
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of cellulose-based fluorescent carbon nanoparticles and their application in trace detection of Pb(II)
Zhang Yun-fei,
Halidan Maimaiti * and
Zhang Bo
* and
Zhang Bo
Institute of Chemistry and Chemical Industry, Xinjiang University, Urumqi 830046, Xinjiang, China. E-mail: m15899160730@163.com
First published on 12th January 2017
Abstract
Nanocrystalline cellulose (NCC) with a particle size of 23.80 ± 0.33 nm was prepared from microcrystalline cellulose by a mixed treatment with acids and ultrasound. The NCC was chlorinated with thionyl chloride, submitted to dehydrating carbonization and oxidization, and then modified with ethylenediamine (EDA) to obtain fluorescent carbon nanoparticles (NCC–EDA). These were characterized by elemental analysis, TEM, BET, FT-IR, XRD, TG-DTA, XPS, UV-Vis, and photoluminescence. It was found that NCC–EDA were vesicle-like porous particles formed through the linking and intertwining effects of EDA on the SOCl2-treated NCC. The surface of NCC–EDA is rich in oxygen-, nitrogen- and sulfur-containing functional groups, and the particle size was 41.45 ± 0.55 nm. This endows the NCC–EDA with good dispersity in water, high absorbance and strong fluorescence. The NCC–EDA are shown to be viable probes in that their fluorescence is selectively quenching by Pb(II). The findings were exploited to design a fluorometric assay for Pb(II) that has a 24 nM detection limit.
1. Introduction
In light of the good optical property and water dispersity of fluorescent carbon nanoparticles (CNPs), especially their low toxicity and desirable biocompatibility relative to quantum dots (QDs),1 the preparation of fluorescent carbon nanomaterials for special applications by using different raw materials and different methods has drawn extensive attention all over the world.2–4 Particularly, the preparation of fluorescent CNPs using environmentally-benign carbohydrates as carbon sources has become the focus of attention in recent years due to the advantages of operation simplicity, short reaction time and high product quantum efficiency. For instance, Sun et al. prepared fluorescent CNPs with relatively high quantum efficiency by treating shaddock peel with a hydrothermal method;5 Chang et al. reported a green approach to the one-pot synthesis of fluorescent CNPs with sp2-bonded carbon atoms from coffee grounds;6 Han et al. synthesized fluorescent CNPs with high quality through low-temperature carbonization and simple filtration using watermelon peel;7 Krysmann et al. obtained fluorescent CNPs via high-temperature pyrolysis by using biomass as starting material;8 Sarka et al. treated roasted peanut shells at 700 °C under nitrogen atmosphere for carbonization, and then carried out oxidization experiment in aqueous solution to obtain cellulose-based fluorescent CNPs;9 Pereira et al. prepared water-soluble bio-based fluorescent CNPs from cellulose nanocrystals using different pyrolysis temperatures;10 Peng et al. treated carbohydrates with concentrated sulfuric acid for dehydrated carbonization, and then executed a series of processes including oxidization with nitric acid and deactivation of amine-terminated compounds to obtain fluorescent CNPs;11 Yang et al. used chitosan as starting material and prepared amino group functionalized fluorescent CNPs.12 In addition, the preparations of fluorescent CNPs using low-molecular-weight carbohydrates such as glucose, sucrose and citric acid have also been reported.13–15It can be seen from the above results that when low-molecular-weight carbohydrates are used as carbon sources, dehydrated carbonization and subsequent modification are considered as the major approaches to prepare fluorescent CNPs; when high-molecular-weight carbohydrates are used, hydrothermal treatment and high-temperature pyrolysis are usually accepted as the proper methods. Then, is it possible to prepare fluorescent CNPs with identical properties by simple acid treatment on high-molecular-weight carbohydrates and subsequent surface modification, but not rely on high temperature pyrolysis? For this purpose, water-soluble cellulose-based fluorescent carbon nanoparticles (NCC–EDA) with relatively high fluorescence quantum yield were prepared by using cellulose, the most abundant organic raw material in nature, as starting material. On this basis, the structure and fluorescence property of the NCC–EDA were studied, and the NCC–EDA were used as fluorescent probe to detect metal ions in water.
2. Experiment
2.1 Materials and methods
Microcrystalline cellulose (MCC, with particle size range of 10–100 μm and moisture content of 6%) of technical grade was purchased from Tianjin Guangfu Fine Chemical Co., Ltd. Thionyl chloride (SOCl2) of analytical grade was purchased from Aladdin Reagent Co., Ltd. Ethylenediamine (EDA) of analytical grade was purchased from Tianjin Zhiyuan Chemical Reagent Co., Ltd. N,N-Dimethylformamide (DMF), tetrahydrofuran (THF) and absolute ethanol (C2H5OH) are all of analytical grade and commercially available.Ready-to-use dialysis bags of Spectrum labs (no. 1: molecular weight cut-off of 100–500; no. 2: molecular weight cut-off of 500–1000) were purchased from Shanghai Toscience Biotechnology Co., Ltd.
2.2 Preparation of product
The preparation process of the NCC–EDA is shown in Scheme 1.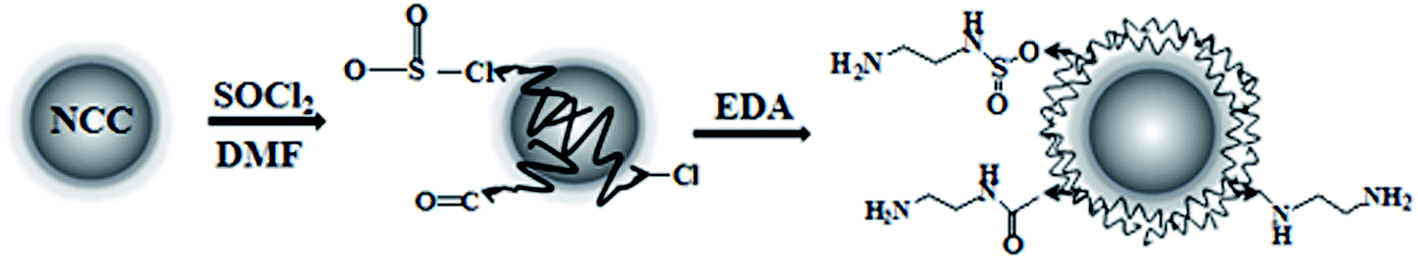 | ||
| Scheme 1 Preparation process of the cellulose based fluorescent nanoparticles. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The supernatant was transferred into no. 2 dialysis bags and dialyzed in water for 72 h to obtain an aqueous dispersion of NCC–EDA. Finally, NCC–EDA particles were obtained by vacuum distillation.
