
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of high antistatic HDPE/polyaniline encapsulated graphene nanoplatelet composites by solution blending
Quan Wanga,
Yuming Wanga,
Qingguo Mengc,
Tinglan Wang*a,
Weihong Guo *ab,
Genhua Wub and
Li Youa
*ab,
Genhua Wub and
Li Youa
aPolymer Processing Laboratory, Key Laboratory for Preparation and Application of Ultrafine Materials of Ministry of Education, School of Material Science and Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: tinglanw@ecust.edu.cn; guoweihong@ecust.edu.cn
bCollaborative Innovation Center for Petrochemical New Materials, Anqing, Anhui 246011, P. R. China
cShanghai Pret Composites CO., LTD., Shanghai 201707, P. R. China
First published on 12th January 2017
Abstract
Graphene nanoplatelets with excellent electrical conductivity in polymer matrices are highly promising for the industrial application of electrical conductive materials, however, poor dispersion results in high contents of graphene nanoplatelets being required for electrical property enhancement. In this study, graphene nanoplatelets (GNP)@polyaniline (PANI) nanocomposites were synthesized by in situ polymerization whereas the compatibility between GNP@PANI nanocomposites and the polymer matrix improved significantly due to graphene nanoplatelet encapsulation with polyaniline. GNP@PANI nanocomposites were utilized to prepare a permanent antistatic high-density polyethylene (HDPE) composite through solution blending and press forming in order for GNP@PANI nanocomposites to be dispersed homogeneously in the HDPE. The dispersion and compatibility of GNP@PANI nanocomposites in the HDPE were verified by morphology characterization, resulting in significant improvement of the electrical properties of the GNP@PANI/HDPE composites. It was observed that surface resistivity (ρs) and volume resistivity (ρv) decreased sharply with a 10 wt% GNP@PANI addition of nanocomposites. The results displayed that in situ polymerization and solution blending were effective methods for a conductive network establishment by addition of only 2 wt% of GNP and 8 wt% of PANI.
Introduction
High-density polyethylene (HDPE) is regarded to be the most widely used polyolefin, combining the advantages of low cost, excellent mechanical properties, insulation capability, thermal stability and ease of processing.1 However, certain problems exist caused by inherent insulation of polymers restricting all application fields, such as the material surface trend for easy accumulation of charges trapping dust and deterioration of product performance and possibly resulting in an explosion.2,3 High-density polyethylene has been widely used in various fields, such as the electronics industry, although electronic equipment requires materials with antistatic properties.4To tackle these problems, it is necessary to improve the antistatic properties of HDPE. Conventionally, antistatic materials are prepared by an amphiphilic surfactant mix into polymers, whereas the antistatic property of the final material is not permanent.5,6 The antistatic property will be weakened in time. The research for novel antistatic agents has attracted the interest of many researchers. Recently, certain conductive fillers as an antistatic agent were added into the polymer matrix in order for composites to obtain the antistatic property, such as carbon black, carbon nanotubes, multi-wall carbon nanotubes, ionic liquids, metal nanoparticles, metal oxide whisker, polyaniline and graphene as final polymers.7–20 The purpose of the term “graphene” utilization was for properties characterization of single two-dimensional carbon layers independent existence.21 Since Novoselov et al. first prepared graphene in 2004, a significant amount of graphene properties and applications have attracted the attention of many researchers. Graphene nanoplatelets (GNP) are shaped as plate-like having dimensions in nanometers and high aspect ratios.22 Many researches have reported that graphene or graphene nanoplatelets were added into polymers, with a desire of excellent electrical properties achievement, including the antistatic property. Although certain properties improved, electrical conductivity of composites was significantly worsened than the inherent properties of graphene nanoplatelets. The main reason for this phenomenon is that graphene nanoplatelets are clustered due to GNP dispersion being poor inside the polymer matrix, resulting in conductive network failure. Jiang et al. prepared HDPE/GNP nanocomposites with a 10 vol% GNP loading and the in-plane and through-plane electrical resistivity of the composites were only about 1010 Ω and 1011 Ω cm approximately, respectively.23 The aggregation of GNP during melt extrusion and the corresponding orientation during injection moulding in both high in-plane and through-plane electrical resistivity.24,25
As it is known, high electrical conductivity of bulk composites is proven significantly difficult to be achieved in comparison to film composites. Many reasons exist regarding the result, such as poor dispersion and compatibility between conductive fillers and polymer matrix and the orientation and the aggregation of the conductive fillers during melt blending. In this study, a novel antistatic agent GNP@PANI nanocomposite was prepared by in situ polymerization. Polyaniline has the advantages of low cost, ease of synthesis and relatively high electrical conductivity.26 Polyaniline was selected firstly for graphene nanoplatelet encapsulation for cost reduction and dispersion and compatibility in high-density polyethylene improvement. In order for the dispersion of the antistatic agent to result in being more uniform in high-density polyethylene, solution blending method was utilized consequently led through press forming for the final composites to be obtained. It was discovered that the corresponding antistatic property was improved, as attributed to good dispersion of GNP@PANI nanocomposites and compatibility between HDPE and antistatic agent. Permanent antistatic composites were successfully prepared by a 10 wt% of GNP@PANI nanocomposite addition only. Fig. 1 is the schematic display of the aforementioned experimental process.
