DOI:
10.1039/C6RA26678A
(Paper)
RSC Adv., 2017,
7, 26361-26369
Inhibition of ADAM17/TACE activity by zinc-chelating rye secalin-derived tripeptides and analogues
Received
11th November 2016
, Accepted 16th April 2017
First published on 17th May 2017
Abstract
“A disintegrin and metalloproteinase 17” (ADAM17), or tumour necrosis factor (TNF)-α converting enzyme, is an upstream target for mitigating TNF-α-mediated inflammation. ADAM17 can be inhibited by chelation of its catalytic site zinc cofactor, which is required for substrate catalysis and structure stabilization. In this study, rye secalin-derived tripeptides (CQV and QCA) and analogues (QCV and QVC) showed zinc-chelating capacity (∼35% at 0.5 μM) and dose-dependently inhibited ADAM17 activity with up to 70% inhibition were observed at 5 μM. Moreover, ADAM17 intrinsic fluorescence emission was quenched by the peptides via the dynamic mechanism, with CQV producing the highest quenching constants. Molecular docking revealed that the tripeptides interacted with ADAM17 active site residues, mostly occupying the S1 and S1′ subsites. CQV had the shortest distance to the zinc cofactor and lowest binding energy. The peptides coordinated zinc through their C-terminal carboxylate anions for QCV, QVC and CQV, and peptide bond carbonyl for CQV. CQV also had more hydrogen bonding with the N, O and H atoms of ADAM17 active site residues but, unlike the other peptides, this did not involve the peptidyl sulfhydryl groups. Interaction with ADAM17 S1′ hydrophobic pockets suggests a possible selectivity of the peptides for ADAM17. Despite their promise as bioactive candidates for controlling inflammation, incubation of THP-1 human monocytic cells with the tripeptide at a concentration that inhibited ADAM17 activity did not result in inhibition of lipopolysaccharide-stimulated TNF-α release.
Introduction
An aberrant immune response to harmful stimuli can result in chronic inflammatory diseases such as rheumatoid arthritis and inflammatory bowel disease, which contribute to the global disease burden.1,2 A popular strategy for treating or managing inflammatory disorders involves the inhibition of tumour necrosis factor (TNF)-α, a pro-inflammatory cytokine whose over-activation is implicated in inflammatory disease onset and progression.3 Different avenues for controlling physiological levels of TNF-α have been studied, and one of them targets the pathway leading to its activation.4 TNF-α is expressed on the cell membrane as an inactive 26 kDa pro-TNF-α, which is then activated via a proteolytic cleavage of the extracellular domain by a zinc-dependent endopeptidase known as “a disintegrin and metalloproteinase 17” (ADAM17) or TNF-α-converting enzyme (TACE).5 This makes ADAM17 a target for controlling inflammatory diseases, and its inhibition is viewed as an upstream target for modulating TNF-α level in the blood.6 Moreover, ADAM17 is a potential target in other chronic diseases notably in cancer, where it is overexpressed leading to the activation of epidermal growth factor receptor (EGFR) signalling pathway by processing EGFR ligands such as transforming growth factor-α.7,8 ADAM17 enzymatic activity can be inhibited by targeting the zinc cofactor present at its catalytic site, which plays a critical role in substrate catalysis and structure stabilization. Several synthetic compounds possessing a zinc-binding ligand inhibit the enzyme, and molecular modelling studies revealed that the ligands coordinate with the catalytic site zinc.9 Attraction of these compounds to the zinc cofactor can also enhance their binding with the enzyme through hydrogen bonds and hydrophobic interactions.9 However, synthetic drug leads have yet to be endorsed for therapeutic usage mostly due to their low bioavailability, lack of clinical efficacy, or potential toxicity.10
Food proteins are natural sources of zinc-chelating peptides,11 which can be explored for ADAM17 inhibition. Moreover, food-derived peptides have exhibited anti-inflammatory activities in different models of inflammation.12 The structural arrangement of amino acids in peptides can be such that it can mimic ADAM17 cleavage sequence (Ala76-Val77) of pro-TNF-α or their neighbouring residues, thereby promoting enzyme–inhibitor interaction. Moreover, some synthetic ADAM17 inhibitors possess a sulfhydryl (SH) group as the zinc-binding ligand, and this can interact with the zinc cofactor leading to disruption of enzymatic activity.13 Notably, cysteinyl SH group is an important zinc-binding ligand especially in endogenous zinc-finger proteins.14 In addition, the C-terminal carboxylate anion, and the carbonyl and imino groups of peptide bonds can also participate in zinc coordination.11,15,16 Therefore, the objectives of this study were to determine the zinc-chelating capacity and ADAM17 inhibitory activity of cysteine-containing rye secalin tripeptides and analogues, and to evaluate the interactions of the peptides with ADAM17 active site using molecular docking and their effect on stimulated TNF-α release in cultured THP-1 human monocytic cells.
Materials and methods
Materials
4-(2-Pyridylazo) resorcinol (PAR), dithiothreitol (DTT) and zinc sulphate heptahydrate (ZnSO4·7H2O) were purchased from Fischer Scientific Co. (Ottawa, ON, Canada); deuterium oxide and recombinant human (rh) ADAM17 expressed in insect cells were purchased from Sigma-Aldrich (Oakville, ON, Canada); and ADAM17 fluorogenic peptide substrate III was purchased from R&D Systems (Minneapolis, MN, USA). Materials for cell culture experiment include THP-1 cells (ATCC, Manassas, VA, USA), RPMI media (Thermo Fisher Scientific, MA, USA), fetal bovine serum (FBS, Seradigm, Providence, UT, USA), lipopolysaccharide (LPS, Enzo Life Sciences, NY, USA) and human TNF-α ELISA kit (R&D Systems, MN, USA).
Peptide production
The signal peptide (f1-19) of rye secalin (UniProtKB/TrEMBL accession number Q9FR41) was removed and the protein chain (f20-455) was then digested in silico with proteinase K (E.C.3.4.21.14) using ExPASy PeptideCutter. Two cysteine-containing tripeptides, CQV (f324-326) and QCA (f343-345), were identified as possible bioactive compounds and then synthesized at 95% purity by GenScript Inc. (Piscataway, NJ, USA). The position of the cysteine residue in CQV was rearranged to obtain the analogues, QCV and QVC, which were also synthesized for structure–function relationship studies.
Determination of zinc-chelating capacity
The tripeptides were evaluated for zinc-chelating capacity as described.17 The assay principle is based on the reaction of 4-(2-pyridylazo) resorcinol with free zinc ions to form a red coordination complex. Solutions of the peptides and reagents were prepared in 40 mM HEPES–KOH buffer (pH 7.5). Then, 250 μL of each sample was mixed with 125 μL of 8 mM DTT and 125 μL of 250 μM ZnSO4·7H2O. Final peptide concentrations were 0.005, 0.05, 0.5 and 5 μM. In the blank experiment, the samples were substituted with an equal volume of the buffer. The mixtures were incubated at 37 °C for 10 min. Thereafter, 25 μL of 2 mM PAR was added to the reaction mixtures to bind free zinc, followed by absorbance measurement at 500 nm. Zinc-chelating capacity was calculated as: [(Ablank − Asample)/Ablank] × 100, where A is absorbance.