000 rpm for 10 min. The supernatant was transferred into no. 2 dialysis bags and dialyzed in water for 72 h to obtain an aqueous dispersion of NCC–EDA. Finally, NCC–EDA particles were obtained by vacuum distillation.2.3 Characterization of product
The aqueous dispersions of NCC and NCC–EDA were respectively dropped onto a copper grid and then dried for observing the morphology and structure of the particles by transmission electron microscope (TEM, Hitachi H-600, Japan). The particle sizes of the NCC and NCC–EDA aqueous dispersions were measured by NANO-S90 laser particle size analyzer. The contents of various elements, such as C, H, O, N and S, in NCC, NCC–Cl and NCC–EDA were determined by FLASHEA-PE2400 elemental analyzer. The specific surface area, pore size and total pore volume of NCC and NCC–EDA were determined by Autosorb-IQ2 surface area and pore size analyzer (Quantachrome, America). FT-IR characterization of starting material and product was performed by Bruker-EQUINOX 55 Fourier transform infrared spectrometer. XRD characterization of NCC, NCC–Cl, and NCC–EDA was carried out by MAC-M18XHF22-SRA X-ray diffractometer. TG-DTA analysis of the starting material and product was performed by PE-DTA/1700 thermal analyzer. The surface state of NCC–EDA was analyzed by ESCALAB 250 X-ray photoelectron spectrometer (XPS Thermo Fisher, America).2.4 Optical property characterization of product
The aqueous dispersion of NCC–EDA was transferred into cuvette after dilution and then measured by UV 3900H UV-Vis spectrophotometer (Hitachi, Japan) for UV-Vis absorption spectra and by Fluorolog-3 fluorescence spectrometer (Horiba, France) for emission spectra at different excitation wavelengths, respectively.2.5 Fluorescence detection of Pb2+ by NCC–EDA
1 mL NCC–EDA aqueous dispersion with a concentration of 0.25 g L−1 was placed in a 100 mL volumetric flask, followed by the addition of phosphate buffer to the scale marking. After the solution in the volumetric flask was shaken well, 1 mL solution containing K+, Na+, Ca2+, Mg2+, Zn2+, Al3+, Fe3+, Cu2+, Pb2+, Mn2+, Ni2+, Co2+, Cd2+, Hg2+ or Cr6+ ions of 30 μM was added respectively. Then, the intensity of the fluorescence emission spectra (F) was measured at room temperature and contrasted with the fluorescence intensity of the blank solution without metal ions (F0) to determine the selectivity of NCC–EDA for Pb2+. Afterwards, a series of Pb2+ ion solutions with different concentrations (0–50 μM) were prepared, and a small amount of the solution was dropped into the dilution of phosphate buffer of NCC–EDA (2 mL phosphate buffer of NCC–EDA diluted with distilled water to 4 mL). After the resulting solution was mixed well, its intensity of fluorescence emission spectra was measured and the sensitivity of NCC–EDA for Pb2+ was determined by observing the change of the light intensity (F). The sensitivity and selectivity measurements were conducted in triplicate.3. Result and discussion
3.1 Morphology, composition and microstructure of NCC–EDA
Fig. 1 shows the TEM images of the aqueous dispersions of NCC and NCC–EDA.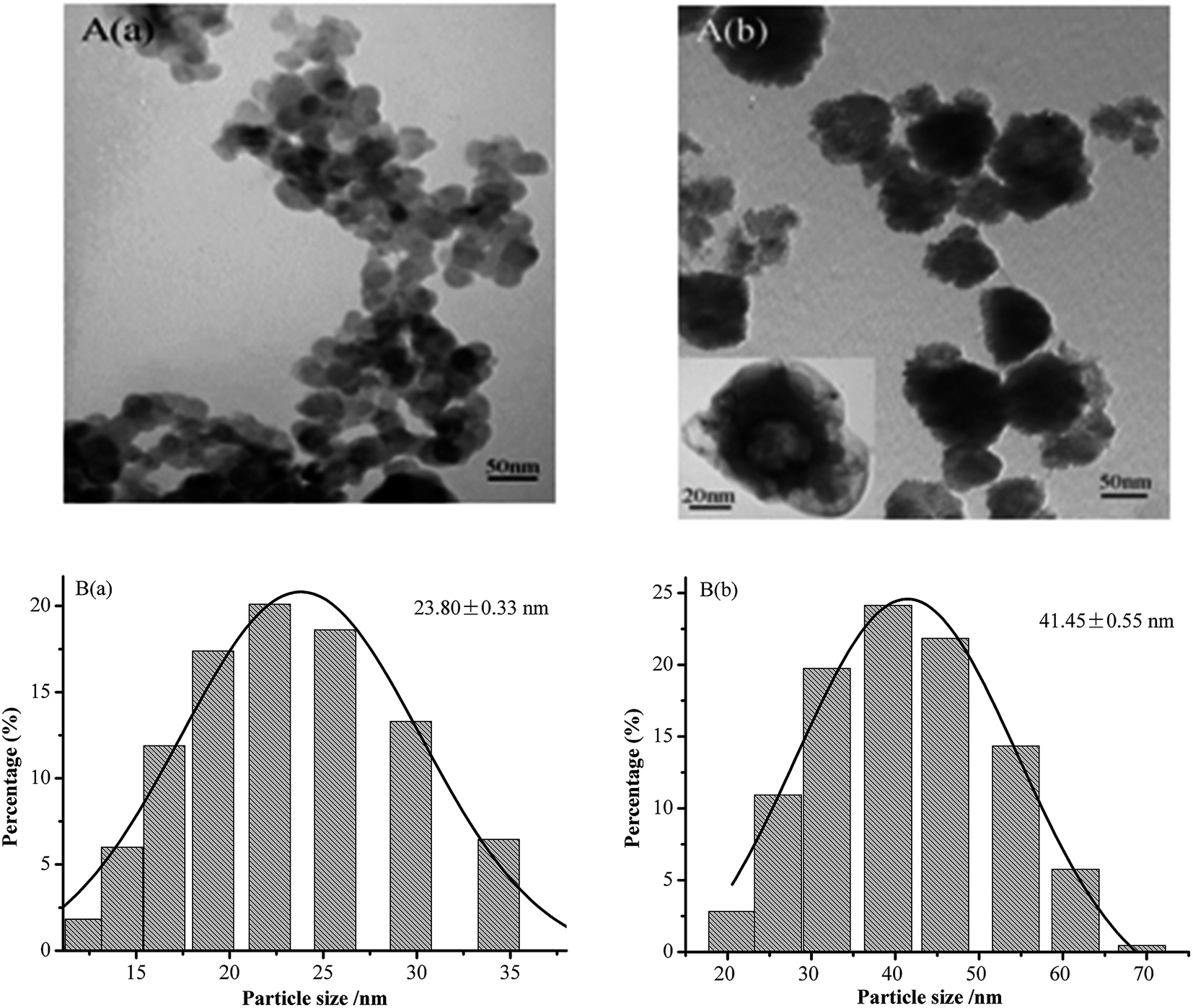 | ||
| Fig. 1 TEM images (A) and size distribution (B) of (a) NCC and (b) NCC–EDA. | ||
As can be seen, the NCC exists in the form of spherical particles with particle sizes of 23.80 ± 0.33 nm, and the formation process includes hydrolysis, dehydrated carbonization, and sulfonation of microcrystalline cellulose under the actions of mixed acids (H2SO4 + HCl) and ultrasound. However, the NCC–EDA modified by EDA are vesicle-like spherical particles (which can be clearly seen from the TEM image magnified by 200000 times (see inset)). They are formed by the linking and intertwining effects of EDA on the SOCl2-treated NCC, and their particle size is 41.45 ± 0.55 nm, about twice the NCC. In addition, NCC–EDA particles can be well dispersed in water instead of aggregating together because of EDA modification.
Table 1 lists the compositions and testing results of the surface characteristics for NCC, NCC–Cl and NCC–EDA.
| Sample | Elemental analysis, w% | S (m2 g−1) | R (nm) | V (cm3 g−1) | |||||
|---|---|---|---|---|---|---|---|---|---|
| C | H | N | S | O | Cl | ||||
| a By difference, C% + H% + N% + S% + O% + Cl% = 100%. | |||||||||
| NCC | 42.22 | 6.34 | 0 | 0.27 | 51.17 | 0 | 2.163 | 19.799 | 7.386 × 10−3 |
| NCC–Cl | 38.15 | 5.41 | 2.79 | 2.26 | 35.26 | 16.13 | — | — | — |
| NCC–EDA | 42.33 | 7.12 | 13.12 | 0.92 | 36.11 | 0.4 | 25.318 | 36.815 | 1.136 × 10−2 |
As the elemental analysis results (Table 1) show, NCC contains a small amount of S, probably due to the introduction of sulfate groups generated by the reaction between sulfuric acid and cellulose. After treatment with SOCl2, the contents of C, H and O in NCC–Cl all decrease, while those of S and N both increase and Cl appears. These indicate that the intermediates from the reaction of NCC with SOCl2 contain both R–Cl and R–SO2Cl, whereas the increase of the N content would probably be due to the remaining DMF in the system. After the intermediates react with EDA, the contents of C, H, and N in the product significantly increase, those S and Cl both decrease, while the O content remains almost the same, indicating that EDA has replaced most of the –Cl and –SO2Cl groups and is linked to NCC. Testing results of the surface characteristics of NCC and NCC–EDA show that the specific surface area, pore size and total pore volume of NCC–EDA are larger than those of NCC, demonstrating that NCC–EDA are particles with porous structure, which is consistent with the TEM results. This type of structure was probably formed by the cross-linking of EDA on the surface of NCC.
To determine the microstructure of the product, FT-IR, XRD and TG-DTA measurements were performed and the results are shown in Fig. 2 and 3.
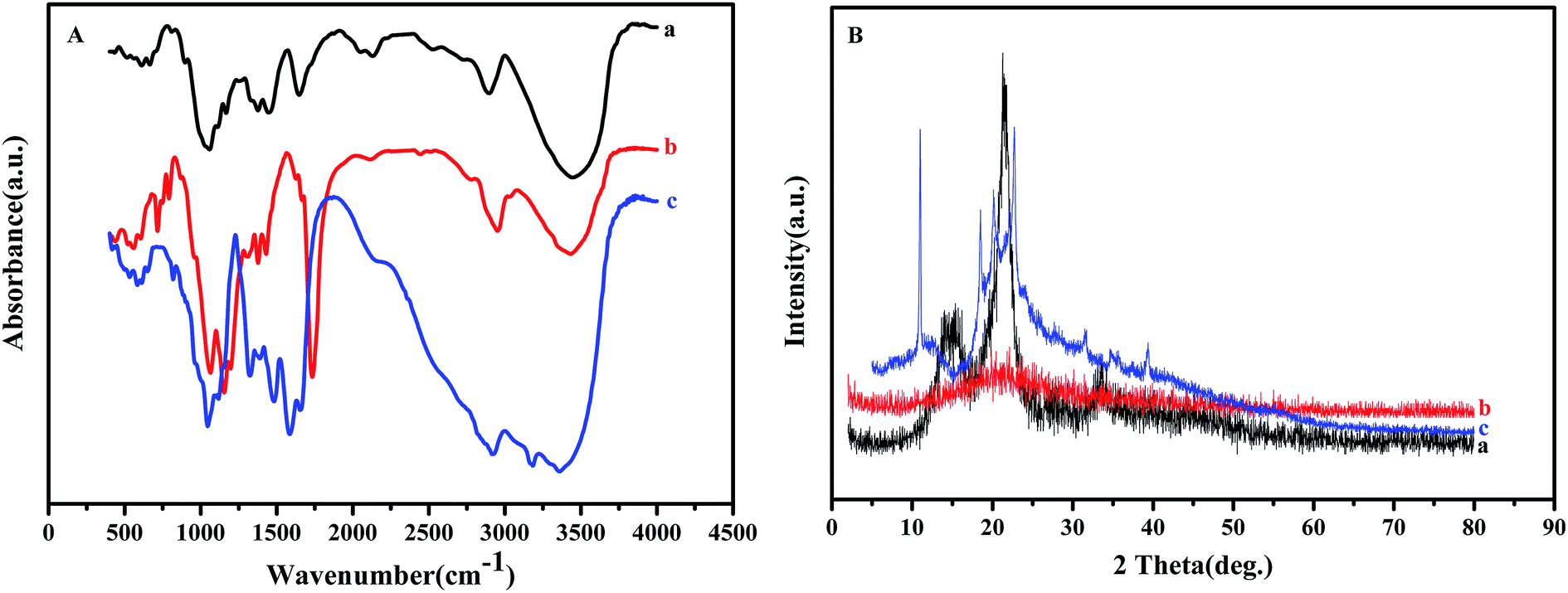 | ||
| Fig. 2 FT-IR (A) and XRD (B) results of (a) NCC, (b) NCC–Cl and (c) NCC–EDA. | ||
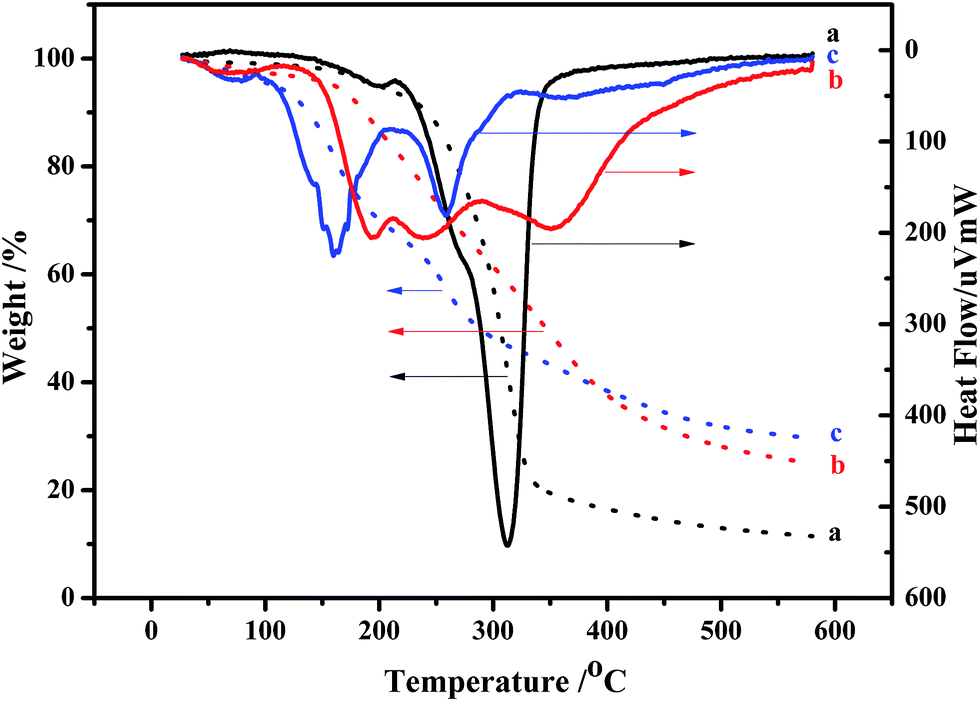 | ||
| Fig. 3 TG-DTA results of (a) NCC, (b) NCC–Cl and (c) NCC–EDA. | ||
Fig. 2(A) shows the FT-IR spectra of NCC, NCC–Cl and NCC–EDA. The FT-IR spectrum of NCC (curve a) exhibits distinct spectral characteristics of cellulose.16 To be specific, the peak at 3445 cm−1 corresponds to the stretching vibration band of the hydroxyl group in cellulose; the peak at 2896 cm−1 is attributed to the stretching vibration band of methylene group (–CH2–); a series of peaks between 1058 and 1167 cm−1 represent the bending vibration band of the ether bond (C–O–C) in cellulose; the peak at 1647 cm−1 is indicative of the absorption band of the hydrogen bonding between the hydroxyl groups and free water; the band between 1444 and 1337 cm−1 reveals the existence of the crystalline structure of cellulose.
