
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation of diazenyl-protected N-heterocycles – a new entry to functionalized lactams†
Martina Petrovićab,
Dina Scarpib,
Martin Niegerc,
Nicole Jung*ad,
Ernesto G. Occhiatob and
Stefan Bräse *ad
*ad
aInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: braese@kit.edu
bUniversità degli Studi di Firenze, Dipartimento di Chimica “U. Schiff”, Via della Lastruccia 13, 50019 Sesto Fiorentino, FI, Italy
cDepartment of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtasen aukio 1), FIN-00014, Finland
dInstitute of Organic Chemistry, Karlsruhe Institute of Technology, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
First published on 31st January 2017
Abstract
Functionalized lactams are an important class of heterocycles since they are useful intermediates in organic synthesis and show biological activity in diverse therapeutic applications. In the herein presented study, a strategy for the synthesis of N-aryldiazenyllactams, offering the direct access to protected lactam derivatives is described. After the formation of triazenes from diazonium salts and commercially available N-heterocycles, oxidation (directed CH activation) with periodate under ruthenium catalysis furnished N-diazenyllactams in one step. To demonstrate the suitability of the resulting lactams for further functionalizations, the alkylation of a N-diazenyllactam is presented for one example.
Introduction
Functionalized lactams are a highly important class of heterocycles in materials science and medicinal chemistry as their core structure is not only part of amide-based materials but also of different well established therapeutics. It has been shown in the past that lactams can be synthesized from the corresponding ketones via the well-known Beckmann rearrangement or from the corresponding heterocyclic amines by oxidation. Although the oxidizing methods are known with any N-substitution, most strategies rely on N-protected amines. In most of the cases, the amines are protected as amides, carbamates (Boc, Cbz) or sulfonamides but groups based on nitrogen as e.g. diazenyls are known as well (Fig. 1 for examples with piperidine).1–4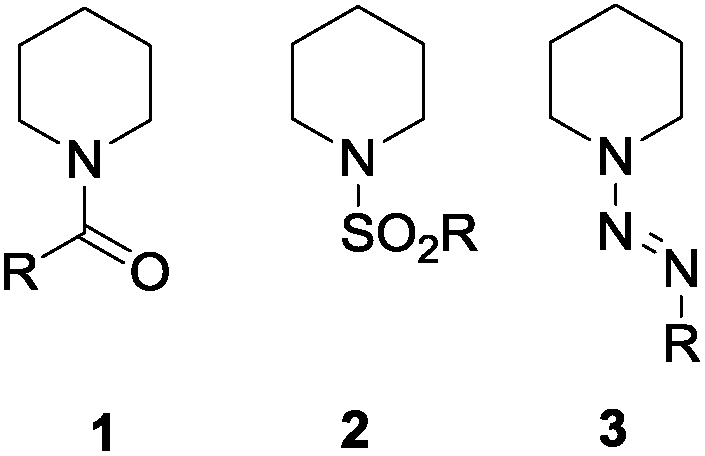 | ||
| Fig. 1 Suitable protecting groups for CH activation of piperidine: amides/carbamates 1, sulfonamides 2,1 diazenyls 3.2–4 | ||
Being modified by one of these protecting groups, oxidation of the amines with ring sizes from four to eight-membered rings have been reported. In most of the oxidation procedures, ruthenium catalysis has been used, but other methods including the use of manganese5,6 as an oxidant or electrochemical oxidation7 have also been described. Although the oxidation of N-derived cyclic amines to the corresponding lactams are known, several challenges still have not been mastered yet. For the oxidation of four-membered N-heterocycles for example, only very few reactions based on N-carbon-5 and N-oxygen-8–11 derived heterocycles have been shown so far. In addition, the oxidation of N-diazenyl-protected cyclic heterocycles (piperidines), has been shown for only a couple of compounds producing the corresponding lactams in very low yields (up to 25%).6 The lack of tools for the latter conversion inspired us to search for novel oxidation protocols.
