
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous electrokinetic concentration and separation of proteins on a paper-based analytical device†
Zhi-Yong Wu *a,
Biao Maa,
Song-Fang Xiea,
Kun Liub and
Fang Fanga
*a,
Biao Maa,
Song-Fang Xiea,
Kun Liub and
Fang Fanga
aResearch Center for Analytical Sciences, Chemistry Department, College of Sciences, Northeastern University, Shenyang 110819, P. R. China. E-mail: zywu@mail.neu.edu.cn; Fax: +86 24 8368 7659; Tel: +86 24 8368 7659
bResearch Institute of Vacuum and Fluid, College of Mechanical Engineering and Automation, Northeastern University, Shenyang 110819, P. R. China
First published on 17th January 2017
Abstract
In this report, we introduced an electrokinetic strategy for simultaneous concentration and separation of charged analytes in an open and free standing paper fluidic channel. This was achieved by taking advantage of the field amplified sample stacking (FASS) effect. An electric field gradient was developed in the channel due to the conductivity difference between the background electrolyte (BGE) and sample solution. Through adjustment of the electroosmotic flow level and BGE concentration, charged analytes were not only stacked but also separated in the gradient based on their difference in electrophoretic mobility. Proof of concept was demonstrated firstly by two food dyes, brilliant blue and amaranth, and each dye was about 100-fold concentrated within 300 s. Simultaneous stacking and separation of two colored proteins, bovine hemoglobin and cytochrome c, was also demonstrated visually with the proposed method. Sample consumption was reduced to 5 μL by directly loading the sample onto the open channel, and the separation was completed within 30 s. This method is simple, rapid and easy to operate, which is particularly beneficial to the development of sensitive and multifunctional paper-based analytical devices (PADs) for field testing.
Introduction
Paper substrates possess many advantages such as low cost, light weight, high surface area to volume ratio, capillary action, biocompatibility, flexibility and disposability.1,2 In 2007, paper as a substrate material for microfluidic devices was first reported by the Whitesides research group.3 Since then, the field of microfluidic paper-based analytical devices (μPADs) has experienced rapid growth due to their salient advantages such as easy fabrication, portability, low sample consumption, multiple assays and simple to operate.4–8 These advantages make μPADs highly attractive for point of care testing (POCT), especially in resource-limited settings. To date, the application scope of μPADs has involved medical diagnosis,9,10 environmental analysis,11 food safety control12 and drug testing.13Preconcentration and separation of sample components are essential steps in many analyses as well as medical diagnostic tests. Despite a number of assays have been demonstrated on μPADs, the integration of multiple pretreatment units remains a challenge. A few efforts have been exerted to add preconcentration function on μPADs, including liquid evaporation,14 dialysis15 and chemical modifications on the paper substrate.16,17 In addition, some electrokinetic preconcentration techniques developed in capillary electrophoresis and microfluidic system were also demonstrated on μPADs. These techniques include ion concentration polarization (ICP),18–25 isotachophoresis (ITP)26–29 and field amplified sample stacking (FASS),30 which are easy to perform on μPADs thanks to the openness of the paper fluidic channel and the simplicity in surface modification.
Paper electrophoresis is a very traditional separation method, and now it has been mostly replaced by slab gel or capillary electrophoresis. Recently, the potential function on electrophoretic separation is redrawing attention in the new scenario of μPADs, as have been demonstrated for electrophoretic separation of amino acids,31 food dyes,32 metal ions,33 proteins34,35 and nucleic acids.19 However, in most cases, the preconcentration and separation steps were performed independently that could not achieve the sensitive detection of complex and low abundant sample components. An applicable way to address this issue is through the combination of the sample preconcentration and separation in a single step. This may be realized by using the present electrokinetic techniques in microfluidic system, such as isoelectric focusing,36 ICP,37 ITP,38 bipolar electrode focusing39 and conductivity gradient focusing.40 Among these techniques, only ICP was carried to μPADs by Gong and co-workers.19 They demonstrated simultaneous preconcentration and separation of hepatitis B virus DNA fragments in human serum by ICP in a nitrocellulose paper fluidic channel. By local patterning of cation selective nanoporous membrane (Nafion) in the channel, an electric field gradient was induced under applied voltage. They also extended the paper-based ICP effect to preconcentrate and separate the ssDNA and dsDNA from lysed sperm cells in order to analyse the integrity of sperm DNA.41
FASS is believed as the simplest online preconcentration technique in capillary electrophoresis.42–44 In our previous work, this technique was first introduced to a μPAD composed of glass fiber paper and up to 1000-fold enrichment factor was obtained.30 Inspired by the aforementioned work by Gong et al.,19 we envisioned that the electric field gradient as developed in the paper-based FASS system should not only enable charged species to be stacked, but also have the potential to separate them based on their difference in electrophoretic mobility. Through the optimization of experimental conditions, simultaneous stacking and separation of small molecule food dyes was demonstrated in the paper fluidic channel, and about 100-fold enrichment factor was obtained for each dye within 300 s. Then we demonstrated concentration and separation of macromolecule proteins. This method is low cost, simple, rapid and easy to operate, which is particularly beneficial to the development of sensitive and multifunctional paper-based analytical devices (PADs) for field testing.
Experimental
Chemicals and materials
Tris(hydroxymethyl)aminomethane (Tris), ammonium chloride (NH4Cl), brilliant blue FCF (MW 792.85), amaranth (MW 604.47) and hydroxyethylcellulose (HEC) were purchased from Aladdin Reagent (Shanghai, China). Hydrochloric acid (HCl) and ammonia solution were purchased from Sinopharm Chemical Reagent Co., Ltd. Bovine hemoglobin (BHb, MW ∼ 65 kDa, pI ∼ 7.0) and cytochrome c (Cyt c, from bovine heart, MW ∼ 13 kDa, pI ∼ 10.1) were obtained from Sangon Biotech (Shanghai, China). Deionized water was obtained from the Millipore Milli-Q system (Millipore, Milford, MA). 500 mM Tris–HCl (pH = 8.1) was prepared as the stock background electrolyte. Glass fiber paper (K49, 90 mm in diameter and 250 μm in thickness) was purchased from Dezhou Kejia Environmental Products Co. The glass fiber paper was cut into strips (30 mm long by 3 mm wide) using a paper trimmer. The lids of the centrifuge tube (1.5 mL) were used as the reservoirs. Platinum wires (0.5 mm in diameter) were used as electrodes. A miniature and low cost DC-DC booster converter (GRB12300D, ESSO Latham Electronic Technology Co., Ltd., Shenzhen, China) linked with a USB portable battery power charger (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 mA h, 5 V output, PH50, Romoss Technology Co., Ltd., Shenzhen, China) was taken as the power supply. The booster converter gives a constant output voltage of 300 V.
400 mA h, 5 V output, PH50, Romoss Technology Co., Ltd., Shenzhen, China) was taken as the power supply. The booster converter gives a constant output voltage of 300 V.
Device operation
The paper strip was placed on two reservoirs at a distance of 20 mm. For each side of the strip, 5 mm was immersed into the solution. 150 μL background electrolyte (BGE) was firstly loaded into the anode reservoir, and most of paper channel (from anode side to the edge of the cathode reservoir) was wetted using BGE. Then 150 μL sample solution was loaded into the cathode reservoir, followed by applying the voltage.Detection and quantitation
A smartphone (Meizu Note1, running Android 5.1) was fixed to the eyepiece tube of the stereomicroscope by an attaching clamp and the back camera was adjusted to align with the eyepiece. In order to avoid the interruption of the ambient light, the stereomicroscope was surrounded using a shade cloth. A white LED light was used to illuminate the paper strip to provide a stable light condition. The photo of the smartphone-based imaging system is shown in Fig. S1 (ESI†). The smartphone was used with a professional camera control application (Camera FV-5 Lite, Version 3.15).Images captured from the smartphone were processed and analyzed using ImageJ software (Version 1.49v, National Institutes of Health, Bethesda, MD). For quantitative assessment of the enrichment factor (EF), calibration curves were constructed based on the mean gray values of the standard solutions. The complete details about the construction of calibration curves were provided in Fig. S2 (ESI†). For calculating the color intensity of the stacking band, the region of each band was selected by the color threshold tool of ImageJ. Then the mean gray value of the selected zone was measured as the color intensity for quantitative analysis, as can be seen in Fig. S3 (ESI†). Note that each band intensity was corrected by subtracting the mean gray value of the BGE zone near the band.
Results and discussion
Simultaneous concentration and separation
When a voltage is applied across a paper fluidic channel which connects with a high conductivity BGE and a low conductivity sample solution, an electric field gradient can be developed in the channel. Once charged molecules migrate into the gradient, they stack together because of the reduction in electromigration velocity. This is the central idea for the paper-based field amplified sample stacking (FASS). Previous work was mainly focused on the use of FASS to preconcentrate analytes.30 Here, it is reasonable to assume that with proper condition optimization, analytes with different mobilities should not only be stacked, but also be separated in the electric field gradient, as shown in Fig. 1a. This was experimentally proved with two food dyes (brilliant blue and amaranth) as shown in Fig. 1b. Both of the dyes are negatively charged in aqueous solution. Two narrow stacking bands are clearly separated as expected. The sequence of the two bands is in agreement with the mobilities of the two dyes.45 The dyes were prepared with low conductivity deionized water while 50 mM Tris–HCl was taken as the BGE. To successfully obtain the simultaneous stacking and separation, both the level of electroosmotic flow (EOF) and that of the electric field gradient should be well optimized, as can be seen in the following section.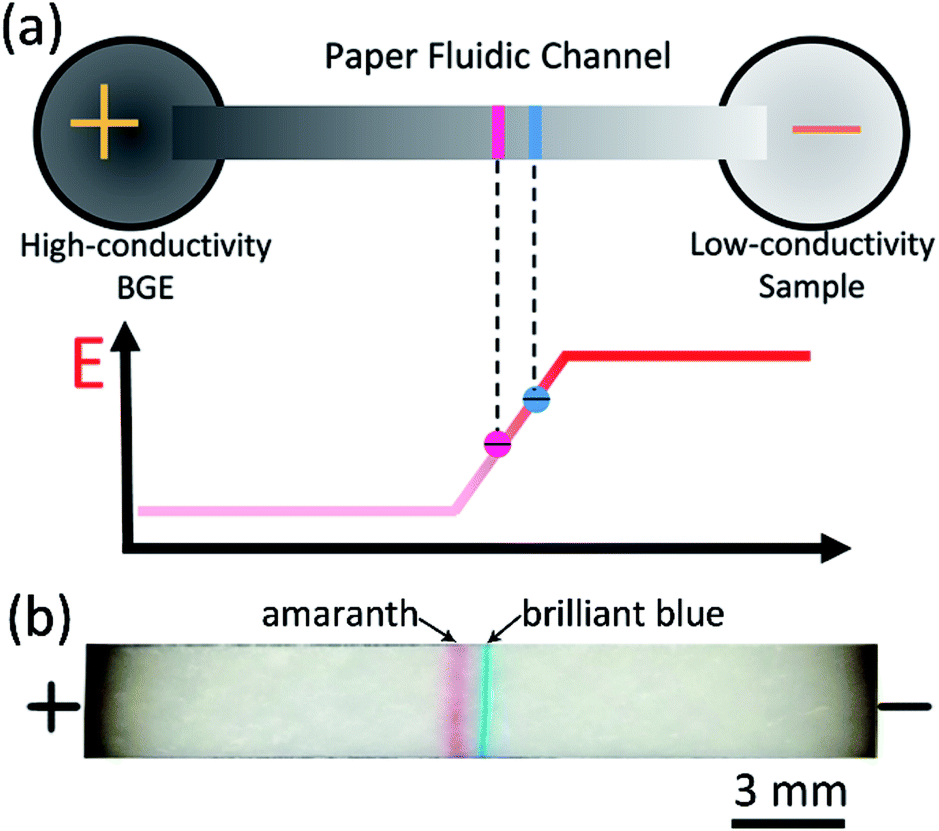 | ||
| Fig. 1 (a) Schematic of paper-based FASS for simultaneous concentration and separation for negatively charged analytes. Stacking is achieved due to reduction of electric field, and separation is achieved because of the difference in electrophoretic mobility. (b) A typical image of the simultaneous concentration and separation of two negatively charged food dyes, brilliant blue and amaranth. | ||
Effect of EOF
As is well known, the negatively charged surface of the glass fiber can generate an EOF in the direction of electric field.46 To investigate the effect of EOF level, BGE of 50 mM Tris–HCl was maintained for each test. Fig. 2 shows the images of channel at representative time points of 30 s and 180 s. Without the suppression of EOF (no HEC in BGE), neither stacking band nor separation was seen. This is because the EOF was too strong to develop a stable electric field gradient in the channel. Adding additive in the BGE is a convenient method to adjust the EOF level, as has been shown in the previous work where a surfactant of polyvinylpyrrolidone (PVP) was used.30 However, PVP was found not as effective for the two food dyes here, and the reason remained unknown to us. Here we used another EOF modifier, hydroxyethylcellulose (HEC).47 By adding 0.2% HEC (m/v) to the BGE, the two stacking bands emerged in the paper channel. When the HEC concentration was increased to 0.4%, the bands became more concentrated and narrower, especially for the amaranth. It should be noted that HEC not only suppresses the EOF, but also increases the viscosity of the solution, which is beneficial to decrease the diffusion of the stacked molecules. When the HEC concentration was above 0.4%, no obvious improvement was observed. Thus, 0.4% HEC was added to each BGE in the following experiments. | ||
| Fig. 2 Images of the stacking bands at 30 s (a) and 180 s (b) with addition of HEC. BGE was 50 mM Tris–HCl, the initial concentration for each dye was 1.0 μg mL−1, voltage was 300 V. The paper fluidic channel was 30 mm long by 3 mm wide. | ||
Effect of BGE concentration
The conductivity difference between BGE and sample is another key factor for a successful FASS operation, which determines the profile of the electric field gradient. Tris–HCl buffers ranging from 25 mM to 200 mM were used to evaluate the effect of BGE concentration on the separation and concentration. As shown in Fig. 3a and b, when the Tris–HCl was 200 mM, a narrow stacking band formed in 30 s, and kept well in 180 s, with slow drifting toward the anode. This phenomenon is in agreement with what was observed in the previous work with fluorescent probes. However, the separation effect was hardly seen. This should be ascribed to that the electric field gradient was so sharp that the two dyes stacked together. With decrease of the Tris–HCl concentration from 200 to 25 mM, the two dyes were found well separated. Fig. 3c shows the intensity distributions along the paper fluidic channels at 180 s, from which we can obtain the band peak intensity and the separation resolution (SR). The SR values were 1.4 and 0.9 for the BGE of 25 and 50 mM Tris–HCl, respectively. The lower the BGE concentration, the better the separation, which implies that the electric field gradient level can be tuned by the BGE concentration. Although the BGE of 25 mM Tris–HCl gave a better SR, the stacking efficiency is correspondingly reduced. Considering the compromise between separation performance and stacking efficiency, 50 mM Tris–HCl was taken as the optimal BGE in the following section.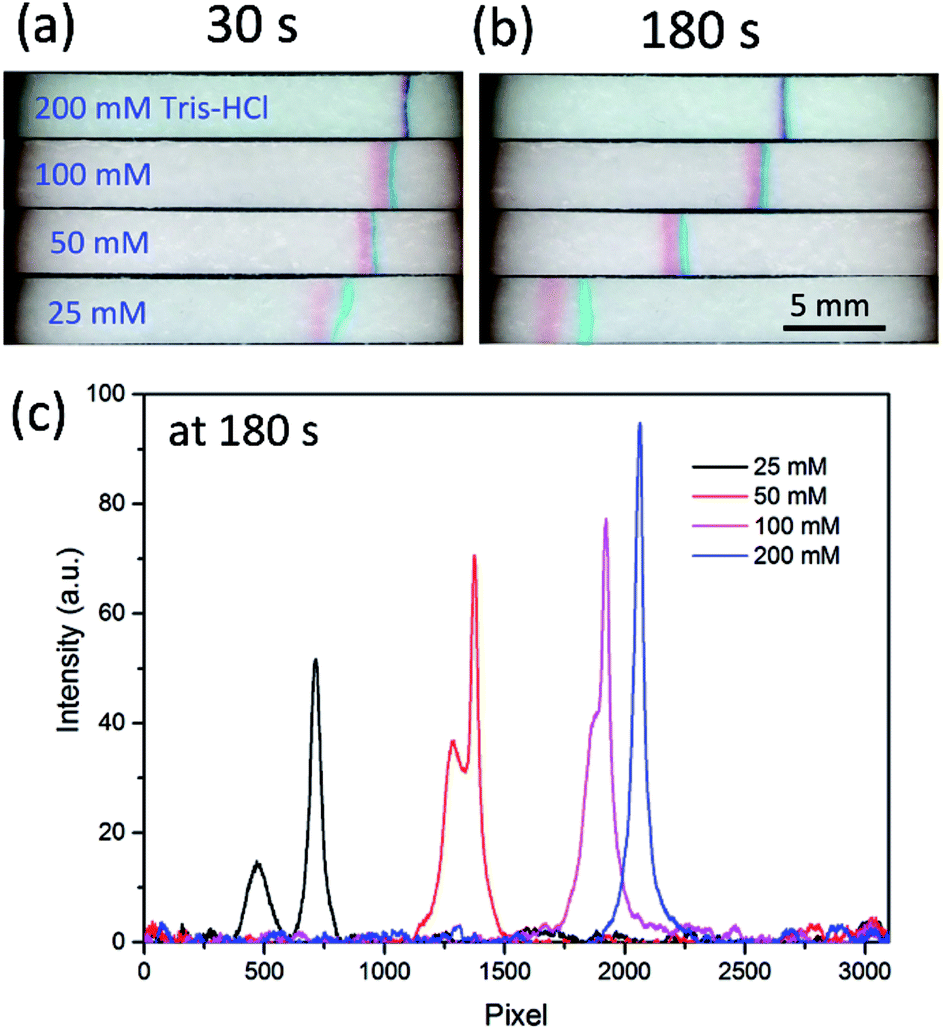 | ||
| Fig. 3 Effect of the BGE (Tris–HCl) concentration. Images of the stacking bands at 30 s (a) and 180 s (b), respectively. (c) The band intensity profile along the channel at 180 s. The other conditions were the same as those in Fig. 2. | ||
Quantitative assessment
Under the optimized conditions, quantitative assessment of the enrichment factor (EF) was performed based the images acquired by the smartphone-based imaging system. Fig. 4a depicts the band intensity as a function of time, and each intensity data was the averaged value of five parallel tests. The initial concentration for each dye was 1.0 μg mL−1, and at this concentration each dye is invisible to naked eyes without stacking (not shown). To determine the EF values for the two dyes, calibration curves were established by measuring the intensities of standard solutions (10–160 μg mL−1), as shown in Fig. S2c and d (ESI†). Without stacking, the limits of detection (3σ) for amaranth and brilliant blue were 21 μg mL−1 and 6.4 μg mL−1, respectively. The EF values for amaranth and brilliant blue were 103 and 98, respectively, which were obtained by comparing the band intensity to the standard reference as indicated in Fig. 4a as dotted lines. Fig. 4b shows a typical stacking/separation process. Note that the light condition and the smartphone imaging parameters were kept constant for the acquired images.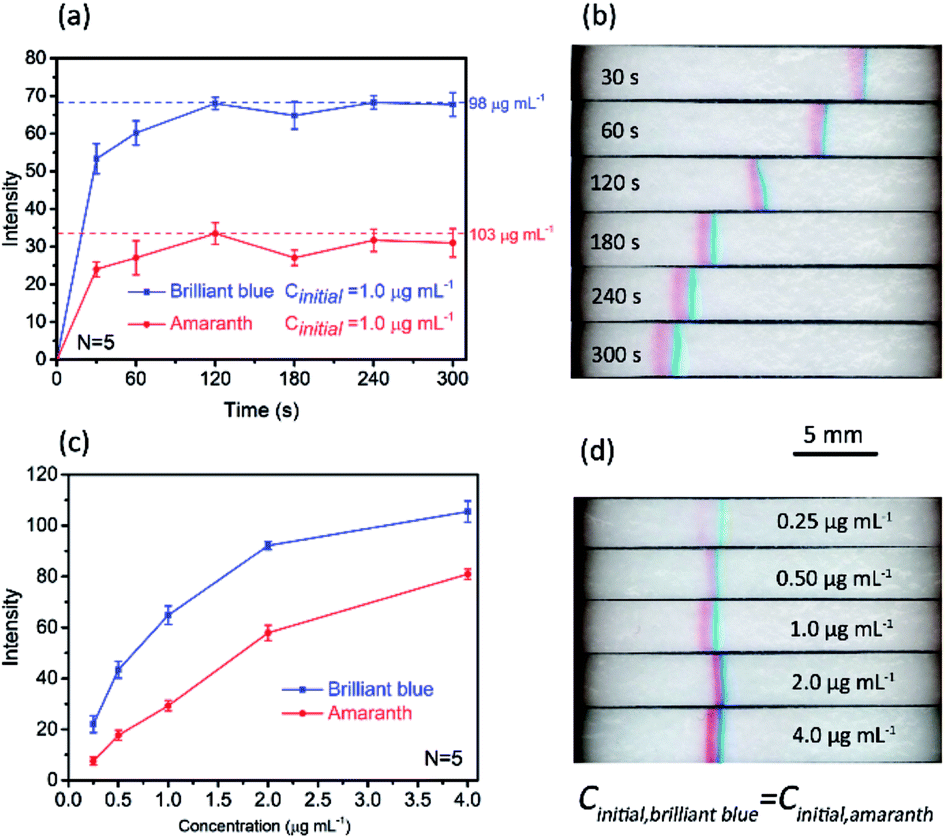 | ||
| Fig. 4 Quantitation of paper-based FASS for sample concentration and separation. (a) Band's intensity development over time. The initial concentration for each dye was 1.0 μg mL−1. (b) A typical stacking/separation process. (c) Band intensity as a function of initial concentration ranging from 0.25 μg mL−1 to 4.0 μg mL−1. (d) Corresponding images of stacking bands with different initial concentrations at 180 s. Error bars in (a) and (c) represent standard deviation for five independent measurements. The other conditions were the same as those in Fig. 2. | ||
To explore the potential of this method on quantitative analysis, the band intensity as a function of initial concentration under a given stacking/separation time was also investigated, as shown in Fig. 4c. The linear dynamic range for brilliant blue was 0.25–4.0 μg mL−1 (linear regression equation: y = 71.62lgx − 149.3, y for the band intensity, x for brilliant blue at the concentration of ng mL−1, correlation coefficient: 0.9961). The linear dynamic range for amaranth was 0.25–2.0 μg mL−1 (linear regression equation: y = 28.1x + 1.82, y for the band intensity, x for amaranth at the concentration of μg mL−1, correlation coefficient: 0.9980). The limits of detection (3σ) for amaranth and brilliant blue were 0.31 μg mL−1 and 0.17 μg mL−1, respectively. This result indicated that limit of detection of sub ppm level can be achieved colorimetrically with this simple paper device. Again, it should be noted that, in Fig. 4c, each data point was the average result over five independent measurements, and all the RSDs were below 7.5%. This precision was relatively high considering the simple setup and manual operation. The images of the stacking bands with different initial concentrations from 0.25 to 4.0 μg mL−1 are shown in Fig. 4d.
Sample consumption
In above experiments, we loaded 150 μL sample solution to the sample reservoir. For some valuable biosamples, this large sample volume may not be available. As the paper fluidic channel is open, it is easy to reduce the sample consumption by direct loading the sample solution onto the channel. To be specific, 150 μL deionized water instead of sample solution was added to the cathode reservoir. After 60 s establishment of the electric field gradient, 5 μL sample solution was loaded onto the paper channel close to the cathode reservoir with pipette. As shown in Fig. S4 (ESI†), the stacking bands develop quickly after 5 s of sample loading. The two bands are eventually separated within 30 s.Simultaneous stacking and separation of proteins
Simultaneous stacking and separation of proteins in the paper fluidic channel is even more attractive either for sensitive online detection or for further analysis by sophisticated instruments. To demonstrate the feasibility, two colored proteins, bovine hemoglobin (BHb) and cytochrome c (Cyt c) were tried with the proposed method. Fig. 5a depicts the stacking process for the two model proteins in which the 150 μL sample solution was loaded in the cathode reservoir. The initial concentration for BHb was 0.1 mg mL−1 and for Cyt c it was 0.2 mg mL−1. With application of voltage, two stacking bands were well stacked and separated in the channel within 60 s, and the SR was 1.1 at t = 300 s. Note that the model proteins were prepared in 200 mM Tris solution with a pH of 10.7, and 20 mM NH4Cl–NH3 with the pH of 10.8 was taken as the BGE. Both the pH of sample media and BGE are greater than the pIs of the proteins to make them negatively charged. The conductivity of 20 mM NH4Cl–NH3 was 2.80 mS cm−1, which is close to that of 50 mM Tris–HCl (2.75 mS cm−1). Whereas the conductivity of 200 mM Tris was only 0.11 mS cm−1, much lower than that of the BGE, meeting the basic requirement of FASS. A low sample volume of 5 μL was also implemented as aforementioned. Note that 200 mM Tris instead of water was added to cathode reservoir here. As shown in Fig. 5b, the two proteins were stacked within 10 s, and they were separated at 30 s with a SR of 0.9. Here the initial concentration for each protein was 2.5 mg mL−1 for clear observation of the effect. | ||
| Fig. 5 Paper-based FASS for simultaneous concentration and separation of proteins. (a) A stacking process in which the protein sample (150 μL) was added to the cathode reservoir. The initial concentrations for BHb and Cyt c were 0.1 mg mL−1 and 0.2 mg mL−1, respectively. (b) Demonstration of small volume protein sample stacking process by loading 5 μL sample onto the paper channel instead of the reservoir. The initial concentration for each protein was 2.5 mg mL−1. The proteins were prepared with 200 mM Tris (pH = 10.7). 20 mM NH4Cl–NH3 (pH = 10.8) with 0.4% HEC was used as the BGE. The other conditions were the same as those in Fig. 2. | ||
Conclusions
In this work, we successfully demonstrated simultaneous sample concentration and separation on a simple straight paper fluidic channel by FASS. This method is low cost and rapid, which is applicable to both small molecule food dyes and macromolecule proteins. This work shows the potential of paper-based FASS on the development of sensitive and multifunctional μPADs. A portable power supply together with a smartphone-based imaging system makes this device suitable for on-site analysis. The proposed method may also find its application in protein analysis coupled with other advanced analytical devices such as liquid chromatography or mass spectrometry as a rapid and low cost offline sample pretreatment method.Acknowledgements
This research was financially supported by National Natural Science Foundation of China (21575019, 51376039) and Education Department of Liaoning Province of China (LZ2015036).References
- R. Pelton, TrAC, Trends Anal. Chem., 2009, 28, 925–942 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed. Engl., 2007, 46, 1318–1320 CrossRef CAS PubMed.
- X. Li, D. R. Ballerini and W. Shen, Biomicrofluidics, 2012, 6, 11301–1130113 CrossRef PubMed.
- M. Santhiago, E. W. Nery, G. P. Santos and L. T. Kubota, Bioanalysis, 2014, 6, 89–106 CrossRef CAS PubMed.
- D. M. Cate, J. A. Adkins, J. Mettakoonpitak and C. S. Henry, Anal. Chem., 2015, 87, 19–41 CrossRef CAS PubMed.
- A. M. Lopez-Marzo and A. Merkoci, Lab Chip, 2016, 16, 3150–3176 RSC.
- N. A. Meredith, C. Quinn, D. M. Cate, T. H. Reilly 3rd, J. Volckens and C. S. Henry, Analyst, 2016, 141, 1874–1887 RSC.
- C. Zhao and X. Liu, Biomicrofluidics, 2016, 10, 024119 CrossRef PubMed.
- S. T. Sanjay, M. Dou, J. Sun and X. Li, Sci Rep., 2016, 6, 30474 CrossRef CAS PubMed.
- C. Sicard, C. Glen, B. Aubie, D. Wallace, S. Jahanshahi-Anbuhi, K. Pennings, G. T. Daigger, R. Pelton, J. D. Brennan and C. D. Filipe, Water Res., 2015, 70, 360–369 CrossRef CAS PubMed.
- Y. Zhang, P. Zuo and B. C. Ye, Biosens. Bioelectron., 2015, 68, 14–19 CrossRef CAS PubMed.
- M. Su, L. Ge, S. Ge, N. Li, J. Yu, M. Yan and J. Huang, Anal. Chim. Acta, 2014, 847, 1–9 CrossRef CAS PubMed.
- S. Y. Wong, M. Cabodi, J. Rolland and C. M. Klapperich, Anal. Chem., 2014, 86, 11981–11985 CrossRef CAS PubMed.
- R. Tang, H. Yang, J. R. Choi, Y. Gong, J. Hu, S. Feng, B. Pingguan-Murphy, Q. Mei and F. Xu, Talanta, 2016, 152, 269–276 CrossRef CAS PubMed.
- S. A. Byrnes, J. D. Bishop, L. Lafleur, J. R. Buser, B. Lutz and P. Yager, Lab Chip, 2015, 15, 2647–2659 RSC.
- A. Abbas, A. Brimer, J. M. Slocik, L. Tian, R. R. Naik and S. Singamaneni, Anal. Chem., 2013, 85, 3977–3983 CrossRef CAS PubMed.
- M. M. Gong, P. Zhang, B. D. MacDonald and D. Sinton, Anal. Chem., 2014, 86, 8090–8097 CrossRef CAS PubMed.
- M. M. Gong, R. Nosrati, M. C. San Gabriel, A. Zini and D. Sinton, J. Am. Chem. Soc., 2015, 137, 13913–13919 CrossRef CAS PubMed.
- R. J. Yang, H. H. Pu and H. L. Wang, Biomicrofluidics, 2015, 9, 014122 Search PubMed.
- S. I. Han, K. S. Hwang, R. Kwak and J. H. Lee, Lab Chip, 2016, 16, 2219–2227 RSC.
- S. Hong, R. Kwak and W. Kim, Anal. Chem., 2016, 88, 1682–1687 CrossRef CAS PubMed.
- L.-H. Hung, H.-L. Wang and R.-J. Yang, Microfluid. Nanofluid., 2016, 20, 1–9 CrossRef CAS.
- D.-T. Phan, S. A. M. Shaegh, C. Yang and N.-T. Nguyen, Sens. Actuators, B, 2016, 222, 735–740 CrossRef CAS.
- S. H. Yeh, K. H. Chou and R. J. Yang, Lab Chip, 2016, 16, 925–931 RSC.
- B. Y. Moghadam, K. T. Connelly and J. D. Posner, Anal. Chem., 2014, 86, 5829–5837 CrossRef CAS PubMed.
- T. Rosenfeld and M. Bercovici, Lab Chip, 2014, 14, 4465–4474 RSC.
- X. Li, L. Luo and R. M. Crooks, Lab Chip, 2015, 15, 4090–4098 RSC.
- B. Y. Moghadam, K. T. Connelly and J. D. Posner, Anal. Chem., 2015, 87, 1009–1017 CrossRef CAS PubMed.
- B. Ma, Y. Z. Song, J. C. Niu and Z. Y. Wu, Lab Chip, 2016, 16, 3460–3465 RSC.
- L. Ge, S. Wang, S. Ge, J. Yu, M. Yan, N. Li and J. Huang, Chem. Commun., 2014, 50, 5699–5702 RSC.
- C. Xu, M. Zhong, L. Cai, Q. Zheng and X. Zhang, Electrophoresis, 2016, 37, 476–481 CrossRef CAS PubMed.
- L. OuYang, C. Wang, F. Du, T. Zheng and H. Liang, RSC Adv., 2014, 4, 1093–1101 RSC.
- L. Luo, X. Li and R. M. Crooks, Anal. Chem., 2014, 86, 12390–12397 CrossRef CAS PubMed.
- C. L. Chagas, F. R. de Souza, T. M. Cardoso, R. C. Moreira, J. A. da Silva, D. P. de Jesus and W. K. Coltro, Anal. Methods, 2016, 8, 6682–6686 RSC.
- H. Cui, K. Horiuchi, P. Dutta and C. F. Ivory, Anal. Chem., 2005, 77, 1303–1309 CrossRef CAS PubMed.
- W. Ouyang, S. H. Ko, D. Wu, A. Y. Wang, P. W. Barone, W. S. Hancock and J. Han, Anal. Chem., 2016, 88, 9669–9677 CrossRef CAS PubMed.
- C. Eid, G. Garcia-Schwarz and J. G. Santiago, Analyst, 2013, 138, 3117–3120 RSC.
- D. R. Laws, D. Hlushkou, R. K. Perdue, U. Tallarek and R. M. Crooks, Anal. Chem., 2009, 81, 8923–8929 CrossRef CAS PubMed.
- D. W. Inglis, E. M. Goldys and N. P. Calander, Angew. Chem., Int. Ed. Engl., 2011, 50, 7546–7550 CrossRef CAS PubMed.
- R. Nosrati, M. M. Gong, M. C. San Gabriel, A. Zini and D. Sinton, Anal. Methods, 2016, 8, 6260–6264 RSC.
- D.-S. Lian, S.-J. Zhao, J. Li and B.-L. Li, Anal. Bioanal. Chem., 2014, 406, 6129–6150 CrossRef CAS PubMed.
- Y. Y. Tu, M. M. Hsieh and S. Y. Chang, Electrophoresis, 2015, 36, 2828–2836 CrossRef CAS PubMed.
- B. Jung, R. Bharadwaj and J. G. Santiago, Electrophoresis, 2003, 24, 3476–3483 CrossRef CAS PubMed.
- K. S. Lee, M. J. Shiddiky, S. H. Park, D. S. Park and Y. B. Shim, Electrophoresis, 2008, 29, 1910–1917 CrossRef CAS PubMed.
- Y. Oyama, T. Osaki, K. Kamiya, R. Kawano, T. Honjoh, H. Shibata, T. Ide and S. Takeuchi, Lab Chip, 2012, 12, 5155–5159 RSC.
- D. Belder and M. Ludwig, Electrophoresis, 2003, 24, 3595–3606 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The photo of the smartphone-based imaging system (Fig. S1), details about the construction of the calibration curves (Fig. S2), data processing of image (Fig. S3), the stacking and separation process for low sample consumption (Fig. S4). See DOI: 10.1039/c6ra26500f |
| This journal is © The Royal Society of Chemistry 2017 |