Compared with that of NCC, in the FT-IR spectrum of the product obtained by SOCl2 chlorination (curve b), the intensity of the absorption peak of hydroxyl group at around 3400 cm−1 decreases, the peaks corresponding to the absorption bands of –S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –SO2 groups in sulfite appear at 1195 cm−1 and 1311 cm−1 respectively, and the characteristic peaks of the carbon–halogen bond C–Cl show up at 791 cm−1, 753 cm−1, and 716 cm−1. These results are probably attributed to the generation of cellulose sulfonyl chloride R–SO2Cl and HCl through the replacement of hydrogen in the hydroxyl group of NCC by SOCl in SOCl2, and the generation of chlorinated cellulose R–Cl by partial removal of SO2 from R–SO2Cl.17 In addition, the peak corresponding to the stretching vibration absorption of CO and CC bonds appears at 1734 cm−1 in curve (b), probably due to the further oxidization and dehydrated carbonization of NCC caused by excess SOCl2.18,19
O and –SO2 groups in sulfite appear at 1195 cm−1 and 1311 cm−1 respectively, and the characteristic peaks of the carbon–halogen bond C–Cl show up at 791 cm−1, 753 cm−1, and 716 cm−1. These results are probably attributed to the generation of cellulose sulfonyl chloride R–SO2Cl and HCl through the replacement of hydrogen in the hydroxyl group of NCC by SOCl in SOCl2, and the generation of chlorinated cellulose R–Cl by partial removal of SO2 from R–SO2Cl.17 In addition, the peak corresponding to the stretching vibration absorption of CO and CC bonds appears at 1734 cm−1 in curve (b), probably due to the further oxidization and dehydrated carbonization of NCC caused by excess SOCl2.18,19
Compared with curve (b), in the FT-IR spectrum of NCC–EDA (curve c), besides the distinct characteristic peaks of amide group (CONH–) at 3359 cm−1 and 3184 cm−1, the absorption peaks of band I, II and III of amide group appear at 1653 cm−1, 1585.2 cm−1 and 1323.4 cm−1 respectively; meanwhile, the stretching vibration peaks of C–Cl at 791 cm−1 and 716 cm−1, and the stretching vibration peaks of CO and CC bonds at 1734 cm−1 and 1627 cm−1 all disappear, while the absorption peaks at 819 cm−1 and 650 cm−1 corresponding to the O–S bond still exist, demonstrating that amination product has been successfully generated by the reaction of EDA with the intermediates.
Fig. 2(B) shows the XRD patterns of NCC, NCC–Cl and NCC–EDA. NCC exhibits adjacent diffraction peaks at 2θ between 14.2° and 15.6° corresponding to (101) crystal plane; meanwhile, a sharp diffraction peak at 21.5° corresponding to (002) crystal plane, and a diffraction peak at 33.4° corresponding to (040) crystal plane also appear (curve a). These diffraction characteristics belong to a crystalline structure of typical cellulose I, which indicates that the cellulose in NCC is in highly-ordered aggregation state via strong hydrogen bonding between hydroxyl groups.20 However, after treatment with SOCl2, the degree of crystallinity of the intermediates is fundamentally changed and the product turns into amorphous structure completely (curve b). This change can be attributed to the following two aspects: on the one hand, NCC is further degraded by the penetration and corrosion of acidic modification reagent; on the other hand, the intramolecular hydrogen bonding between NCC molecular chains is significantly weakened due to the introduction of the chloride ion and the intramolecular dehydration of cellulose.
Although the crystalline region of NCC is severely destroyed after chlorination, the degree of freedom of molecules will increase, which can enhance the possibility of the recombination of molecular chains. Therefore, after EDA treatment, the diffraction peaks for (101) and (040) planes completely disappear, and NCC–EDA exhibit diffraction peaks corresponding to the structure of cellulose III at 2θ = 11.2°, 18.4°, 20.2° and 22.7°. These demonstrate that EDA treatment leads to the recrystallization of the completely disrupted cellulose structure and promotes the disordered region to gradually form the structure of cellulose III (curve c).21 Moreover, the modification reagent may penetrate into the interior of the cellulose crystalline structure, which can change the cellulose crystal form and enlarge the interplanar spacing; meanwhile, EDA molecules bonded onto the cellulose surfaces may cross-link with each other and form new diffraction planes. As can be seen from curve (c), the degree of crystallinity of NCC–EDA decreases relative to that of NCC; the peak position of (101) plane shifts to smaller angle, and new diffraction peaks appear in the range of 2θ = 31.5–39.4°.
Fig. 3 shows the TG-DTA measurement results of NCC, NCC–Cl and NCC–EDA.
In general, MCC is observed to decompose in the temperature range of 250–420 °C, and the maximum weight loss peak starts at 350 °C.22 However, due to the smaller particle size of NCC relative to that of MCC, higher surface area can be exposed to heat.23 Additionally, the sulfate end groups from acid hydrolysis for NCC extraction have higher susceptibility to oxidative degradation.24 As a result, the NCC prepared herein decomposes at significantly lower temperatures of 150–350 °C, the major loss starts at around 215 °C and ends at about 350 °C, and the maximum degradation temperature of NCC decreases to 312 °C (curve a).
Due to the increase of sulfate groups, the weight loss of NCC–Cl starts at about 130 °C (curve b), which is earlier than that of NCC. Combined with the analysis results of XRD, it can be determined that the structure of the reaction intermediates between NCC and SOCl2 has already loosened, thus decreasing the degree of crystallinity. However, at 600 °C, the weight loss ratio of NCC–Cl is lower than that of NCC, and the content of remaining carbon is 25%, indicating the occurrence of oxidation, dehydrated carbonization in the cellulose during SOCl2 treatment.25,26
The major weight loss of NCC–EDA takes place in two stages (curve c). In the first stage, the weight loss occurs between 107–204 °C, which is caused by the pyrolysis of EDA groups linked to the cellulose surfaces; in the second stage, the weight loss occurs between 225–320 °C, which corresponds to the weight loss peak of the main chain of cellulose. The major weight loss peaks of NCC–EDA start earlier than those of NCC, but the undecomposed residue accounts for 29.3% at 600 °C. This can be attributed to the following two aspects: on the one hand, the main chain of NCC–EDA with the structure of cellulose III obtained through chlorination, oxidation, dehydrated carbonization and ethylenediamine processing has turned into cellulose II with stronger hydrogen bonding after heating,27 which requires more energy to accomplish its complete pyrolysis; on the other hand, the oxygen-containing groups doped with sulfur, chlorine and nitrogen, which are introduced into NCC–EDA by the chlorination of thionyl chloride and the deactivation of ethylenediamine, may play a role of flame retardant.28
To further confirm the above results, the surface state of the NCC–EDA was analyzed by XPS and the results are shown in Fig. 4.
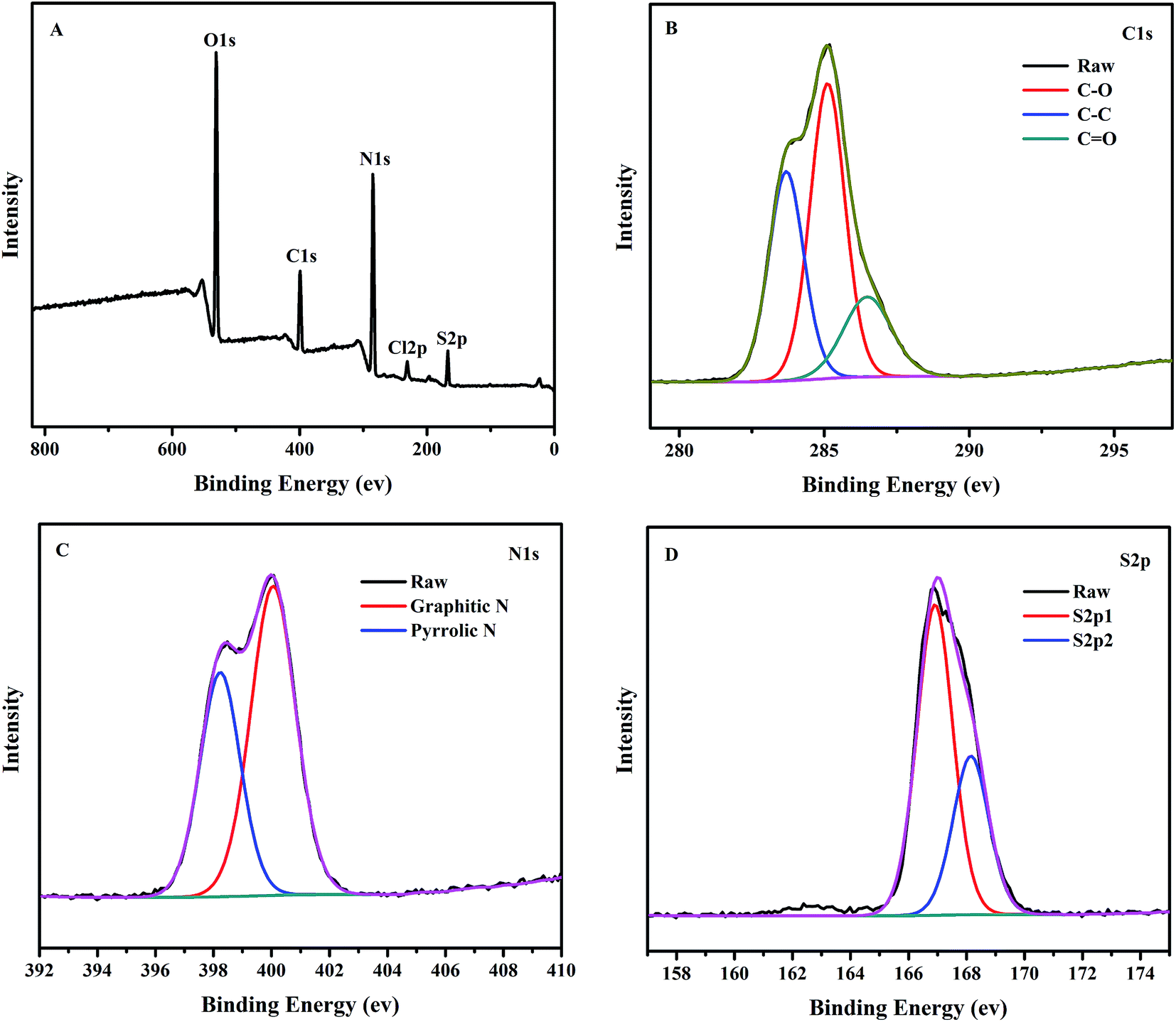 | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) of the NCC–EDA. | ||
As can be seen from Fig. 4(A), five characteristic peaks of NCC–EDA appear at 531.0 eV, 399.8 eV, 285.0 eV, 231.7 eV and 168.1 eV, which correspond to O1s, C1s, N1s, S2p and Cl2p, respectively. This indicates that NCC–EDA is mainly composed of five elements including carbon, nitrogen, oxygen, sulfur and chlorine, respectively with a content of 52.41%, 13.75%, 29.08%, 4.11%, and 0.65%. The XPS results basically agree with the elemental analysis result above, demonstrating that the surface of NCC–EDA has a large number of functional groups containing oxygen, nitrogen and sulfur, together with a small amount of chlorine probably from incomplete substitution of –SO2Cl by EDA.
Fig. 4(B) shows the high-resolution XPS spectrum of C1s. C1s peak splits into three adjacent peaks at 283.6 eV, 285.1 eV and 286.5 eV, which correspond to C–C/CC, C–S and C–N bonds,29,30 respectively. In the high-resolution XPS spectrum of N1s (Fig. 4(C)), N1s peak splits into two adjacent peaks at 398.2 eV and 400.1 eV, which correspond to amide bond (CONH–) and aminolysis positive charge –NH3+, respectively. This result is consistent with the FT-IR result, demonstrating that EDA has been successfully linked to the NCC surface. In the high-resolution XPS spectrum of S2p (Fig. 4(D)), S2p peak splits into two adjacent peaks at 166.9 eV and 168.1 eV, which are characteristic peaks representing some sulfur-containing oxides (–C–SOx– (x = 2, 3, 4)).29 Combined with the analysis results above, it can be deduced that the two peaks are respectively from the sulfate group introduced by the reaction between H2SO4 and the hydroxyl groups in NCC, and the intermediate sulfonyl chloride groups generated by the reaction between NCC and SOCl2.
3.2 Optical property of the NCC–EDA
Fig. 5 shows the absorption and fluorescence emission spectra of the NCC–EDA aqueous dispersion. It can be seen that the NCC–EDA exhibits excellent absorption and fluorescence emission abilities.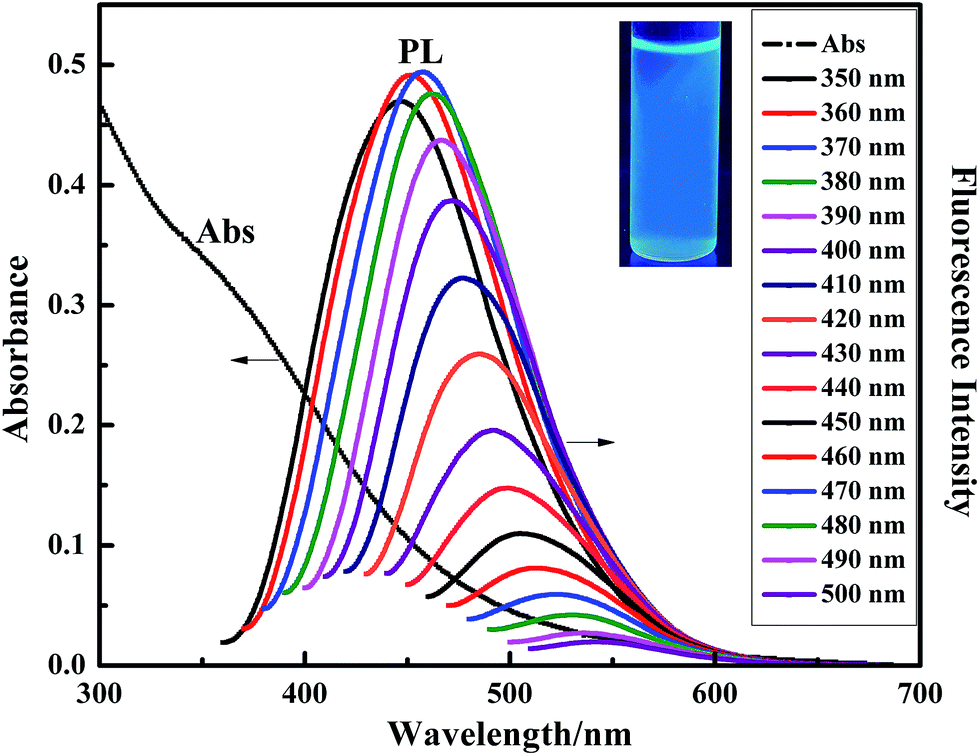 | ||
| Fig. 5 Adsorption and fluorescence emission spectra of the NCC–EDA. | ||
As the UV-Vis absorption spectrum presents, NCC–EDA has absorption in both ultraviolet and visible light regions, and exhibits a distinct absorption band at about 340 nm. When the aqueous dispersion of NCC–EDA is excited by the incident light of 350–500 nm in wavelength, fluorescence emission peaks appear between 448–550 nm, which can be attributed to the surface modification of cellulose nanoparticles.31,32 From the aforementioned experimental results, it can be known that, during the preparation of NCC–EDA, oxygen-containing groups doped with sulfur and nitrogen atoms have been successfully introduced into NCC–EDA due to the chlorination of thionyl chloride and the deactivation of ethylenediamine. These groups can not only generate chromophores on the surface of NCC–EDA, but also act as energy trap sites to trap excitons and induce transitions, thus emitting fluorescence. In addition, with the increase of excitation wavelength, the fluorescence intensity increases first and then decreases; meanwhile gradual redshift can be observed. The optimal excitation wavelength is achieved at 360 nm, and the strongest emission peak appears at about 452 nm. The fluorescence spectral characteristics of NCC–EDA are basically the same as those of other typical luminescent carbon nanoparticles, which depend on the excitation wavelength and have spectral characteristics of multiple excitation and multiple emission.32–35 Moreover, the fluorescence quantum yield of the NCC–EDA aqueous dispersion is determined as 36.82% by using quinine sulfate as reference. Hence, it is easy to observe the strong and stable blue-green fluorescence emitted by the NCC–EDA aqueous dispersion under the UV lamp of 365 nm in wavelength (see inset of Fig. 5).