Proton nuclear magnetic resonance (1H NMR) spectroscopy
Peptides were mixed with ZnSO4·7H2O at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in deuterium oxide and allowed to stand for 10 min at room temperature before analysis. To study zinc–peptide interactions, 1H NMR spectra of the peptides and their zinc complexes were recorded using Bruker Avance 300 spectrometer (Billerica, MA, USA) comprising of a 5 mm auto-tuning broadband probe with a Z-gradient, at an operating frequency of 300.1 Hz, temperature was stabilized at 25 °C, and a total of 64 scans were performed. The spectra were processed using Bruker TopSin version 1.3 and calibrated based on the residual non-deuterated solvent (δH 4.80). Assignment of protons was done using homonuclear correlation spectroscopy (1H–1H COSY) data.
:10 in deuterium oxide and allowed to stand for 10 min at room temperature before analysis. To study zinc–peptide interactions, 1H NMR spectra of the peptides and their zinc complexes were recorded using Bruker Avance 300 spectrometer (Billerica, MA, USA) comprising of a 5 mm auto-tuning broadband probe with a Z-gradient, at an operating frequency of 300.1 Hz, temperature was stabilized at 25 °C, and a total of 64 scans were performed. The spectra were processed using Bruker TopSin version 1.3 and calibrated based on the residual non-deuterated solvent (δH 4.80). Assignment of protons was done using homonuclear correlation spectroscopy (1H–1H COSY) data.
ADAM17 intrinsic fluorescence emission
ADAM17 intrinsic fluorescence emission was measured in the absence and presence of the peptides using Nova spectrofluorometer (SPEX Industries, NJ, USA) at excitation and emission bandwidths of 5 nm. ADAM17 (final concentration, 0.02 ng μL−1) and tripeptides (final concentration, 0, 0.005, 0.05, 0.5, 1.5, 2.5 and 5 μM) were mixed in a quartz cuvette and equilibrated at 37 °C for 2 min. ADAM17 was substituted with Tris–HCl buffer (25 mM, pH 8.0) in blank experiments. Fluorescence intensity was recorded at the excitation and emission wavelengths of 280 and 300–400 nm, respectively.
Determination of fluorescence quenching constant
Fluorescence quenching constant was determined using the Stern–Volmer equation: F0/F = 1 + kqτ0[Q] = 1 + KSV[Q], where F0 and F are the maximum fluorescence intensities in the absence and presence of the peptides, respectively; KSV is the Stern–Volmer quenching constant; and [Q] is the concentration of the quencher (peptides). The slope of the F0/F versus [Q] plot was taken to be KSV. To determine the quenching mechanism, the bimolecular quenching constant, kq, was calculated from the Stern–Volmer equation as: kq = KSV/τ0, where τ0 is the fluorescence lifetime in the absence of a quencher, and is usually 10−8 s for biological macromolecules.18
ADAM17 inhibition assay
ADAM17 inhibitory activity was determined at 0.005, 0.05, 0.5 and 5 μM peptide concentrations prepared in 25 mM Tris buffer (pH 8.0). In a 96-well microplate, 25 μL of 0.4 ng μL−1 rh ADAM17 was mixed with equal volumes of the peptide solutions and 10 μM ZnSO4·7H2O. The control experiment contained all assay components except the peptides, which were substituted with an equal volume of buffer. The reaction mixtures were equilibrated at 37 °C for 5 min, followed by the addition of 25 μL of 40 μM ADAM17 fluorogenic peptide substrate, Mca-PLAQAV-Dpa-RSSSR-NH2. Fluorescence intensity was measured at excitation and emission wavelengths of 320 nm and 405 nm, respectively, every 30 s for 5 min. ADAM17 enzymatic activity was determined as the rate of release (R) of the fluorescent reaction product. Percentage enzyme inhibition was calculated as: (R0 − R/R0) × 100, where R0 and R are the reaction rates in the absence and presence of the peptides, respectively.
Molecular docking
Molecular docking was performed using the Accelrys Discovery Studio software 2.5 (DS 2.5). The tripeptide structures were generated with DS 2.5, and energy was minimized with the CHARMm program. A crystal structure of the catalytic domain of ADAM17 (1bkc, Protein Data Bank) was used for docking. A binding site with a radius of 10 Å and coordinates, x: 4.263; y: −4.2; and z: 5.617, was created in the subunit I of ADAM17. Automated molecular docking was performed using the partial flexibility CDOCKER tool of the DS 2.5 software in the presence of zinc cofactor. Evaluation of the molecular docking was done according to the scores and binding energy values in order to obtain the best peptide poses. DS 2.5 software was also used to identify hydrogen bonds as well as hydrophobic, hydrophilic and coordination interactions with amino acid residues in the enzyme active site.
THP-1 cell culture and LPS-induced TNF-α release
THP-1 cells were maintained in RPMI media containing 10% FBS and incubated at 37 °C, 95% humidity, and 5% CO2. To examine TNF-α release, cells were plated in 24-well plates. One h after plating, cells were incubated with media containing 2 μg ml−1 LPS and 5 μM tripeptides as indicated for 3 h. Thereafter, media was collected and spun at 10000 × g for 10 min. Levels of released TNF-α were determined in the supernatant using ELISA selective for human TNF-α.
Statistical analysis
All experiments (except peptide production, NMR and docking) were done in triplicate, and results were expressed as mean ± standard deviation. Significant differences (P < 0.05) between results were determined by one-way analysis of variance followed by a Holm–Sidak multiple comparison test. Statistical analyses were done using SigmaPlot 12.1 (Systat Software, San Jose, CA, USA) or Prism (GraphPad Software, La Jolla, CA, USA).
Results and discussion
Rye proteins as a source of bioactive peptides
Rye (Secale cereale L.) is a staple food in the human diet and accounts for 1% of the world's total cereal production.19 The major rye proteins are secalins including γ-40k, γ-75k, ω and high molecular weight secalins, which are among the plant storage proteins known as prolamins.20 Similar to prolamins in other cereal grains such as wheat and barley, secalins are regarded to be of poor nutritional value due to their low content of the essential amino acids lysine, tryptophan and threonine.21 Moreover, prolamins are constituents of gluten, which can evoke autoimmune response in the intestine of people with celiac disease.22 Valorization of prolamins can be achieved through enzymatic processing and fermentation to produce peptides that can be bioactive and potentially useful for human health promotion.23,24 This process can also deactivate antigenic epitopes in the cereal proteins since the smallest currently known antigenic gluten peptides are nonapeptides.25 Moreover, cereal grain proteins are thought to be sources of peptides (e.g. lunasin, VPP, IPP, LQP, LLP, etc.) that possess activities for combating chronic diseases such as cancer, diabetes and cardiovascular diseases.19,24–27
Bioactive peptide discovery can be facilitated by the use of bioinformatics, which can aid the identification of sustainable food protein precursors of peptides, proteases for their release, and prediction of their biological activities.28 For instance, ACE-inhibitory peptide sequences have been identified in rye secalins using this approach.29 In silico hydrolysis of rye gluten proteins resulted in the release of tripeptides, QCA and CQV, by proteinase K from f343-345 and f324-326 of rye secalin, respectively. The protease has potential for use in food processing considering its broad cleavage specificity at wide pH and temperature ranges,30 to ensure the deactivation of antigenic sequences. The secalin tripeptides used in this study are not part of the antigenic sequences of gluten known to be involved in celiac disease. Analysis of secalin with ZincExplorer revealed several possible zinc-binding sites, each with scores of <0.4 (0 for no binding and 1 for strong binding), but only QCA and CQV were released as small peptides by proteinase K, with zinc-binding cysteine residue scores of 0.36559 and 0.15098, respectively. Moreover, glutamine can also participate in zinc chelation; therefore, the tripeptides were further evaluated for in vitro zinc-chelating capacity at physiological pH and temperature. Food-derived bioactive di- and tripeptides are thought to have strong prospects for translation as active components of functional foods, compared to larger peptides, as they are more stable to gut proteolytic deactivation due to their lower number of scissile bonds, and can be transported across the intestine into the blood via dedicated transporters.31
Zinc-chelating capacity of the tripeptides
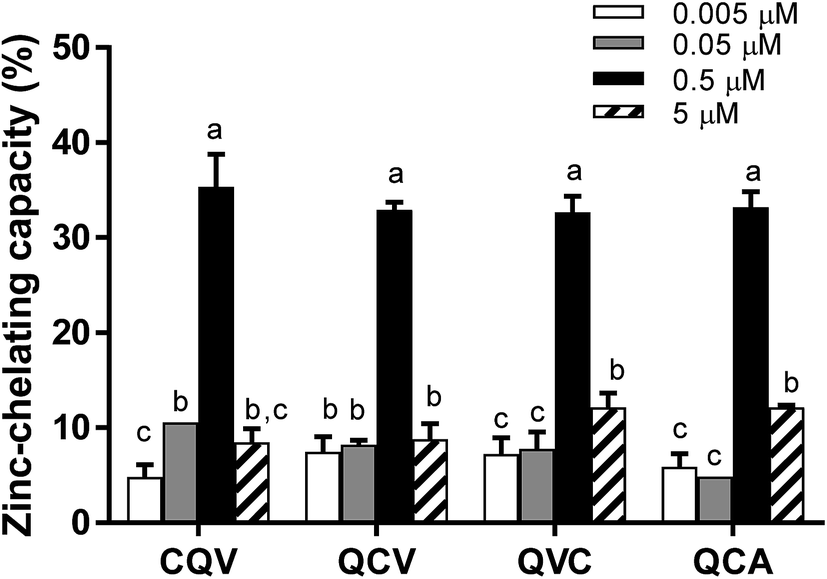
The tested tripeptides exhibited a dose-dependent increase in their zinc-chelating capacity, with the highest activity observed at 0.5 μM (Fig. 1). However, zinc chelating capacity dropped by 63–75% at 5 μM. This is possibly due to increased proximity of the peptides in solution, which can facilitate intermolecular interactions leading to the depletion of the zinc-binding sites. The arrangement of amino acid residues in a peptide chain can influence their chemical behaviour and biological activities,32 including zinc-chelating capacity.11 For instance, the presence of a histidine residue at the N-terminal position enhanced the metal-chelating capacity of peptides.33 In our study, structural differences (composition and sequence arrangement) did not significantly alter the zinc-chelating capacity of the secalin tripeptides and analogues. Other di-, tri- and tetrapeptides derived from sesame and rapeseed proteins using trypsin and Alcalase, respectively, were reported to have 34–82% zinc-chelating capacity at higher concentrations, with tripeptides NCS (sesame) and NSM (rapeseed) exhibiting the highest zinc chelation.34,35 Based on zinc chelation, the cysteine-containing tripeptides are expected to inhibit the catalytic activity of ADAM17 by interacting with its zinc cofactor.
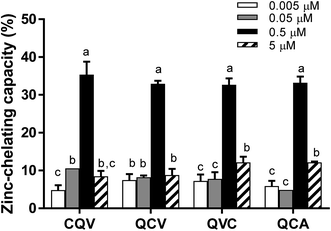 |
| | Fig. 1 Zinc-chelating capacity of secalin-derived tripeptides and analogues. Peptides were mixed with ZnSO4 followed by incubation for 10 min, addition of 4-(2-pyridylazo) resorcinol to react with free zinc, and absorbance measurement at 500 nm. Bars in each chart of a group of peptides with different letters represent significantly different mean values with P < 0.001. | |
NMR characterization of the peptide and their complexes
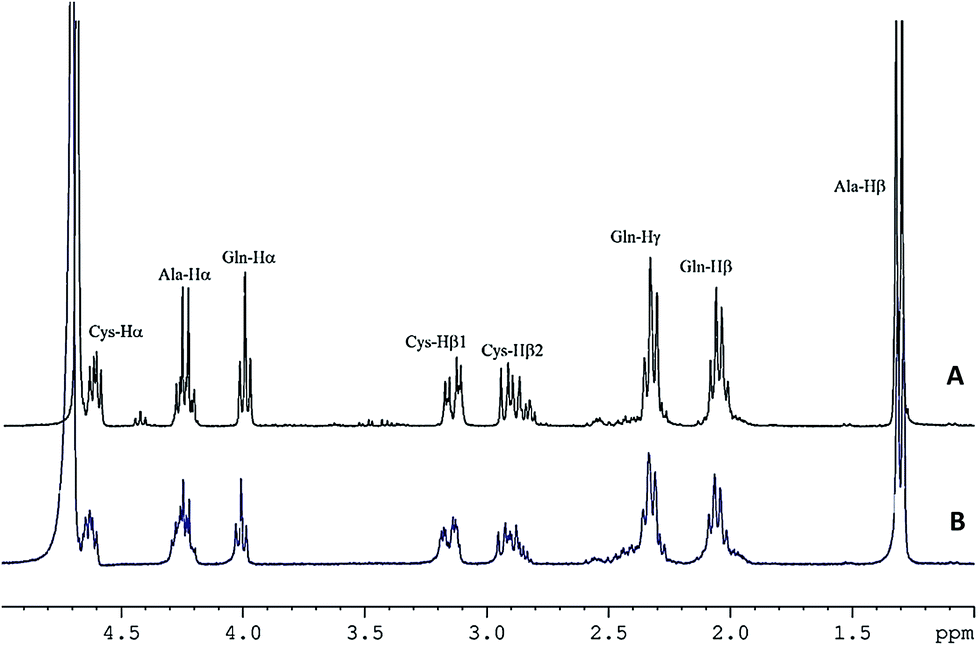
NMR spectra of the peptides and their complexes were obtained to confirm their binding and to gain insight on the peptidic ligands that participated in zinc chelation. 1H NMR spectra effectively showed that complexes were formed and that their proton signals overlapped with those of the unbound peptides. For instance, in the 1H NMR spectrum of QCA (Fig. 2A), protons of the methylene group of cysteine appeared as doublet doublets at δ 2.91 ppm (Cys-Hβ1, J = 8.7, 14.4 Hz) and 3.15 ppm (Cys-Hβ2, J = 5.1 and 14.4 Hz). In the Zn–QCA complex (Fig. 2B), the protons had similar chemical shifts but they appeared as multiplets, hence coupling constants could not be determined. The methine proton of cysteine in QCA appeared at δ 4.62 (Cys-Hα, dd, J = 5.1 and 8.7 Hz) compared to δ 4.63 ppm (Cys-Hα, m) in the free and Zn–QCA, respectively, which suggests a change in proton environment in the presence of zinc. However, ionic strength and pH of the chelating assay could not be used in the NMR study. The labile protons of SH, NH and COOH were not detected due to their exchange with deuterium from the solvent, and it was therefore not possible to identify these zinc-binding sites.
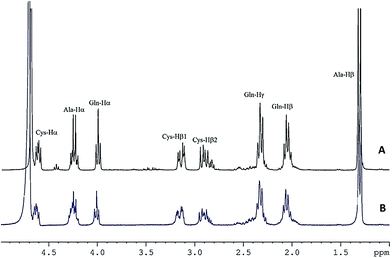 |
| | Fig. 2 1H NMR spectra of (A) QCA and (B) Zn–QCA at peptide–zinc molar ratio of 1:10 in deuterium oxide. Proton signal assignment was done using 1H–1H COSY. | |
Effect of the tripeptides on ADAM17 fluorescence emission
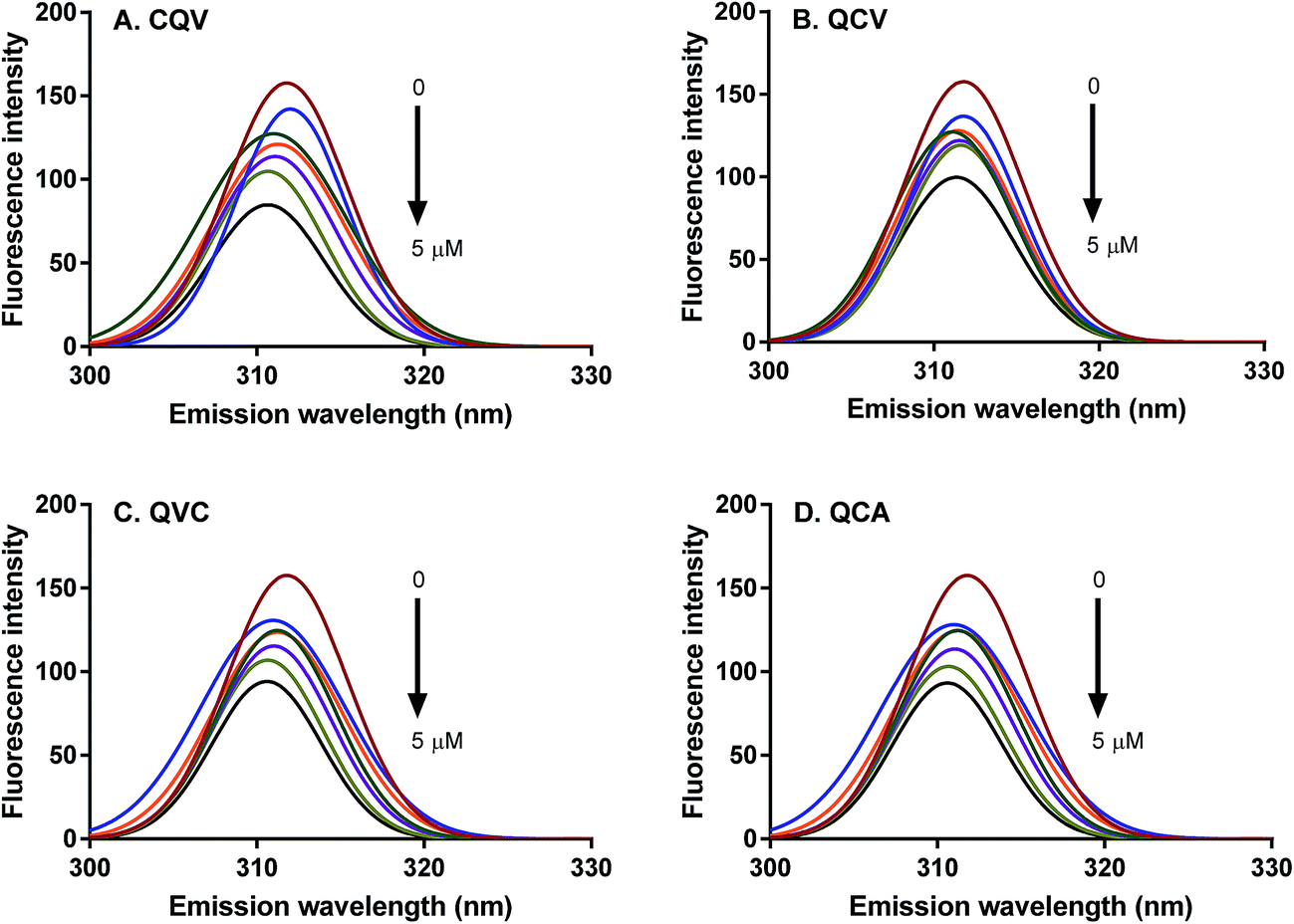
Proteins intrinsically emit fluorescence due to their aromatic amino acid residues, tryptophan, tyrosine and phenylalanine.36 The fluorescence emission spectra of these residues provide information on their microenvironment, the conformational state of proteins, and binding interactions between proteins and their ligands. ADAM17 region starting from the catalytic to the cysteine-rich domains (Arg215-Asn671) contains three tryptophan and many tyrosine residues, which are the protein fluorophores that absorb at 280 nm.37,38 The fluorescence emission of these fluorophores, particularly tryptophan, is dependent on the polarity of their environment. The wavelength of maximum fluorescence emission (λmax) of ADAM17 was observed at 310 nm (Fig. 3); a similar emission wavelength of 316 nm was previously reported for ADAM17 catalytic domain, and indicates that the fluorophores are buried in hydrophobic cores.39 In the presence of the tripeptides, no substantial shift was observed in ADAM17 λmax, which suggests that microenvironments of tryptophan and tyrosine residues were largely unperturbed, hence, conformational changes such as protein unfolding did not occur in the presence of the peptides.40 However, ADAM17 fluorescence emission was dose-dependently quenched by 9–40% in the presence of the peptides (Fig. 3), with CQV producing the highest effect as shown by the magnitude of the quenching constant, KSV (Table 1). The occurrence of quenching is evidence that the peptides were in direct contact with the fluorophore(s) or they were close enough to quench the fluorescence.18 Thus, CQV must have better interaction with ADAM17 despite its structural similarity to analogues QCV and QVC. Therefore, the relative position of amino acid residues in the peptides affected their interaction with ADAM17, since they are likely to be positioned differently on the enzyme structure. Fluorescence quenching occurs by two main mechanisms, which are dynamic (or collisional) and static quenching.18 Dynamic quenching occurs at the excited state of the fluorophore, where it interacts with the quencher and returns to the ground state without photon emission, thereby decreasing the fluorescence intensity. Conversely, static quenching results from complex formation between the quencher and the fluorophore at the ground state, which consequently decreases the number of fluorophores and hence the fluorescence intensity. The type of quenching occurring in a given system can be determined from Stern–Volmer equation by calculating the bimolecular quenching constant (Kq). The maximum scatter collision quenching constant of various quenchers for biomolecules is taken to be less than 2 × 1010 M−1 s−1 for collisional mechanism and greater for static quenching.41,42 Also, both mechanisms can occur concurrently. Linear Stern–Volmer plots were observed in this study, suggesting that the quenching of ADAM17 fluorescence emission by the tripeptides occurred mostly by one mechanism, and that the enzyme fluorophores were accessible to the quenchers. Moreover, the kq values of the peptides ranged from 6.5–9.9 (×106 M−1 s−1) (Table 1), indicating that the peptides quenched ADAM17 fluorescence emission by the dynamic mechanism. This demonstrates that the tripeptides interacted with ADAM17 at the excited state, which can lead to inhibition of enzymatic activity.
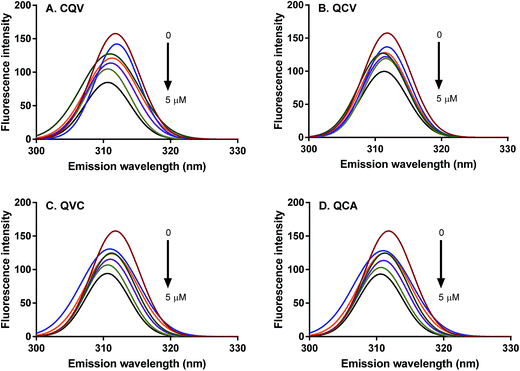 |
| | Fig. 3 Fluorescence emission spectra of ADAM17 showing the quenching effect of tripeptides (A) CQV (B) QCV, (C) QVC and (D) QCA each at 0, 0.005, 0.05, 0.5, 1.5, 2.5 and 5 μM tripeptide concentrations. | |
Table 1 Stern–Volmer fluorescence quenching constants for interaction of rye secalin-derived tripeptides with ADAM17a
| Peptide |
KSV (×103 M−1) |
kq (M−1 s−1) |
| Numbers in each column with different superscript letters represent significantly different mean values with P < 0.001. |
| CQV |
99.7 ± 0.0052a |
9.97 × 106a |
| QCV |
65.0 ± 0.0078b |
6.50 × 106b |
| QVC |
69.8 ± 0.0058b |
6.98 × 106b |
| QCA |
72.0 ± 0.0005b |
7.20 × 106b |
Effect of the tripeptides on ADAM17 enzymatic activity
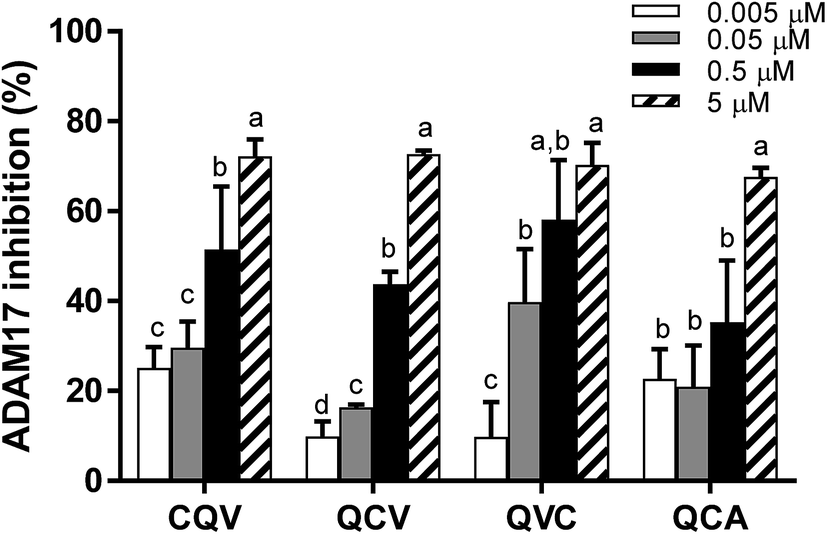
ADAM17 enzymatic activity significantly decreased in the presence of the tripeptides; hence, the enzyme–peptide interaction observed in the fluorescence quenching study resulted in inhibition. As shown in Fig. 4, the peptides dose-dependently inhibited ADAM17, with the highest activity (70%) observed at 5 μM. The inhibitory capacity of the peptides at this concentration does not correlate with the zinc-chelating capacity, which suggests that, although zinc chelation can play a role in ADAM17 inhibition, other molecular interactions with the enzyme are likely involved. This is the first report of ADAM17 inhibitory activity of food protein-derived peptides. TAPI-1, a hydroxamate-containing ADAM17 inhibiting peptide, used as a positive control, inhibited 0%, 19% and 45% of ADAM17 activity at 0.001, 0.01 and 0.1 μM, respectively. However, cysteine-containing endogenous tripeptide, glutathione, was inactive at the concentration (5 μM) at which the food peptides inhibited the activity of ADAM17. The tested tripeptides are small enough to access and occupy ADAM17 active site and have potential as desirable inhibitory molecules because their small sizes may make them bioavailable.43,44 Some studies have demonstrated that enterally administered peptides are transported across enterocytes into the blood stream via peptide transporter, PepT1.45 The peptides can also exert anti-inflammatory activity in the gut in conditions such as inflammatory bowel disease.
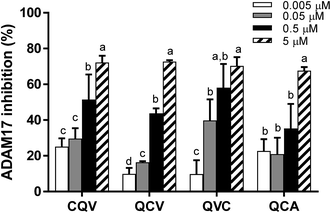 |
| | Fig. 4 ADAM17 inhibitory activity of rye secalin-derived tripeptides and analogues at 0.005–5 μM. Peptide samples were incubated with rh ADAM17 and its fluorogenic peptide substrate in the presence of ZnSO4 followed by fluorescence measurement for 5 min. Bars in each chart of a group of peptides with different letters represent significantly different mean values with P < 0.001. | |
Structure–function relationship of the tripeptides
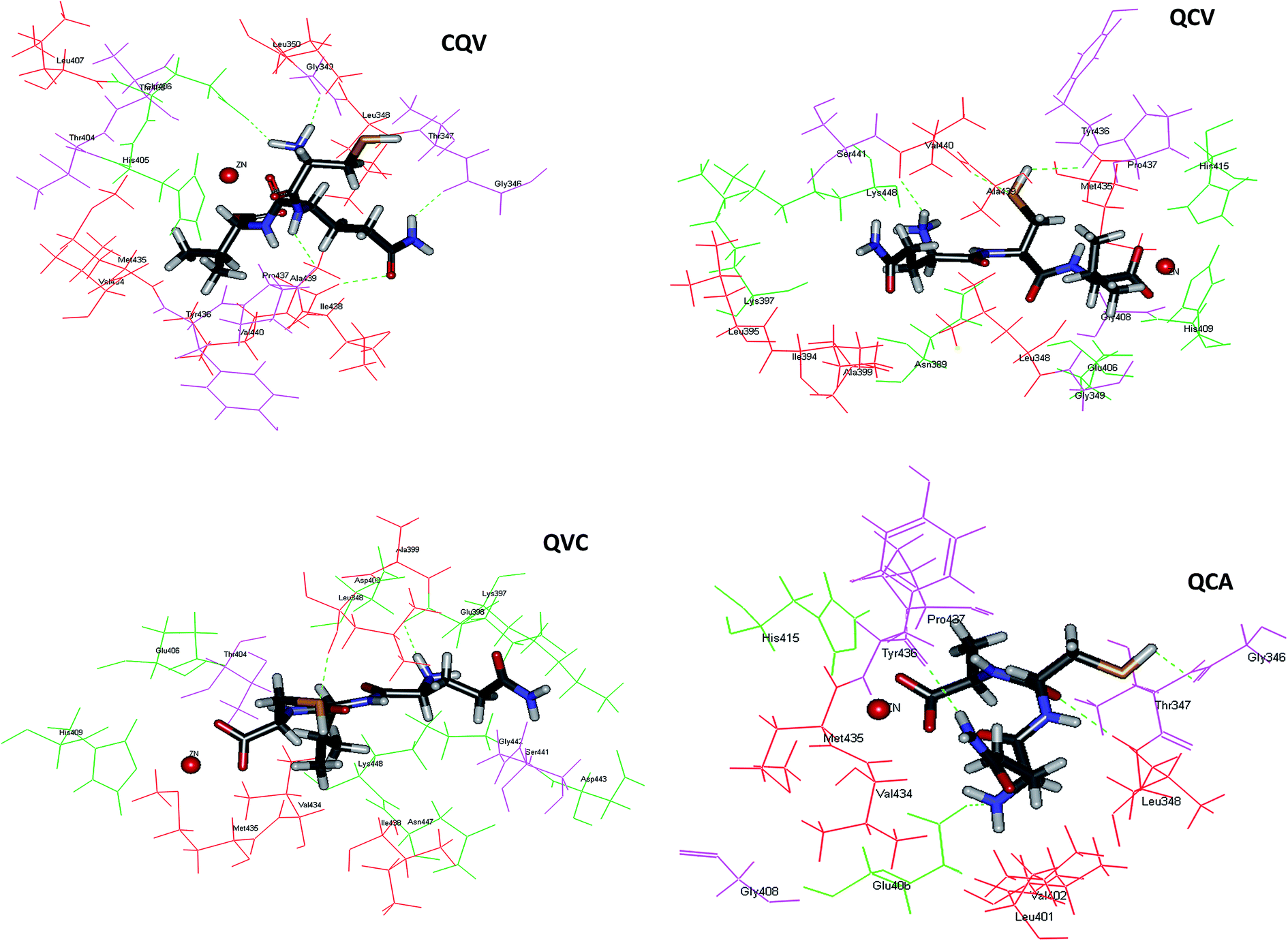
Application of food-derived peptides as ADAM17 inhibitors would be facilitated by the understanding of their structure–function relationship. The mode(s) of interaction of the tripeptides with ADAM17 was elucidated by molecular docking. The peptides were stabilized in the active site of the enzyme by a combination of zinc coordination, hydrogen bonds and hydrophobic interactions, and mainly occupy the S1 and S1′ subsites of the enzyme. These binding modes have been reported for synthetic ADAM17 inhibitors.9 The most preferred conformations of the tripeptides in the enzyme active site are shown in Fig. 5. As shown for synthetic ADAM17 inhibitors bearing carboxylate group as their zinc-binding ligand,46 the C-terminal carboxylate anions of QCA, QCV and QVC in our study served as bidentate ligands. Thus, zinc was in pentagonal coordination geometry with the three catalytic site histidine residues and the two oxygen atoms of the tripeptide carboxylate groups (Fig. 5). The spatial orientation of the C-terminal carboxylate anion of CQV was such that its interaction with zinc was prohibited. Therefore, zinc coordination occurred through the carbonyl oxygen of the peptide bond between glutamine and valine residues. The distance between zinc and tripeptide donor atoms ranged from 2.145 to 2.339 Å, with CQV maintaining the closest interaction and having the least binding energy (Table 2). This indicated a more stable zinc coordinate complex with CQV and the proximity explained its pronounced ADAM17 fluorescence quenching effects.
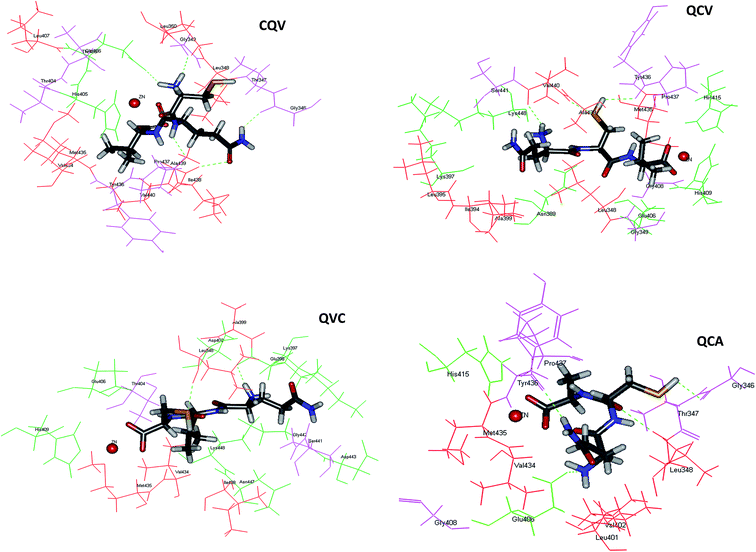 |
| | Fig. 5 Molecular interactions between ADAM17 and the tripeptides after automated docking at ADAM17 active site; ADAM17 hydrophobic and hydrophilic residues are shown in red and green, respectively; hydrogen bonds are represented as green dotted line; the red ball is zinc atom; the four tripeptides are represented in stick models; image were obtained with Accelrys DS Visualizer software. | |
Table 2 Predicted binding energies and coordination distances of the tripeptides with ADAM17 zinc cofactor
| Peptide |
Zinc coordination |
CDocker energy |
| Atom |
Distance (Å) |
| CQV |
O2 |
2.145 |
47.1495 |
| QCA |
O21, 17 |
2.339, 2.236 |
86.2813 |
| QCV |
O17, 23 |
2.320, 2.304 |
66.8699 |
| QVC |
O18, 23 |
2.243, 2.243 |
55.5615 |
ADAM17 is auto-regulated by a mechanism that was suggested to involve its Asp-Asp-Val-Ile (f134-137) motif, which is not structurally related to the secalin tripeptides and analogues. Moreover, matrix metalloproteinases (MMPs) have a prodomain cysteine-switch motif, containing Cys184 for ADAM17, which bind the zinc ion cofactor and is responsible for enzyme auto-inhibition.5 The cysteine-containing tripeptide in this motif (VCG, f183-185) for ADAM17 is not structurally related to the secalin tripeptides, except for the presence of the zinc-binding cysteine residue. The tripeptide cysteine SH moiety, the key zinc ligand, was observed to be located away from the zinc cofactor and therefore did not participate in zinc chelation. However, both the hydrogen and sulphur atom of the SH in QCA, QCV and QVC acted as either hydrogen bond donor or acceptor for binding with the S1′ subsite residues, Gly346, Tyr436, Val440 and Leu348, with similar bond distances (Fig. 5, Table 3). Other hydrogen bond donors and acceptors in QCA, QCV, and QVC include the hydrogen atom of the N-terminal amino groups, the oxygen atom of the peptide bond, and the side chain NH of glutamine amide group (Table 3). CQV had the highest number of hydrogen bond contacts but, unlike the other peptides, its SH did not form hydrogen bonds with the enzyme. The hydrogen bonding between the peptides and Glu406 (Table 3), which serves as a base during substrate catalysis,47 indicated a possible disruption of the highly conserved zinc structural motif and could explain the inhibitory activity. Peptides interactions with ADAM17 Tyr436 and Tyr433 as well as the residues in their vicinity viz. Gly431, Lys432, Val434, Met435, Pro437, Ile438, Ala439 and Val440 can partly explain the enzyme fluorescence emission quenching. Interaction of ADAM17 inhibitors with the S1′ hydrophobic pocket has been proposed to be key to achieving a selective inhibition and possibly eliminating physiological side effects.48,49 Therefore, data from this study provide important insight on peptide affinity and their possible selectivity for the enzyme.
Table 3 Hydrogen bonds observed between ADAM17 and top-ranked pose of the docked tripeptides
| ADAM17 residues in H-bonding |
Number of H-bonds and their corresponding distance (Å) |
| CQV |
QCA |
QCV |
QVC |
| GLY346:O:H46 |
1(2.04159) |
|
|
|
| GLY346:O:H40 |
|
1(2.49284) |
|
|
| LEU348:HN:S22 |
|
|
|
1(2.22821) |
| LEU348:HN:O11 |
|
1(2.38225) |
|
|
| GLY349:O:H41 |
1(1.66522) |
|
|
|
| GLU398:O:H40 |
|
|
|
1(1.88054) |
| GLU406:OE2:H42 |
1(1.86224) |
|
|
|
| GLU406:OE2:H36 |
|
1(1.80084) |
|
|
| TYR436:O:H46 |
|
|
1(2.27011) |
|
| TYR436:O:H38 |
|
1(2.25238) |
|
|
| PRO437:O:H44 |
1(2.12058) |
|
|
|
| ALA439:HN:O14 |
1(2.45995) |
|
|
|
| VAL440:HN:S15 |
|
|
1(2.18480) |
|
| VAL440:O:H41 |
|
|
1(1.93199) |
|
| Total |
5 |
4 |
3 |
2 |
Effect of the tripeptides on TNF-α release from THP-1 cells
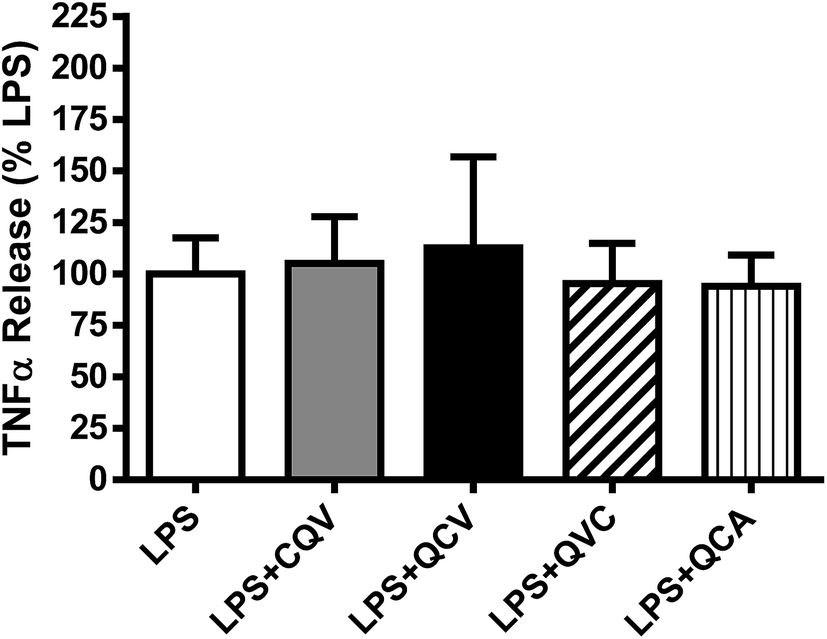
The release of TNF-α from THP-1 cells (human monocytes) exposed to LPS was examined to evaluate the effect of the tripeptides on cellular ADAM17 activity under inflammatory conditions. Incubation of THP-1 cells with LPS increased the TNF-α level in the media. The highest concentration of the tripeptides that inhibited ADAM17 activity by 70% in vitro did not have an effect on media TNF-α level under our experimental conditions (Fig. 6). It is possible that the tripeptides were not bioavailable to exert their effects in the complex extracellular matrix of the cultured monocytes, or that the cells employed compensatory mechanisms to normalize TNF-α processing after the peptide treatments. Besides, it is not certain what fraction of ADAM17 activity is needed to produce sufficient TNF-α release. For instance, blocking ADAM17 expression by 90% with antisense oligonucleotides was found to reduce the level of released TNF-α only by 60% in cultured THP-1 cells,50 which suggests that complete and sustained ADAM17 inhibition may be required to achieve a substantial reduction in TNF-α release from the cells.
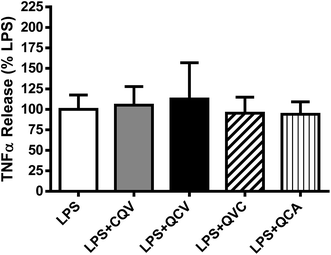 |
| | Fig. 6 Effect of the tripeptides on LPS-stimulated TNF-α release from human monocytic cells. THP-1 cells were incubated with LPS in absence and presence of the tripeptides for 3 h and released TNF-α in the media was determined using an ELISA. No significant differences (P < 0.05) were detected when comparing LPS + peptide groups with the LPS group. TNF-α levels were not detectable in the absence of LPS stimulation. Data reflect three independent experiments. | |
Conclusion
Food protein-derived peptides can have regulatory functions in inflammation. In this study, new rye secalin-derived tripeptides and analogues were found, for the first time, to chelate zinc and inhibit the activity of ADAM17, an enzyme that is involved in the activation of the pro-inflammatory cytokine, TNF-α. Peptides inherently contain heterogeneous group of atoms that were found to interact with ADAM17 through zinc coordination, hydrogen bonds and hydrophobic interactions. The tripeptides did not inhibit LPS-stimulated TNF-α release from cultured human monocytes under our experimental conditions. Future studies are needed to evaluate their cellular distribution, interaction with other MMPs structurally related to ADAM17, and effects on inflammatory signalling pathways and associated oxidative damages at higher treatment doses. The observed ADAM17 inhibitory activities of the tripeptides can play a role in their use as anti-inflammatory compounds in developing functional food products.
Conflict of interest
The authors do not have any existing conflict of interest in this study.
Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery Grant Program (RGPIN 435865-2013).
References
- H. El-Gabalawy, L. C. Guenther and C. N. Bernstein, J. Rheumatol., 2010, 37, 2–10 Search PubMed.
- H. Maradit-Kremers, P. J. Nicola, C. S. Crowson, K. V. Ballman and S. E. Gabriel, Arthritis Rheum., 2005, 52, 722–732 CrossRef PubMed.
- L. C. R. Silva, L. C. M. Ortigosa and G. Benard, Immunotherapy, 2010, 2, 817–833 CrossRef CAS PubMed.
- F. C. Nelson and A. Zask, Expert Opin. Invest. Drugs, 1999, 8(4), 383–392 CrossRef CAS PubMed.
- M. Gooz, Crit. Rev. Biochem. Mol. Biol., 2010, 45, 146–169 CrossRef CAS PubMed.
- J. Arribas and C. Esselens, Curr. Pharm. Des., 2009, 15, 2319–2335 CrossRef CAS PubMed.
- N. B. Merchant, I. Voskresensky, C. M. Rogers, B. Lafleur, P. J. Dempsey, R. Graves-Deal, F. Revetta, A. C. Foutch, M. L. Rothenberg, M. K. Washington and R. J. Coffey, Clin. Cancer Res., 2008, 14, 1182–1191 CrossRef CAS PubMed.
- G. Sinnathamby, J. Zerfass, J. Hafner, P. Block, Z. Nickens, A. Hobeika, A. A. Secord, H. K. Lyerly, M. A. Morse and R. Philip, Clin. Exp. Immunol., 2011, 163, 324–332 CrossRef CAS PubMed.
- P. R. Murumkar, R. Giridhar and M. R. Yadav, Expert Opin. Drug Discovery, 2013, 8, 157–181 CrossRef CAS PubMed.
- M. L. Moss, L. Sklair-Tavron and R. Nudelman, Nat. Clin. Pract. Rheumatol., 2008, 4, 300–309 CrossRef CAS PubMed.
- M. C. Udechukwu, S. A. Collins and C. C. Udenigwe, Food Funct., 2016, 7, 4137–4144 CAS.
- S. Chakrabarti, F. Jahandideh and J. Wu, BioMed Res. Int., 2014, 2014, 1–12 CrossRef PubMed.
- U. K. Bandarage, T. Wang, J. H. Come, E. Perola, Y. Wei and B. G. Rao, Bioorg. Med. Chem. Lett., 2008, 18, 44–48 CrossRef CAS PubMed.
- S. S. Krishna, Nucleic Acids Res., 2003, 31, 532–550 CrossRef CAS PubMed.
- J. Sanchiz, C. Kremer, M. H. Torre, G. Facchin, E. Kremer, E. E. Castellano and J. Ellena, J. Mol. Struct., 2006, 797, 179–183 CrossRef CAS.
- I. Sóvágó, C. Kállay and K. Várnagy, Coord. Chem. Rev., 2012, 256, 2225–2233 CrossRef.
- U. Jakob, M. Eser and J. C. A. Bardwell, J. Biol. Chem., 2000, 275, 38302–38310 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, 2006 Search PubMed.
- H. J. Jeong, J. R. Lee, J. B. Jeong, J. H. Park, Y. Cheong and B. O. de Lumen, Nutr. Cancer, 2009, 61, 680–686 CrossRef CAS PubMed.
- C. Gellrich, P. Schieberle and H. Wieser, Cereal Chem., 2003, 80, 102–109 CrossRef CAS.
- P. R. Shewry and N. G. Halford, J. Exp. Bot., 2002, 53, 947–958 CrossRef CAS PubMed.
- K. Kaukinen and M. Mäki, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 80–82 CrossRef PubMed.
- F. Bamdad, J. Wu and L. Chen, J. Cereal Sci., 2011, 54, 20–28 CrossRef CAS.
- M. Ortiz-Martinez, R. Winkler and S. Garcia-Lara, J. Proteomics, 2014, 111, 165–183 CrossRef CAS PubMed.
- Y. Hu, A. Stromeck, J. Loponen, D. Lopes-Lutz, A. Schieber and M. G. Gänzle, J. Agric. Food Chem., 2011, 59, 11983–11989 CrossRef CAS PubMed.
- A. Cavazos and E. Gonzalez de Mejia, Compr. Rev. Food Sci. Food Saf., 2013, 12, 364–380 CrossRef CAS.
- C. G. Rizzello, L. Nionelli, R. Coda and M. Gobbetti, Nutr. Cancer, 2012, 64, 111–120 CrossRef CAS PubMed.
- C. C. Udenigwe, Trends Food Sci. Technol., 2014, 36, 137–143 CrossRef CAS.
- J. Loponen, Agric. Food Sci., 2004, 13, 39–45 CrossRef CAS.
- W. Ebeling, N. Hennrich, M. Klockow, H. Metz, H. D. Orth and H. Lang, Eur. J. Biochem., 1974, 47, 91–97 CrossRef CAS PubMed.
- A. Dhaval, N. Yadav and S. Purwar, Int. J. Pept. Res. Ther., 2016, 22, 377–398 CrossRef CAS.
- B. Ismail and Z. Gu, Midwest Dairy Foods Research Center, 2010, 55–79 Search PubMed.
- H. Chen, K. Muramoto, F. Yamauchi, K. Fujimoto and K. Nokihara, J. Agric. Food Chem., 1998, 46, 49–53 CrossRef CAS PubMed.
- C. Wang, B. Li and J. Ao, Food Chem., 2012, 134, 1231–1238 CrossRef CAS PubMed.
- N. Xie, J. Huang, B. Li, J. Cheng, Z. Wang, J. Yin and X. Yan, Food Chem., 2015, 173, 210–217 CrossRef CAS PubMed.
- A. P. Demchenko, Ultraviolet Spectroscopy of Proteins, Springer Verlag, New York, 1981 Search PubMed.
- M. L. Moss, S. L. Jin, M. E. Milla, D. M. Bickett, W. Burkhart, H. L. Carter, W. J. Chen, W. C. Clay, J. R. Didsbury, D. Hassler, C. R. Hoffman, T. a. Kost, M. H. Lambert, M. a. Leesnitzer, P. McCauley, G. McGeehan, J. Mitchell, M. Moyer, G. Pahel, W. Rocque, L. K. Overton, F. Schoenen, T. Seaton, J. L. Su and J. D. Becherer, Nature, 1997, 385, 733–736 CrossRef CAS PubMed.
- F. Schmid, Encycl. Life Sci., 2001, pp. 1–4 Search PubMed.
- J. D. Leonard, F. Lin and M. E. Milla, Biochem. J., 2005, 387, 797–805 CrossRef CAS PubMed.
- R. He, R. E. Aluko and X. R. Ju, PLoS One, 2014, 9, e91051 Search PubMed.
- M. R. Eftink and C. A. Ghiron, Anal. Biochem., 1981, 114, 199–227 CrossRef CAS PubMed.
- J. Jayabharathi, K. Jayamoorthy, V. Thanikachalam and R. Sathishkumar, Spectrochim. Acta, Part A, 2013, 108, 146–150 CrossRef CAS PubMed.
- R. C. Newton, K. a. Solomon, M. B. Covington, C. P. Decicco, P. J. Haley, S. M. Friedman and K. Vaddi, Ann. Rheum. Dis., 2001, 60(Suppl 3), iii25–iii32 CAS.
- G. Wagner and S. Laufer, Med. Res. Rev., 2006, 26, 1–62 CrossRef CAS PubMed.
- M. Segura-Campos, L. Chel-Guerrero, D. Betancur-Ancona and V. M. Hernandez-Escalante, Food Rev. Int., 2011, 27, 213–226 CrossRef CAS.
- Z. Guo, P. Orth, S. C. Wong, B. J. Lavey, N. Y. Shih, X. Niu, D. J. Lundell, V. Madison and J. A. Kozlowski, Bioorg. Med. Chem. Lett., 2009, 19, 54–57 CrossRef CAS PubMed.
- K. Maskos, C. Fernandez-Catalan, R. Huber, G. P. Bourenkov, H. Bartunik, G. a. Ellestad, P. Reddy, M. F. Wolfson, C. T. Rauch, B. J. Castner, R. Davis, H. R. Clarke, M. Petersen, J. N. Fitzner, D. P. Cerretti, C. J. March, R. J. Paxton, R. a. Black and W. Bode, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3408–3412 CrossRef CAS.
- P. D. Brown, Expert Opin. Invest. Drugs, 2000, 9, 2167–2177 CrossRef CAS PubMed.
- J. J.-W. Duan, L. Chen, Z. R. Wasserman, Z. Lu, R.-Q. Liu, M. B. Covington, M. Qian, K. D. Hardman, R. L. Magolda, R. C. Newton, D. D. Christ, R. R. Wexler and C. P. Decicco, J. Med. Chem., 2002, 45, 4954–4957 CrossRef CAS PubMed.
- T. P. Condon, S. Flournoy, G. J. Sawyer, B. F. Baker, T. K. Kishimoto and C. F. Bennett, Antisense Nucleic Acid Drug Dev., 2001, 11, 107–116 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ae
*ae