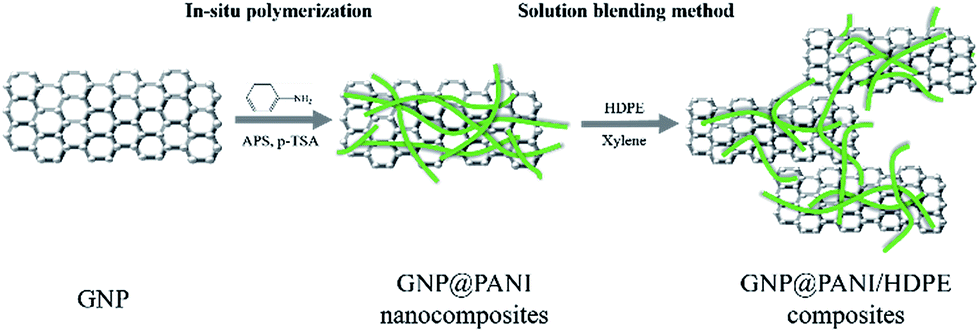 | ||
| Fig. 1 Experimental process. | ||
Experimental
Materials
High density polyethylene (HDPE), under code 60550AG, was purchased from the Lanzhou Petrochemical limited company (China), presenting a melting flow rate of 7.5 g/10 min at 2.16 kg/190 °C measured by the GB/T3682-2000 test method. The average molecular weight (Mw) and the polydispersity of the HDPEs were 407![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 5.2, respectively. The HDPE-g-MAH (MAPE) was purchased from the Nantong Sunny Polymer New Material Technology limited company (China) with a grafting degree of 0.5–1.0 wt% and melting flow rate of 4.0–10.0 g/10 min at 2.16 kg/190 °C measured by the ASTM D-1238 test method, under the code of Fine-Blend™ CMG5804. Graphene nanoplatelets were purchased from the Suzhou Geruifeng Nanotechnology limited company (China). The aniline was purchased from Shanghai Aladdin Biochemical Technology limited company (China). The ammonium persulfate was purchased from the Shanghai Taitan Technology limited company (China). The p-toluenesulfonic acid monohydrate was purchased from Shanghai Macklin Biochemical Technology limited company (China).
000 g mol−1 and 5.2, respectively. The HDPE-g-MAH (MAPE) was purchased from the Nantong Sunny Polymer New Material Technology limited company (China) with a grafting degree of 0.5–1.0 wt% and melting flow rate of 4.0–10.0 g/10 min at 2.16 kg/190 °C measured by the ASTM D-1238 test method, under the code of Fine-Blend™ CMG5804. Graphene nanoplatelets were purchased from the Suzhou Geruifeng Nanotechnology limited company (China). The aniline was purchased from Shanghai Aladdin Biochemical Technology limited company (China). The ammonium persulfate was purchased from the Shanghai Taitan Technology limited company (China). The p-toluenesulfonic acid monohydrate was purchased from Shanghai Macklin Biochemical Technology limited company (China).
Synthesis of GNP@PANI nanocomposites
GNP@PANI nanocomposites were synthesized by in situ polymerization. The procedure was executed as follows: 2 g of GNP were added in 200 mL of 0.5 M p-toluenesulfonic acid aqueous solution under constant stirring for GNP dispersion formation inside an ice-bath. Then 8 g of the aniline monomer were added into the GNP dispersion. 9.8 g of ammonium persulfate (APS) dissolved in 100 mL of 0.5 M p-toluenesulfonic acid were added into the aforementioned mixture for in situ polymerization initiation. The reaction was executed under constant stirring for 8 h inside an ice-bath. The resulting dark green floccule was filtered and washed with deionized water, ethanol and xylene several times until the filtrate resulted in being colourless.Preparation of GNP@PANI/HDPE composites
GNP@PANI/HDPE composites were prepared by solution blending. The GNP@PANI nanocomposites were dispersed in xylene under constant stirring and ultrasonic frequency for 60 min. Following heating to 125 °C, both HDPE and MAPE were rapidly added into the dispersion and the mixture was constantly being stirred for 5 h. Consequently, the mixture was added slowly into 500 mL of low temperature ethanol for the reaction product to be obtained. Finally, the resulting product was filtrated, washed with ethanol three times and dried at 95 °C for 24 h. The MAPE was considered to improve the HDPE compatibility with GNP@PANI due to the rule of polarity nearness and loading preservation at 5 wt% in all samples. The resulting samples were prepared by hot-pressing at 150 °C and 10 MPa for sheets with suitable sizes to be obtained. A series of composites containing 0, 5, 10, 15 and 20 wt% GNP@PANI were prepared by the same procedure. The content of GNP in GNP@PANI was approximately 20 wt%.Characterizations
Fourier transform infrared spectra (FTIR) measurements were conducted on a Nicolet 6700 FTIR spectrometer. All samples were dispersed in KBr and then pressed into sheets for testing.Raman spectroscopy measurements were conducted on an Invia Raman Microscope. All powdered samples were provided for testing.
X-ray diffraction (XRD) pattern measurements were conducted on a D8 advance X-ray diffraction device. All of the samples were ground under 300 mesh for testing.
The surface resistivity (ρs) and volume resistivity (ρv) were tested by a ZC-90 megohmmeter. The GNP@PANI nanocomposites samples were prepared by hot-pressing at 150 °C and 10 MPa for circular plates with a diameter of 10 cm and a thickness of 2 mm to be obtained. The ZC-90 megohmmeter was preheated for half an hour before testing. The temperature and relative humidity were 25 °C and 65% (65% RH), respectively. The instrument was zeroed before the 500 V voltage application. The latter parameters are referred in the GB/T1410-1989 standard.
The morphologies of samples were analysed by field emission scanning electron microscopy (FE-SEM, Hitachi S4800, 30 kV). The GNP, PANI and GNP@PANI nanocomposites were dispersed in ethanol and consequently dropped into the copper network for testing. Previous to the fracture surface morphology study, the specimens were quenched in liquid nitrogen and subsequently the surface of each sample was sputtered with a thin layer of gold.
The morphologies and the internal structure were analysed by transmission electron microscopy (TEM, JEOL JEM-1400, 200 kV). All samples were dispersed in ethanol and consequently dropped into the copper network for testing. Previous to the GNP@PANI/HDPE composite study, the specimens were quenched in liquid nitrogen and subsequently severed into ultra-thin slices previous to copper dropping.