3.3 Trace detection of Pb2+ ions by NCC–EDA as fluorescent probe
Fluorescent nanoparticles are similar to semiconductor quantum dots, which can change the recombination efficiency of electron–hole pair on the surface and result in the enhancement or quenching of fluorescence through interaction with the targets, thus realizing the qualitative or quantitative analysis of the targets.36 The structure and property of the fluorescence nanoparticles prepared by different methods are significantly different, so that their specific fluorescence responses to ions or other substances may be different.To test the fluorescence responsivity of NCC–EDA synthesized in this paper to metal ions, the effects of 15 common metal ions (K+, Na+, Ca2+, Mg2+, Zn2+, Al3+, Fe3+, Cu2+, Pb2+, Mn2+, Ni2+, Co2+, Cd2+, Hg2+, Cr6+) on the fluorescence intensity of NCC–EDA were studied, and the results are shown in Fig. 6(A).
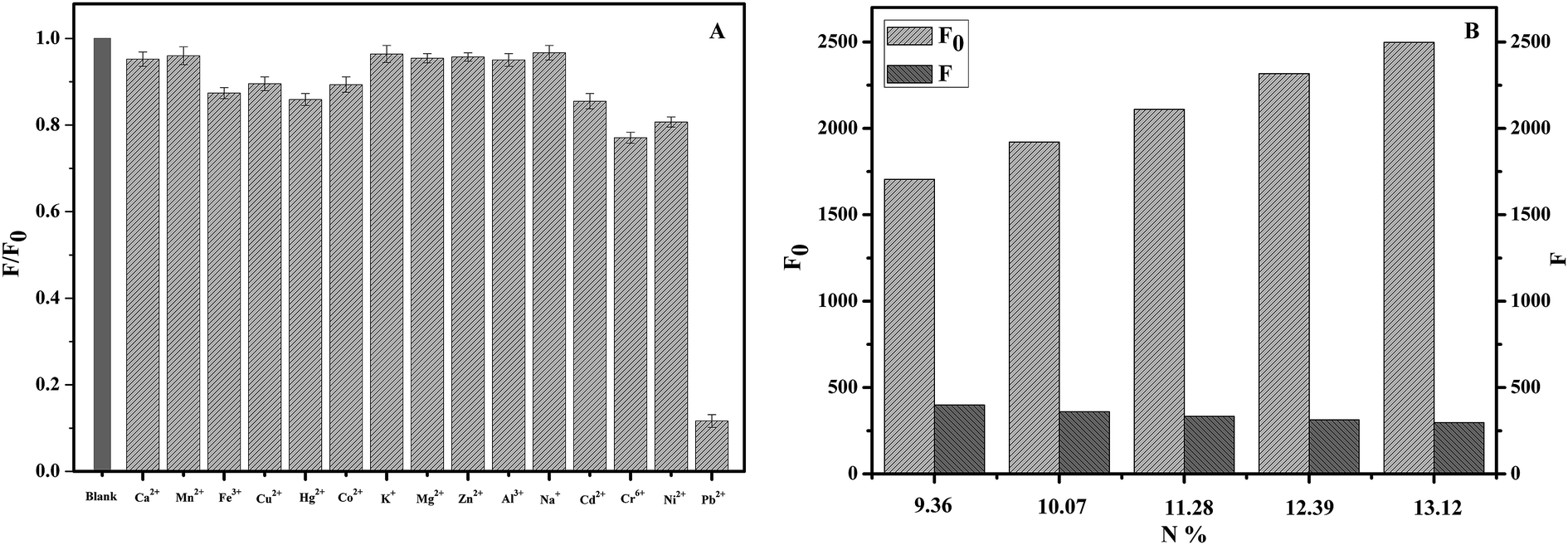 | ||
| Fig. 6 (A) Selectivity of the NCC–EDA as a probe for metal ions in phosphate buffer; (B) effect of Pb2+ on the fluorescence quenching of NCC–EDA with different amino content (y-axis represents the ratio of the fluorescence intensity of NCC–EDA with the addition of Pb2+ (F) to that of NCC–EDA without the addition of Pb2+ (F0); pH = 7; content of the NCC–EDA, 2.5 μg mL−1; concentration of metal ions, 30 μM). | ||
As can be seen, the presence of Ca2+, Mn2+, K+, Mg2+, Zn2+, Al3+ and Na+ does not cause the fluorescence intensity of the NCC–EDA aqueous dispersion to drop. In the presence of Fe3+, Cu2+, Hg2+, Co2+, Cd2+, Cr6+ and Ni2+, the fluorescence intensity slightly decays but not to a large extent. In the presence of Pb2+, the intensity decreases significantly. These indicate that Pb2+ ions have certain selective fluorescence quenching effect on the fluorescence of NCC–EDA.
For clear the mechanism of the cellulose-based fluorescent carbon nanoparticles to Pb2+, a batch of NCC–EDA with different amino content (N content) was synthesized and used to examine the effect of Pb2+ on the fluorescence quenching of it (Fig. 6(B)). Within the scope of the study, the fluorescence intensity of the NCC–EDA was increased with the amino content, however, after the presence of Pb2+, the fluorescence intensity of the NCC–EDA drops with the increase of amino content in the product. The results shows that the effect of Pb2+ ions on the fluorescence quenching of NCC–EDA is significant, the fluorescence quenching associated with the surface characteristics of NCC–EDA and the existence of a large number of oxygen-containing functional groups doped with N and S on the surface. These functional groups can result in the fluorescence quenching of NCC–EDA via strong chelation with Pb2+. In addition, excessive EDTA, a strong chelating agent, is added into the test solutions to chelate with the Pb2+ ions in the solution completely. It is found that the fluorescence intensity of the solution almost recovers to the intensity in the absence of Pb2+ ions. Thus it can be further confirmed that the fluorescence quenching of the NCC–EDA aqueous dispersion is caused by the action between Pb2+ and the amino functional groups on the surface of NCC–EDA.
Fig. 7 demonstrates the influence of pH values of the test solution and reaction time of the NCC–EDA with Pb2+ ions on the fluorescence intensity change ratio (F/F0) of the NCC–EDA.
