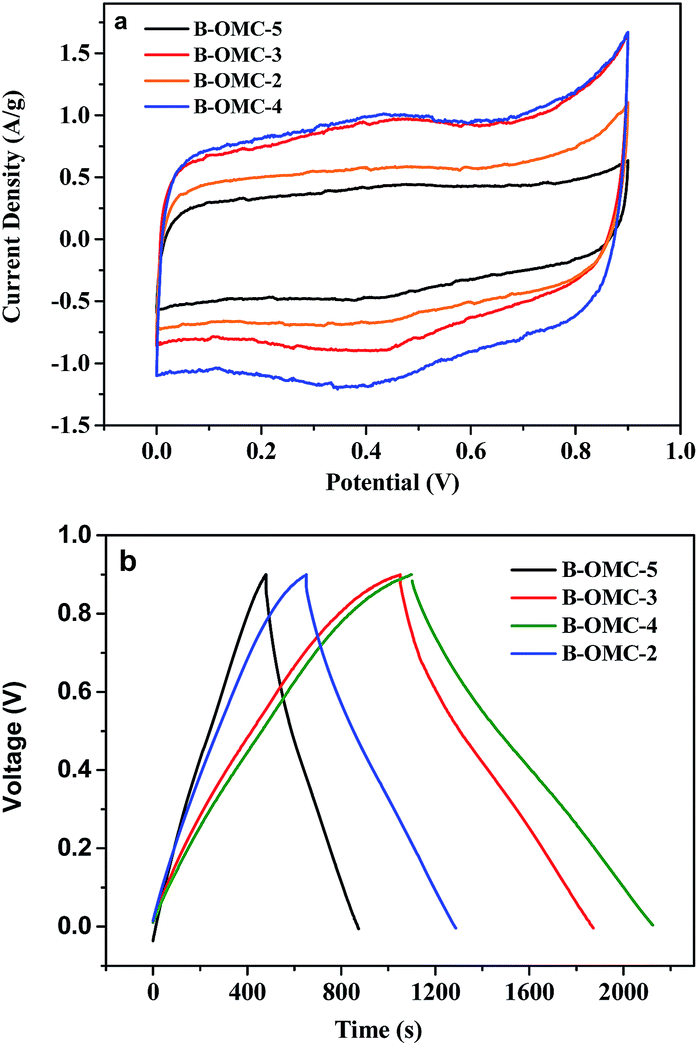 | ||
| Fig. 8 CV curves (a) and GC curves (b) of the B-OMC-4 prepared by using F127 as a template via the EISA method under the different pH and calcined at 600 °C under N2, recorded in 3 M H2SO4 with a scan rate of 5 mV s−1. | ||
4. Conclusions
By using HCl as acidity regulator, boron-doped mesoporous carbon (B-OMCs) were synthesized via solvent evaporation induced self-assembly (EISA) method. The effect of HCl addition on the mesostructure and character of mesoporous carbons were studied. Both double-layer hydrogen bonding and electrostatic Coulomb forces act as the driving force for self-assembly of B-OMCs under HCl conditions. The results show that along with the increment of HCl content, pore size of B-OMCs decreases, while the surface area, pore order and boron content of B-OMCs increases first, and then drop off gradually. When pH = 4, the obtained B-OMC have the well-ordered mesoporous structure, highest surface area (690 m2 g−1) and boron content (1.96 wt%). Besides, it also possesses excellent electrochemical and capacitance performance (200 F g−1).Acknowledgements
The work was supported by the National Natural Science Foundation of China (51303054), National Key Basic Research Program of China (973 Program) (no. 2013CB035505), Shanghai Leading Academic Discipline Project (B502).References
- A. Corma, Chem. Rev., 1997, 97, 2373–2420 CrossRef CAS PubMed.
- K. Ariga, A. Vinu, Q. M. Ji, O. Ohmori, J. P. Hill, S. Acharya, J. Koike and S. Shiratori, Angew. Chem., Int. Ed., 2008, 47, 7254–7257 CrossRef CAS PubMed.
- M. Vallet-Regi, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef CAS PubMed.
- E. Jo, M. C. Lim, H. N. Kim, H. J. Paik, Y. R. Kim and U. Jeong, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 89–95 CrossRef CAS.
- N. Wickramaratne and M. Jaroniec, RSC Adv., 2012, 2, 1877–1883 RSC.
- D. S. Yuan, X. L. Yuan, S. L. Zhou, W. J. Zou and T. X. Zhou, RSC Adv., 2012, 2, 8157–8163 RSC.
- Y. S. Shin, G. Fryxell, W. Y. Um, K. Parker, S. V. Mattigod and R. Skaggs, Adv. Funct. Mater., 2007, 17, 2897–2901 CrossRef CAS.
- R. Li, Z. D. Wei, X. L. Gou and W. Xu, RSC Adv., 2013, 3, 9978–9984 RSC.
- L. J. Yang, S. J. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Z. Wang, Q. Wu, J. Ma, Y. W. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- Y. Jeong and T. C. M. Chung, Carbon, 2010, 48, 2526–2537 CrossRef CAS.
- Y. Y. Shao, X. Q. Wang, M. Engelhard, C. M. Wang, S. Dai, J. Liu, Z. G. Yang and Y. H. Lin, J. Power Sources, 2010, 195, 4375–4379 CrossRef CAS.
- X. C. Zhao, A. Q. Wang, J. W. Yan, G. Q. Sun, L. X. Sun and T. Zhang, Chem. Mater., 2010, 22, 5463–5473 CrossRef CAS.
- Y. Hirshiyama, H. Irumano, Y. Kaburagi and Y. Soneda, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63(24), 303–306 Search PubMed.
- L. R. Radovic, M. Karra, K. Skokova and P. A. Thrower, Carbon, 1998, 36, 1841–1854 CrossRef CAS.
- D. H. Zhong, H. Sano, Y. Uchiyama and K. Kobayashi, Carbon, 2000, 38, 1199–1206 CrossRef CAS.
- X. L. Zhai, Y. Song, J. Q. Liu, P. Li, M. Zhong, C. Ma, H. Q. Wang, Q. G. Guo and L. J. Zhi, J. Electrochem. Soc., 2012, 159, E177–E182 CrossRef CAS.
- T. Wang, C. X. Zhang, X. Sun, Y. X. Guo, H. Guo, J. Tang, H. R. Xue, M. Z. Liu, X. X. Zhang, L. Zhu, Q. Q. Xie and J. P. He, J. Power Sources, 2012, 212, 1–12 CrossRef CAS.
- M. Enterria, M. F. R. Pereira, J. I. Martins and J. L. Figueiredo, Carbon, 2015, 95, 72–83 CrossRef CAS.
- N. Wickramaratne and M. Jaroniec, RSC Adv., 2012, 2, 1877–1883 RSC.
- W. Li, Q. Yue, Y. H. Deng and D. Y. Zhao, Adv. Mater., 2013, 25, 5129–5152 CrossRef CAS PubMed.
- C. D. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS PubMed.
- B. Yang, C. Guo, S. Chen, J. H. Ma, J. Wang, X. F. Liang, L. Zheng and H. Z. Liu, J. Phys. Chem. B, 2006, 110, 23068–23074 CrossRef CAS PubMed.
- K. Hirano and M. Asami, React. Funct. Polym., 2013, 73, 256–269 CrossRef CAS.
- M. O. Abdalla, A. Ludwick and T. Mitchell, Polymer, 2003, 44, 7353–7359 CrossRef CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- J. Song, Y. Zhang and Y. J. Liu, RSC Adv., 2015, 5, 20734–20740 RSC.
- J. Gao, X. Su and L. Xia, Int. J. Polym. Mater., 2005, 54, 949–961 CrossRef CAS.
- S. L. Ding, S. J. Zheng, M. J. Xie, L. M. Peng, X. F. Guo and W. P. Ding, Microporous Mesoporous Mater., 2011, 142, 609–613 CrossRef CAS.
- D. H. Zhong, H. Sano, Y. Uchiyama and K. Kobayashi, Carbon, 2000, 38, 1199–1206 CrossRef CAS.
- D. W. Wang, F. Li, Z. G. Chen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- S. D. Gardner, C. S. K. Singamsetty, G. L. Booth, G. R. He and C. U. Pittman Jr, Carbon, 1995, 33, 587–595 CrossRef CAS.
- T. Kwon, H. Nishihara, H. Itoi, Q. H. Yang and T. Kyotani, Langmuir, 2009, 25, 11961–11968 CrossRef CAS PubMed.
- T. H. Ko, W. S. Kuo and Y. H. Chang, Polym. Compos., 2000, 21, 745–750 CrossRef CAS.
- Y. Zhang, S. Shen and Y. Liu, Polym. Degrad. Stab., 2013, 98, 514–518 CrossRef CAS.
- X. S. Du, Z. Z. Yu, A. Dasari, J. Ma, Y. Z. Meng and Y. W. Mai, Chem. Mater., 2006, 18, 5156–5158 CrossRef CAS.
- S. W. Kuo, C. L. Lin and F. C. Chang, Macromolecules, 2002, 35, 278–285 CrossRef CAS.
- D. W. Wang, F. Li, Z. G. Chen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- C. D. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS PubMed.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- L. D. Huffman, M. M. Rosenblatt and K. S. Suslick, J. Am. Chem. Soc., 1998, 120, 6183–6184 CrossRef.
- R. T. Mayes, C. Tsouris, J. O. Kiggans, S. M. Mahurin, D. W. DePaoli and S. Dai, J. Mater. Chem., 2010, 20, 8674–8678 RSC.
- M. J. Xie, Y. F. Xia, J. Y. Liang, L. H. Chen and X. F. Guo, Microporous Mesoporous Mater., 2014, 197, 237–243 CrossRef CAS.
- M. J. Xie, H. H. Dong, D. D. Zhang, X. F. Guo and W. P. Ding, Carbon, 2011, 49, 2459–2464 CrossRef CAS.
- D. Liu, L. J. Xia, D. Y. Qu, J. H. Lei, Y. Li and B. L. Su, J. Mater. Chem. A, 2013, 1, 15447–15458 CAS.
- C. D. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS PubMed.
- D. W. Wang, F. Li, Z. G. Chen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- X. Zhao, A. Wang, J. Yan, G. Sun, L. Sun and T. Zhang, Chem. Mater., 2010, 22, 5463–5473 CrossRef CAS.
- X. Zhao, Q. Zhang, B. Zhang, C.-M. Chen, J. Xu, A. Wang, D. S. Su and T. Zhang, RSC Adv., 2013, 3, 3578 RSC.
| This journal is © The Royal Society of Chemistry 2017 |