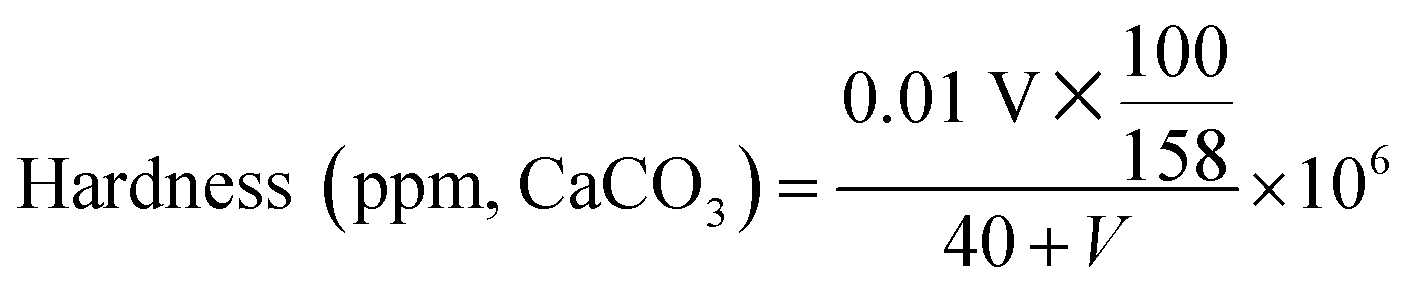DOI:
10.1039/C6RA26629K
(Paper)
RSC Adv., 2017,
7, 9338-9346
Investigation of pseudo-polyanion formation between polyvinylpyrrolidone and sodium dodecanoate in aqueous solution by capillary electrophoresis, conductometry, tensiometry and calcium stability
Received
11th November 2016
, Accepted 26th January 2017
First published on 31st January 2017
Abstract
There is a lack of information about association between nonionic polymers and weak electrolyte type anionic surfactants, therefore the complexation between polyvinylpyrrolidone (PVP) and sodium dodecanoate (SD) is investigated by capillary electrophoresis for the first time, in assistance with conductometry, tensiometry and calcium stability. The experimental results show that values of critical aggregation concentration (cac) and polymer saturation point (psp) of the system determined by several methods are consistent with each other. It is firstly found that the PVP–SD complex is actually PVP–SD pseudo-polyanions with different charge densities corresponding to different pH levels. It is also found that H-bonding and counterion bridging between PVP and bound SD micelles play important roles in the self-assembling of the PVP–SD pseudo-polyanions, and the pH-dependent capillary electrophoresis behavior of the pseudo-polyanions is ascribed to sodium bridging being replaced by H-bonding at lower pH.
1 Introduction
Mixtures of nonionic polymers and anionic surfactants in aqueous solution have been widely applied in different fields1–4 including pharmaceutical, cosmetics, enhanced oil recovery and nanoscience, and thus are of great theoretical and practical significance. The systems were mainly investigated by several methods including tensiometry,5,6 conductometry,7 viscometry,8 potentiometry,9 NMR,10–12 fluorescence spectroscopy,13 light scattering,14 surfactant ion-selective electrodes15 and isothermal titration calorimetry.16 Polyelectrolyte-like complexes,17,18 or polymer-bound surfactant micelles along with polymer chains, were found to form via counterion bridging within the surfactant concentration between the critical aggregation concentration (cac) and polymer saturation point (psp) based on the solution behavior. Recently, both Oztekin19 and our team20 applied electrophoresis capillary (CE) to study the polyvinylpyrrolidone (PVP)–sodium dodecylsulfate (SDS) complex, and we directly proved the formation of PVP–SDS pseudo-polyanions, in which counterion bridging played an important role. Therefore, application of CE and the experimental results presented a positive and significant effect on the study of interaction between polymers and surfactants.
There were many reports concerning on association interaction between nonionic polymers such as PVP or polyethyleneglycol (PEG) and strongly ionizable anionic surfactants such as SDS,19,20 sodium dodecylsulfonate21,22 or sodium dodecylbenzenesulfonate.23 It was found in our research of PVP–SDS complex20 that electrophoretic mobility of the complex decreased with pH reduction, and the complexation between PVP and the bound SDS micelles became stronger as pH decreased to slightly acidic condition even though SDS is a strong electrolyte. Such association researches also involved in weak electrolyte type surfactants such as anionic phosphates24,25 and carboxylates.26 For example, a few researches on PEG–sodium dodecanoate (SD) system were reported by Zanette team24,26,27 and Blokhus28 mainly by using conductometry, which revealed that SD did associate with PEG but the interaction was weaker than PEG–SDS. The interaction of SD with other nonionic polymers including polyacrylamide29 and ethyl(hydroxyethyl)cellulose30–32 was investigated by tensiometry and conductometry, and the attention in the former was paid to hydrogen bond formation between PAM and SD, and the later concerned about association parameters of SD with the polymer such as cac and psp and the synergetic effect with another anionic surfactant including SDS. What's more, the interests in association between nonionic polymers and carboxylates were also extended to different carbon chain length,28,33 cyclic cholate,34,35 unsaturated oleate36,37 and dimeric carboxylate.38 Though association between nonionic polymers and weak electrolyte type surfactants covers a large field of application, there was still a lack of research on the association strength and mechanism between polymers and carboxylates due to the complicated and pH-dependent solution behavior of weak electrolyte type surfactants. Inspired by the abnormally strong PVP–SDS complexation with pH reduction,20 it is predictable that the PVP–SD association would encounter great impact of pH since SD exists in different proportion of protonated/ionized species at different pH levels in aqueous solution.
Capillary electrophoresis (CE), a high performance separation and analytical technique, has many advantages such as high resolution and sensitivity, good repeatability, small sample volume, efficient and rapid, and possible to realize full automation. CE was thus widely applied in the field of medicine39 and biology40 especially in the separation and analysis of complicated biological macromolecules. Among the reported CE detections, polymers and surfactants were widely adopted as additives in different capillary electroseparation methods containing capillary gel electrophoresis41 and micellar electrokinetic chromatography42 for better analyzation or separation of different types of mixtures. But there was almost no research utilizing CE to directly study polymer–anionic surfactant complexation for the purpose of colloidal and interfacial phenomena rather than separations except Oztekin and Erim19 preliminary employed CE to determine cac of PVP–SDS system. Based on the advantage of CE for distinguishing species with different charge types or densities, the pH-dependent PVP–SD complexation is investigated in this paper by separating and detecting the PVP–SD complex in assistance with conductometry, tensiometry and calcium stability.