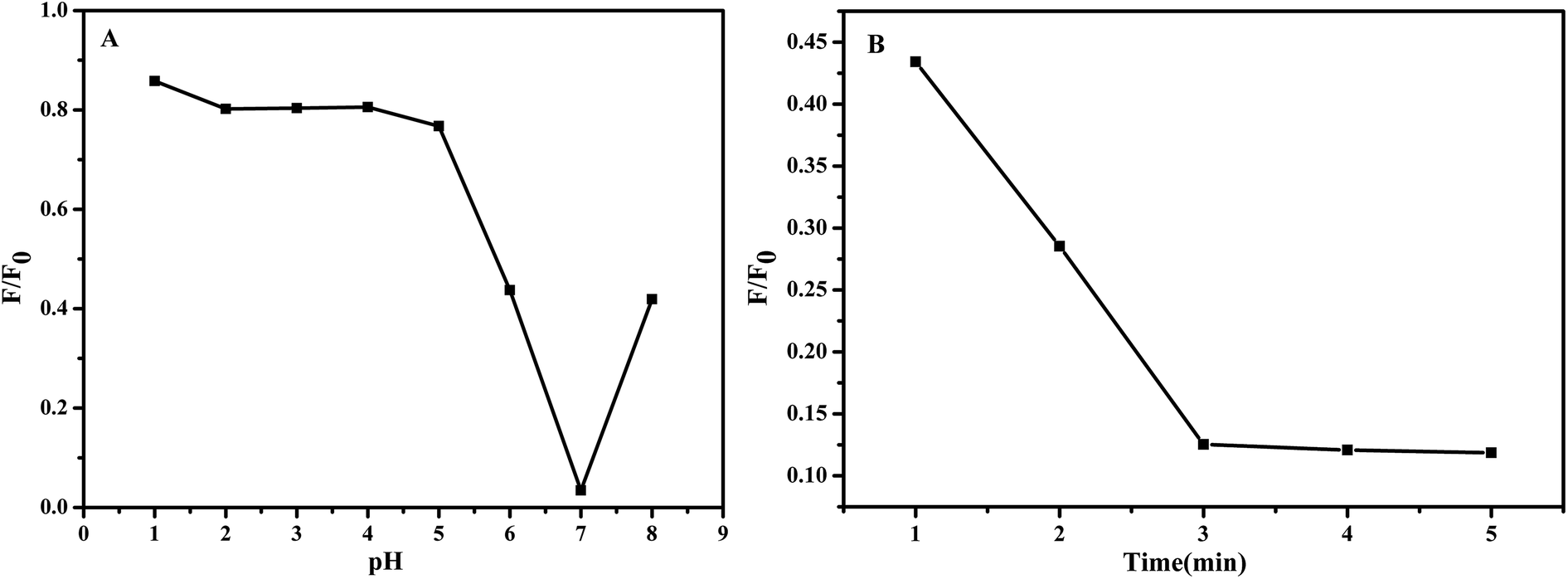 | ||
| Fig. 7 Influence of (A) pH values of the test solution and (B) reaction time of the NCC–EDA with Pb2+ ions on the fluorescence intensity change ratio (F/F0) of the NCC–EDA aqueous dispersion. | ||
As can be seen from Fig. 7(A), in the range of 1–8, the adjustment of pH value of the NCC–EDA aqueous dispersion has significant effect on the chelation of the NCC–EDA for Pb2+ ions. At lower pH values (pH < 5), There may be a protonation occurs between the active sites of NCC–EDA with H+, positive charges repel Pb2+, the binding ability of the active sites of NCC–EDA toward Pb2+ ions will decline, the fluorescence quenching of the NCC–EDA aqueous dispersion is limited; by contrast, when the pH value is higher than 7, Pb2+ may form hydroxide precipitates, the Pb2+ that can form chelate with NCC–EDA and make its fluorescence quenching will be reduced; when pH = 7, the fluorescence intensity of the NCC–EDA exhibits a larger decrease. This shows that the NCC–EDA and Pb2+ ions can form stable chelate under neutral condition. To achieve optimum chelation and prevent the hydroxide precipitation of Pb2+ ions, pH = 7 was selected as the optimum value for chelation of Pb2+ ions. Furthermore, the reaction between NCC–EDA and Pb2+ ions is fast, the fluorescence intensity of the NCC–EDA aqueous dispersion has a significant reduction within 3 min (Fig. 7(B)). This suggests that the nanoprobes with quick response prepared in this work hold great promise for real-time tracking of Pb2+ in environmental system.
Fig. 8(A) displays the dependence of fluorescence intensity change ratio (F/F0) of the NCC–EDA on the concentration of Pb2+ ions as well as the corresponding fluorescence spectra (see inset) after adding different concentrations of Pb2+ ions into the NCC–EDA aqueous dispersion. It can be seen that with the increase of Pb2+ concentration, F/F0 gradually decreases. When Pb2+ concentration is above 30 μM, the value of F/F0 tends to level off with further increase of Pb2+ concentration, which may be due to that the absorption of NCC–EDA toward Pb2+ ions has reached the saturated state.
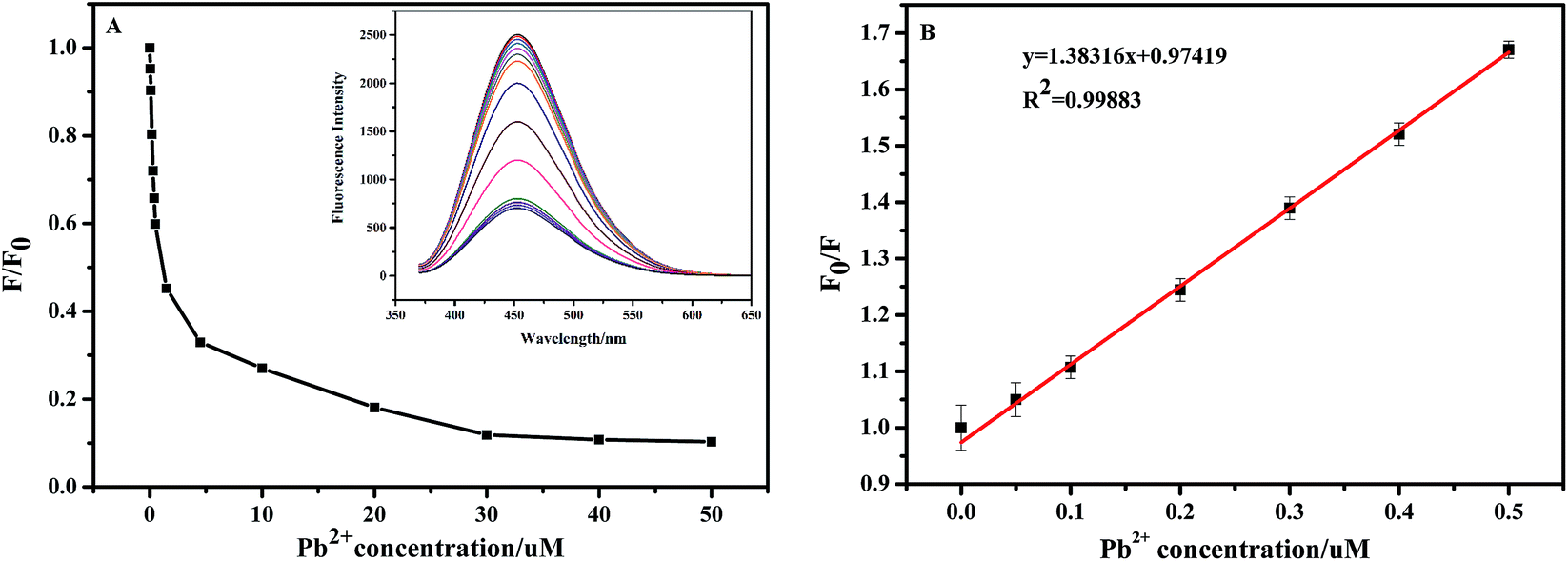 | ||
| Fig. 8 (A) The dependence of F/F0 on the concentration of Pb2+ in the range of 0–50 μM (inset: the corresponding fluorescence response of the NCC–EDA dispersion upon the addition of various concentrations of Pb2+); (B) Stem–Volmer plot for the fluorescence quenching efficiency of the NCC–EDA at lower Pb2+ concentrations of 0.0187–0.5 μM. | ||
The fluorescence quenching efficiency of NCC–EDA at lower Pb2+ concentrations (0.0187–0.5 μM) fits the Stern–Volmer equation: F0/F = l + Ksv[Q], in which Ksv is the Stern–Volmer quenching constant and [Q] represents the Pb2+ concentration. Within this low concentration range, F0/F is linear with Pb2+ concentration (see Fig. 8(B)), and the regression equation is F0/F = 1.38316x + 0.97419 (R2 = 0.99883), with the lowest detection limit of 24 nM.
A comparison was made between our present work and those reported in literature for determination of Pb(II), which was shown in Table 2. As can be seen, the present work has high sensitivity, good linear response range and comparable detection limit to that found in other studies. This result demonstrates that the NCC–EDA synthesized in this experiment can be used as fluorescent probe to detect the lead ions in aqueous solutions.
4. Conclusion
Cellulose-based fluorescent nanoparticles (NCC–EDA) with relatively high fluorescence quantum yield were prepared in this paper. The prepared NCC–EDA were vesicle-like porous particles formed by the linking and intertwining effects of EDA on the SOCl2-treated NCC. During preparation of the NCC–EDA particles, oxygen-, sulfur- and nitrogen-functional groups are introduced via the chlorination with thionyl chloride and subsequent deactivation by using ethylenediamine. This generates chromophores on the surface of the NCC–EDA and sites to trap excitons and to induce transitions. Hence, aqueous dispersion of NCC–EDA that possess strong absorbance and fluorescence.In the presence of trace Pb2+ ions, the prepared NCC–EDA showed selective fluorescence quenching, and the detection limit of Pb2+ ions in water was as low as 24 nM. It was believed that the NCC–EDA hold great promise for real-world sensor applications.
Acknowledgements
The authors are grateful for the financial support of the National Science Foundation of China (21663028).References
- S. C. Ray, A. Saha, N. R. Jana and R. Sarkar, J. Phys. Chem. C, 2009, 113, 18546–18551 CAS.
- J. Schill, A. P. H. J. Schenning and L. Brunsveld, Macromol. Rapid Commun., 2015, 36, 76–83 CrossRef PubMed.
- X. C. Sun, J. Mater. Chem. A, 2016, 4, 103893 Search PubMed.
- P. Zuo, D. Xiao, M. Gao, J. Peng, R. Pan, Y. Xia and H. He, Microchim. Acta, 2014, 181, 1309–1316 CrossRef CAS.
- W. Lu, X. Qin, S. Liu, G. Chang, Y. Zhang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2012, 84, 5351–5357 CrossRef CAS PubMed.
- P. C. Hsu, Z. Y. Shih, C. H. Lee and H. T. Chang, Green Chem., 2012, 14, 917–920 RSC.
- J. Zhou, Z. Sheng, H. Han, M. Zou and C. Li, Mater. Lett., 2012, 66, 222–224 CrossRef CAS.
- M. J. Krysmann, A. Kelarakis and E. P. Giannelis, Green Chem., 2012, 14, 3141–3145 RSC.
- M. Saxena and S. Sarkar, Diamond Relat. Mater., 2012, 24, 11–14 CrossRef CAS.
- D. R. D. S. Souza, J. P. D. Mesquita, R. M. Lago, L. D. Caminhas and F. V. Pereira, Ind. Crops Prod., 2016, 1, 121–128 CrossRef.
- H. Peng and J. Travassejdic, Chem. Mater., 2009, 21, 5563–5565 CrossRef CAS.
- Y. Yang, J. Cui, M. Zheng, C. Hu, S. Tan, Y. Xiao, Q. Yang and Y. Liu, Chem. Commun., 2011, 48, 380–382 RSC.
- H. Li, X. He, Y. Liu, H. Huang, S. Lian, S. T. Lee and Z. Kang, Carbon, 2011, 49, 605–609 CrossRef CAS.
- J. Zhang, New J. Chem., 2010, 11, 1336–1339 Search PubMed.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, V. Georgakilas and E. P. Giannelis, Chem. Mater., 2008, 20, 4539–4541 CrossRef CAS.
- A. Alemdar and M. Sain, Bioresour. Technol., 2008, 99, 1664–1671 CrossRef CAS PubMed.
- K. Oka, Synthesis, 1981, 9, 661–681 CrossRef.
- L. K. Ibrahim, S. M. Hussin and Y. M. Al-obaidi, Int. J. Mater. Chem. Phys., 2015, 1, 99–109 Search PubMed.
- M. Z. Fan, D. Dai and B. Huang, Fourier Transform-Materials Analysis, 2012, vol. 3, pp. 45–68 Search PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 36, 3358–3393 CrossRef PubMed.
- M. Lewin and L. G. Roldan, J. Polym. Sci., Part C: Polym. Symp., 1971, 36, 213–229 CrossRef.
- S. Spoljaric, A. Genovese and R. A. Shanks, Composites, Part A, 2009, 40, 791–799 CrossRef.
- R. S. Cheng and J. S. China, J. South China Univ. Technol., Nat. Sci., 2007, 35, 91–98 CAS.
- K. A. Iyer, A. M. Flores and J. M. Torkelson, Polymer, 2015, 75, 78–87 CrossRef CAS.
- M. Roman and W. T. Winter, Biomacromolecules, 2004, 5, 1671–1677 CrossRef CAS PubMed.
- D. Y. Kim, Y. Nishiyama, M. Wada and S. Kuga, Cellulose, 2001, 8, 29–33 CrossRef CAS.
- A. Sarko and R. Muggli, Macromolecules, 1974, 7, 486–494 CrossRef CAS.
- S. Gaan and G. Sun, J. Anal. Appl. Pyrolysis, 2007, 78, 371–377 CrossRef CAS.
- C. Wang, RSC Adv., 2014, 4, 54060–54065 RSC.
- X. Jin, X. Sun, G. Chen, L. Ding, Y. Li, Z. Liu, Z. Wang, W. Pan, C. Hu and J. Wang, Carbon, 2015, 81, 388–395 CrossRef CAS.
- R. Liu, D. Wu, S. Liu, K. Koynov, W. Knoll and Q. Li, Angew. Chem., Int. Ed., 2009, 48, 4598–4601 CrossRef CAS PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. Shiral Fernando, P. Pathak, M. J. Mezinai, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and M. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- C. Zhu, J. Zhai and S. Dong, Chem. Commun., 2012, 48, 9367–9369 RSC.
- D. Sun, R. Ban, P. H. Zhang, G. H. Wu, J. R. Zhang and J. J. Zhu, Carbon, 2013, 64, 424–434 CrossRef CAS.
- F. Yan, Y. Zou, M. Wang, X. Mu, N. Yang and L. Chen, Sens. Actuators, B, 2014, 192, 488–495 CrossRef CAS.
- D. Zhu, X. Li, X. Liu, J. Wang and Z. Wang, Biosens. Bioelectron., 2012, 31, 505–509 CrossRef CAS PubMed.
- Y. L. Hung, T. M. Hsiung, Y. Y. Chen, Y. F. Huang and C. C. Huang, J. Phys. Chem. C, 2010, 114, 16329–16334 CAS.
- S. S. Wee, Y. H. Ng and S. M. Ng, Talanta, 2013, 116, 71–76 CrossRef CAS PubMed.
- M. Mališić, A. Janošević, B. Š. Paunković, I. Stojković and G. Ćirić-Marjanović, Electrochim. Acta, 2012, 74, 158–164 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |