
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sonochemical one pot synthesis of novel spiroacridines catalyzed by magnetically functionalized Fe3O4 nanoparticles with chitosan as a reusable effective catalyst†
Hossein Naeimi* and
Sepideh Lahouti
Department of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan, 87317, I.R. Iran. E-mail: naeimi@kashanu.ac.ir; Fax: +98 3155912397; Tel: +98 3155912388
First published on 12th January 2017
Abstract
A facile one-pot and three-component economical synthesis of spiroacridine using acenaphthoquinone, different anilines and dimedone in the presence of Fe3O4@CS–SO3H NPs catalyst under ultrasonic irradiation is described. Firstly, the Fe3O4@CS NPs were prepared from reaction of chitosan with Fe3O4 NPs. Afterwards, treatment of Fe3O4@CS NPs with chlorosulfonic acid leads to the formation of Fe3O4@CS–SO3H NPs. Compared to the conventional methods, the ultrasound procedure showed several advantages including mild reaction conditions, high yield of products, short reaction times, and reusability of the catalyst. The core–shell structure and the composition of the produced magnetic nanocatalyst were analyzed using FT-IR, XRD, VSM, SEM and EDX techniques.
1. Introduction
Ultrasound irradiation has been considered to be a clean and beneficial protocol in organic synthesis in the last three decades.1 The role of sonochemistry in the development of “benign by-design” synthetic procedures is clear from the description: low level of wastage, inherent safety, material and energy saving, with an optimized use of nonrenewable resources, and a preferential operation of renewable ones.2 The application of ultrasound in organic synthesis has been increasing because of its advantages such as shorter reaction times, milder reaction condition, and higher yields in analogy with the classical methods.3–5 Spiro compounds having cyclic structures fused at a central carbon are of recent interest because of their interesting conformational features and their structural implications on biological systems.6–8The synthesis of spiro-heterocycles presents an interesting synthetic challenge to organic chemists because of its structural rigidity and complexity. Nitrogen inclosing spiro-heterocycles display remarkable biological properties, and are also frequently observed in natural products such as paraherquamide A, spirotryprostatin A, spirotryprostatin B, mitraphylline, pteropodine, and isopteropodine (Fig. 1).9
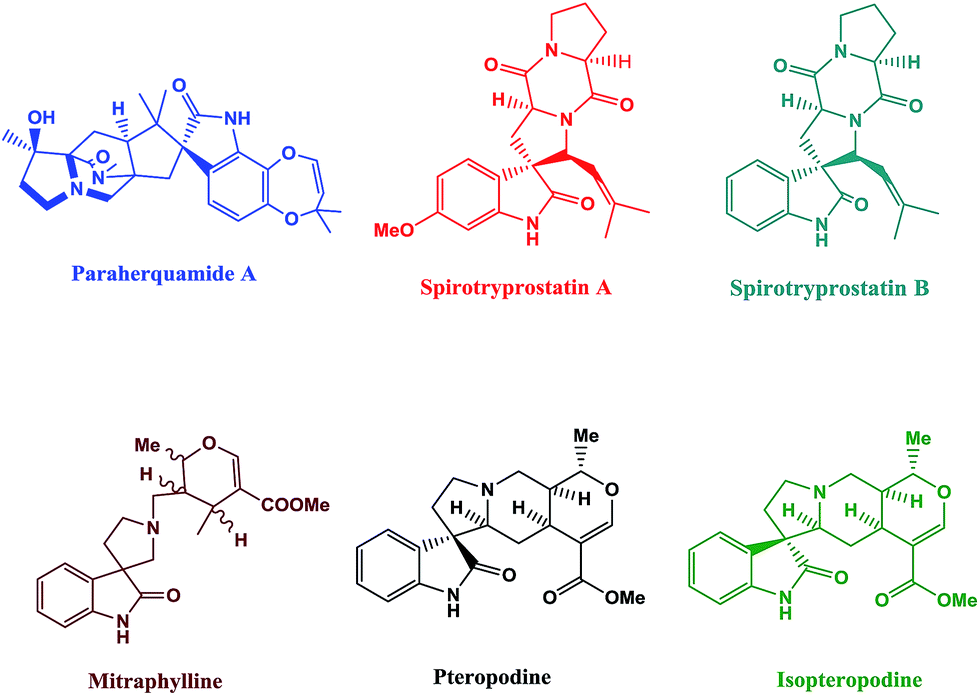 | ||
| Fig. 1 Natural products possessing spiro-centres. | ||
Generally, many synthetic analogs containing an oxindole also display antiparasitic, antibacterial, antimicrobial, anti-mycobacterial, antitubercular, anticancer, analgesic and anti-inflammatory, and anti-HIV activities.10–13 Spirocyclic systems including one carbon atom common to two rings are structurally interesting.14 The attendance of the sterically forced spiro structure in various natural products also adds to the interest in the investigations of spiro compounds.15 Spiro compounds represent an important class of naturally happening substances characteristic by their highly pronounced biological properties. Similarly, functionalized spiro cycloalkyloxindoles are found in a diversity of natural products and bioactive molecules.16 Nanomaterials collected together result in multifunctional nanostructures having innovative industrial and biomedical applications.17
In recent decades, scrambles have been prepared combine different types of nanoparticles (NPs) in order to attain superior composite material properties.18 The magnetic iron(III) oxides NPs (Fe3O4 and γ-Fe2O3) have absorbed consideration because of their magnetic property.19–21 Functionalization and modification of the surface of the Fe3O4 nanoparticles with different biocompatible and biodegradable polymers have been widely tested because the introduction of new substance could also bring new functions for magnetite.22–25 Chitosan is a partially acetylated glucosamine biopolymer with many useful features such as hydrophilicity, biocompatibility, and biodegradability.26 Chitosan, due to its high amino content, has been found to possess good absorption capacity for many heavy metal ions through complexation with the amine groups. It has been widely used as biosorbent for removing different metal ions from wastewater. Furthermore, due to magnetic properties, it can easily be separated from the sorption system by using magnetic field.27–29
We report herein a clean synthesis of spiroacridine derivatives from a wide of acenaphthoquinone, different anilines and dimedone compounds in the presence of Fe3O4@CS–SO3H NPs under ultrasonic irradiation. The high yields, and the possibility of easy recovering of the catalyst are the most advantages of this method (Scheme 1).
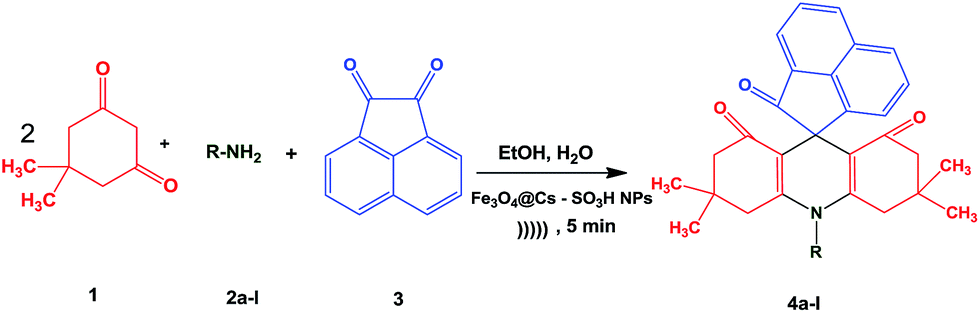 | ||
| Scheme 1 Various synthesized spiroacridine in the presence of Fe3O4@CS–SO3H NPs. | ||
2. Experimental section
2.1. Materials
The chemicals used in this work were obtained from Fluka and Merck Chemical Company and were used without purification.2.2. Apparatus
FT-IR spectra were obtained with potassium bromide pellets in the range 400–4000 cm−1 with a Perkin-Elmer 550 spectrometer. 1H NMR and 13C NMR spectra were recorded with a Bruker DRX-400 spectrometer at 400 and 100 MHz respectively. NMR spectra were reported as parts per million (ppm) downfield from tetramethylsilane as internal standard. The abbreviations used are: singlet (s), doublet (d), triplet (t) and multiplet (m). Nanostructures were characterized using a Holland Philips Xpert X-ray powder diffraction (XRD) diffractometer (CuK, radiation, k = 0.154056 nm), at a scanning speed of 2° min−1 from 10° to 100° (2θ). A BANDELIN ultrasonic HD 3200 with probe model KE 76, with the diameter of 6 mm, was used to produce ultrasonic irradiation and homogenizing the reaction mixture. Piezoelectric crystal of this kind of probe normally works in the range of 700 kHz, but by using some proper clamps, the output frequency of piezoelectric crystal have controlled and reduced to 20 kHz. Therefore, the induced frequency of probe to the reaction mixture is equal to 20 kHz. A thermal method was used for the calibration of ultrasonic power. Melting points are determined in open capillaries using an Electrothermal Mk3 apparatus and are uncorrected. The purity determination of the substrates and reaction monitoring were accomplished by TLC on silica-gel polygram SILG/UV 254 plates (from Merck Company). FE-SEM images of the products were visualized by a Sigma ZEISS, Oxford Instruments Field Emission Scanning Electron Microscope. The magnetic properties of nanoparticles have been measured with a vibrating sample magnetometer (VSM, PPMS-9T) at 300 K in Iran (University of Kashan).2.3. General information
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300000) is dissolved in 25 mL of 0.05 M acetic acid solution; to which FeCl2·4H2O (1.29 g, 0.5 mmol) and FeCl3·6H2O (3.51 g, 0.25 mmol) are added. The resulting solution is mechanically stirred for 6 h at 80 °C, under N2 atmosphere. Consequently, 6 mL of 25% NH4OH is injected drop wise into the reaction mixture with constant stirring. After 40 min, the mixture is cooled to room temperature and chitosan coated over magnetic nanoparticles were separated by magnetic decantation and washed three times with distilled water, then ethanol, and finally dried under vacuum at room temperature.
000–300000) is dissolved in 25 mL of 0.05 M acetic acid solution; to which FeCl2·4H2O (1.29 g, 0.5 mmol) and FeCl3·6H2O (3.51 g, 0.25 mmol) are added. The resulting solution is mechanically stirred for 6 h at 80 °C, under N2 atmosphere. Consequently, 6 mL of 25% NH4OH is injected drop wise into the reaction mixture with constant stirring. After 40 min, the mixture is cooled to room temperature and chitosan coated over magnetic nanoparticles were separated by magnetic decantation and washed three times with distilled water, then ethanol, and finally dried under vacuum at room temperature.2.4. Preparation of sulfonated chitosan encapsulated nano iron oxide Fe3O4@CS–SO3H NPs
Fe3O4@CS NPs (10 mg) is dispersed in dry CH2Cl2 (2 mL) and sonicated for 20 min, then chlorosulfonic acid (0.3 mL) is added drop-wise over a period of 10 min, at room temperature under N2 atmosphere. Subsequently, the mixture is mechanically stirred for 15 min until HCl gas evolution is stopped. Finally, functionalized magnetic Fe3O4@CS–SO3H NPs is separated by an external magnet, washed several times with dry CH2Cl2, until a neutral pH level is achieved, then dried under vacuum at room temperature (Scheme 2).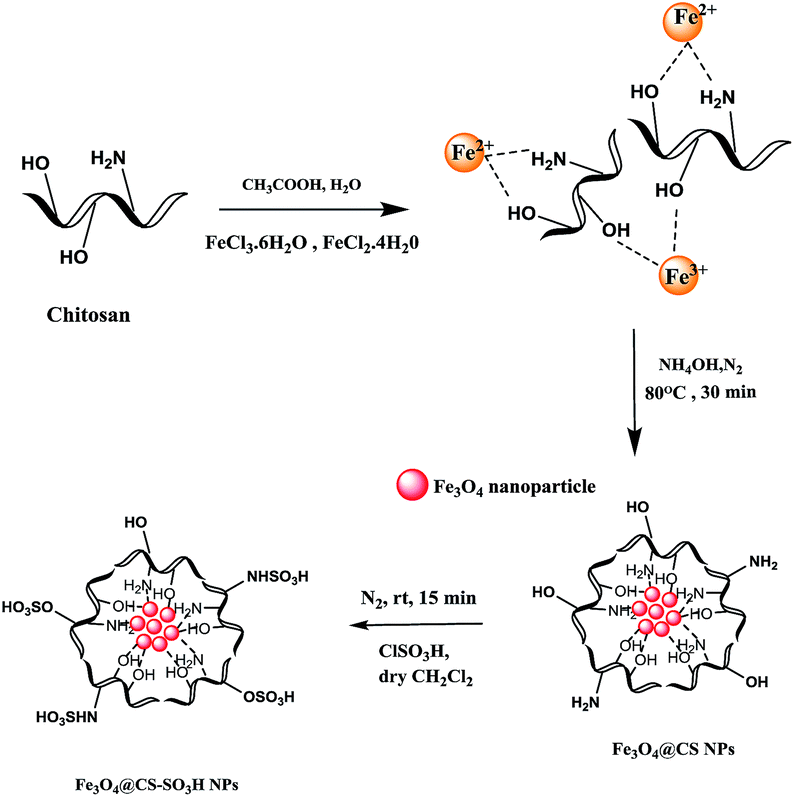 | ||
| Scheme 2 Preparation steps of Fe3O4@CS–SO3H NPs. | ||
In order to determine the acid strength of catalyst, a 0.08 M solution of sodium hydroxide was used for measurement of total surface acidity for the Fe3O4@CS–SO3H NPs. Then, 0.02 g of solid was suspended in this solution for 18 h, and the excess amount of base was titrated against the hydrochloric acid using phenolphthalein as an indicator.
2.5. General procedure for the synthesis of spiroacridines catalyzed by Fe3O4@CS–SO3H NPs
:1, v/v) (5 mL) was stirred for 60 min. After the reaction was completed, the catalyst was separated by an external magnet. The reaction mixture was cooled to room temperature. Then, the precipitated product was filtered and washed ethanol to afford the pure product.:1, v/v) (5 mL) were added into a 25 mL round bottomed flask. The reaction mixture was sonicated under 40 W for the period of time (the reaction was monitored by TLC). The reaction mixture was filtered and the precipitate washed with ethanol. The crude solid product was recrystallized from ethanol to give pure spiroacridine as a target product. Then, all of the products were identified by IR, 1H NMR, 13C NMR and C.H.N analyses.2.6. Representative spectral data
3. Results and discussion
3.1. Preparation and characterization of Fe3O4@CS–SO3H NPs
Fe3O4@CS–SO3H NPs was prepared according to a modified procedure in the literature (Scheme 1). The Fe3O4 MNPs were prepared by the well-known Massart's method30 which consists of Fe(III) and Fe(II) co-precipitation with molar ratio 2:1. Then, the synthesized magnetic nanoparticles were modified with chitosan as a natural polymer. The resulting solution is mechanically stirred for 6 h at 80 °C under N2 atmosphere. The NH4OH is injected drop wise into the reaction mixture with constant stirring for 30 min. Fe3O4@CS NPs is dispersed in dry CH2Cl2 in an ultrasonic bath for 20 min, then chlorosulfonic acid is added. The magnetically heterogeneous organocatalyst, Fe3O4@CS–SO3H NPs, is characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), vibrating sample magnetometer (VSM) and Fourier transform infrared (FT-IR) spectroscopy.
 | ||
| Fig. 2 IR spectrum of (a) chitosan (CS), (b) Fe3O4@CS NPs, (c) Fe3O4@CS–SO3HNPs. | ||
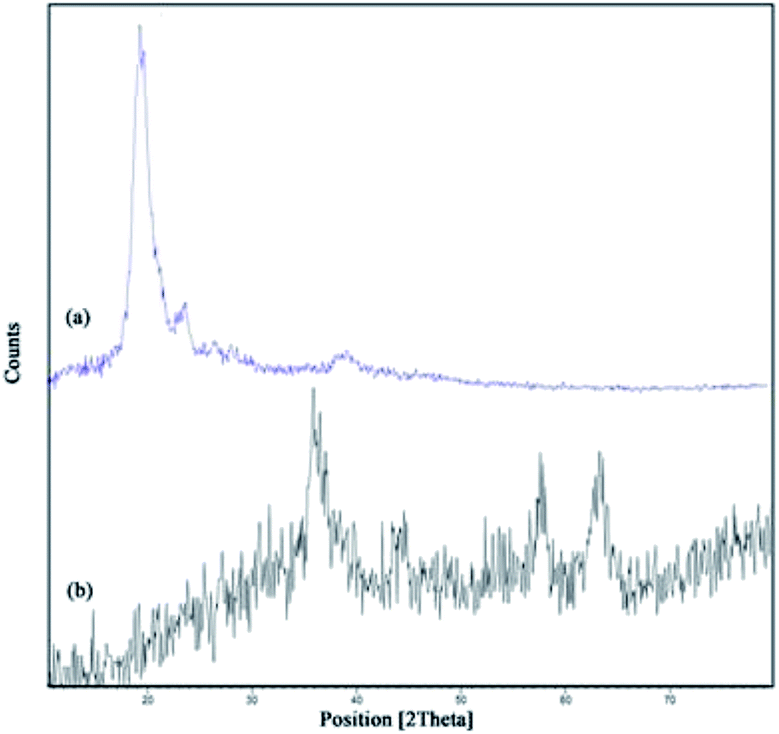 | ||
| Fig. 3 XRD pattern of (a) chitosan, (b) Fe3O4@CS–SO3H NPs. | ||
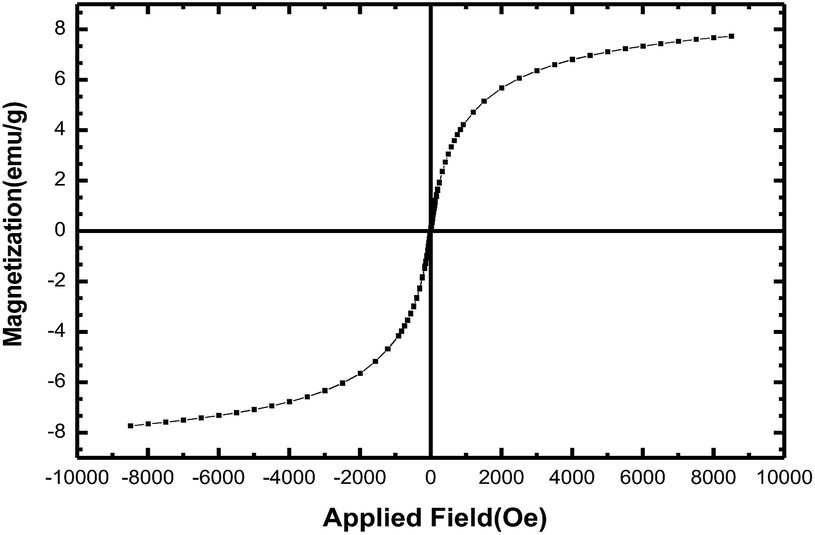 | ||
| Fig. 4 Magnetization versus applied for Fe3O4@CS–SO3H NPs. | ||
 | ||
| Fig. 5 SEM image of Fe3O4@CS–SO3H NPs. | ||
 | ||
| Fig. 6 The energy dispersive X-ray (EDX) of Fe3O4@CS–SO3H NPs. | ||
Furthermore, the total surface acidity for the Fe3O4@CS–SO3H NPs was determined by sodium hydroxide titration. In this measurement procedure, the pH of Fe3O4@CS–SO3H NPs catalyst was 1.02 that can be equal to loading the 2.14 mmol (H+) g−1.
3.2. Procedures for the synthesis of spiroacridines
After the characterization of the catalyst, in order to optimize the reaction parameters, we investigated the reaction of dimedone (2 mmol), aniline (1 mmol), acenaphthoquinone (1 mmol) in the presence of Fe3O4@CS–SO3H NPs. The model reaction was checked in the presence of different amount of Fe3O4@CS–SO3H NPs as a catalyst at a range of various temperatures and also under ultrasonic irradiation in various solvents. The results are shown in Table 1. As this table indicates, when the reaction was carried out under harsh heating conditions, it gave low yields of products and took longer reaction times, while the same reaction was carried out under ultrasonic irradiation at room temperature to provide excellent yields of products in short reaction times. Thus, the best results were obtained when the reaction was performed using 1 mg of the catalyst in EtOH/H2O under ultrasonic irradiation (Table 1, entry 3).
|
||||
|---|---|---|---|---|
| Entry | Cat. (mg) | Solvent | Heating conditiona | Ultrasonic irradiation |
| Time (h)/Yieldb, % | Time (min)/Yieldb, % | |||
| a Under reflux at boiling points of different solvents.b Isolated yields. | ||||
| 1 | 0.3 | EtOH/H2O | 2/59 | 10/78 |
| 2 | 0.5 | EtOH/H2O | 2/63 | 10/78 |
| 3 | 1 | EtOH/H2O | 1/68 | 5/93 |
| 4 | 1.5 | EtOH/H2O | 1/68 | 5/93 |
| 5 | 1 | CH3CN | 2/48 | 20/57 |
| 6 | 1 | H2O | 2/— | 20/— |
| 7 | — | EtOH/H2O | 4/trace | 35/42 |

In continuation of this method, the role of ultrasonic irradiation and the effect of various powers of ultrasonic irradiation on the reaction and the reaction under without ultrasound were examined (Table 2). As can be seen in this table, the reaction on the without ultrasound (silent) at room temperature was resulted any product even continued at 150 min. While, replacement of the same reaction under ultrasonic condition at various power were obtained the target product in good to excellent yields (Table 2, entry 1 vs. entries 2–6). These results show that the sonication certainly affected the reaction system. The probable explanation for the positive connotation of irradiation is that the ultrasonic irradiation could increase the number of active cavitation bubbles of which are expected to result in higher maximum collapse temperature and speeded respective reaction. We employed various conditions and it was found that the reaction gave satisfying result in the presence of 1 mg of Fe3O4@CS–SO3H NPs under ultrasonic irradiation with the power of 40 W in aqueous ethanol.
We examined condensation reaction under ultrasonic irradiation without catalyst. When the reaction was examined without catalyst, the products were obtained in a moderate to good yields (Table 1). The results show that the sonication certainly affected the reaction system.
We also applied Fe3O4@CS–SO3H NPs in synthesis spiroacridines derivatives from various aromatic amines under similar condition as represented in Table 3. In general, the reactions are clean and high-yielding. Several groups, such as Cl, Br, NO2, OCH3, OH and CH3, are compatible under the reaction conditions. Interestingly, a variety of aromatic amines, including ortho, meta and para-substituted aryl amines, participated well in this reaction and gave the corresponding products in a good to excellent yield (Table 3). The influence of electron-withdrawing and electron-donating substituents on the aromatic ring of amines upon the reaction yields was investigated. These results show that aromatic amines with electron-releasing groups reacted faster than those with electron-withdrawing groups.
| Entry | Product | R (amine) | Time (min) | Yieldb (%) |
|---|---|---|---|---|
| a Dimedone (2 mmol), aniline (1 mmol), acenaphthoquinone (1 mmol) and Fe3O4@CS–SO3H NPs (1 mg).b Isolated yields. | ||||
| 1 | 4a | 4-NO2–C6H4 | 8 | 85 |
| 2 | 4b | 4-OH–C6H4 | 5 | 93 |
| 3 | 4c | 4-Cl–C6H4 | 6 | 87 |
| 4 | 4d | 4-Br–C6H4 | 6 | 89 |
| 5 | 4e | 4-Me–C6H4 | 5 | 91 |
| 6 | 4f | 4-OMe–C6H4 | 5 | 93 |
| 7 | 4g | 2-OH–C6H4 | 5 | 90 |
| 8 | 4h | 2-NO2–C6H4 | 8 | 83 |
| 9 | 4i | 2-Cl–C6H4 | 6 | 80 |
| 10 | 4j | 3-NO2–C6H4 | 9 | 76 |
| 11 | 4k | 2,4-Me2–C6H3 | 5 | 92 |
| 12 | 4l | Phenyl | 7 | 88 |
| 13 | 4m | 4-H2N–C6H4 | 8 | 90 |
| 14 | 4n | Thiazolyl | 80 | N.R. |
The proposed reaction mechanism for synthesis of spiroacridines catalyzed by Fe3O4@CS–SO3H NPs is depicted in Scheme 3. Initially, the Fe3O4@CS–SO3H NPs catalyst protonates the carbonyl group of acenaphthoquinone (1), which then condenses with CH acidic group of dimedone (2) through a fast Knoevenagel condensation to afford A derivative. Then, dimedone (2) participates in a Michael addition with compound A to form intermediate B, the –NH group of amine which subsequently undergoes an intramolecular attack of the carbonyl group, resulting in a cyclization reaction that affords C. Finally, intermediate C loses a molecule of H2O to lead to the formation of acridine D.
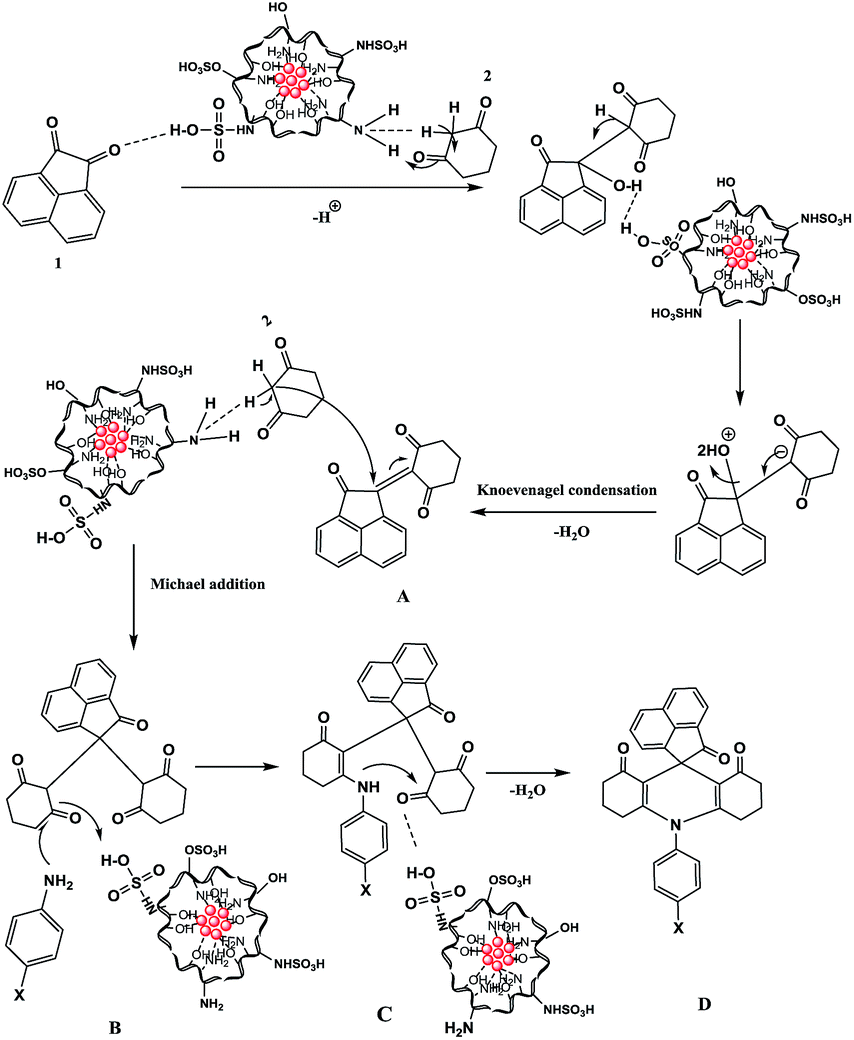 | ||
| Scheme 3 Plausible mechanism for the synthesis of spiroacridine. | ||
For practical applications of such heterogeneous system, the reusability is one of the important properties of the catalyst. The reusability of used catalyst was investigated using the reaction of dimedone, aniline and acenaphthoquinone in the presence of Fe3O4@CS–SO3H NPs as a catalyst under sonication condition. The catalyst was isolated by simple filtration, washed exhaustively with water–acetone and dried. The results in five consecutive runs (98%, 98%, 97%, 96% and 96%) indicated the yields remained similar with no detectable loss of yield and catalytic activity under sonication condition (Fig. 7).
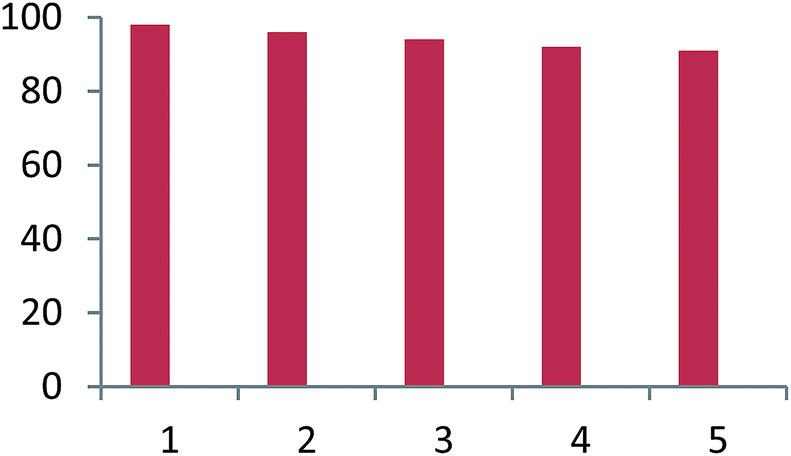 | ||
| Fig. 7 Reusability of catalyst for synthesis of spiroacridine. | ||
4. Conclusions
In this paper, we have described the synthesis of spiroacridine derivatives using dimedone and acenaphthoquinone with substituted aromatic amines, under the action of ultrasonic irradiation, in the presence of Fe3O4@CS–SO3H NPs catalyst. The desired products were obtained in high to excellent yields and high purities with short reaction times. The prepared magnetic catalyst could be easily recovered by simple filtration and the efficiency of the catalyst remains unaltered after several times. Also, we have presented that compared to heating method; ultrasound irradiation can speed up the reaction and is more suitable and efficient.Acknowledgements
The authors are grateful to the University of Kashan for supporting this work by Grant no. 159148/66.References
- J. T. Li, Y. Yin and M. X. Sun, Ultrason. Sonochem., 2010, 17, 363–366 CrossRef CAS PubMed
.
- Y. Zou, Y. Hu, H. Liu and D. Shi, ACS Comb. Sci., 2011, 14, 38–43 CrossRef PubMed
- M. Mamaghani and S. Dastmard, Ultrason. Sonochem., 2009, 16, 445–447 CrossRef CAS PubMed
- S. J. Ji, Z. L. Shen, D. G. Gu and X. Y. Huang, Ultrason. Sonochem., 2005, 12, 161–163 CrossRef CAS PubMed
- A. R. Khosropour, Ultrason. Sonochem., 2008, 15, 659–664 CrossRef CAS PubMed
- K. Takahashi, B. Witkop, A. Brossi, M. A. Maleque and E. X. Albuquerque, Helv. Chim. Acta, 1982, 65, 252–261 CrossRef CAS
- A. Longeon and M. Guyot, Experientia, 1990, 46, 548–550 CrossRef CAS
- J. Kobayashi, M. Tsuda, K. Agemi, H. Shigemori, M. Ishibashi and T. Sasaki, Tetrahedron, 1991, 47, 6617–6622 CrossRef CAS
- P. Maloo, T. Kanchan Roy, D. M. Sawant, R. T. Pardasani and M. M. Salunkhe, RSC Adv., 2016, 6, 41897–41906 RSC
- D. M. James, H. B. Kunze and D. J. Faulkner, J. Nat. Prod., 1991, 54, 1137–1140 CrossRef CAS
- T. Okita and M. Isobe, Tetrahedron, 1994, 50, 11143–11152 CrossRef CAS
- P. Rosemmond, M. M. Hossemi and C. Bub, Liebigs Ann. Chem., 1994, 2, 151–158 CrossRef
- M. J. Kornet and A. P. Tnio, J. Med. Chem., 1976, 19, 892–898 CrossRef CAS PubMed
- N. Lashgari and G. Mohammadi Ziarani, ARKIVOC, 2012, 41, 277–320 Search PubMed
- K. Jadidi, R. Ghahremanzadeh and A. Bazgir, Tetrahedron, 2009, 65, 2005–2009 CrossRef CAS
- R. Ranjith Kumar, S. Perumal, P. Senthilkumar, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2009, 44, 3821–3829 CrossRef CAS PubMed
- N. Kostevsek, E. Locatelli, C. Garrovo, F. Arena, I. Monaco, I. Petrov Nikolov, S. Sturm, K. Z Rozman, V. Lorusso, P. Giustetto, P. Bardini, S. Biffi and M. C. Franchini, Chem. Commun., 2016, 52, 378–381 RSC
- L. Obeid, D. Talbot, S. B. Jaafar, V. Dupuis, S. Abramson, V. Cabuil and M. Welschbillig, J. Colloid Interface Sci., 2013, 410, 52–58 CrossRef CAS PubMed
- L. M. Murillo, J. S. Margarida, M. T. F. Telling, J. P. R. L. L. Parra, S. Landsgesell, R. I. Smith and H. N. Bordallo, J. Alloys Compd., 2014, 584, 514–519 CrossRef
- A. Singh and S. K. Sahoo, Drug Discovery Today, 2014, 19, 474–481 CrossRef CAS PubMed
- X. N. Zhao, H. C. Hu, F. J. Zhang and Z. H. Zhang, Appl. Catal., A, 2014, 482, 258–265 CrossRef CAS
- G. Li, Y. Jiang, K. Huang, P. Ding and J. Chen, J. Alloys Compd., 2008, 466, 451–456 CrossRef CAS
- X. Li, S. Zhang, B. Yang, C. Lv, X. Jia and Z. Hu, RSC
Adv., 2016, 6, 86531–86539 RSC
- A. Pourjavadi, A. Motamedi, S. H. Hosseini and M. Nazari, RSC Adv., 2016, 6, 19128–19135 RSC
- M. N. Shaikh, M. A. Aziz, A. Helal, M. Bououdina, Z. H. Yamani and T. J. Kim, RSC Adv., 2016, 6, 41687–41695 RSC
- J. Qu, G. Liu, Y. Wang and R. Hong, Adv. Powder Technol., 2010, 21, 461–467 CrossRef CAS
- C. Yuwei and W. Jianlong, Chem. Eng. J., 2011, 168, 286–292 CrossRef
- X. Chen, H. Yang and N. Yan, Chem.–Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed
- B. Karami, K. Eskandari and A. Ghasemi, Turk. J. Chem., 2012, 36, 601–614 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26386k |
| This journal is © The Royal Society of Chemistry 2017 |