It has been shown by us12–16 and others17–34 that diazenyl-substituted amines – triazenes – can serve as very valuable starting materials and intermediates. They are stable towards many bases, but can be cleaved under mild acidic conditions to release the free amine or amine-derivative in a simple manner.14 In addition, triazenes can also be used as versatile linkers for the immobilization of amines or anilines in solid phase syntheses. This allows the transfer of triazene chemistry developed in solution to methods that allow combinatorial chemistry on solid phases.
In this report we show that diazenyl-protected N-heterocycles can be oxidized by Ruthenium catalysts in good yields to produce the corresponding functionalized lactams.
Results and discussion
Diazenyl protection of N-heterocycles is a common tool in organic synthesis and has been used not only in solution but also on solid phases. We were interested in the diazenyl protecting group as a valuable derivatization that could allow the modification of cyclic amines and the synthesis of interesting chemical entities. Therefore, we synthesized a set of N-diazenyl heterocycles that have been investigated towards their potential to form functionalized lactams with a diazenyl unit (Fig. 2). The required N-diazenyl heterocycles 3a–d (Fig. 2), among which the quite rare azetinyldiazenes (3a, Fig. 2),35–39 were prepared by standard procedures40 from the corresponding amines and diazonium salts in good yields. We have chosen different ring sizes from 4 to 7, substituted piperidines and various arenes to investigate scope and limitations. All of the N-diazenyl heterocycles are stable and in a few cases even crystalline materials. From the NMR analyses of these compounds resulted that only pyrrolidine derivative 3b exhibits restricted rotation noticeable in the broad NMR signal for the nitrogen-adjacent CH2 groups (for selected X-ray structures of triazenes 3b and 3c{5} see ESI†).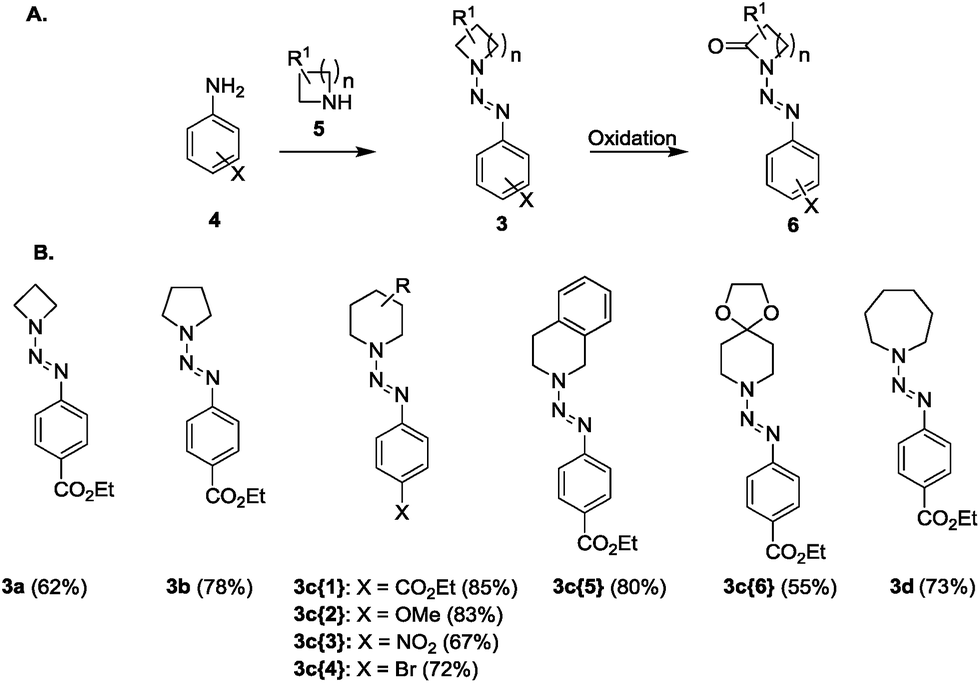 | ||
| Fig. 2 General synthetic outline (A) and building blocks that have been prepared for the oxidation (B). | ||
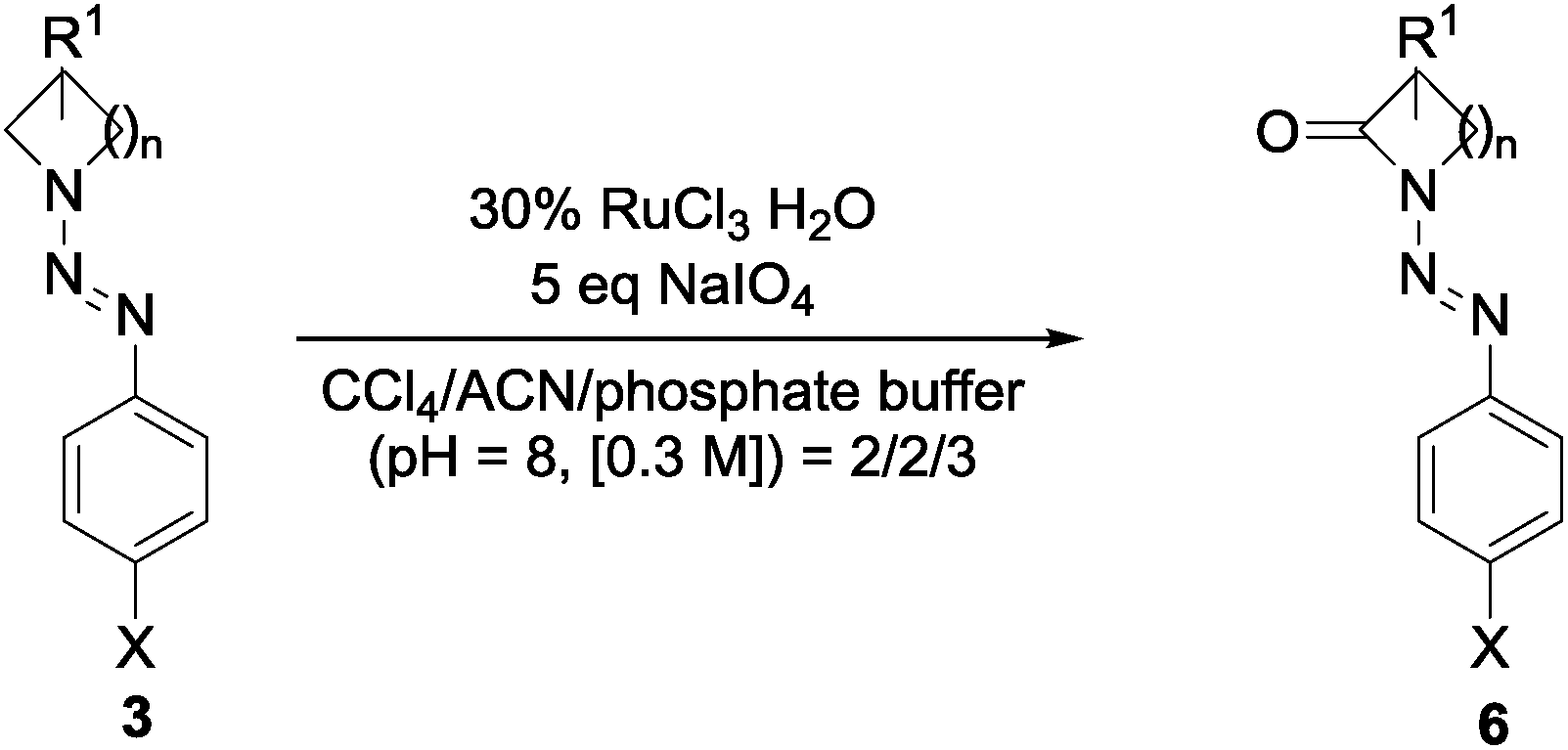
Aiming for the identification of an efficient oxidation strategy for the generated N-diazenyl amines 3a–d, compound 3c{1} was chosen as a model system to screen for the best reaction conditions. Preliminary experiments based on formerly published oxidation strategies41–44 revealed ruthenium based catalytic systems being the most promising choice (additional data referring to experiments with other catalytic systems are given in Table S1 in the ESI†). In Table 1, a summary of the different ruthenium-based experiments with RuCl3 and RuO2 as a pre-catalyst is given. It has been shown that the choice of the solvent has high influence on the outcome of the reaction and in accordance with previously published results for similar conversions,45 a CCl4/acetonitrile/H2O mixture gave the highest yields in the oxidation. The yield of 44% for the synthesis of compound 6c{1} has been further improved by the use of phosphate buffer instead of water giving a yield of 52% for the model system. Reducing the amount of catalyst and oxidant to 15% and 2.5 eq. respectively caused a slight decrease of the obtained yields (entry 3) while the use of another ruthenium-based pre-catalyst (RuO2 etc.) had no detectable effect on the outcome of the oxidation procedure (entries 2 and 6, Table 1).
| Entry | Pre-catalyst | Oxidant | Solvent | Yield [%] | ||
|---|---|---|---|---|---|---|
| 1 | RuCl3·H2O | 30% | NaIO4 | 5 eq. | CCl4/ACN/buffer | 52 |
| 2 | RuCl3·H2O | 30% | NaIO4 | 5 eq. | CCl4/ACN/H2O | 44 |
| 3 | RuCl3·H2O | 15% | NaIO4 | 2.5 eq. | CCl4/ACN/H2O | 39 |
| 4 | RuCl3·H2O | 45% | NaIO4 | 5 eq. | EtOAc/ACN/H2O | 16 |
| 5 | RuCl3·H2O | 30% | NaIO4 | 5 eq. | DCM/H2O | 10 |
| 6 | RuO2·H2O | 30% | NaIO4 | 5 eq. | CCl4/ACN/H2O | 47 |
Triazenes 3 were thus oxidized to the lactams 6 in yields ranging from 10 to 65%, i.e. much higher than those reported with KMnO4.5,6 While all of the results given in Table 2 have been gained in reactions using 1 mmol of the starting material, the reaction to give 3c{1} (entry 3, Table 2) has been repeated on a 2 mmol scale to prove the results with a higher amount of material. The obtained yield (61%) even surpasses the results that have been gained in smaller scale (52%) which was assigned to a better handling during the purification on silica gel. Under the herein described conditions, the obtained arenes are not degraded,46 and also the triazene is not oxidized. The stability of the target compounds under the conditions of the oxidation procedure has been proven for at least 24 h. Only with the four-membered ring compound 3a the oxidation yield was very low (10%). As far as we know, diazenyl β-lactams 6a have not been reported before and synthetic procedures for their preparation haven't been accomplished so far.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Triazene | n | R1 | X | Product | Yield [%] |
| a Upscaling of the reaction to 2 mmol scale. | ||||||
| 1 | 3a | 1 | H | CO2Et | 6a | 10 |
| 2 | 3b | 2 | H | CO2Et | 6b | 65 |
| 3 | 3c{1} | 3 | H | CO2Et | 6c{1} | 52 (61a) |
| 4 | 3c{2} | 3 | H | OMe | 6c{2} | 38 |
| 5 | 3c{3} | 3 | H | NO2 | 6c{3} | 49 |
| 6 | 3c{4} | 3 | H | Br | 6c{4} | 25 |
| 7 | 3c{5} | 3 | –(CH)4– | CO2Et | 6c{5} | 47 |
| 8 | 3c{6} | 3 | –OCH2CH2O– | CO2Et | 6c{6} | 59 |
| 9 | 3d | 4 | H | CO2Et | 6d | 47 |
The structure of the target triazenes has been confirmed by standard techniques as well as X-ray crystallography for one selected example 6c{1} (Fig. 3, details of the crystallographic studies of 6c{1} and triazenes 3b and 3c{5} are given in the ESI†) by which we found that the lactams crystallize as s-trans compounds.
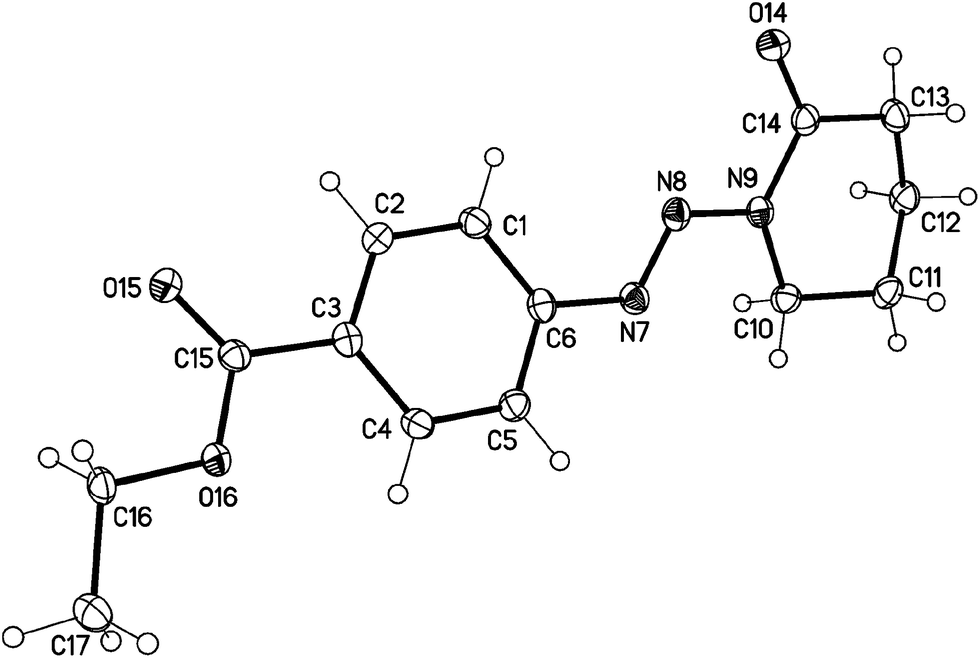 | ||
| Fig. 3 Molecular structure of the N-diazenyllactam 6c{1} (displacement parameters are drawn at 50% probability level). | ||
As a preliminary investigation of the potential of this protecting group, we carried out the alkylation of lactam 6c{2} under strong basic conditions, using LiHMDS as the base and MeI as the electrophile. Gratifyingly, the reaction provided the 3-methyl substituted lactam 7 in good yield (59%) without decomposition or degradation of the protecting group (Scheme 1). The removal of the diazenyl protecting group can be achieved via standard conditions for the cleavage of triazenes14 and has been shown in quantitative conversion for the alkylated heterocycle 7 (see ESI†).
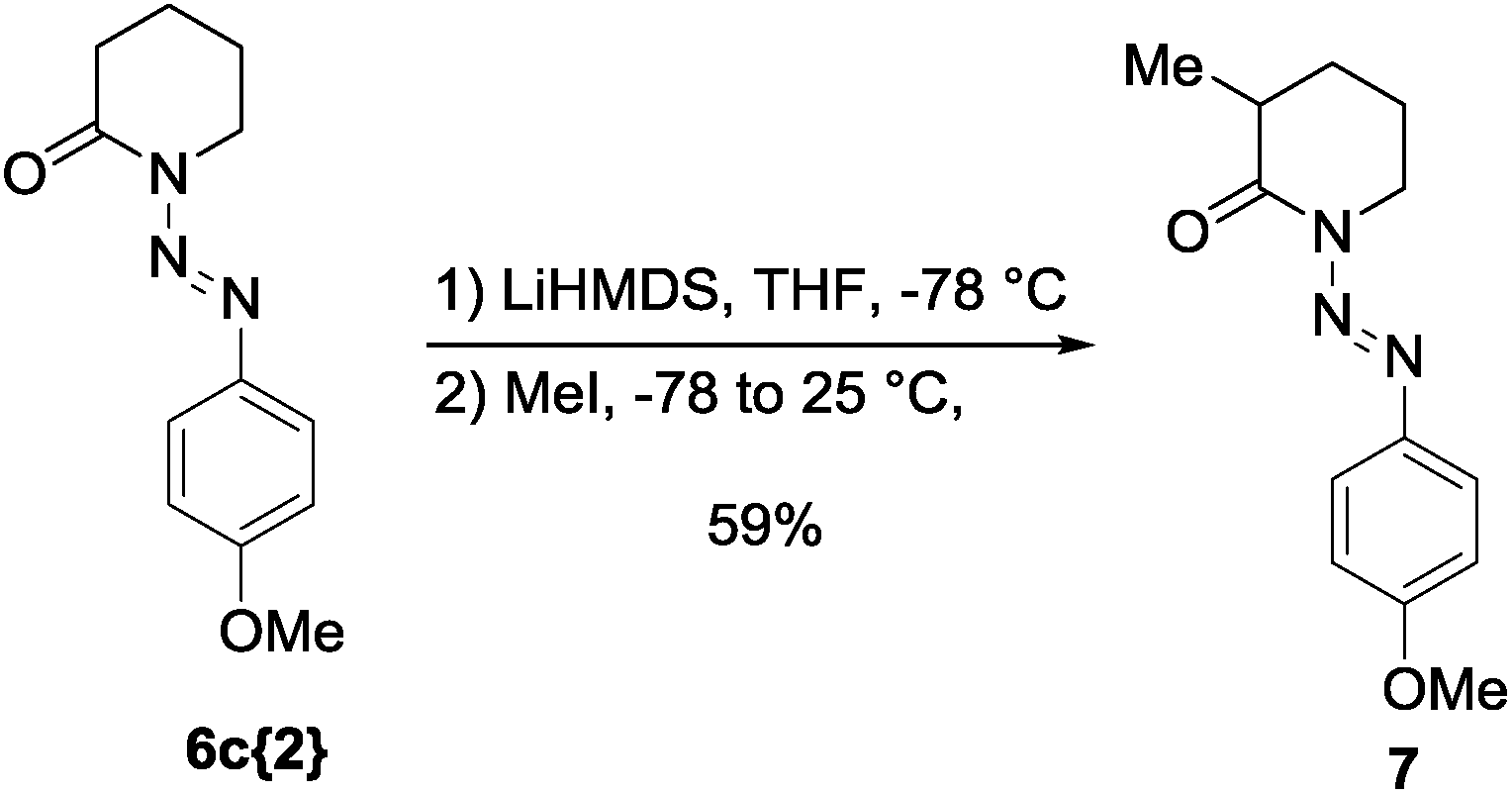 | ||
| Scheme 1 Alkylation of lactam 6c{2} to give α-alkylated heterocycle 7. | ||
Conclusions
The herein described procedure presents as far as we know the first strategy for the synthesis of functionalized lactams with diazenyl unit. The conversion of triazenes (and azo compounds) by Ru-catalyzed oxidation to give N-diazenyl protected lactams allows e.g. the synthesis of 4- and 7-membered diazenyl heterocycles that are reported for the first time. The scope of the reaction has been demonstrated by an α-alkylation of an exemplarily chosen amide allowing the application of the herein presented oxidation for diverse modifications of diazenyl protected heterocycles.Acknowledgements
This work was supported by the Helmholtz program Biointerfaces in Technology and Medicine (BIFTM). M. P. thanks the European Commission for a Marie Curie fellowship (FP7-PEOPLE-2012-ITN, Project: ECHONET “Expanding Capability in Heterocyclic Organic Synthesis”, no. 316379).Notes and references
- T. A. Zenina, I. V. Gavrish, D. S. Melkumyan, T. S. Seredenina and S. B. Seredenin, Bull. Exp. Biol. Med., 2005, 140, 194–196 CrossRef CAS PubMed.
- N. A. Lack, P. Axerio-Cilies, P. Tavassoli, F. Q. Han, K. H. Chan, C. Feau, E. LeBlanc, E. T. Guns, R. K. Guy, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2011, 54, 8563–8573 CrossRef CAS PubMed.
- R. S. Munuganti, E. Leblanc, P. Axerio-Cilies, C. Labriere, K. Frewin, K. Singh, M. D. Hassona, N. A. Lack, H. Li, F. Ban, E. Tomlinson Guns, R. Young, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2013, 56, 1136–1148 CrossRef CAS PubMed.
- K. Jehle, L. Cato, A. Neeb, C. Muhle-Goll, N. Jung, E. W. Smith, V. Buzon, L. R. Carbo, E. Estebanez-Perpina, K. Schmitz, L. Fruk, B. Luy, Y. Chen, M. B. Cox, S. Brase, M. Brown and A. C. Cato, J. Biol. Chem., 2014, 289, 8839–8851 CrossRef CAS PubMed.
- J. H. Markgraf and C. A. Stickney, J. Heterocycl. Chem., 2000, 37, 109–110 CrossRef CAS.
- A. Gescher, C. P. Turnbull and M. F. G. Stevens, J. Chem. Soc., Perkin Trans. 1, 1977, 2078–2083 RSC.
- C. Li, C.-C. Zeng, L.-M. Hu, F.-L. Yang, S. J. Yoo and R. D. Little, Electrochim. Acta, 2013, 114, 560–566 CrossRef CAS.
- M. L. M. Pennings and D. N. Reinhoudt, J. Org. Chem., 1983, 48, 4043–4048 CrossRef CAS.
- P. A. Van Elburg and D. N. Reinhoudt, Heterocycles, 1987, 26, 437–445 CrossRef CAS.
- P. A. Van Elburg and D. N. Reinhoudt, Recl. Trav. Chim. Pays-Bas, 1988, 107, 381–387 CrossRef CAS.
- P. A. Van Elburg, D. N. Reinhoudt, S. Harkema and G. J. Van Hummel, Tetrahedron Lett., 1985, 26, 2809–2812 CrossRef CAS.
- S. Brase and T. Muller, Sci. Synth., 2007, 31, 1845–1872 Search PubMed.
- N. Jung and S. Brase, Sci. Synth., 2010, 41, 613–640 Search PubMed.
- R. Lazny, J. Poplawski, J. Kobberling, D. Enders and S. Brase, Synlett, 1999, 1304–1306 CrossRef CAS.
- R. Lazny, M. Sienkiewicz and S. Brase, Tetrahedron, 2001, 57, 5825–5832 CrossRef CAS.
- V. Zimmermann, F. Avemaria and S. Brase, J. Comb. Chem., 2007, 9, 200–203 CrossRef CAS PubMed.
- M. Barbero, I. Degani, N. Diulgheroff, S. Dughera and R. Fochi, Synthesis, 2001, 2180–2190 CrossRef CAS.
- S. P. Cummings, P. E. Fanwick, J. Savchenko and T. Ren, J. Organomet. Chem., 2013, 745–746, 93–97 CrossRef CAS.
- H. A. Dabbagh, A. Teimouri, A. N. Chermahini and R. Shiasi, Spectrochim. Acta, Part A, 2007, 67, 437–443 CrossRef PubMed.
- Y. Fang, C. Wang, S. Su, H. Yu and Y. Huang, Org. Biomol. Chem., 2014, 12, 1061–1071 CAS.
- A. Khazaei, M. Kazem-Rostami, A. R. Moosavi-Zare, M. Bayat and S. Saednia, Synlett, 2012, 23, 1893–1896 CrossRef CAS.
- D. B. Kimball and M. M. Haley, Angew. Chem., Int. Ed., 2002, 41, 3338–3351 CrossRef CAS PubMed.
- K. Knepper and R. E. Ziegert, T1 and T2 – Versatile Triazene Linker Groups, in Linker Strategies in Solid-Phase Organic Synthesis, ed. P. J. H. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- D. K. Koelmel, N. Jung and S. Braese, Aust. J. Chem., 2014, 67, 328–336 CrossRef CAS.
- R. Lazny, A. Nodzewska and P. Klosowski, Tetrahedron, 2004, 60, 121–130 CrossRef CAS.
- A. S. Monteiro, J. Almeida, G. Cabral, P. Severino, P. A. Videira, A. Sousa, R. Nunes, J. D. Pereira, A. P. Francisco, M. J. Perry and E. Mendes, Eur. J. Med. Chem., 2013, 70, 1–9 CrossRef CAS PubMed.
- K. Nishiwaki, T. Ogawa and K. Matsuo, Angew. Chem., Int. Ed., 2002, 41, 484–486 CrossRef CAS PubMed.
- K. Nishiwaki, T. Ogawa, K. Shigeta, K. Takahashi and K. Matsuo, Tetrahedron, 2006, 62, 7034–7042 CrossRef CAS.
- M. d. J. Perry, E. Carvalho, E. Rosa and J. Iley, Eur. J. Med. Chem., 2009, 44, 1049–1056 CrossRef CAS PubMed.
- C. Picherit, F. Wagner and D. Uguen, Tetrahedron Lett., 2004, 45, 2579–2583 CrossRef CAS.
- S. Recnik, J. Svete and B. Stanovnik, Z. Naturforsch., B: Chem. Sci., 2004, 59, 380–385 CAS.
- M. K. Rofouie, M. Salahinejad, J. B. Ghasemi and A. Aghaei, Magn. Reson. Chem., 2013, 51, 269–274 CrossRef CAS PubMed.
- C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2012, 51, 7242–7245 CrossRef CAS PubMed.
- A. Zarei, L. Khazdooz, H. Aghaei, G. Azizi, A. N. Chermahini and A. R. Hajipour, Dyes Pigm., 2014, 101, 295–302 CrossRef CAS.
- M. H. Akhtar and A. C. Oehschlager, Tetrahedron, 1970, 26, 3245–3263 CrossRef CAS.
- J. Fu and M. Barra, Trends Org. Chem., 2012, 16, 1–8 CAS.
- J. Fu, K. Lau and M. Barra, J. Org. Chem., 2009, 74, 4654 CrossRef CAS.
- J. Fu, K. Lau and M. Barra, J. Org. Chem., 2009, 74, 1770–1773 CrossRef CAS PubMed.
- R. Tabone and M. Barra, Dyes Pigm., 2010, 88, 180–186 CrossRef.
- M. Döbele, S. Vanderheiden, N. Jung and S. Bräse, Angew. Chem., Int. Ed., 2010, 49, 5986–5988 CrossRef PubMed.
- Y. Zhao, J. Q. L. Ang, A. W. T. Ng and Y.-Y. Yeung, RSC Adv., 2013, 3, 19765–19768 RSC.
- Y. Wang, Y. Kuang and Y. Wang, Chem. Commun., 2015, 51, 5852–5855 RSC.
- F. Minisci, C. Punta, F. Recupero, F. Fontana and G. F. Pedulli, J. Org. Chem., 2002, 67, 2671–2676 CrossRef CAS PubMed.
- G. Blay, L. Cardona, B. García, C. L. García and J. Pedro, Tetrahedron Lett., 1997, 38, 8257–8260 CrossRef CAS.
- P. H. J. Carlsen, T. Katsuki, V. S. Martin and K. B. Sharpless, J. Org. Chem., 1981, 46, 3936–3938 CrossRef CAS.
- G. Veeresa and A. Datta, Tetrahedron Lett., 1998, 39, 119–122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: cif-files (3b, 3c{5} and 6c{1}). CCDC 1510209 (3b), CCDC 1510210 (3c{5}) and 1510211 (6c{1}). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26546d |
| This journal is © The Royal Society of Chemistry 2017 |