
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atmospheric chemical reaction mechanism and kinetics of 1,2-bis(2,4,6-tribromophenoxy)ethane initiated by OH radical: a computational study†
Qi Yu a,
Hong-Bin Xie*a,
Tianchi Lia,
Fangfang Maa,
Zihao Fua,
Zhongyu Wanga,
Chao Lib,
Zhiqiang Fua,
Deming Xiaa and
Jingwen Chena
a,
Hong-Bin Xie*a,
Tianchi Lia,
Fangfang Maa,
Zihao Fua,
Zhongyu Wanga,
Chao Lib,
Zhiqiang Fua,
Deming Xiaa and
Jingwen Chena
aKey Laboratory of Industrial Ecology and Environmental Engineering, Ministry of Education, School of Environmental Science and Technology, Dalian University of Technology, Dalian 116024, China. E-mail: hbxie@dlut.edu.cn
bState Environmental Protection Key Laboratory of Wetland Ecology and Vegetation Restoration, School of Environment, Northeast Normal University, Changchun 130117, China
First published on 31st January 2017
Abstract
The unexpected diverse effect of alternatives for banned chemicals has stimulated scientific and public concern on their environmental risk. As an alternative of polybrominated diphenyl ethers (PBDEs), 1,2-bis(2,4,6-tribromophenoxy) ethane (BTBPE) is currently one of the most commonly applied novel brominated flame retardants with wide market prospects. Due to its frequent and high detection in the atmosphere, revealing the atmospheric fate of BTBPE is of great significance. Here, the mechanism and kinetics of the ·OH-initiated atmospheric reaction of BTBPE have been investigated by combined quantum chemical calculations and kinetics modeling. The results indicate that ·OH addition and hydrogen abstraction pathways in the initiated reactions, are competitive with a rate constant ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the intermediates formed would react with O2/NO to finally form peroxy radicals and OH-BTBPE which tends to be more toxic. The calculated overall reaction rate constant is 1.0 × 10−12 cm3 per molecule per s, translating into 11.8 days atmospheric lifetime of BTBPE. This clarifies that BTBPE as a substitute for PBDEs still has atmospheric persistence.
:1, and the intermediates formed would react with O2/NO to finally form peroxy radicals and OH-BTBPE which tends to be more toxic. The calculated overall reaction rate constant is 1.0 × 10−12 cm3 per molecule per s, translating into 11.8 days atmospheric lifetime of BTBPE. This clarifies that BTBPE as a substitute for PBDEs still has atmospheric persistence.
1. Introduction
Synthetic chemicals have been considered as significant risk sources to human and ecological health according to the United Nations Environment Programme.1,2 Currently, a series of laws and regulations, such as the Stockholm Convention3 and REACH (Registration, Evaluation, Authorization and Restriction of Chemicals),4 have been set up to restrict or ban the use of some hazardous chemicals. To meet the market demand, alternatives for those banned chemicals have been largely and rapidly introduced into markets, lacking a comprehensive risk assessment. However, if we looked back, it was not hard to find that some alternatives, especially those that have similar electronic structure properties to the banned chemicals, have led to unexpected diverse effects.5 For instance, hydrochlorofluorocarbons, as alternatives of chlorofluorocarbons, have been found to have ozone destruction and global warming effects.6,7 4,4′-Methylenediphenol, 4-hydroxyphenyl sulfone and 4,4′-(hexafluoroisopropylidene)diphenol, as substitutes of bisphenol A (BPA), have shown similar or even greater estrogenic and/or antiandrogenic activities than BPA.5,8 Therefore, to avoid this regrettable substitution, a comprehensive environmental risk assessment for alternatives, especially those that have similar electronic structure properties to the banned chemicals, has to be performed.As an alternative for polybrominated diphenyl ethers (PBDEs),9 1,2-bis(2,4,6-tribromophenoxy)ethane (BTBPE) has similar electronic structure to PBDEs (Fig. 1). BTBPE has been included in the 2007 OECD list of high production volume chemicals and detected in various environmental matrixes, e.g. indoor dust, air, sewage sludge, water, snow pits and even in Arctic.10–25 To probe the environmental risk of BTBPE, studies on the transformation and fate of BTBPE in the environment have recently come out,11,26–28 concerning whether the transformation of BTBPE can form toxic compounds such as polybrominated dibenzo-p-dioxins and polybrominated dibenzofurans (PBDD/Fs) like transformations of other halogenated flame retardants,28–31 besides the routine toxicological investigation. Zhang et al. found that debromination and ether bond cleavage are the main phototransformation pathways for BTBPE in aqueous solution, which leads to the formation of toxic bromophenols.27 Altarawneh et al. observed that thermal decomposition of BTBPE can generate several congeners of brominated diphenyl ethers and their OH/OCHCH2 substituents, which serve as direct precursors for the formation of polybrominated dibenzo-p-dioxins.28 These studies indicated that the transformation of BTBPE could increase environmental risk, due to the formation of more toxic products. However, information on the atmospheric fate and transformation of BTBPE is scarce although BTBPE has been frequently and highly detected in the atmosphere.11,14–19
 | ||
| Fig. 1 (A) The structures of BTBPE and PBDEs. (B) PBE1PBE/6-31+G(d,p)-optimized global minimum of BTBPE with important bond lengths in angstroms (Å) and angles in degrees (°). | ||
As a semivolatile compound, BTBPE can exist in the gas-phase and particle in the atmosphere.11,14–19 The concentration of BTBPE in particles were detected to be 1–2 times than that in gas-phase in the Great Lakes atmosphere.19 Therefore, in principle, BTBPE can undergo both gas-phase and heterogeneous transformation. Since the gas-particle partition of BTBPE in the atmosphere is under dynamic equilibrium, part of BTBPE in particles will transfer to gas-phase once gaseous BTBPE is transformed. Therefore, gas-phase transformation could be also important although more BTBPE was detected in particles.19 In addition, the information on gaseous transformation of BTBPE should provide a foundation for the future study on heterogeneous one of BTBPE. Considering ·OH initiated oxidation is a dominating removal pathway for PBDEs and most organic pollutants in the atmosphere,32–34 we supposed such oxidation also plays an important role in BTBPE gas-phase transformation.
In this study, we investigated the reaction mechanism and kinetics of BTBPE initiated by ·OH using a combination of quantum chemistry calculations and kinetic modeling. The study includes the reaction mechanism and kinetics of BTBPE + ·OH and subsequent reactions consisting of isomerization, dissociation, and bimolecular reactions with O2/NO. This study is of significance for understanding the fate and environmental risk of BTBPE and other alternatives with similar structures to banned chemicals.
2. Computational details
2.1. Ab initio electronic structure calculations
All the electronic structure and energy calculations were carried out with Density Functional Theory (DFT) method within the GAUSSIAN 09 program package.35 The advantage of DFT lies in its competitive accuracy/cost ratio. However, before being used to predict accurate kinetics for the target system, specific DFT methods need to go through an assessment step by a comparison with higher-level theoretical approaches (e.g., CCSD(T)) or, if available, accurate experimental results. After carefully testing DFT methods on our target system, PBE1PBE and M06-2X functional were selected.36,37 The details for the test were presented in (ESI†). Exactly, all reaction pathways except H-abstractions in ·OH initiated reactions were investigated by PBE1PBE functional, while the H-abstractions in ·OH initiated reactions were investigated by M06-2X functional. The geometry optimizations and harmonic frequency calculations of the reactants, products, intermediates and transition states were performed using the PBE1PBE/M06-2X method with the 6-31+G(d,p) basis set. Transition states were verified to connect designated local minimum using intrinsic reaction coordinate (IRC) calculations at the theoretical level for corresponding geometry optimizations. Single-point energy calculations were performed at PBE1PBE/6-311++G(3df,2pd) or M06-2X/6-311++G(3df,2pd) level with zero-point correction at the theoretical level for corresponding geometry optimizations.2.2. Kinetics calculations
All reaction rate constants were calculated by the MultiWell-2014.1 master equation code.38–41 The MultiWell program employs Rice–Ramsperger–Kassel–Marcus (RRKM) theory to compute energy-dependent microcanonical rate constants for reactions based on sums and densities of states for the PBE1PBE/6-31+G(d,p) or M06-2X/6-31+G(d,p) structures and the PBE1PBE/6-311++G(3df,2pd) or M06-2X/6-311++G(3df,2pd) barrier heights.42 Tunneling correction has been included in the microcanonical rate constant calculation by using one dimensional unsymmetrical Eckart barrier.43 The energy-grained master equation was solved over 2000 grains of 10 cm−1 each, carried on to 85000 cm−1 for the continuum component of the master equation. N2 gas was used as the buffer gas, and the collision transfer was treated using the conventional exponential-down model with ΔEd = 200 cm−1. The Lennard-Jones parameters for intermediates were calculated from an empirical method proposed by Gilbert and Joback et al.44,45 The rate constants for the barrierless entrance pathways were calculated by the restricted Gorin model when the experimental rate constant for a target system or its similar one is available or by the long-range transition state theory when there is no related experimental data.46,47 This scheme had been successfully used in our previous study.48 The details for the reaction rate constant calculations with the restricted Gorin model and long-range transition state theory were presented in the ESI.†
2.3. Global minimum search
The reactant BTBPE has conformational degrees of freedom that may influence the calculated reaction mechanism and kinetics. Here, global minimum of BTBPE was selected as its starting reactant. Ab initio molecular dynamics (AIMD) was used to generate reasonable gas-phase conformations for BTBPE by employing the BLYP functional on def2-SVP basis set with dispersion correction.49 In order to sample a large number of different conformations in a short time, the simulation temperature was set to 500 K. The total length of AIMD simulation was 5000 time steps with each step of 2 fs. AIMD simulations were performed within TURBOMOLE program package.50 We selected the conformations from the AIMD run as the starting point to obtain different local minimum structures at PBE1PBE/6-31+G(d,p) level. Single-point energy calculations were further performed at PBE1PBE/6-311++G(3df,2pd) level. The conformer with the lowest Gibbs free energy (G) value was identified as the global minimum. All structures of the conformers and relative Gibbs free energies values are presented in Table S1 of ESI.†3. Results and discussion
3.1. Global minimum of BTBPE
The global minimum of BTBPE was depicted in Fig. 1B. It exhibits an approximate C2h symmetry, with root-mean-square deviation (RMSD) value 0.006 to corresponding strict C2h symmetry structure. Therefore, the approximate symmetrical sites of BTBPE could have similar reactivity toward ·OH due to their similar chemical environment. It deserves mentioning that strict C2h symmetry structure was identified with one imaginary frequency.3.2. Initial reactions with ·OH
Generally, the reactions of organic pollutants with ·OH mainly proceed via either addition of ·OH to unsaturated bonds or H-abstraction. Therefore, ·OH may add to π bond of phenyl group or abstract H-atom from ethyl group and phenyl group in the reaction of BTBPE with ·OH. Totally, BTBPE has 2 phenyl groups and 8H-atoms, therefore, it has 12 different addition positions and 8H-abstraction sites for the attack by ·OH. The possible addition and H-abstraction pathways for the reactions of BTBPE with ·OH are depicted in Fig. 2, and the optimized geometries for some important transition states and intermediates are presented in Fig. 3. Each reaction pathway proceeds through a pre-reactive complex and H-abstraction pathways also proceed through a post-reactive complex. It deserves mentioning that the C–Br bond is broken when ·OH add to CBr site to form IM2 + Br, IM6 + Br, IM2′ + Br and IM6′ + Br. However, C–Br bond is kept although CBr bond length become longer (from 1.875 Å to 2.055 Å) during ·OH addition to CBr site to form IM4 and IM4′. The calculated relative energy (to the reactants) of pre-reactive complex (ΔERC) and post-reactive complex (ΔEPC), activation energy values (ΔEa) and activation free energy values (ΔG‡) are listed in Table 1. | ||
| Fig. 2 Addition and H-abstraction pathways for the reaction of BTBPE with ·OH. The symbols “TSm/TSm-H” and “IMm/IMm-H” denote the transition states and intermediates involved in the reaction, respectively, in which m denotes different reaction sites. | ||
 | ||
| Fig. 3 PBE1PBE/6-31+G(d,p) or M06-2X/6-31+G(d,p)-optimized geometries for some important transition states and intermediates involved in the reaction of BTBPE + ·OH. The distances are in angstroms (Å). | ||
| Pathways | Speciesa | Sitesa | ΔERCb | ΔEab | ΔG‡b | ΔEPCb |
|---|---|---|---|---|---|---|
| a The species and sites correspond to the reaction pathways showed in Fig. 2.b ΔERC, ΔEa and ΔEPC were calculated at 0 K, and ΔG‡ was calculated at 298 K. | ||||||
| Addition to phenyl group | IM1 | CO | −0.5 | 0.9 | 9.7 | — |
| IM1′ | CO | −0.5 | 0.9 | 9.7 | — | |
| IM2 | CBr | −0.9 | 4.2 | 13.9 | — | |
| IM2′ | CBr | −0.9 | 4.2 | 13.9 | — | |
| IM6 | CBr | −0.9 | 4.2 | 13.9 | — | |
| IM6′ | CBr | −1.2 | 3.9 | 13.4 | — | |
| IM3 | CH | −1.2 | −0.8 | 7.6 | — | |
| IM3′ | CH | −1.2 | −0.8 | 7.6 | — | |
| IM5 | CH | −1.2 | −0.8 | 7.6 | — | |
| IM5′ | CH | −1.2 | −0.8 | 7.6 | — | |
| IM4 | CBr | −0.9 | 3.2 | 10.6 | — | |
| IM4′ | CBr | −0.9 | 3.2 | 10.5 | — | |
| H-abstraction from ethyl group | IM7-1 | CH | −3.4 | −1.0 | 8.7 | −25.1 |
| IM7-2 | CH | −3.4 | −1.0 | 8.7 | −25.1 | |
| IM7′-1 | CH | −3.4 | −1.0 | 8.7 | −25.1 | |
| IM7′-2 | CH | −3.4 | −1.0 | 8.7 | −25.1 | |
| H-abstraction from phenyl group | IM3-H | CH | −1.9 | 5.8 | 14.4 | −5.9 |
| IM3′-H | CH | −1.9 | 5.8 | 14.4 | −5.9 | |
| IM5-H | CH | −1.9 | 5.8 | 14.4 | −5.9 | |
| IM5′-H | CH | −1.9 | 5.8 | 14.4 | −5.9 | |
It can be seen from Table 1 that the ΔEa values for additions occurring at the CH sites of 2,4,6-tribromophenoxy group via transition states TS3, TS3′, TS5 and TS5′ to form IM3, IM3′, IM5 and IM5′, and H-abstractions from –O–CH2–CH2–O– via transition states TS7-1, TS7′-1, TS7-2 and TS7′-2 to form IM7-1 + H2O, IM7′-1 + H2O, IM7-2 + H2O and IM7′-2 + H2O, are comparable and much lower than those of other pathways. Therefore, initial reaction of BTBPE with ·OH mainly form IM3, IM3′, IM5, IM5′, IM7-1, IM7′-1, IM7-2 and IM7′-2.
Based on ΔEa values for all reaction pathways, we discuss the reactivity trend of addition sites and H-abstraction sites of BTBPE toward ·OH. The reaction sites with the higher ΔEa values will have the lower reactivity toward ·OH. Therefore, the reactivity of addition sites follows the order of the CH sites > CO sites > CBr sites of 2,4,6-tribromophenoxy group of BTBPE. We noted that previous study found that ·OH additions to CBr site tend to have a higher Ea value than those of addition to CH sites of bromophenoxy group in the reaction of PBDEs with ·OH.33,34,51,52 This agrees with our findings. The C–H of 2,4,6-tribromophenoxy group has lower H-abstraction reactivity than that of –O–CH2–CH2–O– group. According to the C–H bond dissociation energy in C6H6 (112.9 ± 0.5 kcal mol−1) and CH3OCH3 (95.3 kcal mol−1),53 we speculate that CH bond dissociation energy in 2,4,6-tribromophenoxy group is higher than that in –O–CH2–CH2–O– group, which can explain lower H-abstraction reactivity in 2,4,6-tribromophenoxy group than that in –O–CH2–CH2–O– group. It deserves mentioning that variation tendency of ΔEa is consistent with that of ΔG‡ (Table 1), indicating that the entropy effect can't change the energy order of different pathways. Details of activation enthalpy values (ΔH‡) and activation entropy (ΔS‡) for all pathways were shown in Table S2.† In addition, approximate symmetrical sites have (almost) the same ΔEa values toward ·OH, e.g. 1 and 1′, 2, 6, 2′ and 6′, 3, 5, 3′ and 5′, 4 and 4′, 7-1, 7-2, 7-1′ and 7-2′, respectively. Therefore, in the following discussion, only one site (1, 2, 3, 4, 7-1 and 3-H) among all symmetrical sites was considered.
3.3. Kinetics
The master equation method was used to calculate the reaction rate constants for the reaction of BTBPE + ·OH, including pathways to form IM1, IM2, IM3, IM4, IM7-1 and IM3-H. Moreover, due to the barrierless pre-reactive complexes and post-reactive complex, we performed the long-range transition state theory to calculate the formation rate constant of pre-reactive complexes and post-reactive complex for the reaction BTBPE + ·OH. The overall reaction rate constant was calculated to be sum of all sites by assuming that approximate symmetrical sites have the same reaction rate constants. The values of kOH and Γ for the reaction of BTBPE with ·OH at 298 K are presented in Table 2. To further evaluate the effect of temperature (T) and pressure (P) on kOH, k(T,P), kOH as a function of T and P, was calculated for the overall reaction and two favorable pathways forming IM3 + IM3′ + IM5 + IM5′ and IM7-1 + IM7-2 + IM7′-1 + IM7′-2 at temperature range 270–330 K and pressure range 81–122 kPa (shown in Fig. S1†). As seen in Fig. S1,† k(T,P) presents a negative temperature dependence but no obvious pressure dependence for both the overall reaction and considered two favorable pathways. The overall rate constant was calculated to be 1.0 × 10−12 cm3 per molecule per s at 298 K and 101 kPa, which is about 20 times higher than that of (5.2 × 10−14 cm3 per molecule per s) of 2,2′,4,4′,5-pentabrominated diphenyl ether (BDE99).52 The calculated branching ratio Γ value for the addition products IM3 + IM3′ + IM5 + IM5′ is 73.5% and H-abstraction products IM7-1 + IM7-2 + IM7′-1 + IM7′-2 is 24.9% at 298 K, which are much higher than those for other addition and H-abstraction pathways. Therefore, CH site addition of 2,4,6-tribromophenoxy group and H-abstraction from –O–CH2–CH2–O– group are the most favorable pathways for the reaction BTBPE + ·OH.| Pathways | Speciesa | Sitesa | kOHb | Γb |
|---|---|---|---|---|
| a The species and sites correspond to the reaction pathways showed in Fig. 2 and Table 1.b kOH and Γ were calculated at 298 K and 101 kPa. | ||||
| Addition to phenyl group | IM1 + IM1′ | CO | 1.4 × 10−14 | 1.4% |
| IM2 + IM2′ + IM6 + IM6′ | CBr | 0 | 0% | |
| IM3 + IM3′ + IM5 + IM5′ | CH | 7.4 × 10−13 | 73.5% | |
| IM4 + IM4′ | CBr | 1.9 × 10−15 | 0.2% | |
| H-abstraction from ethyl group | IM7-1 + IM7′-1 + IM7-2 + IM7′-2 | CH | 2.5 × 10−13 | 24.9% |
| H-abstraction from phenyl group | IM3-H + IM3′-H + IM5-H + IM5′-H | CH | 3.4 × 10−16 | 0% |
| Overall | 1.0 × 10−12 | 100% | ||
3.4. Subsequent reactions of primary intermediates
The chemically activated intermediates/products produced in the initial reaction can subsequently isomerize, dissociate or react with atmospheric O2. Here, we focus primarily on the further transformation of the intermediates with higher Γ values i.e. intermediates from addition IM3, IM3′, IM5 and IM5′ and from H-abstraction IM7-1, IM7-2, IM7′-1 and IM7′-2. In addition, based on the consideration of approximate molecular symmetry as mentioned above, only the transformations of IM3 and IM7-1 were investigated.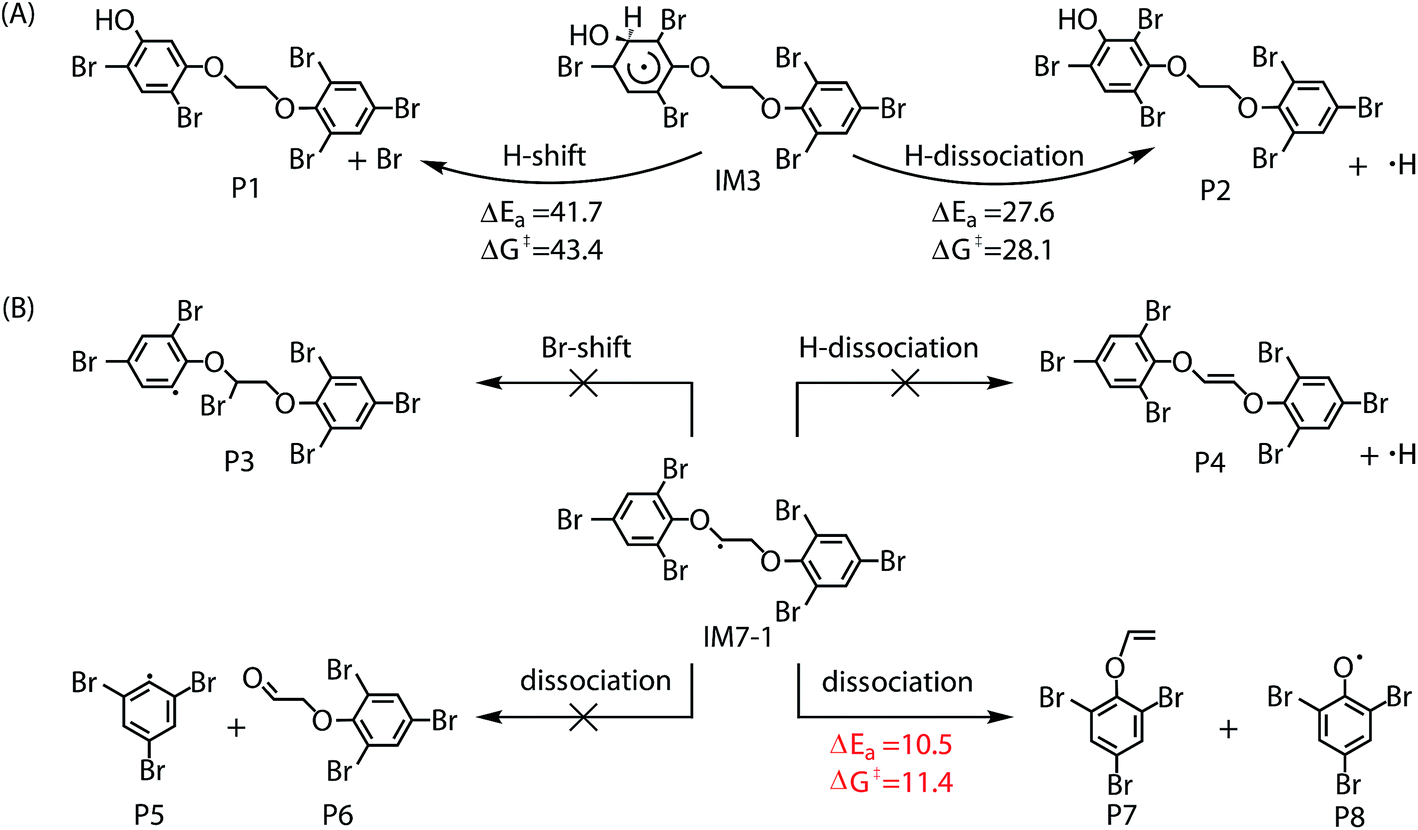 | ||
| Fig. 4 Subsequent self-isomerization and dissociation pathways for IM3 (A) and IM7-1 (B). | ||
 | ||
| Fig. 5 Subsequent reaction pathways for IM3 (A) and IM7-1 (B) with O2. | ||
 | ||
| Fig. 6 PBE1PBE/6-31+G(d,p)-optimized geometries for some important transition states and intermediates involved in the reaction of IM3/IM7-1 + O2. The distances are in angstroms (Å). | ||
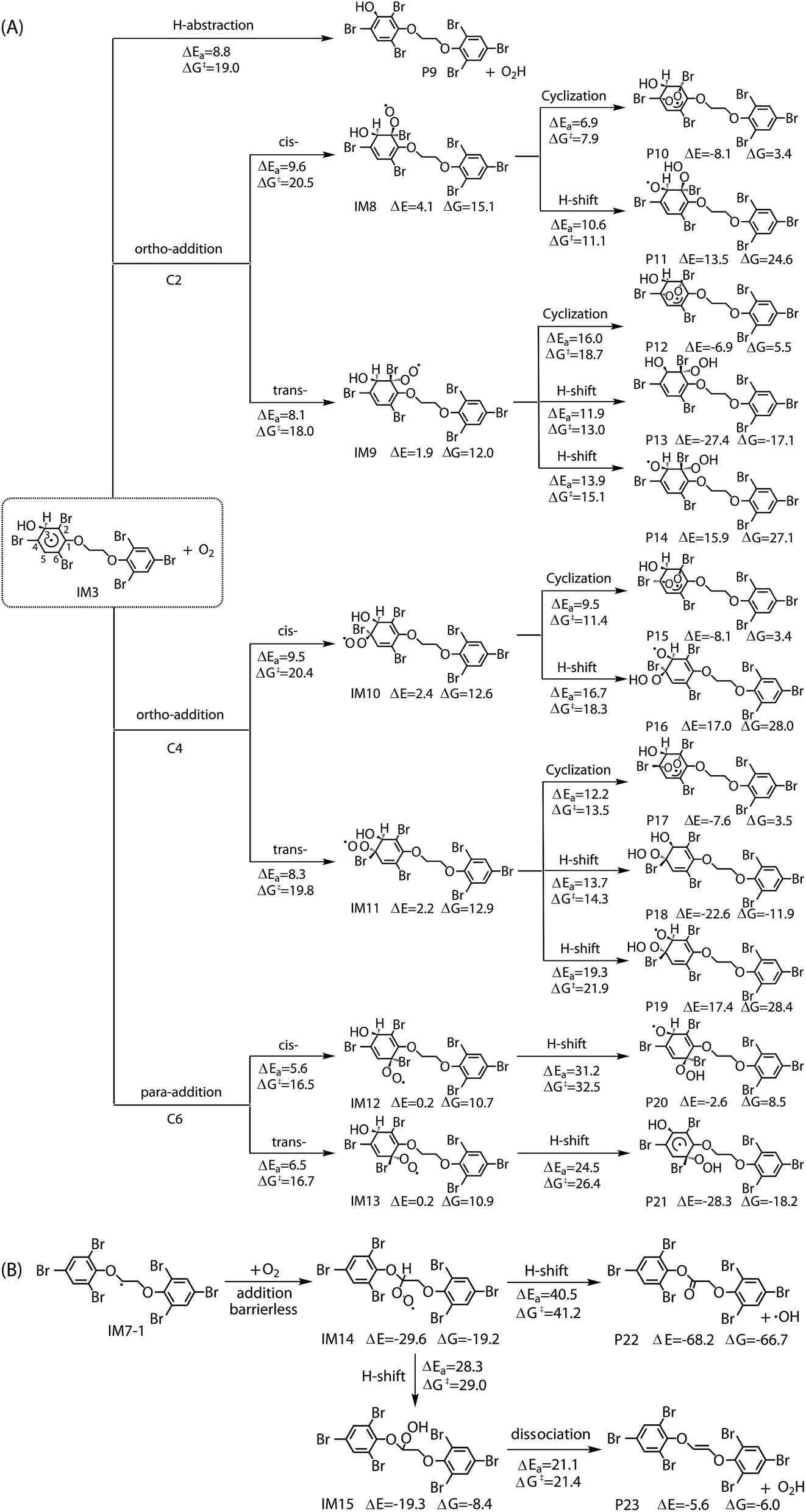
The reaction of IM3 with O2 can proceed via direct H-abstraction to form hydroxylated BTBPE (OH-BTBPE) and O2H or addition to form peroxyl radicals as other aromatic –OH adducts (e.g. benzene-OH and toluene-OH adducts).54,55 Fig. 5A depicts the H-abstraction and addition pathways for the reaction IM3 + O2 with corresponding ΔEa and ΔG‡ values, and relative energy (ΔE) and relative free energy values (ΔG) (to IM3 + O2) of intermediates and products. For the H-abstraction process, O2 could favorably abstract H atom at C site connecting –OH to form OH-BTBPE (P9) and O2H radicals. The ΔEa is 8.8 kcal mol−1, corresponding to rate constant 2.2 × 10−21 cm3 per molecule per s. However, H-abstraction at other sites could lead to thermodynamically unfeasible diradical products. The O2 addition to IM3 should occur at the sites with high spin distribution, i.e. the ortho-carbon atoms (C2 and C4) and the para-carbon (C6) of the –OH position (shown in Fig. S2†), like cases of the reactions benzene-OH adducts + O2 and BDE-OH adducts + O2 since the triplet O2 is diradical.33,55 Also, O2 can be added to the aromatic rings from the same or opposite side of the phenyl ring (cis- and trans-) with respect to the –OH group. A common feature for such additions is that ΔG values of all formed intermediates (IM8-IM13) are >0. ΔEa value for para-addition at C6 site is lower than those of the other additions, indicating that para-addition at C6 site is most favorable.
The activated peroxy radicals (IM8–IM13) formed in the O2 addition pathway could further transform, forming oxy hydroperoxide radical (finally leading to epoxy radicals) via H-shift and bicyclic radicals via cyclization, respectively.54,55 The bicyclic radicals will exclusively recombine with O2 to produce bicyclic peroxy radicals, then the bicyclic peroxy radicals are expected to react with NO to form the bicyclic alkoxy radicals which undergo ring cleavage finally as the reactions of other aromatics such as toluene.54,56 However, some intermediates including IM9, IM11, IM12 and IM13 need to overcome high reaction barriers, and the others including IM8 and IM10 are thermodynamically unfeasible for further reactions. Therefore, due to higher energy of peroxy radicals relative to that of reactants IM3 + O2 and hardly further transformation, the O2 addition reaction could proceed reversibly, which is similar to the case for the reaction of the monoaromatic hydrocarbon-OH adducts such as benzene-OH, toluene-OH and chlorobenzene-OH with O2.54,55,57 As a result, O2-addition rate constants are effectively assumed as the formation rate constant of products (P10–21). The calculated overall addition rate constant (Table S3†) is 1.3 × 10−22 cm3 per molecule per s, in which the trans-addition to C6 site forming P21 dominates the overall rate constant. In addition, the reaction rate constants for H-abstraction pathways, forming OH-BTBPE and O2H radical, are higher than those for addition pathways. Thus, H-abstraction mainly contributes to final products for the further transformation of IM3 in the presence of O2, analogous to the reaction of OH adduct of PBDEs with O2 to form OH-PBDEs.52 Since high abundance of atmospheric O2, the favorable H-abstraction pathway of IM3 with O2 should have atmospheric importance even though its reaction energy barrier is relatively high. This also can be supported by the fact that pseudo-first order rate constant (∼10−2 s−1) of the H-abstraction is much higher than that (∼10−4 s−1) of collision controlled ·OH-molecule reactions that are of significance for the transformation of atmospheric pollutants.
Similar to the reactions of typical C-center radicals with O2, O2 can barrierlessly add to C7 site of IM7-1 to form the intermediate peroxy radicals (Fig. 5B). Peroxy radicals have two isomeric forms (IM14 and IM14′ in Fig. 6) owing to the different attacking direction of O2. IM14 and IM14′ can easily interconvert with the approximate barrier 3 to 4 kcal mol−1. For IM14, there are two possible H-transfer pathways. One is H-transfer from C7 site to OO site, which is followed by HO–O bond capture to form P18; the other is H-transfer from C7′ site to OO site to form oxy hydroperoxide radical (IM15), which can finally form P23 via HOO–C bond capture. The overall reaction energy barrier for latter pathway is much lower than that of former one, indicating the H-transfer from C7′ site to OO site to finally form P23 is most favorable for IM14. Based on the energetic information of the most favorable pathway, the calculated rate constant of IM7-1 + O2 is 2.2 × 10−12 cm3 per molecule per s. In addition, IM14 is an exclusive product with 100% product branching ratio. This should result from the fact that IM14 adduct lies in a deep well. We also note that an adduct formed in the reaction of other C-center radicals such as NH2CHCH2OH with O2,58 can easily further transform to fragmental products, which is different from IM7-1 + O2. The steric or electronic effect of Br near the reactive center could be a possible reason for the difference.
3.5. Fate of primary intermediates
To discuss the final fate of primary intermediates (IM3 and IM7-1) formed by BTBPE + ·OH in the atmosphere, we evaluate the competition of their different transformation pathways, including self-isomerization/dissociation, bimolecular reactions with O2/NO. To effectively compare the competition of these pathways, bimolecular rate constants for IM3/IM7-1 + O2 and IM3/IM7-1 + NO were multiplied by the concentration of O2 (4.92 × 1018 molecules cm−3) and NO (∼5 ppb, 2.69 × 1010 molecules cm−3), respectively, to obtain theirs pseudo-first order rate constants. The reaction rate constant for the reaction of IM3 with NO is assumed in the order of 10−14 cm3 per molecule per s based on the reaction of typical aromatic-OH adducts with NO,59 and IM7-1 + NO in the order of 10−12 cm3 per molecule per s based on the reaction rate constant of ·CH2OCH3 with NO.60 In addition, to evaluate final yield of products, a comprehensive product branching ratio was determined by calculating the product of branching ratio of IM3 and IM7-1 in the ·OH initiated reaction and branching ratio of products of their subsequent reactions, respectively. The rate constants of all pathways and the comprehensive product branching ratio were listed in Table S3† and Fig. 7, respectively. The main fate of IM3 and IM7-1 was presented in Fig. 7A and B, respectively. | ||
| Fig. 7 Summary for the fate of primary intermediates IM3 (A) and IM7-1 (B). The width of the arrows indicates the relative importance of the reaction pathway. Percentage under different products indicates their respective final yield. | ||
For IM3, as can be seen in Fig. 7A, the pseudo-first order rate constant (∼10−2 s−1) with O2 is much higher than that (∼10−4 s−1) with NO and its unimolecular self-isomerization/dissociation rate constant (∼10−8 s−1). The yield of the OH-BTBPE forming from oxidation of BTBPE is about 72.3%, which indeed is the main product. Similar to IM3, the pseudo-first order rate constant (∼107 s−1) of IM7-1 with O2 is much higher than that (∼10−2 s−1) with NO and its unimolecular self-dissociation rate constant (∼104 s−1). It deserves mentioning that unimolecular self-dissociation rate constant of IM7-1 is much higher than that of reaction of IM7-1 with NO, which is different from the case of IM3. The yield of peroxy radicals from the reaction IM7-1 + O2 is estimated to be 24.8%. Based on above discussions, we can conclude that OH-BTBPE and peroxy radicals, are the main fate of primary intermediates formed by BTBPE + ·OH in the atmosphere. In addition, pseudo-first order rate constant of IM3 and IM7-1 with O2 is higher than that (∼10−6 s−1, [OH] is 9.7 × 105 molecules cm−3) of the initial reaction BTBPE + ·OH, indicating the reaction of ·OH with BTBPE is the rate determining step in ·OH initiated reaction of BTBPE.
3.6. Conclusions and atmospheric implications
The atmospheric oxidation mechanism and kinetics of BTBPE initiated by ·OH was investigated by a combined quantum chemical method and kinetics modeling. The initial oxidation proceeds via the ·OH addition and hydrogen abstraction pathways to form intermediates (IM7-1 and IM3), which can further react with O2 to finally form peroxy radicals and OH-BTBPE. The Daphnia Magna LC50 (48 h) values for OH-BTBPE are predicted to be obviously higher than those for BTBPE (details presented in Table S4†). Therefore, formed OH-BTBPE tends to be more toxic than their parent compound BTBPE, which could increase the environmental risk of BTBPE emission and deserve further investigation. This is similar to previous findings that most hydroxylated brominated aromatics are of higher toxicity than themselves, for instance, OH-PBDEs and bromophenol have been proved to be more toxic than PBDEs and bromobenzene, respectively.61,62 The calculated overall reaction rate constant is 1.0 × 10−12 cm3 per molecule per s, translating 11.8 days atmospheric lifetime (τ = 1/kOH[OH]) of BTBPE. Although the lifetime of BTBPE is much lower than that (around hundreds days) of the banned PBDEs (pentaBDE, octaBDE and decaBDE), the substitute of PBDEs by BTBPE still has atmospheric persistence.52,63–65 In addition, it deserves mentioning that heterogeneous reactions of BTBPE on atmospheric particles will affect its overall atmospheric lifetime since BTBPE is a semivolatile compound. Therefore, the effect of other particles such as black carbon, mineral dust on the lifetime of BTBPE deserves future investigation. Our study lays a good foundation for the future study on heterogeneous reactions of BTBPE.Acknowledgements
We thank Prof. John R. Barker (University of Michigan) for providing the MultiWell-2014.1 program. The study was supported by the National Natural Science Foundation of China (21477015, 21325729), Major International (Regional) Joint Research Project (21661142001) and the Fundamental Research Funds for the Central Universities.References
- A. Bergman, J. Heindel, S. Jobling, K. Kidd and R. T. Zoeller, State of the science of endocrine disrupting chemicals, 2012, http://www.unep.org/pdf/9789241505031_eng.pdf Search PubMed.
- United Nations Environment Programme (UNEP), DTI/1639/GE, Global chemicals outlook-Towards sound management of chemicals, ISBN: 978-92-807-3320-4, 2013, https://wedocs.unep.org/rest/bitstreams/14105/retrieve Search PubMed.
- Stockholm Convention, The list of POPs in the Stockholm Convention, http://chm.pops.int/TheConvention/ThePOPs/ListingofPOPs/tabid/2509/Default.aspx Search PubMed.
- Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC (OJ L 396, 30.12.2006).
- J. B. Zimmerman and P. T. Anastas, Science, 2015, 347, 1198–1199 CrossRef CAS PubMed.
- K. Kim, Z. Shon, H. T. Nguyen and E. Jeon, Atmos. Environ., 2011, 45, 1369–1382 CrossRef CAS.
- B. O. Bolaji and Z. Huan, Renewable Sustainable Energy Rev., 2013, 18, 49–54 CrossRef CAS.
- D. Chen, K. Kannan, H. Tan, Z. G. Zheng, Y. L. Feng, Y. Wu and M. Widelka, Environ. Sci. Technol., 2016, 50, 5438–5453 CrossRef CAS PubMed.
- E. Hoh, L. Y. Zhu and R. A. Hites, Environ. Sci. Technol., 2005, 39, 2472–2477 CrossRef CAS PubMed.
- Organisation for Economic Co-operation and Development (OECD), The 2007 OECD list of high production volume chemicals, Environment, Health and Safety Publications, Series on Testing and Assessment, No. 112, 2009, http://www.oecd.org_officialdocuments_publicdisplaydocumentpdf__cote=env_jm_mono(2009)40%26doclanguage=en Search PubMed.
- A. Covaci, S. Harrad, M. A. Abdallah, N. Ali, R. J. Law, D. Herzke and C. A. de Wit, Environ. Int., 2011, 37, 532–556 CrossRef CAS PubMed.
- H. M. Stapleton, J. G. Allen, S. M. Kelly, A. Konstantinov, S. Klosterhaus, D. Watkins, M. D. Mcclean and T. F. Webster, Environ. Sci. Technol., 2008, 42, 9455–9456 CrossRef CAS.
- M. J. L. Guardia and R. C. Hale, Environ. Int., 2015, 79, 106–114 CrossRef PubMed.
- A. Salamova and R. A. Hites, Environ. Sci. Technol., 2011, 45, 8698–8706 CrossRef CAS PubMed.
- H. Qi, W. Li, L. Liu, W. Song, W. Ma and Y. Li, Sci. Total Environ., 2014, 491–492, 60–66 CrossRef CAS PubMed.
- K. Vorkamp, R. Bossi, F. F. Riget, H. Skov, C. Sonne and R. Dietz, Environ. Pollut., 2015, 196, 284–291 CrossRef CAS PubMed.
- K. Arinaitwe, D. C. Muir, B. T. Kiremire, P. Fellin and H. Li, Environ. Sci. Technol., 2014, 48(3), 1458–1466 CrossRef CAS PubMed.
- M. Shoeib, L. Ahrens, L. M. Jantunen and T. Harner, Atmos. Environ., 2014, 99, 140–147 CrossRef CAS.
- Y. Ma, A. Salamova, M. Venier and R. A. Hites, Environ. Sci. Technol., 2013, 47, 11457–11464 CrossRef CAS PubMed.
- E. D. Schreder and M. J. L. Guardia, Environ. Sci. Technol., 2014, 48, 11575–11583 CrossRef CAS PubMed.
- E. F. Davis, S. L. Klosterhaus and H. M. Stapleton, Environ. Int., 2012, 40, 1–7 CrossRef CAS PubMed.
- R. Yang, H. Wei, J. Guo and A. Li, Environ. Sci. Technol., 2012, 46, 3119–3126 CrossRef CAS PubMed.
- K. Vorkamp and F. F. Rigét, Chemosphere, 2014, 111, 379–395 CrossRef CAS PubMed.
- C. A. D. Wit, D. Herzke and K. Vorkamp, Sci. Total Environ., 2010, 408, 2885–2918 CrossRef PubMed.
- A. Möller, Z. Xie, M. Cai, G. Zhong, P. Huang, M. Cai, R. Sturm, J. He and R. Ebinghaus, Environ. Sci. Technol., 2011, 45, 6793–6799 CrossRef PubMed.
- H. Hakk and R. J. Letcher, Environ. Int., 2003, 29, 801–828 CrossRef CAS PubMed.
- Y. N. Zhang, J. Chen, Q. Xie, Y. Li and C. Zhou, Chemosphere, 2016, 150, 453–460 CrossRef CAS PubMed.
- M. Altarawneh and B. Z. Dlugogorski, Environ. Sci. Technol., 2014, 48, 14335–14343 CrossRef CAS PubMed.
- M. Altarawneh and B. Z. Dlugogorski, Environ. Sci. Technol., 2013, 47, 5118–5127 CrossRef CAS PubMed.
- M. Altarawneh and B. Z. Dlugogorski, Chemosphere, 2014, 114, 129–135 CrossRef CAS PubMed.
- M. Altarawneh and B. Z. Dlugogorski, J. Phys. Chem. A, 2014, 118, 9338–9346 CrossRef CAS PubMed.
- J. D. Raff and R. A. Hites, J. Phys. Chem. A, 2006, 110, 10783–10792 CrossRef CAS PubMed.
- H. Cao, M. He, D. Han, J. Li, M. Li, W. Wang and S. Yao, Environ. Sci. Technol., 2013, 47, 8238–8247 CAS.
- J. Zhou, J. Chen, C. H. Liang, Q. Xie, Y. N. Wang, S. Zhang, X. Qiao and X. Li, Environ. Sci. Technol., 2011, 45, 4839–4845 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- G. P. Wood, A. Sreedhara, J. M. Moore and B. L. Trout, J. Phys. Chem. A, 2014, 118, 2667–2682 CrossRef CAS PubMed.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J. R. Barker, Int. J. Chem. Kinet., 2009, 41, 748–763 CrossRef CAS.
- J. R. Barker, Int. J. Chem. Kinet., 2001, 33, 232–245 CrossRef CAS.
- J. R. Barker, N. F. Contributors Ortiz, J. M. Preses, L. L. Lohr, A. Maranzana, P. J. Stimac, T. L. Nguyen and T. J. D. Kumar, MultiWell-2014.1 Software, Universityof Michigan, Ann Arbor, MI, 2014 Search PubMed.
- J. R. Barker and N. F. Ortiz, Int. J. Chem. Kinet., 2001, 33, 246–261 CrossRef CAS.
- P. J. Robinson and K. A. Holbrook, Unimolecular Reactions, John Wiley & Sons, New York, 1972 Search PubMed.
- C. Eckart, Phys. Rev., 1930, 35(11), 1303–1309 CrossRef CAS.
- R. G. Gilbert and S. C. Smith, Theory of unimolecular and recombination reactions, Blackwell Scientific Publications, Carlton, Australia, 1990 Search PubMed.
- K. G. Joback and R. C. Reid, Chem. Eng. Commun., 2007, 57, 233–243 CrossRef.
- J. R. Barker, N. F. Ortiz, J. M. Preses, L. L. Lohr, A. Maranzana, P. J. Stimac, T. L. Nguyen and T. J. Dhilip Kumar, MultiWell Program Suite User Manual, (MultiWell-2014.1), http://aoss-research.engin.umich.edu/multiwell/ Search PubMed.
- Y. Georgievskii and S. J. Klippenstein, J. Chem. Phys., 2005, 122, 194103 CrossRef PubMed.
- H. B. Xie, F. F. Ma, Y. Wang, N. He, Q. Yu and J. W. Chen, Environ. Sci. Technol., 2015, 49, 13246–13255 CrossRef CAS PubMed.
- C. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- H. Cao, M. He, D. Han, Y. Sun and J. Xie, Atmos. Environ., 2011, 45, 1525–1531 CrossRef CAS.
- H. Cao, D. Han, M. Li, X. Li, M. He and W. Wang, J. Phys. Chem. A, 2015, 119, 6404–6411 CrossRef CAS PubMed.
- Y. R. Luo, Handbook of bond dissociation energies in organic compounds, Crc Press Inc., 2003, pp. 39–69 Search PubMed.
- I. Suh, R. Zhang, L. T. Molina and M. J. Molina, J. Am. Chem. Soc., 2003, 125, 12655–12665 CrossRef CAS PubMed.
- D. R. Glowacki, L. M. Wang and M. J. Pilling, J. Phys. Chem. A, 2009, 113, 5385–5396 CrossRef CAS PubMed.
- R. Wu, S. Pan, Y. Li and L. Wang, J. Phys. Chem. A, 2014, 118, 4533–4547 CrossRef CAS PubMed.
- R. Wu, S. Wang and L. Wang, Chemosphere, 2014, 111, 537–544 CrossRef CAS PubMed.
- H. B. Xie, C. Li, N. He, C. Wang, S. Zhang and J. Chen, Environ. Sci. Technol., 2014, 48, 1700–1706 CrossRef CAS PubMed.
- R. Koch, R. Knispel, M. Elend, M. Siese and C. Zetzsch, Atmos. Chem. Phys., 2007, 7, 2057–2071 CrossRef CAS.
- M. Rissanen, M. Ihlenborg, T. T. Pekkanen and R. S. Timonen, J. Phys. Chem. A, 2015, 119, 7734–7741 CrossRef CAS PubMed.
- A. R. Zota, J. S. Park, Y. Wang, M. Petreas, R. T. Zoeller and T. J. Woodruff, Environ. Sci. Technol., 2011, 45, 7896–7905 CrossRef CAS PubMed.
- J. Jiang, Y. Gao, S. Y. Pang, Q. Wang, X. Huangfu, Y. Liu and J. Ma, Environ. Sci. Technol., 2014, 48, 10850–11088 CrossRef CAS PubMed.
- European Union, Directive 2002/95/EC of the European Parliament and of the Council on the restriction of the use of certain hazardous substances in electrical and electronic equipment, Off J Eur Union, 2003, vol. 37, pp. 19–23.
- European Court of Justice, Cases C-14/06 and C-295/06, Judgement of the Court, 1 April 2008, Directive 2002/95/EC and Commission Decision 2005/717/EC, 2008Error! Hyperlink reference not valid. http://curia.europa.eu, accessed, July 2010.
- I. Liagkouridis, A. P. Cousins and I. T. Cousins, Sci. Total Environ., 2015, 524, 416–426 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of methods and rate calculation, Tables S1–S4, Fig. S1 and S2. See DOI: 10.1039/c6ra26700a |
| This journal is © The Royal Society of Chemistry 2017 |