2 Experimental
2.1 Materials
Polyvinylpyrrolidone (PVP, MW = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from International Specialty Products Inc., used without further purification. Sodium dodecanoate (SD) was prepared by neutralization of dodecanoic acid (Aldrich) with sodium hydroxide in molar ratio of 1:1. Sodium borate, sodium hydroxide, methanol and other reagents in analytical reagent grade were purchased from Sinopharm Chemical Reagent Ltd. Corp. Ultrapure millipore water (18.2 MΩ cm) was used throughout the text.
000) was purchased from International Specialty Products Inc., used without further purification. Sodium dodecanoate (SD) was prepared by neutralization of dodecanoic acid (Aldrich) with sodium hydroxide in molar ratio of 1:1. Sodium borate, sodium hydroxide, methanol and other reagents in analytical reagent grade were purchased from Sinopharm Chemical Reagent Ltd. Corp. Ultrapure millipore water (18.2 MΩ cm) was used throughout the text.
2.2 Methods
Sample preparation. SD solutions at different concentrations of SD (cSD) with/without PVP (2 g L−1) were prepared using a buffer solution of sodium borate (25 mM)∥sodium hydroxide (35 mM) as solvent at different pH levels that was adjusted by hydrochloric acid and monitored with a pH meter (FE20, Mettler-Toledo, Shanghai, China).
Conductometry. Conductivity measurements of the above SD solutions with/without PVP at different pH levels were carried out at 30 ± 0.05 °C by using a conductivity meter (FE30, Mettler-Toledo, Shanghai, China) with cell constant 1.023. The conductometer is calibrated with KCl solution.
Tensiometry. Surface tension (γ) of the above SD solutions with/without PVP at different pH levels was measured at 30 ± 0.05 °C by drop-volume method using a capillary with the external diameter of 3.150 mm. Each measurement was repeated at least three times at the same condition (the error of drop volume is less than ±0.0002 mL) and the average was taken as the final result. In order to obtain the critical concentrations cac, cmc and psp, γ was calculated according to Young–Laplace equation and plotted as a function of SD concentration.
Capillary electrophoresis (CE). The above solutions of SD without PVP were used as running buffers and the corresponding SD solutions with 2 g L−1 of PVP at different pH levels were injected as samples into a CE system (P/ACE MDQ, Beckman Coulter, USA) with a UV visible detector at anode by 20 kPa nitrogen pressure for 2 s. The effective length of the fused silica capillary (75 μm × 60 cm, Yongnian Photoconductive Fiber Factory, Hebei, China) to the detector was 50 cm. The migration times of samples in the corresponding running buffers were detected under the separating voltage of 10 kV at 30 °C. The migration time of methanol was also tested under the same condition as reference. The capillary was flushed with water, 0.1 M sodium hydroxide, water and running buffer each for 10 min at the beginning of each day. It was also washed with water, 0.1 M sodium hydroxide, water and running buffer each for 5 min between runs to raise the reproducibility of the results.
Calcium stability. A series of 25 mM of SD solutions with PVP (2 g L−1) corresponding to different pH levels were prepared according to the procedure of “sample preparation”, and the effect of pH on calcium stability of the solutions was tested by the method outlined by Wilkes and Wickert.43 A 40 mL of the solutions pipetted in a 100 mL beaker was titrated under magnetic stirring with a 1 wt% solution of calcium acetate at room temperature (25 °C). The degree of developed turbidity, which just obscured typewriter print on paper fastened to the other side of the beaker and viewed horizontally through the solution, was taken as the end point of the titration. And the test for another series of 25 mM of SD solutions without PVP was run similarly as a comparison. The results of these tests were interpreted into ppm of solution hardness represented by CaCO3 according to eqn (1):| |
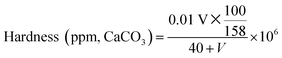 | (1) |
where V is the consumption volume of calcium acetate solution (mL).
3 Results and discussion
3.1 pH-dependent double breakpoints and ionizations of the PVP–SD complex
As a typical method to investigate polymer–surfactant interaction, conductivity measurements were at first carried out for SD and PVP–SD in the buffer solution corresponding to different pH levels (as shown in Fig. 1) to evaluate the effect of H+ and Na+ on the PVP–SD association. The cmc values of SD were determined by fitting the data below and above the breakpoint to two straight lines. On solving the two regression equations of lines simultaneously, one could get the point of intersection corresponding to cmc value of surfactant at particular concentration.44 And thus the cmc data of SD solution corresponding to different pH levels are presented in Table 1. It could be seen from conductivity (κ) versus cSD plots in Fig. 1 that the cmc value of SD decreases with pH reduction within certain range (10–8.7) above its pKa since too low a pH value would result in vesiculation and following phase separation of dodecanoic acids. The effect of pH reduction on micellization of aqueous SD solution could be ascribed to promoting hydrolysis equilibrium of SD towards dodecanoic acids and thus increasing hydrophobicity of SD-relevant species, which is certainly in favor of micellization. Meanwhile, as shown in Fig. 1, two breakpoints emerge from the κ–cSD plots of PVP–SD solutions in the presence of 2 g L−1 of PVP rather than one in SD solutions free of polymer. Fitting the experimental points below the first breakpoint and above the second breakpoint to two straight lines and connecting the two breakpoints by the third auxiliary straight line,24,26–28 cac and psp data corresponding to different pH levels are obtained by the similar method as cmc and collected in Table 1. As literature reported, the first break represents the concentration at which surfactant molecules undergo a polymer-induced aggregation and is known as cac, which is normally lower than cmc of the surfactant.44 And the second break occurs when all interaction sites on the polymer are occupied by surfactant molecules, which is called psp45 and represents the saturation of polymer by the surfactants. Above psp regular micelles should appear and be in dynamic equilibrium with the polymer–surfactant aggregates. Accordingly, the PVP–SD complex is believed to form between the two breakpoints, which means SD does associate with PVP in the experimental pH range as literature reported.28 The κ–cSD plots of PVP–SD solutions show that cac value decreases with pH reduction, and the reason is almost same as that for cmc. And psp value is found increasing with pH reduction, it is maybe analyzed as H-bonding is always stronger than ion–dipole force, so that Na+ ions being partly replaced by H-bonds in the PVP–SDS complex would decrease the charge density of SD molecule and thus decrease both repulsion between head groups of SD species and surface area of per surfactant molecule at surface of the bound SD micelles. The above reasons lead more and more SD species binding onto PVP chains. Moreover, the H-bonding which is stronger than the van der Waals force makes the complex more compact and stable than that bridged by Na+ ions. It is clear that pH reduction causes both cac decreasing and psp increasing, and thus H-bonding replacing sodium bridging in the PVP–SD association makes SD concentration range broadened for the complexation of PVP–SD, which is evidently favourable for forming the PVP–SD complex. Previous literatures suggested SDS monomers would bind on PVP through ion–dipole interaction on the positive centres – nitrogen atoms of the pyrrolidone46 but it is in fact infeasible due to great steric hindrance. Herein the bound SD micelles would bind on the PVP chain through sodium bridging or H-bonding on the negative centres – oxygen atoms of the pyrrolidone and coexists with hydrophobic force forever. The higher pH condition in this buffer system would further weaken the positive nature of nitrogen atoms on the pyrrolidone and thus weaken the interaction between monomer SD and PVP. Therefore the PVP-bound SD micelle is thus believed more stable than the PVP–SD (molecule) complex.
 |
| | Fig. 1 Conductivity (κ)–cSD plots of SD (□) and PVP (2 g L−1)–SD (○) in the buffer solution corresponding to different pH values of 10.0 (A), 9.0 (B) and 8.7 (C) with the same added Na+ concentration of 85 mM at 30 °C. | |
Table 1 Relevant parameters obtained from the conductivity curves of PVP (2 g L−1)–SD and SD in the buffer solution corresponding to different pH levels at 30 °Ca
| pH |
Critical concentration (mM) |
S1 |
S2 |
S3 |
α12 |
α13 |
| cac |
cmc |
psp |
| All the concentration of Na+ in the buffer solution is 85 mM except for pH 9.0* of 50 mM. |
| 10.0 |
6.6 |
14.6 |
22.3 |
0.0570 |
0.0413 |
0.0303 |
0.725 |
0.531 |
| 9.0* |
6.2 |
11.5 |
22.0 |
0.0492 |
0.0405 |
0.0326 |
0.824 |
0.663 |
| 9.0 |
5.5 |
11.0 |
25.2 |
0.0620 |
0.0406 |
0.0283 |
0.655 |
0.456 |
| 8.7 |
5.0 |
10.0 |
31.6 |
0.0527 |
0.0403 |
0.0252 |
0.764 |
0.478 |
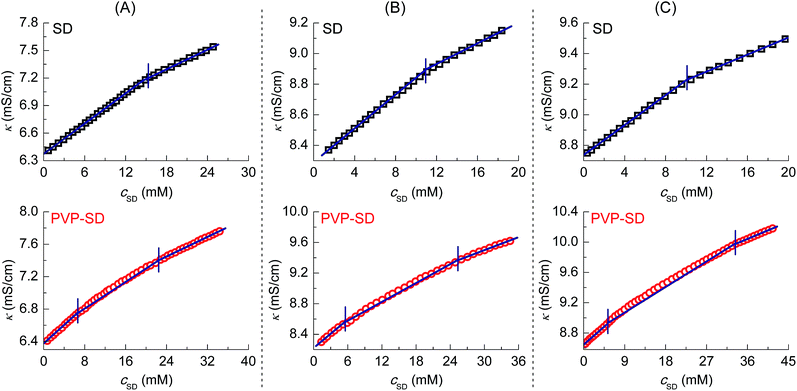
Shown as the κ–cSD plots of PVP–SD solutions in Fig. 1, the slopes of each three lines S1, S2 and S3 are used to calculate the average ionization degree α12 of the PVP–SD complex (α12 = S2/S1) and the average ionization degree α13 of the normal SD micelle above c2 (α13 = S3/S1),14 and all the data are listed in Table 1. The α12 value is found much greater than the α13 value, illustrating more Na+ ions diffused from the PVP–SD complex than from the normal SD micelle, which comes down to the competitive interaction of PVP and SD ions. At the first stage to decrease pH from 10 to 9, the lower ionization degree (Table 1) is mainly due to the fact that more and more SD species binding onto PVP chains by sodium bridging that would depress diffusion of sodium ions, whereas at the second stage to decrease pH from 9 to 8.7, ionization degree returning to a greater value in response to the lower pH comes down to the directionality and saturability of H-bonds to force more and more sodium ions moving into diffused electric double layer. As in the case of changing the concentration of added counterions at the same pH level, the influence of counterion concentration on the polymer–surfactant association is shown in Fig. 2. The cmc value of SD solution decreases with adding Na+ ions (Fig. 2A) and the range of cac and psp in the buffer solution with 85 mM of Na+ is broader than that with 50 mM of Na+ (Fig. 2B) (detail data see Table 1 ), which means Na+ does favor to formation of the SD micelles and the PVP–SD pseudo-polyanions and consequently results in a stronger PVP–SD association. In conclusion, both H+ and Na+ will positively affect the formation of PVP–SD complex at some extent by H-bonding or counterion bridging as PVP–SDS does.20
 |
| | Fig. 2 Conductivity (κ)–cSD plots of SD (A) and PVP (2 g L−1)–SD (B) in the buffer solution at pH 9 with the added Na+ concentration of 85 mM (□) and 50 mM (○) at 30 °C. | |
3.2 Double breakpoints and molecular interaction parameter [Γ∞] of the PVP–SD complex confirmed by tensiometry
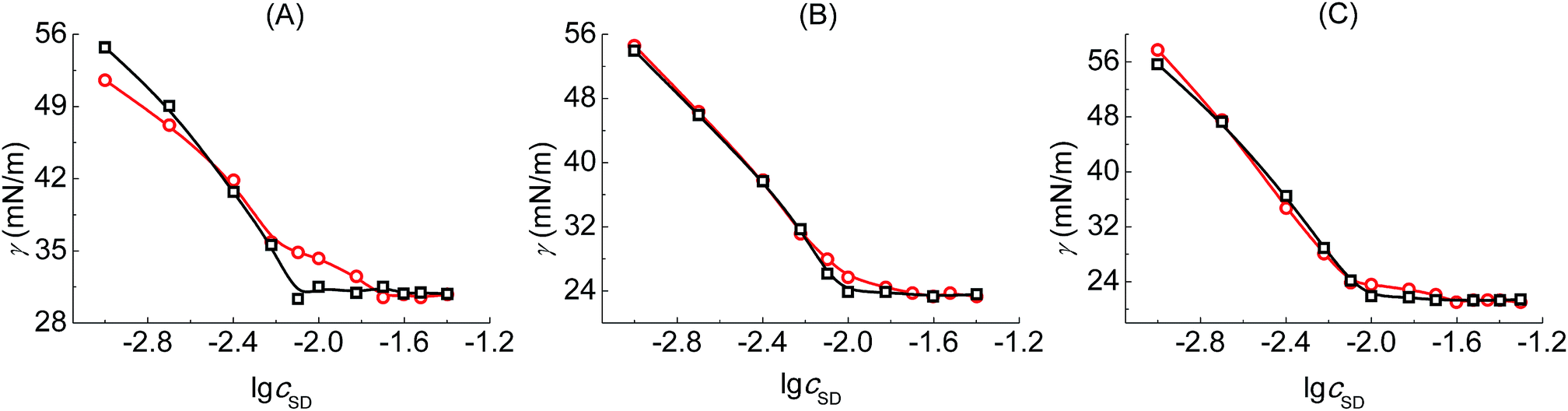
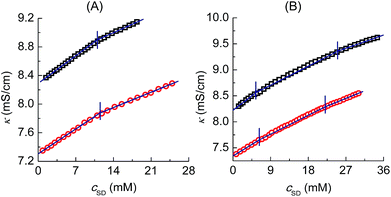
Tensiometry is used to further judge whether there is formed the PVP–SD complex. Fig. 3 shows the γ–lgcSD plots of SD solutions and PVP–SD solutions corresponding to different pH values of 10.0, 9.0 and 8.7. Based on the γ–lgcSD plots, it can be clearly concluded that the effect of PVP at different pH levels on the aggregation of SD is similar with the κ–cSD plots of the corresponding systems, in which double breakpoints appear, one before cmc and the other after cmc. The first breakpoint occurs at the starting binding of SDS to the polymeric chains and hence it refers to the cac. The second breakpoint definitely refers to the psp where SD molecules bind with PVP saturated and the free SD micelles begin to form. The surface tension of PVP–SD solution beyond psp keeps constant and equal to that of SD solution beyond cmc. The occurrence of double breakpoints can be explained by polymer–surfactant association as PVP–SDS does.5 The critical concentrations (cac and psp) and the values of surface tension at cmc (γcmc) obtained from the γ–lgcSD plots of SD solutions and PVP–SD solutions at different pH levels are presented in Table 2. The value of γcmc decreases with pH reduction since the hydrolyzation of SD becomes stronger with the increasing of H+ and the surfactant molecules arrange more compact with less repulsion between the head groups at surface of aqueous solution.
 |
| | Fig. 3 γ–lgc plots of SD solutions (○□) and PVP (2 g L−1)–SD (□○) solutions corresponding to different pH values of 10.0 (A), 9.0 (B) and 8.7 (C) at 30 °C. | |
Table 2 Relevant parameters obtained from the surface tension curves of PVP (2 g L−1)–SD and SD solutions corresponding to different pH levels at 30 °C
| pH |
SD solution |
PVP–SD solution |
[Γ∞] |
γcmc (mN m−1) |
| cmc (mM) |
cac (mM) |
psp (mM) |
| Marked the [Γ∞] value of PVP–SDS system in aqueous solution.20 |
| 10.0 |
13.0 |
6.0 |
20.0 |
0.389 |
30.6 |
| 9.0 |
10.0 |
6.0 |
20.0 |
0.555 |
23.4 |
| 8.7 |
10.0 |
8.0 |
25.0 |
0.833 |
21.3 |
| — |
— |
— |
— |
0.416a |
— |
The specific saturation capacity of clusterization [Γ∞]47 was raised as a molecular interaction parameter describing saturated association of the bound surfactant micelles on polymer chain. The [Γ∞] values are calculated from [Γ∞] = (psp − cmc) × Mn × 10−3/ρ (mol mol−1 monomer) and listed in Table 2, which means the molar saturated aggregation capacity of the micellar surfactant molecules on per molar polymer unit, where Mn is the molecule weight of VP monomer, that is, 111; and ρ is the mass concentration (g L−1) of PVP. It can be seen that the [Γ∞] value in lower pH solution is greater than that in higher pH solution, which further proves the PVP–SD association would be promoted by elevating H+. Comparing the [Γ∞] values of PVP–SD solutions with that of PVP–SDS solutions as listed in Table 2, SD molecules would associate with PVP weaker than SDS at higher pH but stronger than SDS at lower pH due to the weak electrolyte nature of SD.
3.3 pH-dependent charge densities of the PVP–SD pseudo-polyanions detected by CE
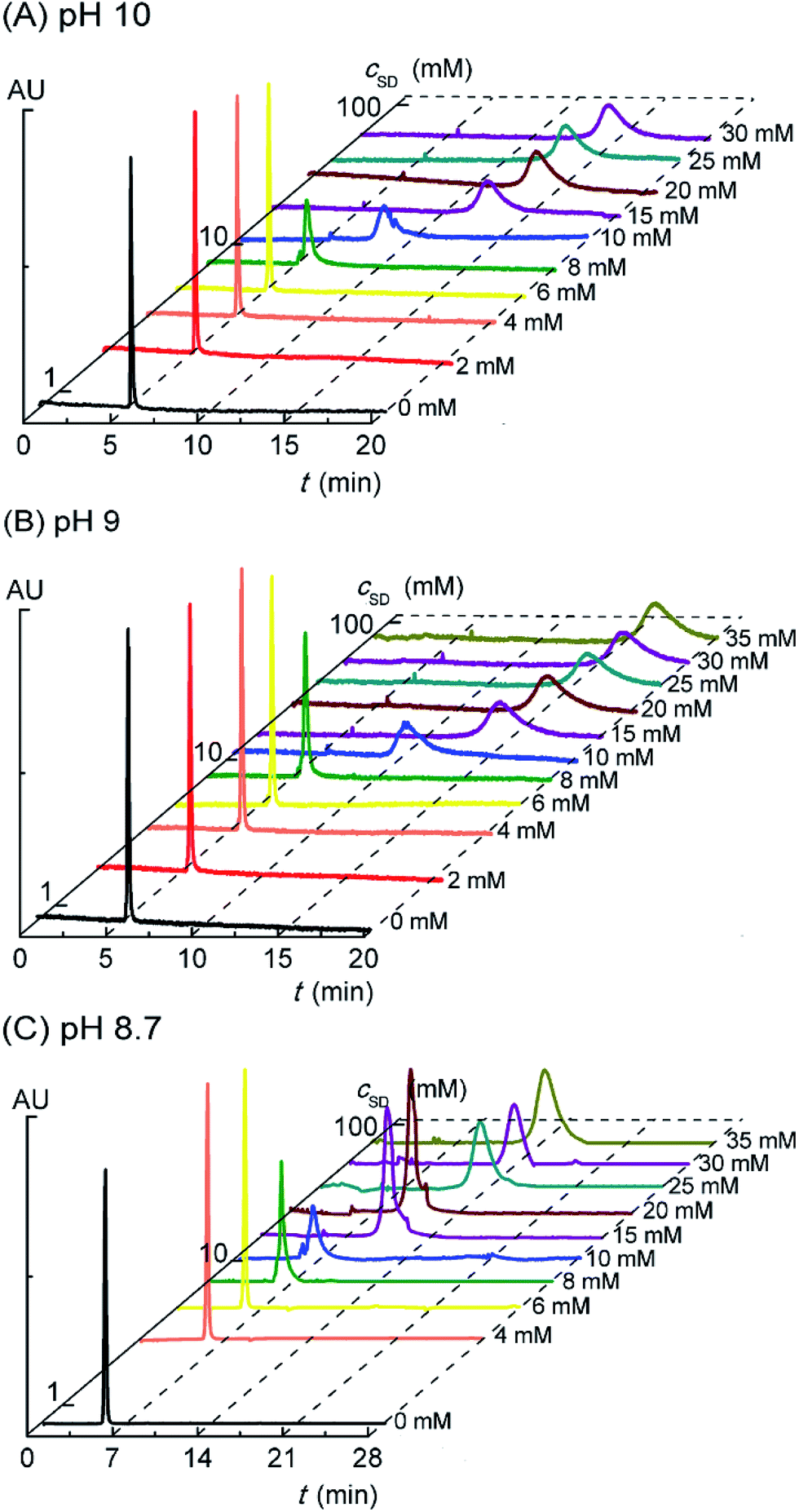
In the previous report,20 we have found formation of the PVP–SDS pseudo-polyanions by capillary electrophoresis (CE) that could separate and detect analytes with different charge types or charge densities. Based on the strong UV absorption of PVP at 210 nm and weak UV absorption of SD, it is rational to explore the PVP–SD association by detecting the migration times of PVP with increasing cSD in electropherograms. What's more, the charge type and the variation of charge density of PVP–SD complex in response to cSD increasing or pH reduction could be confirmed by CE. Fig. 4A shows the electropherograms of PVP–SD solutions corresponding to different SD concentrations in the case of pH 10.0. The migration time of PVP alone is 5.3 min in accordance with that of methanol (5.2 min), which means PVP in the buffer solution free of SD is almost electrically neutral. The PVP-relevant peak stays unchanged until cSD reaching and exceeding 8 mM, which means PVP does not associate with SD below 8 mM. The PVP-relevant peak begins to migrate above 8 mM of SD and the migration time is longer and longer than ever as cSD increasing. Considering the sample was injected at anode and all emerging peaks are observed behind the control PVP migrating peak, the PVP–SD complex is speculated to be the PVP–SD pseudo-polyanions. The PVP–SD complex is negatively charged because it is composed of PVP chains with the bound SD micelles linked by the bound Na+ counterions in the Stern layer of the latter, which is coexisted with the diffused Na+ counterions in the Gouy–Chapman layer of the latter. It is the first and direct evidence of the negatively charged PVP–SD pseudo-polyanions obtained by CE here similar to our previous work about PVP–SDS.20 The PVP–SD complex is named the PVP–SD pseudo-polyanions instead of the PVP–SD polyanions because it is not a real linear polyelectrolyte but the PVP bound SD micelles formed by dint of supramolecular interaction; therefore it behaves like a polyanion in water but without the exact polyanion structure. Why the charge densities of the PVP–SD pseudo-polyanions increase with increasing cSD is due to more surfactants binding onto the polymer chain in the form of negatively charged PVP-bound SD micelles. The migration time reaches maximum at 20 mM, indicating that the saturation of PVP associated by SD causes the migration stopped. These deduced conclusions are in good agreement with the clustering process of PVP–SD found between the double critical concentrations acquired by the aforementioned conductivity and surface tension experiments. What can be further deduced from the electropherograms is that once PVP begins to associate with the bound SD micelles, all the polymer chains simultaneously interact with them. It exactly agrees with Nikas and Blankschtein48 that if PVP chain is once bounded by the bound SDS micelles, every one of all the polymer chains would be attached by the first one, then a second, then a third, and so on, until the maximum number, n, is reached. The rest of Fig. 4 shows the electropherograms of PVP–SD solutions corresponding to different pH levels in the case of pH 9.0 (Fig. 4B) and pH 8.7 (Fig. 4C), which has the similar variation tendencies with Fig. 4A. In the tested pH range of electrophoresis, PVP does associate with SD, and the obtained critical concentrations are basically consistent with that acquired by conductometry and tensiometry. It is clear from Fig. 4 that all the electrophoresis peaks corresponding to different cSD values and different pH levels have different migration times, which means that the PVP–SD complex is actually PVP–SD pseudo-polyanions with broad distribution patterns and different charge densities. Furthermore, every electrophoresis peak corresponding to a given cSD and pH value is of polydispersity in contrast to neutral PVP, which means that even the PVP–SD complex corresponding to the given cSD and pH value is actually a family of the PVP–SD pseudo-polyanions with different charge densities, too. That is caused by the polydispersity of both molecular weights of PVP and amounts of the bound SD micelles associating with per PVP chain.
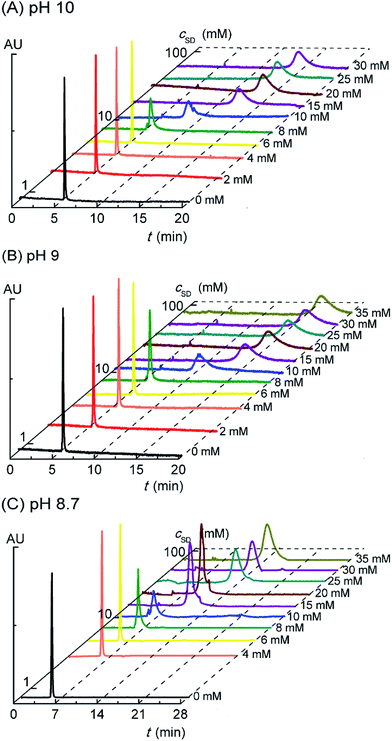 |
| | Fig. 4 Electropherograms of PVP (2 g L−1)–SD solution at pH of 10.0 (A), 9.0 (B) and 8.7 (C) at 30 °C. | |
Effective electrophoretic mobility, μe of the charged PVP-bound SD micelle was calculated from
| |
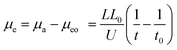 | (2) |
where
μa is apparent mobility of the negatively charged analyte,
μeo is equal to the electroosmotic mobility inside the capillary,
U is the applied voltage (kV),
L is the capillary total length (cm), and
L0 is the length of capillary between injection and detection points (cm),
t0 is the migration time of the control sample methanol (s), and
t is the migration time of PVP–SD pseudo-polyanions (s). As the sample is injected into the capillary at anode,
μe values of the PVP–SD complex are acquired negative according to
eqn (2), which means the PVP–SD complex electronegative. A larger absolute value of
μe means a lower apparent electrophoretic mobility of the PVP–SD pseudo-polyanions in comparison to neutral PVP. The plots of
μe varying with
cSD at different pH levels are shown in
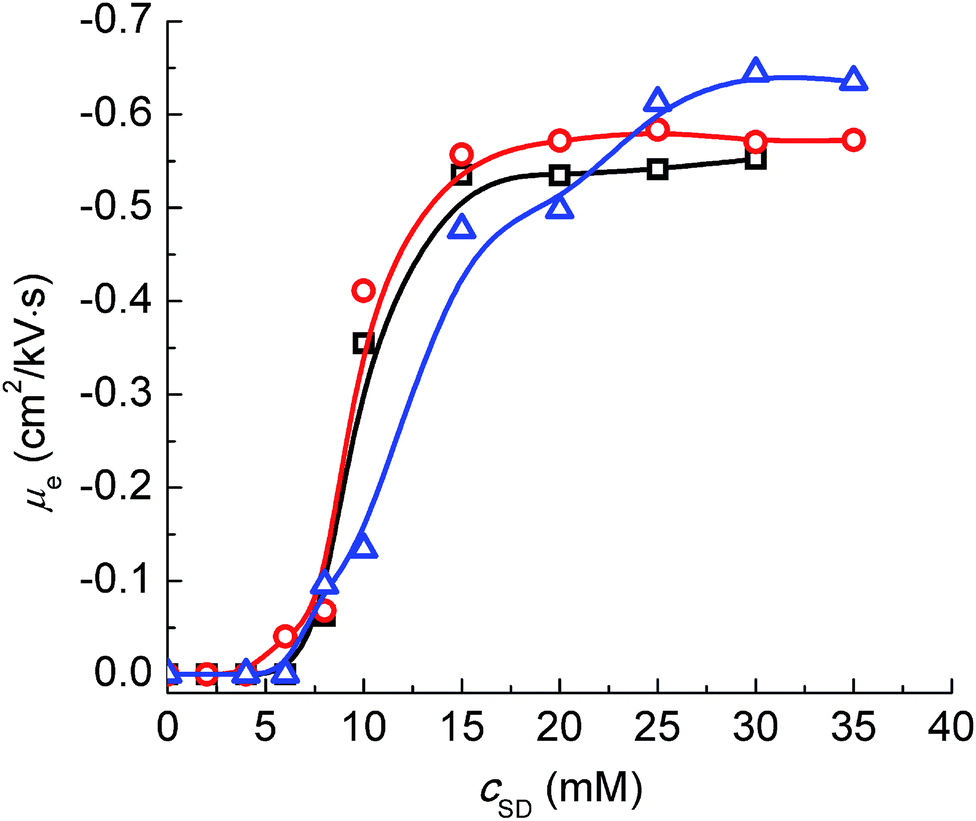
Fig. 5, which indicates the change of
μe of the PVP–SD pseudo-polyanions corresponding to different pH levels has similar tendency but H-bonding exerts a greater influence on
μe than sodium bridging does.
μe values of the PVP–SD pseudo-polyanions become more negative with the increase of
cSD,
viz., the charge density of the PVP–SD pseudo-polyanions gradually increases as
cSD is high enough. Comparing the
μe values at different pH levels as peaks of the PVP–SD pseudo-polyanions stay unchanged above psp, it further demonstrates that the PVP–SD complexation would be promoted by elevating H
+ and the corresponding PVP–SD pseudo-polyanions possess higher charge density. The result is owed to sodium bridging between the head groups of SD and the electronegative centers of PVP chains being partly replaced by H-bonding that is stronger than ion–dipole force. Therefore H-bonding plays an important role in the PVP–SD complexation similar to the PVP–SDS system at some extent.
20 Based on the previous report of the p
Ka value of 7.5 for dodecanoic acid,
49 it could be deduced that the percentage of ionized dodecanoic acid is separately 99.7% and 94.1% at pH of 10.0 and 8.7. Therefore, the greater value of
μe or charge density of the PVP–SD pseudo-polyanions at pH 8.7 is owing to the enhanced hydrolysis equilibrium of SD and thus increasing hydrophobicity of SD species, which is in favor of micellization and further associating with PVP. On the other hand, due to the closer packing and stronger interaction of H-bonding than sodium bridging, the sodium bridging is partly replaced by H-bonding, which makes SD associated more compact with PVP, and thus more Na
+ ions are forced moving into the diffused electric double layer with pH reduction. All in all, H-bonding exerts stronger and stronger influence upon the PVP–SD complexation when changing pH values from 10.0 to 8.7, and
Fig. 6 illustrates the relationship between the mechanism of H-bonding influencing on the PVP–SD complexation and a longer migration time of the PVP–SD pseudo-polyanions on the electropherograms corresponding to pH 8.7 than pH 10.0.
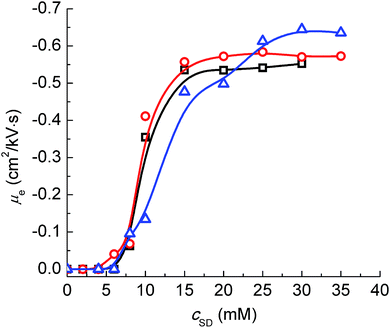 |
| | Fig. 5 Effective electrophoretic mobility (μe) of the PVP–SD complex corresponding to different pH levels of 10.0 (□), 9.0 (○) and 8.7 (Δ) at 30 °C. | |
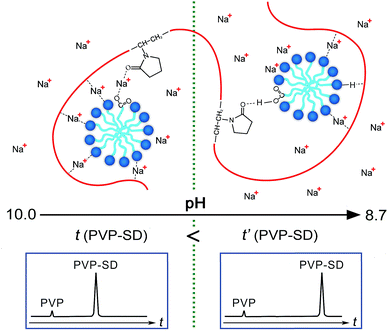 |
| | Fig. 6 Schematic representation of the pH-dependent complexation in the PVP–SD pseudo-polyanions and its influence on the migration time variation in electropherograms. | |
Much importance has been attached to the solubility of metal soaps offered to industry for wetting and detergent operations. Calcium stability of PVP–SD solutions at different pH values from 10.0 to 8.7 at 25 °C was tested to estimate the influence of PVP on SD by the action of hard water and to further verify the prospected pseudo-polyanion structure. The results of these tests, interpreted into ppm of solution hardness represented by CaCO3, are shown in Fig. 7. Based on the experiment results of conductivity, tensiometry and CE, it is predictable that the complexation pattern between PVP and SD should promote Ca2+ ions complexing preferentially with the anionic head group of SD and PVP simultaneously. It could be seen that calcium stability of PVP–SD solution at higher pH is somewhat higher than that of SD solution, meaning that the PVP–SD complexation benefits for dispersing or preventing the precipitation of lime soaps in comparison to SD alone. The ability of PVP to increase the tolerance of soap solutions for calcium hardness clearly indicates that the possible mechanism is preferential Ca2+ bridging between PVP and ionized SD via counterion bridging. This is attributed to solubility product principle, which indicates that Ca2+ ions are easy bridging with ionized SD in comparison with Na+ and preferentially resulting in the formation of stable PVP–Ca2+-ionized SD structure under the protection of PVP chain rather than the hydrophobic reverse lime soap micelles of SD alone. Additionally, it also has positive contribution to calcium stability that the addition of PVP results in cac much less than cmc of SD as shown in Table 2, since the less saturated SD concentration in PVP–SD mixture hinder the formation of lime soap. As for calcium stability of PVP–SD solution at lower pH is appreciably higher than that of SD solution, it depends on the co-effects that H-bonding should further force more Na+ ions moving into diffused electric double layer with pH reduction and that the Ca2+ bridging should reduce the chances of Ca2+ ions to complex with free SD. The experimental results of calcium stability keep consistence with the prediction depending on the PVP–SD pseudo-polyanion structure confirmed by conductometry, tensiometry and CE, and the relationship between the pH-dependent calcium stability with the Ca2+-induced pseudo-polyanion variation is schematically presented in Fig. 7, which provides another solid support for the aforementioned PVP–SD pseudo-polyanion structure. Therefore, the experimental results in Fig. 7 provide not only some useful information about the pseudo-polyanions to prevent the agglomeration and precipitation of lime soap that has practical utility in such detergent operations as laundering and shampooing, but also the possibility of the pseudo-polyanions to be used as microreactor for synthesizing metal nanoparticles by the precursor metal ions acting as the bridging ions in the aforementioned pseudo-polyanions.
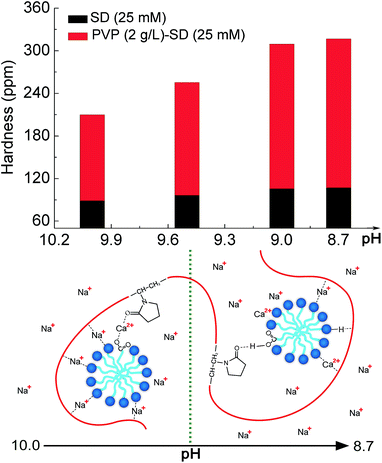 |
| | Fig. 7 Schematic illustration of the Ca2+-induced and pH-dependent PVP–SD pseudo-polyanion structure and its influence on the pH-dependent calcium stability in aqueous solution. | |
4 Conclusions
In view of a lack of complexation information of nonionic polymer and carboxylate for the complicated and pH-dependent solution behavior of weak electrolyte type surfactants, PVP–SD complexation at different pH levels is investigated by capillary electrophoresis (CE) for the first time, in assistance with conductometry, tensiometry and calcium stability in this paper. The experimental results show that values of critical aggregation concentration (cac) and polymer saturation point (psp) of the system determined by several methods are consistent with each other. All of the four methods found PVP does associate with SD and the complexation becomes stronger and stronger with pH reduction, and even stronger than the PVP–SDS complexation as pH is below 9. It is firstly reported by CE that the PVP–SD complex is actually the PVP–SD pseudo-polyanions with different charge densities corresponding to different pH levels. What's more, counterion and H-bond between PVP and the bound SD micelles plays an important role, that is, sodium bridging and H-bonding between the head groups of SD and the electronegative centers of PVP chains to construct the PVP–SD pseudo-polyanions, which is also supported by the calcium stability experiment. Therefore, the pH-dependent capillary electrophoresis behavior of the pseudo-polyanions is ascribed to sodium bridging being partly replaced by H-bonding at lower pH. The better calcium stability of PVP–SD pseudo-polyanions comes down to sodium bridging being partly replaced by calcium bridging, and further being partly replaced by H-bonding at lower pH. This work not only fills the research gap of the complexation mechanism between PVP and SD in the field of colloid and interface science, but also provides some hints on the application of the pH-dependent complex in laundering detergents, personal care products, cosmetics and toiletries, and even microreactor for synthesizing metal nanoparticles.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21276113), Jiangsu Provincial Post Graduate Innovation Plan (CXZZ13_0745) and Doctor Candidate Foundation of Jiangnan University (JUDCF13019).
References
- L. E. Zerpa, N. V. Queipo, S. Pintos and J. L. Salager, J. Pet. Sci. Eng., 2005, 47, 197–208 CrossRef CAS.
- S. Qi, S. Roser, K. J. Edler, C. Pigliacelli, M. Rogerson, I. Weuts, F. Van Dycke and S. Stokbroekx, Pharm. Res., 2013, 30, 290–302 CrossRef CAS PubMed.
- C. Wanawongthai, A. Pongpeerapat, K. Higashi, Y. Tozuka, K. Moribe and K. Yamamoto, Int. J. Pharm., 2009, 376, 169–175 CrossRef CAS PubMed.
- L. M. Qi, J. Li and J. M. Ma, Adv. Mater., 2002, 14, 300–303 CrossRef CAS.
- B. M. Folmer and B. Kronberg, Langmuir, 2000, 16, 5987–5992 CrossRef CAS.
- J. Dey, N. Sultana, S. Kumar, V. K. Aswal, S. Choudhury and K. Ismail, RSC Adv., 2015, 5, 74744–74752 RSC.
- E. Bayat and R. Sadeghi, Colloids Surf., A, 2013, 436, 260–269 CrossRef CAS.
- H. J. Liu and M. T. Hai, J. Chem. Eng. Data, 2010, 55, 354–357 CrossRef CAS.
- S. M. Ghoreishi, Y. Li, D. M. Bloor, J. Warr and E. Wyn-Jones, Langmuir, 1999, 15, 4380–4387 CrossRef CAS.
- Z. S. Gao, R. E. Wasylishen and J. C. T. Kwak, J. Phys. Chem., 1991, 95, 462–467 CrossRef CAS.
- S. S. Hou, J. K. Tzeng and M. H. Chuang, Soft Matter, 2010, 6, 409–415 RSC.
- Y. X. Luan, A. X. Song and G. Y. Xu, Soft Matter, 2009, 5, 2587–2595 CAS.
- C. K. Chee, S. Rimmer, I. Soutar and L. Swanson, Soft Matter, 2011, 7, 4705–4714 RSC.
- A. P. Romani, M. H. Gehlen and R. Itri, Langmuir, 2005, 21, 127–133 CrossRef CAS PubMed.
- G. B. Li, H. M. Ma and J. C. Hao, Soft Matter, 2012, 8, 896–909 RSC.
- M. Prasad, R. Palepu and S. P. Moulik, Colloid Polym. Sci., 2006, 284, 871–878 CAS.
- D. P. Norwood, E. Minatti and W. F. Reed, Macromolecules, 1998, 31, 2957–2965 CrossRef CAS.
- E. Minatti, D. P. Norwood and W. F. Reed, Macromolecules, 1998, 31, 2966–2971 CrossRef CAS.
- N. Oztekin and F. B. Erim, J. Surfactants Deterg., 2013, 16, 363–367 CrossRef CAS.
- Y. F. Wu, J. Chen, Y. Fang and M. Zhu, J. Colloid Interface Sci., 2016, 479, 34–42 CrossRef CAS PubMed.
- R. Sadeghi and S. Shahabi, J. Chem. Thermodyn., 2011, 43, 1361–1370 CrossRef CAS.
- Z. Hou, Z. Li and H. Wang, Colloid Polym. Sci., 1999, 277, 1011–1018 CAS.
- Z. S. Hou, Z. P. Li and H. Q. Wang, J. Dispersion Sci. Technol., 1999, 20, 1507–1516 CrossRef CAS.
- D. Zanette, C. F. Lima, A. A. Ruzza, A. T. N. Belarmino, S. D. Santos, V. L. A. Frescura, D. M. O. Marconi and S. J. Froehner, Colloids Surf., A, 1999, 147, 89–105 CrossRef CAS.
- J. C. Brackman and J. B. F. N. Engberts, J. Colloid Interface Sci., 1989, 132, 250–255 CrossRef CAS.
- D. Zanette, V. Soldi, A. P. Romani and M. H. Gehlen, J. Colloid Interface Sci., 2002, 246, 387–392 CrossRef CAS PubMed.
- D. Zanette and V. L. Frescura, J. Colloid Interface Sci., 1999, 213, 379–385 CrossRef CAS PubMed.
- A. M. Blokhus and K. Klokk, J. Colloid Interface Sci., 2000, 230, 448–451 CrossRef CAS PubMed.
- G. Y. Xu, G. Z. Li, F. Li, H. Z. Mao, M. X. Liu and H. M. Su, Acta Chim. Sin., 1995, 53, 837–841 CAS.
- A. Dal Bo, B. Schweitzer, A. C. Felippe, D. Zanette and B. Lindman, Colloids Surf., A, 2005, 256, 171–180 CrossRef.
- S. D. Modolon, A. C. Felippe, T. E. Fizon, L. da Silva, M. M. D. Paula and A. G. Da-Bo, Carbohydr. Polym., 2014, 111, 425–432 CrossRef CAS PubMed.
- A. G. Dal-Bo, R. Laus, A. C. Felippe, D. Zanette and E. Minatti, Colloids Surf., A, 2011, 380, 100–106 CrossRef CAS.
- X. Xin, G. Y. Xu, D. Wu, Y. M. Li and X. R. Cao, Colloids Surf., A, 2007, 305, 138–144 CrossRef CAS.
- A. C. Felippe, B. Schweitzer, A. G. Dal Bo, R. Eising, E. Minatti and D. Zanette, Colloids Surf., A, 2007, 294, 247–253 CrossRef CAS.
- D. Zanette, A. C. Felippe, B. Schweitzer, A. Dal Bo and A. Lopes, Colloids Surf., A, 2006, 279, 87–95 CrossRef CAS.
- V. I. Petrenko, M. V. Avdeev, V. M. Garamus, L. A. Bulavin and P. Kopcansky, Colloids Surf., A, 2015, 480, 191–196 CrossRef CAS.
- X. Xin, G. Y. Xu, D. Wu, H. J. Gong, H. X. Zhang and Y. J. Wang, Colloids Surf., A, 2008, 322, 54–60 CrossRef CAS.
- N. Kumar and R. Tyagi, J. Dispersion Sci. Technol., 2015, 36, 1601–1606 CrossRef CAS.
- G. Hanrahan and F. A. Gomez, Chemometric methods in capillary electrophoresis, John Wiley & Sons, Hoboken, 2010 Search PubMed.
- L. Holland, Anal. Bioanal. Chem., 2015, 407, 6909–6910 CrossRef CAS PubMed.
- J. P. Hsu, C. H. Huang and S. Tseng, Soft Matter, 2013, 9, 11534–11541 RSC.
- S. Q. Hu, G. X. Wang, W. B. Guo, X. M. Guo and M. Zhao, J. Chromatogr. A, 2014, 1342, 86–91 CrossRef CAS PubMed.
- B. G. Wilkes and J. N. Wickert, Ind. Eng. Chem., 1937, 29, 1234–1239 CrossRef CAS.
- S. Chauhan, R. Singh, K. Sharma and K. Kumar, J. Surfactants Deterg., 2015, 18, 225–232 CrossRef CAS.
- J. Q. Chen, H. J. Xue, Y. F. Yao, H. Yang, A. M. Li, M. Xu, Q. Chen and R. S. Cheng, Macromolecules, 2012, 45, 5524–5529 CrossRef CAS.
- A. Dan, S. Ghosh and S. P. Moulik, J. Phys. Chem. B, 2008, 112, 3617–3624 CrossRef CAS PubMed.
- Y. Fang, K. Cai, L. Y. Zong, X. F. Liu and M. S. He, Chem. Res. Chin. Univ., 2004, 25, 888–891 CAS.
- Y. J. Nikas and D. Blankschtein, Langmuir, 1994, 10, 3512–3528 CrossRef CAS.
- J. R. Kanicky, A. F. Poniatowski, N. R. Mehta and D. O. Shah, Langmuir, 2000, 16, 172–177 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Wangsong Wang
* and
Wangsong Wang