The dielectric constant, dielectric loss and AC conductivity were detected by a broadband dielectric spectrometer (Novocontrol Concept 40). The samples were converted into circular plates with a diameter of 3 cm and a thickness of 2 mm.
Tensile and flexural tests were performed on a universal test machine (CMT4024-20KN, Shenzhen Sans Co., Ltd) in room temperature according to the GB/T1040.2-2006:1993 standard with the dumbbell sample dimensions being 150 × 10 × 4 mm and according to the GB/T9341-2000 standard with the sample dimensions being 80 × 10 × 4 mm, respectively. A notch impact test was performed according to the GB/T1843-1996 standard with the sample dimensions being 80 × 10 × 4 mm.
Results and discussion
FTIR
In Fig. 2 the FTIR spectra of pure GNP, pure PANI and GNP@PANI nanocomposites are presented. The FTIR spectra of pure GNP exhibited the main bands at 3444 cm−1 and 1638 cm−1 indicating the stretching vibration of the O–H bond at the edge of the GNP plane and vibration of the graphitic double bonds, respectively.27 In the FTIR spectra of the pure PANI, the weak bands at 3425 cm−1 and 2919 cm−1 were assigned to the N–H and C–H bonds stretching, respectively. The bands at 1563 cm−1, 1477 cm−1, 1299 cm−1 and 1122 cm−1 were assigned to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the quinoid ring, the stretching vibration of the CC in benzenoid ring, the stretching of the C–N secondary aromatic amine and the stretching of the CN (–NquinoidN–), respectively.28 In the FTIR spectra measurements of the GNP@PANI nanocomposites, following GNP encapsulation by aniline, the bands at 3436 cm−1, 2927 cm−1, 1564 cm−1 and 1483 cm−1 were shifted in positions and with enhanced intensities caused by strong molecular interactions between the GNP and PANI, which were assigned to N–H bond stretching, C–H bond stretching, stretching vibration of the CC in quinoid ring and the benzenoid ring. The bands of stretching vibration of CC shifted to a higher wave number in the GNP@PANI nanocomposites demonstrating the π–π interaction existence and hydrogen bonding between graphene nanoplatelets and PANI. As it was observed in the FTIR spectra of GNP@PANI nanocomposites, the band at 1638 cm−1 disappeared because the unstable graphitic double bonds reduced during aniline addition to graphene nanoplatelets dispersion and aniline in situ polymerization of aniline. The result indicated that both GNP and PANI existed simultaneously and the GNP@PANI nanocomposites were successfully prepared.
C in the quinoid ring, the stretching vibration of the CC in benzenoid ring, the stretching of the C–N secondary aromatic amine and the stretching of the CN (–NquinoidN–), respectively.28 In the FTIR spectra measurements of the GNP@PANI nanocomposites, following GNP encapsulation by aniline, the bands at 3436 cm−1, 2927 cm−1, 1564 cm−1 and 1483 cm−1 were shifted in positions and with enhanced intensities caused by strong molecular interactions between the GNP and PANI, which were assigned to N–H bond stretching, C–H bond stretching, stretching vibration of the CC in quinoid ring and the benzenoid ring. The bands of stretching vibration of CC shifted to a higher wave number in the GNP@PANI nanocomposites demonstrating the π–π interaction existence and hydrogen bonding between graphene nanoplatelets and PANI. As it was observed in the FTIR spectra of GNP@PANI nanocomposites, the band at 1638 cm−1 disappeared because the unstable graphitic double bonds reduced during aniline addition to graphene nanoplatelets dispersion and aniline in situ polymerization of aniline. The result indicated that both GNP and PANI existed simultaneously and the GNP@PANI nanocomposites were successfully prepared.
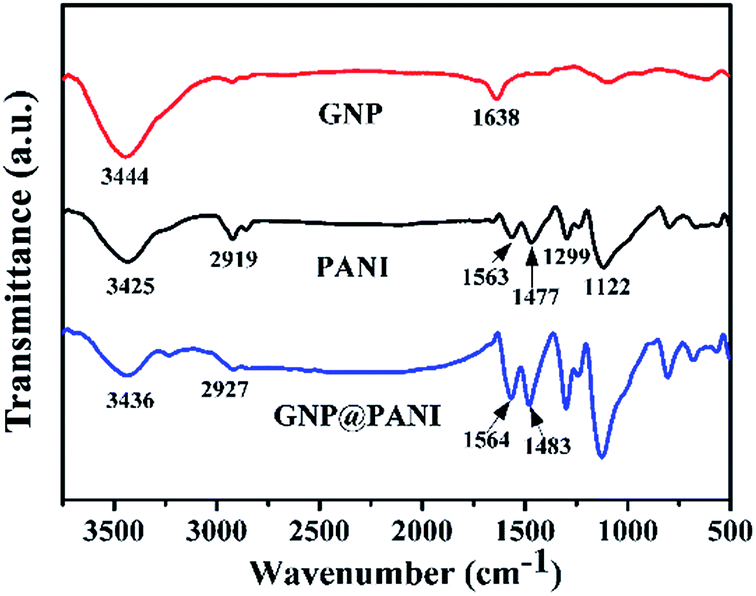 | ||
| Fig. 2 FTIR spectra of GNP, PANI and GNP@PANI. | ||
Raman
In Fig. 3 the Raman spectra of pure GNP, pure PANI and GNP@PANI nanocomposites are presented. Graphene distinguishing from bulk graphite and structural defects detection can be characterized by Raman spectroscopy.29 In the Raman spectra of pure GNP, the D, G and 2D bands were signified, corresponding to 1358 cm−1, 1580 cm−1 and 2725 cm−1 values respectively. The slight D band was assigned to vibrations of sp3 hybridized carbon atoms which were disordered, forming few defects and the sharp G band was assigned to vibrations of the sp2 hybridized carbon atoms in the basal plane.30 The sharp 2D band was assigned to the number of stacked graphene layers in the platelets.31 In the Raman spectra of GNP@PANI nanocomposites, the D band was disappeared, because in Raman spectroscopy the surface of the material can only be detected, whereas the surface of graphene nanoplatelets was encapsulated by polyaniline, therefore the weak D band was disappeared. The same statement is true for the 2D band that became weakened. The G band shifted from 1580 cm−1 to 1588 cm−1 and the G band almost fused with the sharpest band of polyaniline. The result demonstrated that graphene nanoplatelets and polyaniline co-existed and a great interaction formed between the latter.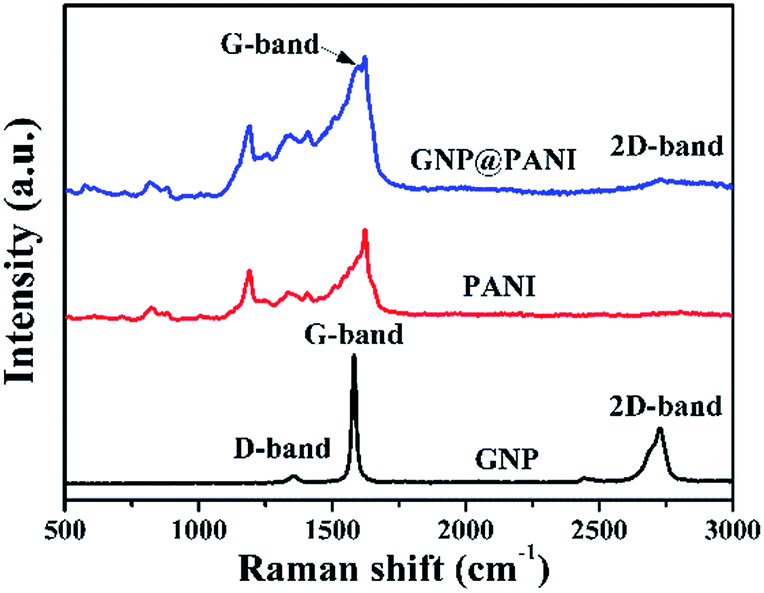 | ||
| Fig. 3 Raman spectra of GNP, PANI and GNP@PANI. | ||
XRD
In Fig. 4 the XRD patterns of pure GNP, pure PANI and GNP@PANI nanocomposites are presented. In the XRD pattern for pure GNP, a single high intense diffraction peak can be observed at 26.41° as 2θ with an inter-spacing of 3.35 Å, corresponding to the XRD peak of the (002) crystalline plane of the graphene nanoplatelet. The peaks at 42.20°, 44.35° and 56.61° as 2θ were assigned to the (100), (101) and (004) planes, respectively.32 In the XRD pattern for pure PANI, three distinctive peaks can be observed at 15.01°, 20.17°, 25.34° as 2θ corresponding to (011), (020) and (200) crystalline plane of PANI, respectively.33 In the XRD pattern for GNP@PANI nanocomposites, peaks corresponding to both GNP and PANI are visible. This result demonstrated that graphene nanoplatelets and polyaniline existed also, whereas an interaction was created between graphene nanoplatelets and polyaniline.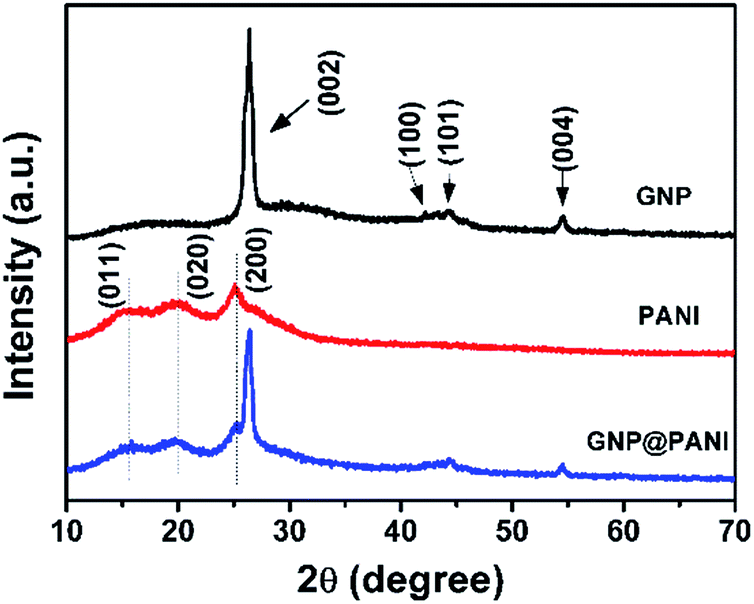 | ||
| Fig. 4 XRD pattern of GNP, PANI and GNP@PANI. | ||
Surface and volume resistivity
In Fig. 5 the histogram of GNP@PANI/HDPE surface resistivity and volume resistivity of GNP@PANI/HDPE composites with various contents in GNP@PANI nanocomposites. Pure HDPE without any antistatic agents displayed a significantly high surface and volume resistivity, corresponding to the ρs = 2.82 × 1016 Ω per sq. and ρv = 6.61 × 1016 Ω cm, respectively. Both surface and volume resistivity decreased as the content of GNP@PANI nanocomposites increased. Both exact surface (ρs) and volume (ρv) resistivity of GNP@PANI/HDPE composites are presented in Table 1. When the content of GNP@PANI nanocomposites was 10 wt%, both surface and volume resistivity were significantly lower than the corresponding resistivity properties of pure HDPE, decreased to ρs = 6.36 × 1011 Ω per sq. and ρv = 4.17 × 1012 Ω cm. Moreover, when the antistatic agent content was 20 wt%, both surface (ρs) and volume (ρv) resistivity decreased to the minimum value of 2.88 × 106 Ω per sq. and 2.09 × 107 Ω cm, respectively. According to the military handbook DOD-HDBK-263, the ρs value of the antistatic material is between 109 and 1014 Ω per sq.34–36 The electrostatic dissipative material having the ρs values at 105 to 1012 Ω per sq. and 104 to 1011 Ω per sq. were defined by the industry standards ANSI/EIA-541-1988 (USA) and ANSI/ESD S541-200 (USA), respectively.37 According to the aforementioned three standards, GNP@PANI/HDPE composites could be referred as antistatic materials when the corresponding content of antistatic agent exceeded 10 wt%. The results displayed that the addition of GNP@PANI nanocomposites can improve the antistatic property of the composites significantly. The explanation of this result was that the conductive network formed inside composites as the content of GNP@PANI was 10 wt%. The result displayed that the excellent dispersion of GNP@PANI nanocomposites in the non-polar polymer matrix was achieved by solution blending. In other words, the bulk antistatic composites contained only 2 wt% of graphene nanoplatelets.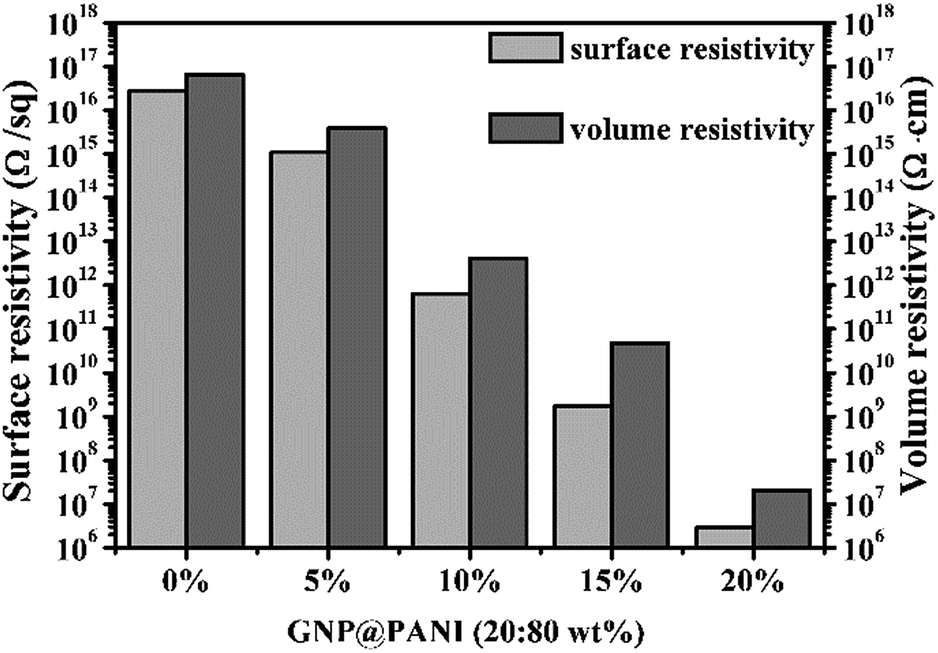 | ||
| Fig. 5 Surface and volume resistivity of GNP@PANI/HDPE composites with various contents in antistatic agents. | ||
| GNP@PANI (wt%) | ρs (Ω per sq.) | ρv (Ω cm) |
|---|---|---|
| 0 | 2.82 × 1016 | 6.61 × 1016 |
| 5 | 1.12 × 1015 | 3.89 × 1015 |
| 10 | 6.46 × 1011 | 4.17 × 1012 |
| 15 | 1.74 × 109 | 4.79 × 1010 |
| 20 | 2.88 × 106 | 2.09 × 107 |
FE-SEM and TEM
In Fig. 6 the morphology and structure of pure GNP, pure PANI, GNP@PANI nanocomposites, pure HDPE and GNP@PANI/HDPE composites characterized by FE-SEM and TEM is presented. As it can be observed in Fig. 6a, the GNP was quite thin with a smooth surface and certain GNP were aggregated and corrugated with a diameter of few micrometers. As it can be observed in Fig. 6b, the GNP was transparent and no further substances adhered to the surface. As it can be observed in Fig. 6c and d, pure PANI nanorods exhibited diameters of 80–130 nm and lengths between 0.5 and 1 μm in the absence of GNP. Regarding GNP@PANI nanocomposites in Fig. 6e, the PANI was encapsulated onto the GNP surface, during GNP addition into the in situ polymerization reaction solution. The nanocomposites were quite thick and the surface was tough relatively. As it can be observed in Fig. 6f, a lot of granular and rod-like PANIs attached to the surface of GNP providing an increase for translucent flaky nanocomposite formation. The results displayed that graphene nanoplatelets can be successfully encapsulated with polyaniline by in situ polymerization. As it can be observed in Fig. 6g, i and k, both pure HDPE and 20 wt% GNP@PANI/HDPE composites were quenched in liquid nitrogen and consequently gold sputtered previous to observation. Through comparison from Fig. 6g and i, pure HDPE was a homogeneous phase and the dispersion of GNP@PANI nanocomposites was uniform in the HDPE whereas large scale aggregation did not occur. It could be clearly observed in Fig. 6k that the compatibility between GNP@PANI nanocomposites and HDPE was remarkable. As it can be observed in Fig. 6h, j and l, all three figures confirmed that the dispersion of GNP@PANI nanocomposites was significantly uniform in the polymer matrix and the compatibility between GNP@PANI nanocomposites and HDPE was relatively excellent. | ||
| Fig. 6 FE-SEM micrographs of pure GNP (a), pure PANI (c), GNP@PANI nanocomposites (e), pure HDPE (g), 20 wt% GNP@PANI/HDPE composites (i) and (k); TEM micrographs of pure GNP (b), pure PANI (d), GNP@PANI (f), pure HDPE (h), 20 wt% GNP@PANI/HDPE composites (j) and (l). | ||
Dielectric properties
In Fig. 7 the variation of dielectric constant, dielectric loss and AC conductivity of the frequency for GNP@PANI/HDPE composites with various antistatic agent contents at 25 °C is presented. The dielectric performance of GNP@PANI/HDPE composites with various amounts of GNP@PANI were measured by a broadband dielectric spectrometer. As presented in Fig. 7a, a negligible variation of the dielectric constant exist on the frequency for GNP@PANI/HDPE composites with the content of GNP@PANI nanocomposites being 0 wt%, 5 wt% or 10 wt%. However, when the content of GNP@PANI nanocomposites was 15 wt% or 20 wt%, the dielectric constant increased significantly and consequently decreased sharply as frequency increased. The results indicated that a homogeneous dispersion of GNP@PANI nanocomposites in the HDPE existed due to excellent compatibility between GNP@PANI nanocomposites, whereas the HDPE improved the dielectric constant of composites significantly. The percolation network was established by an alternate existence of the antistatic agent and the insulating polymer layer resulting in instability and easy damage by frequency.38 It was noted that the maximum dielectric constant of 49.88 was 21 times approximately larger than the corresponding constant of pure HDPE. As presented in Fig. 7b, dielectric loss increased along with the content of antistatic agents. Moreover, the conductivity constituted additional critical parameter of antistatic materials. As presented in Fig. 7c, a strong relationship between AC conductivity and the frequency existed. In addition, the AC conductivity increased significantly along with the content of GNP@PANI nanocomposites. The reason for this result was that conductive paths were forming in the polymer matrix as the content of GNP@PANI nanocomposites increased.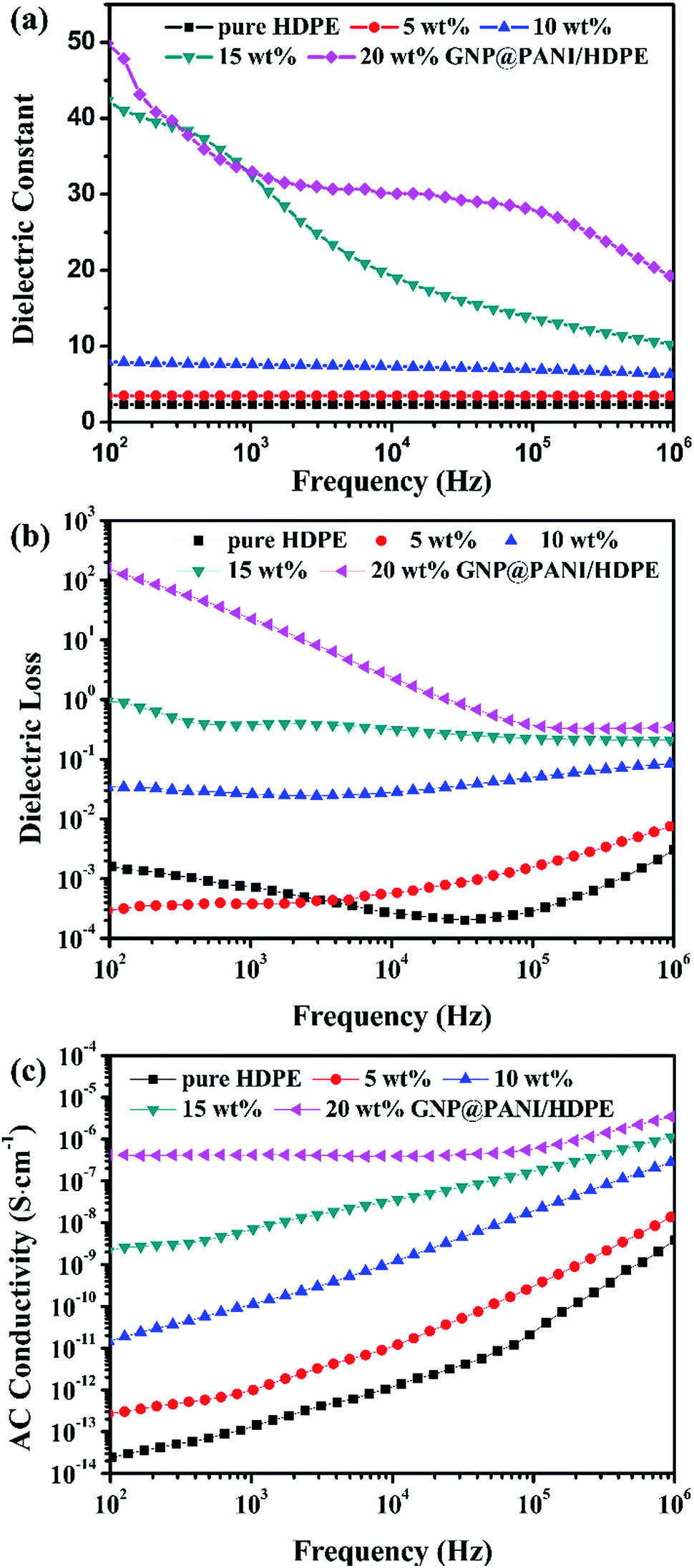 | ||
| Fig. 7 Dielectric frequency dependence (a), dielectric loss (b), and AC conductivity (c) of pure HDPE and GNP@PANI/HDPE composites with various contents. | ||
Mechanical properties
In Fig. 8 the effects of the GNP@PANI nanocomposites content on tensile strength, flexural modulus and notch impact strength are presented. As it can be observed in Fig. 8a, the tensile strength of pure HDPE was 25.73 MPa and when the content of GNP@PANI nanocomposites was 5 wt%, the tensile strength reached the maximum value at 27.38 MPa. When the GNP@PANI nanocomposite contents exceeded 5 wt%, the tensile strength gradually decreased along with the GNP@PANI nanocomposite contents increase. It was worth noting that when the content of GNP@PANI nanocomposites was 10 wt%, the tensile strength was 24.79 MPa, being similar to the pure HDPE value. The reason for this phenomenon is described as follows: when the content of GNP@PANI nanocomposites was low, the nanocomposites dispersion was significantly better than the high content and the composites still remained in the highly continuous phase of HDPE, therefore the graphene nanoplatelets exhibited reinforcement. As it can be observed in Fig. 8b and c, flexural modulus and notch impact strengths of pure HDPE were 0.96 GPa and 16.59 kJ m−2, respectively. The flexural modulus gradually increased as the content of GNP@PANI nanocomposites increased and the notch impact strength sharply decreased as the content of GNP@PANI nanocomposites increased. The reason for this phenomenon was that graphene nanoplatelets were rigid materials, therefore the composite toughness decreased significantly by graphene nanoplatelets addition. Based on aforementioned results, the 10 wt% content of GNP@PANI nanocomposites constituted the optimal solution.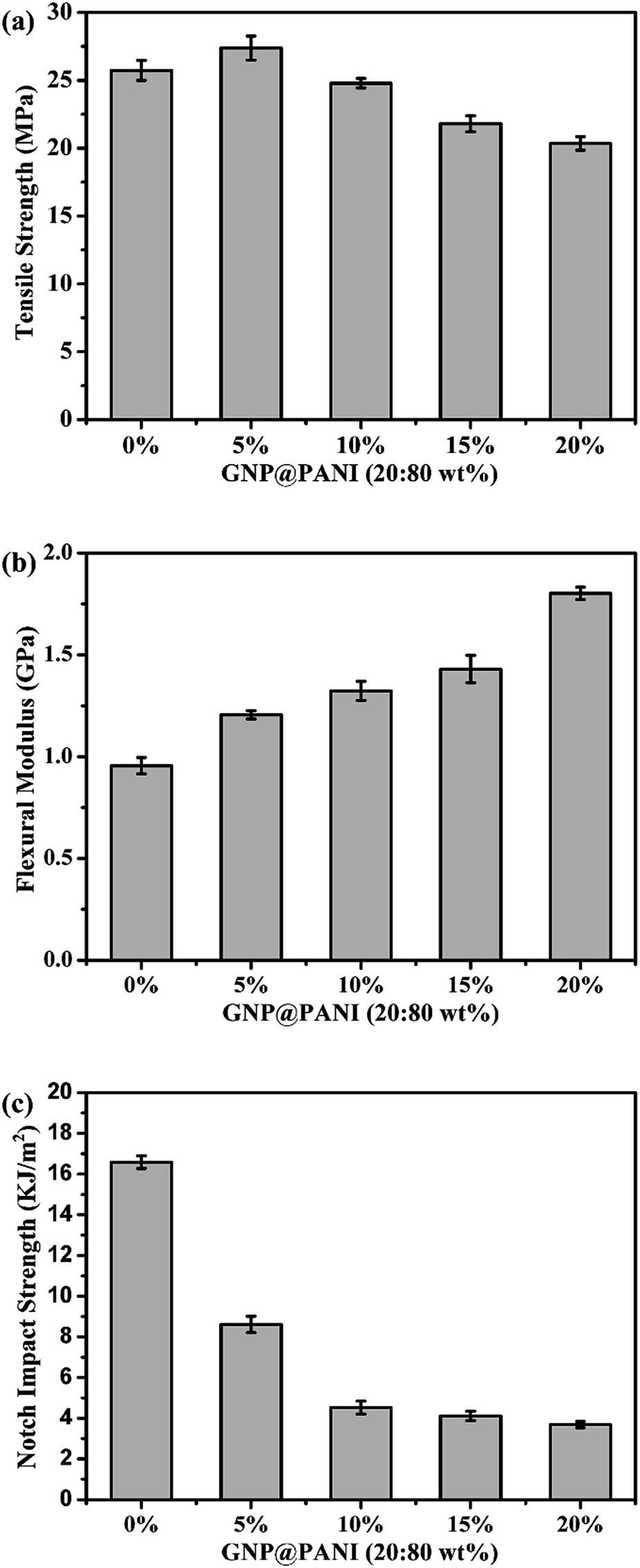 | ||
| Fig. 8 GNP@PANI contents influence on mechanical properties of composites. | ||
Conclusions
In this study a novel antistatic agent and a suitable method for permanent antistatic GNP@PANI/HDPE composites preparation were developed for the orientation and aggregation of nanofillers during processing to be avoid. The antistatic agent of GNP@PANI nanocomposites was successfully prepared by graphene nanoplatelets and aniline through in situ polymerization. Graphene nanoplatelets encapsulated with polyaniline significantly improved the compatibility between GNP@PANI nanocomposites and HDPE. The homogeneous dispersion of GNP@PANI nanocomposites in HDPE was achieved by solution blending. As a result, the surface (ρs) and volume (ρv) resistivity decreased as the content of GNP@PANI nanocomposites increased. Furthermore, permanent antistatic high-density polyethylene composites were prepared by a 10 wt% GNP@PANI nanocomposites addition only due to the established conductive network. Therefore, permanent antistatic high-density composites with excellent mechanical and antistatic properties can be utilized in many industrial applications.Acknowledgements
The authors sincerely acknowledge ‘Shanghai Collaboration Program (CXY-2014-023)’, ‘Scientific and Technological Achievements Transformation Program of Jiangsu Province (SBA2014010034)’ and ‘Ningbo Industrial Major Projects (201601ZD-A01026)’.References
- E. Tarani, A. Wurm, C. Schick, D. N. Bikiaris, K. Chrissafis and G. Vourlias, Thermochim. Acta, 2016, 643, 94–103 CrossRef CAS.
- V. Dudler, M. C. Grob and D. Mérian, Polym. Degrad. Stab., 2000, 68, 373–379 CrossRef CAS.
- C. C. Chang, F. H. Hwang, C. Y. Hsieh, C. C. Chen and L. P. Cheng, J. Coat. Technol. Res., 2013, 10, 73–78 CrossRef CAS.
- I. Krupa, V. Cecen, A. Boudenne, J. Prokeš and I. Novák, Mater. Des., 2013, 51, 620–628 CrossRef CAS.
- Y. Wang, C. Zhang, Z. J. Du, H. Q. Li and W. Zou, Synth. Met., 2013, 182, 49–55 CrossRef CAS.
- J. L. Wang, L. X. Bao, H. B. Zhao and J. X. Lei, Compos. Sci. Technol., 2012, 72, 976–981 CrossRef CAS.
- N. A. Aal, F. El-Tantawy, A. Al-Hajry and M. Bououdina, Polym. Compos., 2007, 29, 125–132 CrossRef.
- C. C. Hu, S. S. Chang and N. Y. Liang, J. Text. Inst., 2016, 107, 976–984 CAS.
- C. S. Li, T. X. Liang, W. Z. Lu, C. H. Tang, X. Q. Hu, M. S. Cao and J. Liang, Compos. Sci. Technol., 2004, 64, 2089–2096 CrossRef CAS.
- M. Sangermano, S. Pegel, P. Poetschke and B. Voit, Macromol. Rapid Commun., 2008, 29, 396–400 CrossRef CAS.
- C. X. Wang, J. C. Lv, Y. Ren, T. Zhi, J. Y. Chen, Q. Q. Zhou, Z. Q. Lu, D. W. Gao and L. M. Jin, Appl. Surf. Sci., 2015, 359, 196–203 CrossRef CAS.
- F. Ying, Y. H. Cui, G. X. Xue, H. Q. Qian, A. H. Li, X. Wang, X. J. Zhang and D. D. Jiang, Polym. Bull., 2016, 73, 2815–2830 CrossRef CAS.
- J. G. Wang, C. Zhang, Z. J. Du, H. Q. Li and W. Zou, RSC Adv., 2016, 6, 31782–31789 RSC.
- Y. S. Lee, S. H. Park, J. C. Lee and K. Ha, Korean J. Chem. Eng., 2016, 33, 1095–1103 CrossRef CAS.
- A. Tsurumaki, F. Bertasi, K. Vezzù, E. Negro, V. D. Noto and H. Ohno, Phys. Chem. Chem. Phys., 2016, 18, 2369–2387 RSC.
- A. M. R. Galletti, C. Antonetti, M. Marracci, F. Piccinelli and B. Tellini, Appl. Surf. Sci., 2013, 280, 610–618 CrossRef.
- Z. W. Zhou, L. S. Chu, W. M. Tang and L. X. Gu, J. Electrost., 2003, 57, 347–354 CrossRef CAS.
- K. Chen, C. X. Xiong, L. B. Lin, L. Zhou, Y. A. Lei and L. J. Dong, Polym. Compos., 2009, 30, 226–231 CrossRef CAS.
- C. R. Martins and M. A. D. Paoli, Eur. Polym. J., 2005, 41, 2867–2873 CrossRef CAS.
- H. Wang, G. Y. Xie, M. H. Fang, Z. Ying, Y. Tong and Y. Zeng, Composites, Part B, 2015, 79, 444–450 CrossRef CAS.
- H. P. Boehm, Angew. Chem., Int. Ed., 2010, 49, 9332–9335 CrossRef CAS PubMed.
- E. Tarani, A. Wurm, C. Schick, D. N. Bikiaris, K. Chrissafis and G. Vourlias, Thermochim. Acta, 2016, 643, 94–103 CrossRef CAS.
- X. Jiang and L. T. Drzal, Composites, Part A, 2011, 42, 1840–1849 CrossRef.
- X. Jiang and L. T. Drzal, Composites, Part A, 2011, 42, 1840–1849 CrossRef.
- X. Jiang and L. T. Drzal, Polym. Compos., 2010, 31, 1091–1098 CAS.
- J. L. Shen, C. Y. Yang, X. W. Li and G. C. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8467–8476 CAS.
- A. K. Das, S. Maiti and B. B. Khatua, J. Electroanal. Chem., 2015, 739, 10–19 CrossRef CAS.
- N. Y. Yuan, F. F. Ma, Y. Fan, Y. B. Liu and J. N. Ding, Composites, Part A, 2012, 43, 2183–2188 CrossRef CAS.
- L. Valentini, S. Bittolo Bon, M. A. Lopez-Manchado, R. Verdejo, L. Pappalardo, A. Bolognini, A. Alvino, S. Borsini, A. Berardo and N. M. Pugno, Compos. Sci. Technol., 2016, 128, 123–130 CrossRef CAS.
- X. R. Song, H. J. Li, X. R. Zeng and L. L. Zhang, Mater. Lett., 2016, 183, 232–235 CrossRef CAS.
- F. Marra, A. G. D'Aloia, A. Tamburrano, I. M. Ochando, G. D. Bellis, G. Ellis and M. S. Sarto, Polymers, 2016, 8, 272 CrossRef.
- N. Parveen, N. Mahato, M. O. Ansari and M. H. Cho, Composites, Part B, 2016, 87, 281–290 CrossRef CAS.
- A. K. Das, S. K. Karan and B. B. Khatua, Electrochim. Acta, 2015, 180, 1–15 CrossRef CAS.
- Department of Defence, Military Handbook DOD-HDBK-263, Washington, DC, USA, 1980 Search PubMed.
- S. Z. Li, J. K. Wang, Y. Y. Li, G. H. Wu, Y. M. Wang, W. Q. Wang and J. H. Guo, J. Appl. Polym. Sci., 2016, 133, 44126 Search PubMed.
- A. N. Zheng, X. Xu, H. N. Xiao, N. Li, Y. Guan and S. Z. Li, Appl. Surf. Sci., 2012, 258, 8861–8866 CrossRef CAS.
- A. N. Zheng, X. Xu, H. N. Xiao, N. Li, Y. Guan and S. Z. Li, Appl. Surf. Sci., 2012, 258, 8861–8866 CrossRef CAS.
- Z. M. Dang, L. Wang, Y. Yin, Q. Zhang and Q. Q. Lei, Adv. Mater., 2007, 19, 852–857 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |