
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas phase conversion of eugenol into various hydrocarbons and platform chemicals†
Anand Mohan Verma and
Nanda Kishore*
and
Nanda Kishore*
Department of Chemical Engineering, Indian Institute of Technology Guwahati, India – 781039. E-mail: nkishore@iitg.ernet.in; mail2nkishore@gmail.com
First published on 12th January 2017
Abstract
The unprocessed bio-oil derived from pyrolysis of lignocellulosic biomass is a mixture of hundreds of oxy-compounds which vitiate the quality of bio-oil. Eugenol is one of the most promising model compounds of the phenolic fraction of unprocessed bio-oil and it comprises two oxy-functionals, namely, hydroxyl and methoxy functionals. In this study, eight reaction pathways are carried out, using eugenol as the model compound, producing many important products viz. toluene, propylbenzene, guaiacol, allylbenzene, 4-propylphenol, isoeugenol, etc. in a gas phase environment by using the B3LYP/6-311+g(d,p) level of theory under the density functional theory (DFT) framework. The thermochemical study of these reactions is also carried out in a wide range of temperatures between 298–898 K with an interval temperature of 100 K and fixed pressure of 1 atm. The direct cleavage of the functional groups of eugenol followed by an atomic hydrogenation reaction to produce lower fraction products is found to be not favourable; however, an atomic hydrogenation reaction prior to the removal of functional groups of eugenol makes these reactions more favourable. The activation energy for the production of guaiacol from eugenol under reaction scheme 2 is reported to be 10.53 kcal mol−1 only, which is the lowest activation energy required amongst all reaction schemes. The reaction scheme 5, i.e., the production of propylcyclohexane from eugenol, is reported to be the most exothermic and spontaneous reaction at all temperature conditions; however, ΔG values increase with increasing temperature and ΔH values decrease with increasing temperature.
Introduction
In the 21st century, the energy requirement and the pollution caused by non-renewable energy resources are the main concerns in front of most of the countries, be it developed or developing country.1–4 The depletion of non-renewable energy resources is also alarming researchers; therefore, most of countries are shifting their energy sources from non-renewable energy resources to renewable energy resources. The renewable resources such as wind energy, solar energy, tidal energy, biomass, geothermal energy, etc. are operating at their peak performances but another concern arises about the sustainability of carbon for transportation fuel and platform chemicals.1,2,4,5 Fortunately, out of all renewable energy resources, biomass is the only renewable energy resource which provides sustainable carbon for transportation fuel and platform chemicals.2,4,6 In addition, the lignocellulosic biomass is cheap, abundant, and easily available in most of the countries, therefore, it is the best choice for renewable energy resource. The lignocellulosic biomass comprises of three fractions viz. cellulose (40–50%), hemicellulose (15–30%), and lignin (15–30%).7 The lignin fraction which comprises of 15–30% of lignocellulosic biomass is observed to be the high energy density fraction compared to other fractions, i.e., cellulose and hemicellulose.8 To convert the lignocellulosic biomass into biofuels, there are three main channels, namely, gasification, pyrolysis/liquefaction, and hydrolysis. Out of these biomass conversion channels, pyrolysis is found to be one of the best channel to transform lignocellulosic biomass into bio-oil.1 However, the raw bio-oil produced from pyrolysis cannot be used directly to the transportation vehicle because it comprises of over 300 oxy-functionals. The presence of high oxygen content in bio-oil is the root of many drawbacks, e.g., low heating value, less stability, low pH, viscous, etc.1,9,10 Therefore, in order to use the bio-oil in the transportation vehicles, it needs to be upgraded through a proper upgradation channel. Though, the unprocessed bio-oil is a great source of many platform chemicals, e.g., 5-HMF, levulinic acid, etc. Out of various pyrolysis derived bio-oil upgradation channels, the hydrodeoxygenation (HDO) process is reviewed to be one of the best channel.1,6,11 Therefore, in this work, eugenol, an oxy-functional bio-oil model compound has been selected for its conversion using density functional theory (DFT). In addition, results on thermochemistry of eugenol conversion are also presented in the range of T = 298–898 K at atmospheric pressure.Literature review
It has been stated that unprocessed bio-oil comprises of hundreds of oxy-functionals. Out of which, eugenol component is one of the most promising model component of the phenolic fraction of bio-oil because it comprises of two oxy-functionals viz. hydroxyl and methoxy functionals; and an alkenyl functional.12–14 Eugenol component is observed as one of the product in many lignocellulosic biomass pyrolytic study.15,16 Therefore, eugenol can be one of the best model compound to study the bio-oil upgradation process. In addition, the number of carbon atoms in the eugenol (C10H12O2) is more than other competitive model components of phenolic fraction of bio-oil, e.g., guaiacol (C7H8O2),17–19 catechol (C6H6O2),17,19,20 phenol (C6H6O),17,20,21 vanillin (C8H8O3),22,23 etc. which can be significant in the production of many specialty or platform chemicals. There has been a very few experimental studies8,12–14,24,25 on eugenol as the bio-oil model compound and only a single combined, i.e., both experimental and computational study25 of eugenol as the bio-oil model compound is available in the literature to the best of authors' knowledge. For instance, Nimmanwudipong et al.13 carried out conversion reactions of eugenol in the presence of Pt/γ-Al2O3 and HY zeolite catalysts; and reported various products, e.g., phenol, guaiacol, veratrole, 4-propylguaiacol, 4-propylphenol, p-cresol, etc. They reported guaiacol and isoeugenol as main products using HY zeolite catalyst, whereas, 4-propylguaiacol as the main product from Pt/γ-Al2O3 catalyst. On the other hand, Zhang et al.14 carried out experiments of eugenol hydrogenation/deoxygenation in the presence of Pd/C and HZSM-5 catalysts; and reported that eugenol first underwent allyl hydrogenation to produce 4-propylguaiacol followed by ring saturation to produce 2-methoxyl-4-propyl-cyclohexanol. Deepa and Dhepe24 carried out hydrodeoxgenation experiments of phenol, guaiacol, and eugenol in the presence of noble metal catalysts; and they schemed that eugenol first underwent saturation of allyl followed by ring saturation; however, guaiacol first underwent the ring saturation. On the other hand, Horáček et al.12 also suggested that eugenol first underwent allyl hydrogenation over zeolite supported platinum catalyst. Chen et al.8 carried out hydrodeoxygenation of eugenol over carbon nanotube (CNT) supported ruthenium catalyst and reported propylcyclohexane as final alkane product with 4-propylguaiacol and 4-propylcyclohexanol as intermediate products. A combined study of eugenol decomposition in vapour phase carried out by Ledesma et al.25 and they reported methane, ethylene, acetylene, propylene, benzene, CO, and 1-butene as the major products.Finally, it can be understood from the literature review that eugenol component has been studied by a very few experimental researchers and only one computational work due to Ledesma et al.25 is available. However, Ledesma et al.25 carried out only one reaction pathway which initiated from the dehydrogenation of hydroxyl functional. Therefore, for the conversion of eugenol to various products, in this numerical study, we have carried out eugenol decomposition in gas phase by eight primary reaction schemes producing various important products such as propylbenzene, guaiacol, methylbenzene, propylcyclohexane, 4-propylphenol, allylbenzene, isoeugenol, and 2-methyl-4-propyl-phenol. In the course of production of such products, there are many important intermediates obtained such as 4-methylguaiacol, 4-propylcyclohexanol, 4-propylcycloguaiacol, p-cresol, etc.
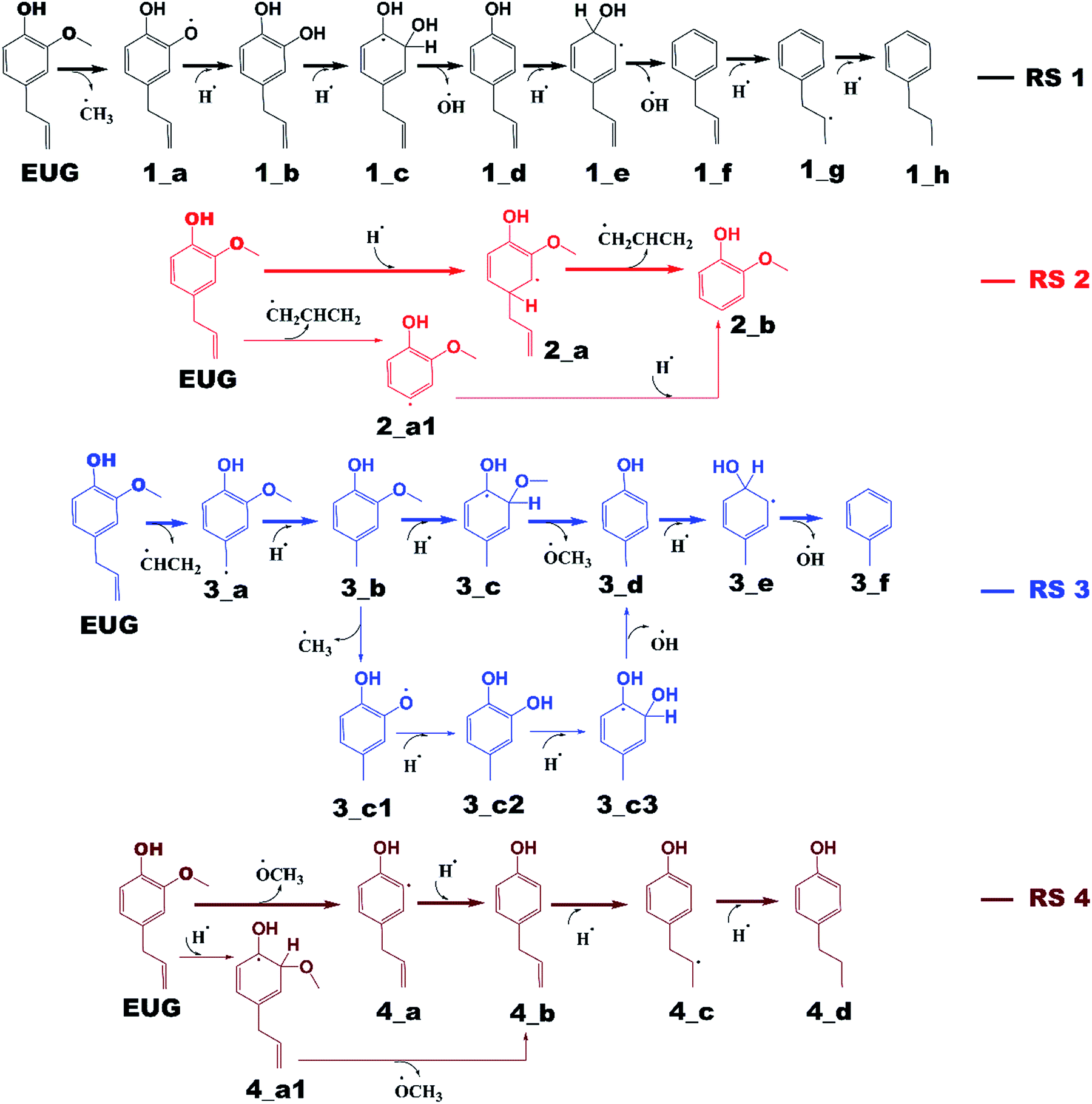
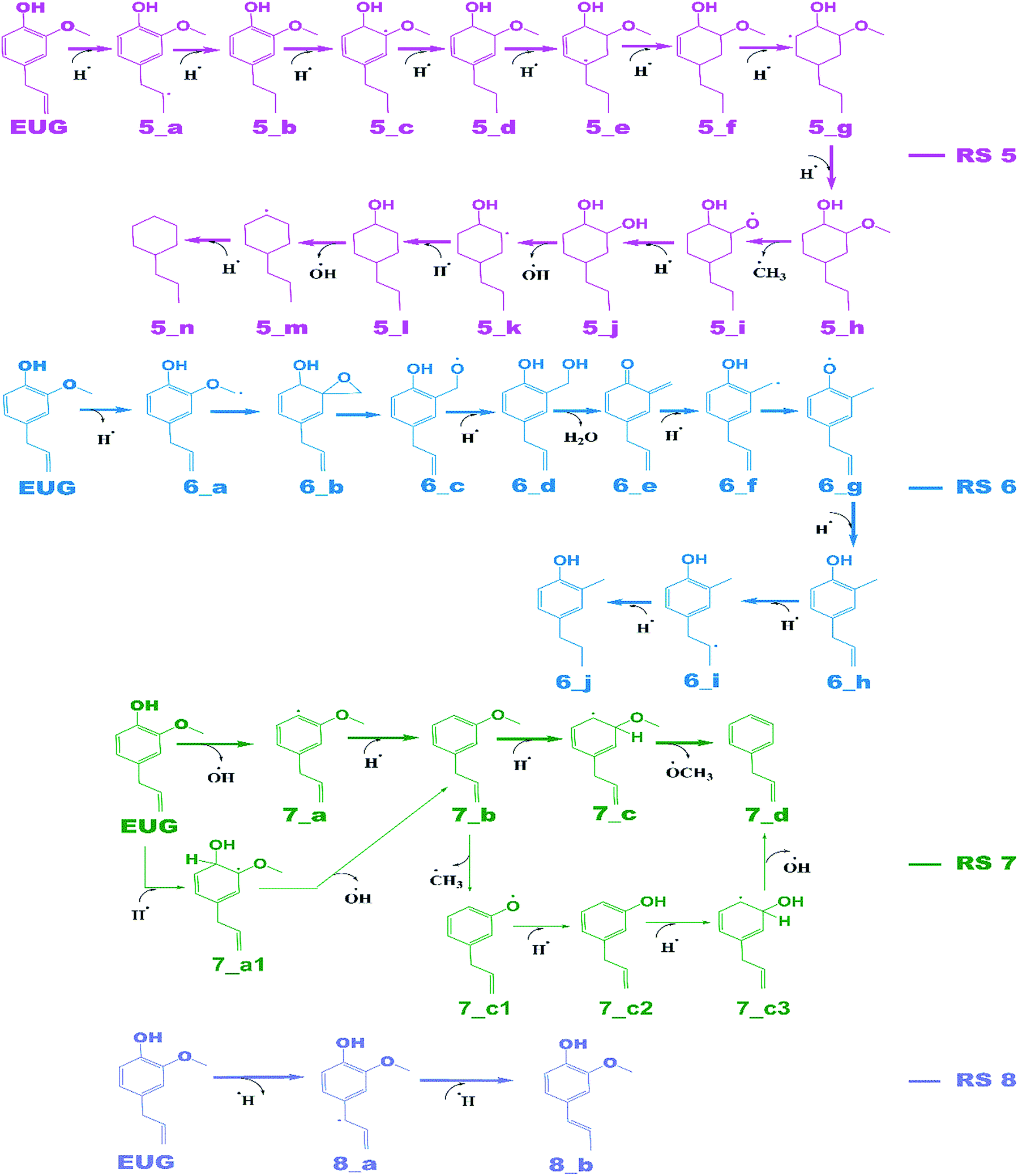
Reaction schemes
The notations in reaction schemes (RSs) and Fig. 1–9 are shown as, in general, X_Y, where X and Y denote the reaction pathway number and structure number in that particular reaction pathway, respectively. For instance, in 4_b, b is the structure of reaction pathway 4. Similarly, the transition state structures have been designated as TSX_Y in all potential energy surface figures, where X is reaction pathway number and Y is the transition state number in a given reaction pathway, e.g., TS3_1 is the first transition state structure in reaction pathway 3.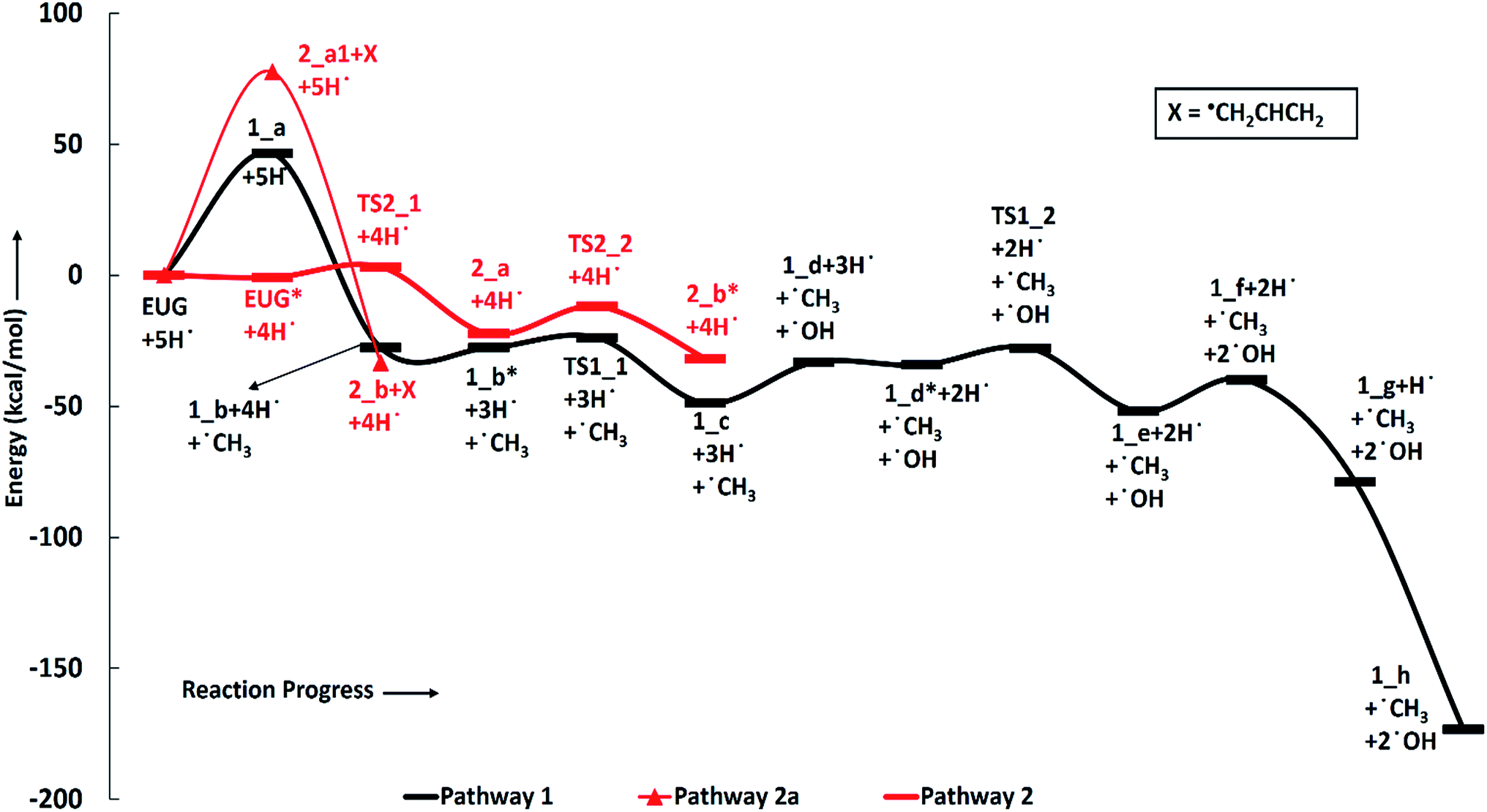 | ||
| Fig. 1 Potential energy surfaces of reaction pathways 1, 2 and 2a. | ||
The reaction scheme 1 presents the demethylation of eugenol followed by single step hydrogenation reaction to produce 4-allylcatechol. Further, 4-allylcatechol undergoes a single step hydrogenation reaction followed by dehydroxylation reaction to produce 4-allylphenol; followed by the similar reactions to produce 4-allylbenzene. Finally, the two step hydrogenation reactions to the double bond of allyl are performed to produce 4-propylbenzene (see RS 1). The reaction scheme 2 describes a secondary reaction scheme as well. Under primary reaction scheme 2, a hydrogen atom is adsorbed on the aromatic carbon of Caromatic–allyl bond followed by removal of allyl group to produce guaiacol, whereas, the secondary reaction scheme is about the cleavage of allyl group of eugenol followed by a single step hydrogenation to produce guaiacol. The third reaction scheme is about the vinyl group removal from eugenol as the first reaction step followed by an atomic hydrogen addition to produce 4-methylguaiacol. Further, 4-methylguaiacol follows two schemes. Under primary reaction scheme 3 (bold blue lines in RS 3), a single step hydrogenation reaction is carried out at the aromatic carbon of Caromatic–OCH3 sigma bond followed by demethoxylation reaction to produce 4-methylphenol. The secondary reaction scheme 3 (regular blue lines in RS 3) is the demethylation reaction of O–CH3 bond of 4-methylguaiacol followed by an atomic hydrogenation to produce 4-methylcatechol; and a single step hydrogenation reaction at the aromatic carbon of Caromatic–OH bond, meta positioned to the methyl group, is carried out followed by dehydroxylation reaction to produce 4-methylphenol. The 4-methylphenol, an intermediate where secondary reaction scheme 3 merges, is further hydrogenated to remove the hydroxyl functional to produce methylbenzene. Similar to the reaction scheme 3, the reaction scheme 4 also has a secondary reaction scheme. Under primary reaction scheme 4 (bold brown lines in RS 4), the eugenol undergoes methoxy group cleavage followed by a single step hydrogenation reaction to produce 4-allylphenol; and two step hydrogenation reactions to the double bond of allyl to produce 4-propylphenol. On the other hand, the secondary reaction scheme 4 (regular brown lines in RS 4) presents a single step hydrogenation reaction to the aromatic carbon of Caromatic–OCH3 bond followed by methoxy group removal and merges to 4-allylphenol. The reaction scheme 5 first carries out allyl hydrogenation reactions to produce 4-propylguaiacol followed by ring saturation reactions. The produced component after ring saturation, 2-methoxy-4-propylcyclohexanol, undergoes demethylation reaction followed by a single step hydrogenation reaction to produce 4-propyl-1,2-cyclohexanediol. This further undergoes hydroxyl cleavage and a single step hydrogenation reaction to produce 4-propylcyclohexanol. Similarly, another hydroxyl cleavage of 4-propylcyclohexanol is carried out followed by an atomic hydrogenation reaction to produce propylcyclohexane. The reaction scheme 6 is about the methoxy group rearrangement to produce 4-allyl-2-(hydroxymethyl)phenol. Further, the produced component undergoes water compound removal followed by two atomic hydrogen addition reactions to yield 4-allyl-2-methylphenol. Finally, the hydrogenation reactions to allyl produce 4-propyl-2-methylphenol. The reaction scheme 7 describes hydroxyl group cleavage and a single step hydrogenation reaction to produce 3-allylanisole which further undergoes an atomic hydrogen addition to Caromatic–OCH3 bond followed by demethoxylation reaction to produce allylbenzene. Similar to reaction schemes 3 and 4, the reaction scheme 7 also includes a secondary reaction scheme in which eugenol undergoes a single step hydrogenation followed by hydroxyl removal to produce 3-allylanisole; which further undergoes demethylation reaction and an atomic hydrogenation reaction to produce 3-allylphenol. Finally, a single step hydrogenation to 3-allylphenol followed by hydroxyl cleavage produces allylbenzene. The reaction scheme 8 is about the dissociation of benzylic hydrogen (see RS 8) followed by an atomic hydrogen addition reaction to the terminal carbon (methylene group) to produce isoeugenol. All above reaction pathways are carried out theoretically in gas phase milieu at B3LYP/6-311+g(d,p) level of theory under density functional theory (DFT).
Computational insights
Recently, density functional theory (DFT) has emerged as one of the best tool for computational quantum chemical calculations because of its ability to provide trustworthy and precise results in less computational time compared to other contemporary theories. The energy under DFT is a functional of electron density; and electron density is function of three spatial variables. On the other hand, the wavefunction theory is a function of 4N variables, where N is number of electrons. Therefore, DFT reduces a lot of computational time and allows one to perform on higher level of theories or on a big molecule in less computational time compared to ab initio methods. DFT has many advantages, however, its functionals are still in open questions for their better performances. Fortunately, Simón and Goodman26 carried out 19 organic reactions using Hartree–Fock theory and different DFT functionals such as generalized-gradient approximation (GGA), hybrid-GGA, and meta-GGA. They reported that B3LYP (Becke-3 parameter-Lee–Yang–Parr) functional is superior for organic reactions amongst other reported functionals and theories, therefore, all geometries and transition state structure calculations are carried out using B3LYP27,28 functional and 6-311+g(d,p)29 basis set under DFT30,31 perspective. To confirm the transition state structures as an actual transition state, minimum structures as real minimum structures, and to get the thermochemical parameters, normal mode vibrational frequency calculations are carried out at the same level of theory as of optimization theory. One and zero imaginary frequencies count in frequency results affirm the structures as real transition state structure and minimum structure, respectively. Further, to link the transition state structure to its two minima, an intrinsic reaction coordinate (IRC)32 calculation has been carried out for each transition state structure using B3LYP/6-311+g(d,p) level of theory. All open-shell calculations are performed under unrestricted-DFT theory with spin multiplicity of 2. Moreover, the electronic energy added with zero-point vibrational energy (ZPVE), ZPVE, and spin multiplicity corresponding to each molecular structure involved in all reaction schemes are provided in ESI.† Further, the xyz coordinates along with electronic energy and spin multiplicity of few structures associated with reaction scheme 1 (RS 1) are also included in ESI.†Further, the transition state structures of many organic homolysis reactions are very difficult to find and, in this case, the bond dissociation energies (BDEs) are better approximations to the activation energies, therefore, BDE calculations have been carried out for such homolysis reactions.22,33 The expression of BDE22,33 calculation is as follows:
| BDE298(R–A) = H298(R) + H298(A) − H298(R–A) | (1) |
The thermochemistry analyses, using vibrational frequency calculations, are carried out at a wide range of temperature of 298–898 K with an interval temperature of 100 K and a fixed pressure of 1 atm. All quantum chemical calculations are carried out in gas phase environment using Gaussian 09 (ref. 34) and Gauss View 5 (ref. 35) software packages.
Results and discussion
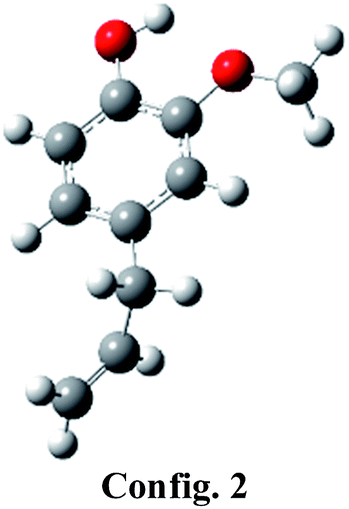
Eugenol component can be represented by various conformers of competitive energetics; however, two eugenol conformers are very close in their energetics and are presented in Table 1. The eugenol structure of config. 1 is 0.39 kcal mol−1 more stable than the eugenol structure of config. 2. The present conformational analysis is under excellent agreement with Olbert-majkut et al.36 because they also reported config. 1 structure as the ground state conformer and according to their simulations, the eugenol structure of config. 2 (see Table 1) is 0.39 kcal mol−1 less stable than the config. 1. Therefore, all calculations, e.g., BDEs, reaction pathways, etc. are carried out using eugenol structure of config. 1.| Structures | 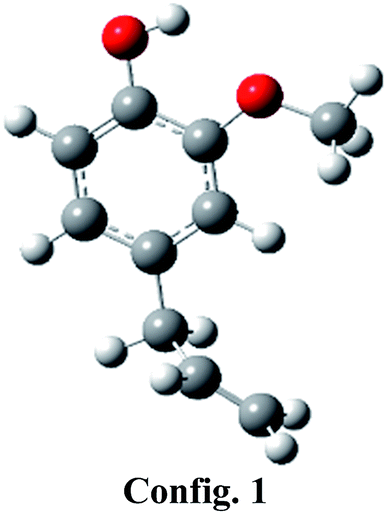 |
 |
| Relative energy | 0 | 0.39 |
Bond dissociation energies (BDEs)
The bond dissociation energies (in kcal mol−1) of eugenol based on the structure of config. 1 are shown in Table 2. As it can be seen from Table 2 that the methyl cleavage, D1, from eugenol structure is found to be the least energy demanding but it slightly varies with the BDE given by Ledesma et al.25 However, the energetics of D1 given by Ledesma et al.25 at B3LYP/6-311+g(d,p) level of theory is also the least energy demanding compared to other cleavages. The variation can be explained by the ground state conformer of eugenol but there is no molecular structure of eugenol reported by Ledesma et al.25 based on which it could have been verified. However, the ground state eugenol structure, in this study, is validated with Olbert-majkut et al.36 and it is in excellent agreement with them. The second most favourable bond cleavage is reported as D8. The bond cleavages D5 and D6 are very competitive to each other in energetics; which are the allyl and vinyl group cleavages from eugenol, respectively. It can be seen that many bond cleavages are in ∼5% error with the literature values but the average deviation between the literature values25 and present values is calculated to be 5.74%. The bond dissociation energies in ascending order of their energetics are laid as D1 < D8 < D5 < D6 < D2 < D3 < D7 < D9 < D4 < D10 < D11 < D12 < D13, respectively.| Bond | BDE (kcal mol−1) | % error | ||
|---|---|---|---|---|
| Ledesma et al.25 | Present (6-311+g(d,p)) | |||
| D1 | 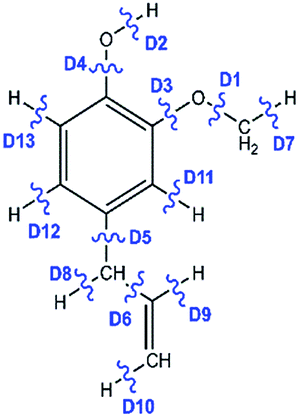 |
52.2 | 47.85 | 9.10 |
| D2 | 87.8 | 82.59 | 6.31 | |
| D3 | 97.2 | 92.39 | 5.21 | |
| D4 | 112 | 108.02 | 3.68 | |
| D5 | 82.1 | 78.14 | 5.07 | |
| D6 | 83.2 | 78.17 | 6.44 | |
| D7 | 102 | 96.23 | 6.00 | |
| D8 | 79.1 | 73.68 | 7.35 | |
| D9 | 112 | 106.15 | 5.51 | |
| D10 | 116 | 110.36 | 5.11 | |
| D11 | 117 | 111.49 | 4.94 | |
| D12 | 118 | 112.13 | 5.23 | |
| D13 | 119 | 113.71 | 4.65 | |
Potential energy surfaces
 | ||
| Fig. 2 The optimized molecular structures corresponding to PESs of pathways 1, 2, 2a, and 3. | ||
| Pathways | Bond dissociation energies/barrier heights (kcal mol−1) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Step 1 | Step 2 | Step 3 | Step 4 | Step 5 | Step 6 | Step 7 | Step 8 | Step 9 | Step 10 | Step 11 | Step 12 | Step 13 | Step 14 | |
| 1 | 47.85 | 75.22 | 3.81 | 16.51 | 6.38 | 5.65 | 39.60 | 96.33 | — | — | — | — | — | — |
| 2 | 3.95 | 10.53 | — | — | — | — | — | — | — | — | — | — | — | — |
| 2a | 78.14 | 112.46 | — | — | — | — | — | — | — | — | — | — | — | — |
| 3 | 78.17 | 86.55 | 4.11 | 9.30 | 6.31 | 13.37 | — | — | — | — | — | — | — | — |
| 3a | Same as path 3 | 46.81 | 74.22 | 4.60 | 17.04 | — | — | — | — | — | — | — | — | |
| 4 | 92.39 | 111.76 | 35.97 | 99.86 | — | — | — | — | — | — | — | — | — | — |
| 4a | 4.48 | 9.40 | Same as path 4 | — | — | — | — | — | — | — | — | — | — | |
| 5 | 39.37 | 96.11 | 22.75 | 67.99 | 52.51 | 76.67 | 36.79 | 97.49 | 73.28 | 100.31 | 89.76 | 97.72 | 87.32 | 96.44 |
| 6 | 96.23 | 14.71 | 0.03 | 102.76 | 32.26 | 58.54 | 27.74 | 80.30 | 39.46 | 96.32 | — | — | — | — |
| 7 | 108.02 | 113.91 | 6.23 | 10.07 | — | — | — | — | — | — | — | — | — | — |
| 7a | 5.17 | 16.70 | 56.41 | 84.29 | 6.73 | 11.98 | — | — | — | — | — | — | — | — |
| 8 | 73.68 | 79.64 | — | — | — | — | — | — | — | — | — | — | — | — |
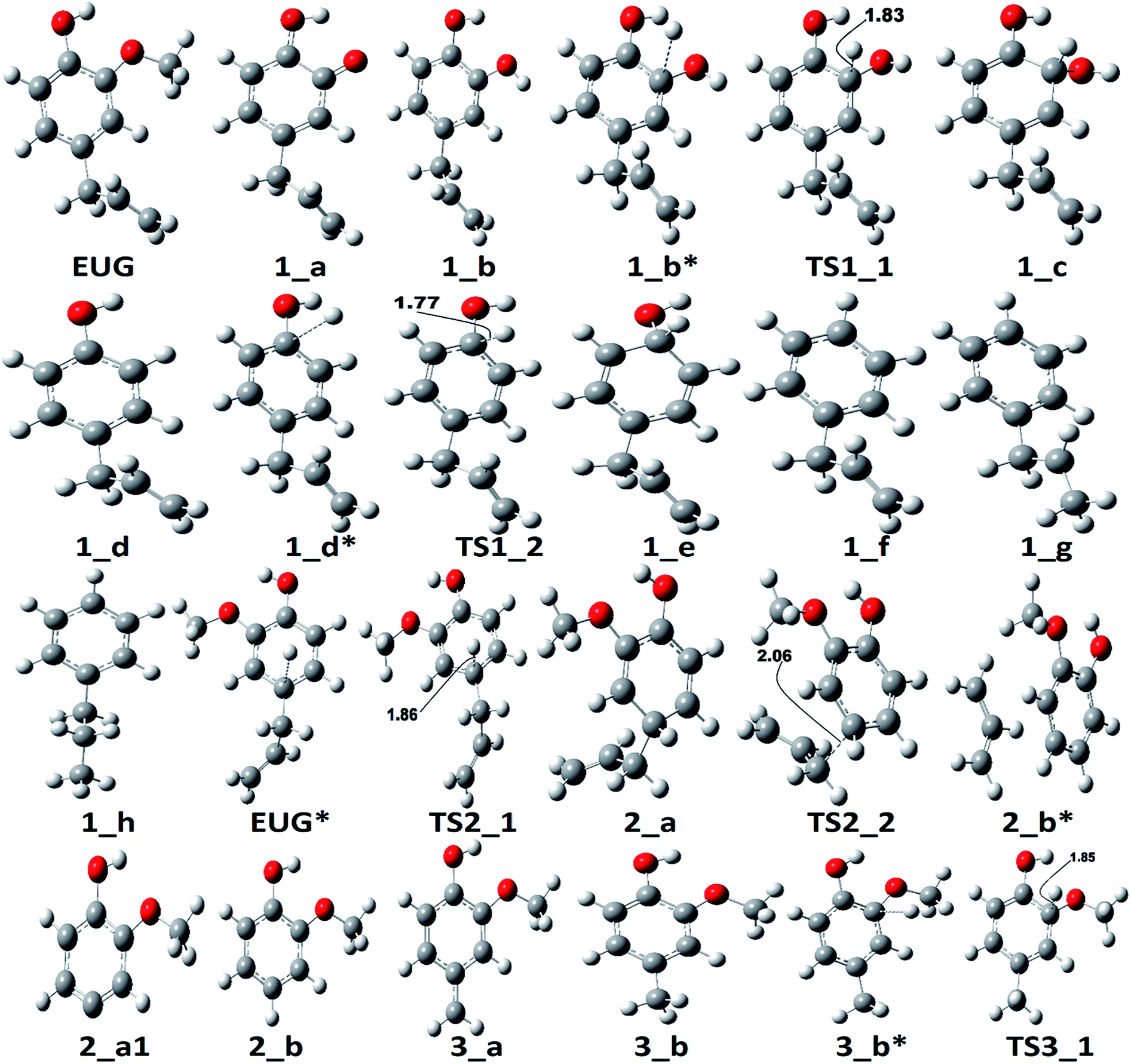
The reaction scheme 1 produces propylbenzene from eugenol. As the BDE study suggests that the methyl group cleavage is most favourable compared to other bond cleavages, therefore, the reaction pathway 1 starts from methyl group cleavage. The produced component after methyl cleavage undergoes a single step hydrogenation reaction to produce 4-allylcatechol. The BDEs of these two reaction steps are calculated to be 47.85 kcal mol−1 and 75.22 kcal mol−1, respectively. Further, a hydrogen atom is adsorbed on the aromatic carbon of Caromatic–OH sigma bond, the one which is meta positioned to allyl group, followed by the hydroxyl group removal reaction to produce 4-allylphenol. The barrier height of the hydrogen addition over aromatic carbon is calculated to be 3.81 kcal mol−1 and since, the transition state for hydroxyl removal from 1_c structure is hard to find, BDE calculation is carried out for this reaction step which is calculated to be 16.51 kcal mol−1. The produced component is recognized as 4-allylphenol. Further, to produce allylbenzene from 4-allylphenol, the similar reaction steps as of 1_b → 1_c → 1_d are carried out as 1_d → 1_e → 1_f. The barrier height of the reaction step 1_d → 1_e is calculated to be 6.38 kcal mol−1 and BDE of the reaction step 1_e → 1_f is calculated to be 5.65 kcal mol−1. The production of allylbenzene is also carried out in the reaction scheme 7 with different mechanism compared to the reaction scheme 1 to compare the activation energy. Nevertheless, the allylbenzene component does not contain any oxy-functional which is prime motive of bio-oil upgradation but it does contain a double bond in allyl group other than the ones in phenyl ring. Therefore, the allylbenzene is further hydrogenated to saturate the double bond of allyl of allylbenzene into single bond by two single step hydrogenation reactions. Nimmanwudipong et al.13 in their experiment suggested to hydrogenate the terminal carbon group first followed by the middle one, therefore, a single step hydrogenation is carried out at the terminal carbon of allyl followed by another single step hydrogenation to saturate the radical on second carbon. These two steps require BDEs of 39.60 kcal mol−1 and 96.33 kcal mol−1, respectively. The imaginary frequencies corresponding to TS1_1 and TS1_2 are calculated to be 762.50i and 933.63i cm−1, respectively.
The second reaction scheme is about the production of guaiacol by deallylation of eugenol. The formation of guaiacol, as major product, has been suggested by Nimmanwudipong et al.13 in their experiment of eugenol conversion over HY zeolite catalyst. Zhang et al.14 also found guaiacol as one of the product in their experiment of eugenol over Pd/C and HZSM-5 catalysts. As it has been pointed out that the guaiacol production, in this study, is carried out by two reaction pathways, i.e., reaction pathways 2 and 2a. It can be seen in RS 2, under primary reaction pathway 2 in bold red color line, that hydrogen atom is adsorbed on the aromatic carbon of Caromatic–allyl bond prior to the removal of allyl group to produce guaiacol, whereas, under secondary reaction pathway 2a in regular red color line in RS 2, the eugenol component first undergoes allyl group cleavage followed by a single step hydrogenation reaction to produce guaiacol. The barrier heights of the hydrogen adsorption on the aromatic carbon of Caromatic–allyl bond and the deallylation reaction under reaction pathway 2 are calculated to be 3.95 kcal mol−1 and 10.53 kcal mol−1, respectively. On the other hand, the BDEs of direct cleavage of the allyl group and a single step hydrogenation to the carbon radical under reaction pathway 2a are calculated to be 78.14 kcal mol−1 and 112.46 kcal mol−1, respectively. It can be seen that the direct cleavage of allyl group is not favourable at all; however, if the hydrogen atom is added on the aromatic carbon of Caromatic–allyl bond prior to allyl cleavage then it is favourable. The activation energies of both reaction pathways 2 and 2a can be written as 10.53 kcal mol−1 and 112.46 kcal mol−1, respectively. Therefore, it can be believed that the allyl group cleavage occurs after the hydrogen atom addition. The imaginary frequencies corresponding to TS2_1 and TS2_2 are calculated to be 749.74i and 585.99i cm−1, respectively.
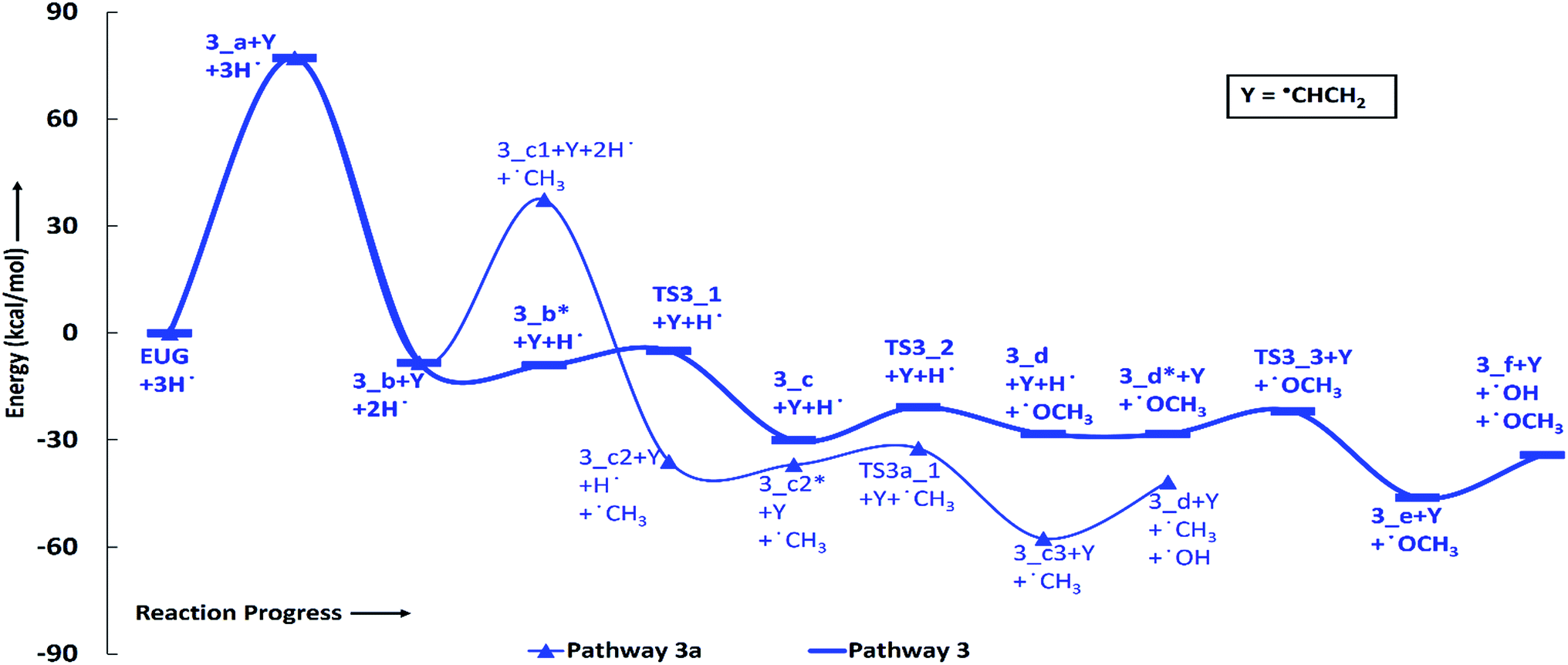 | ||
| Fig. 3 Potential energy surfaces of reaction pathways 3 and 3a. | ||
 | ||
| Fig. 4 The optimized molecular structures corresponding to PESs of pathways 3, 3a, 4, 4a, and 5. | ||
The reaction starts from vinyl group cleavage of eugenol followed by a single step hydrogenation reaction to produce 4-methylguaiacol. The production of 4-methylguaiacol is carried out by Deepa and Dhepe24 and they reported that eugenol first undergoes allyl group saturation followed by methyl group cleavage from propyl group and addition of a hydrogen atom. Similar to the experiment of Deepa and Dhepe,24 Nimmanwudipong et al.13 also suggested the same for the formation of 4-methylguaiacol. Here in reaction scheme 3, another approach has been considered, i.e., the methylene group is cleaved from allyl group of eugenol followed by a single step hydrogenation reaction to produce 4-methylguaiacol; however, the production of 4-methylguaiacol according to Nimmanwudipong et al.13 and Deepa and Dhepe24 will be discussed in the sub-section of reaction scheme 5 as the reaction steps EUG → 5_a → 5_b → 3_a → 3_b.
According to the reaction pathway 3, BDEs of the reaction steps EUG → 3_a → 3_b are calculated to be 78.17 kcal mol−1 and 86.55 kcal mol−1, respectively. The produced component, 4-methylguaiacol, follows two pathways as reaction pathways 3 and 3a. As it has been observed in the subsection of ‘Bond dissociation energies’ that the direct cleavages of methoxy, hydroxyl, and allyl of eugenol require a high BDEs, therefore, the direct cleavages of functional groups may not be favourable. To avoid such cases, other approaches are carried out in reaction pathways 3 and 3a. Under reaction pathway 3, bold blue lines in RS 3, a hydrogen atom is adsorbed on the aromatic carbon of Caromatic–OCH3 sigma bond followed by methoxy group removal. The barrier heights of reaction steps 3_b → 3_c → 3_d are calculated to be 4.11 kcal mol−1 and 9.30 kcal mol−1, respectively. On the other hand, the reaction pathway 3a undergoes demethylation of 4-methylguaiacol followed by a single step hydrogenation reaction requiring bond dissociation energies of 46.81 kcal mol−1 and 74.22 kcal mol−1, respectively, to produce 4-methylcatechol. The 4-methylcatechol further undergoes a hydrogen atom adsorption on aromatic carbon of Caromatic–OH bond, the one which is meta positioned to methyl group, followed by hydroxyl group removal requiring barrier height and BDE of 4.60 kcal mol−1 and 17.04 kcal mol−1, respectively, to produce 4-methylphenol. The secondary reaction pathway merges to the structure of p-cresol, i.e., 3_d, from which the main reaction pathway 3 carries on. Although, p-cresol is an important compound in the industries but, for study purpose, further oxygen removal of p-cresol is carried out to produce toluene which is also a great industrial chemical. The single step hydrogenation reaction is carried out to the aromatic carbon of Caromatic–OH sigma bond which requires a barrier height of 6.31 kcal mol−1 followed by the hydroxyl group removal and BDE of reaction step 3_e → 3_f is calculated to be 13.37 kcal mol−1. The imaginary frequencies corresponding to TS3_1, TS3_2, TS3_3, and TS3a_1 are calculated to be 750.33i, 403.66i, 927.32i, and 722.18i cm−1, respectively.
The rate determining step of reaction pathway 3 is found to be the single step hydrogenation reaction which produces 4-methylguaiacol, i.e., 3_a → 3_b, therefore, the activation energy is 86.55 kcal mol−1. The production of 4-methylphenol from 4-methylguaiacol under reaction pathway 3 seems to be favourable because of less barrier height compared to that of reaction pathway 3a. Therefore, the production of toluene favours the reaction pathway 3 compared to pathway 3a, however, the production of 4-methylguaiacol from eugenol requires a high amount of energy. Nevertheless, another approach will be discussed in the subsection of ‘Reaction pathways 5 and 6’ for the production of 4-methylguaiacol from eugenol according to the mechanism provided by Nimmanwudipong et al.13 and Deepa and Dhepe.24
 | ||
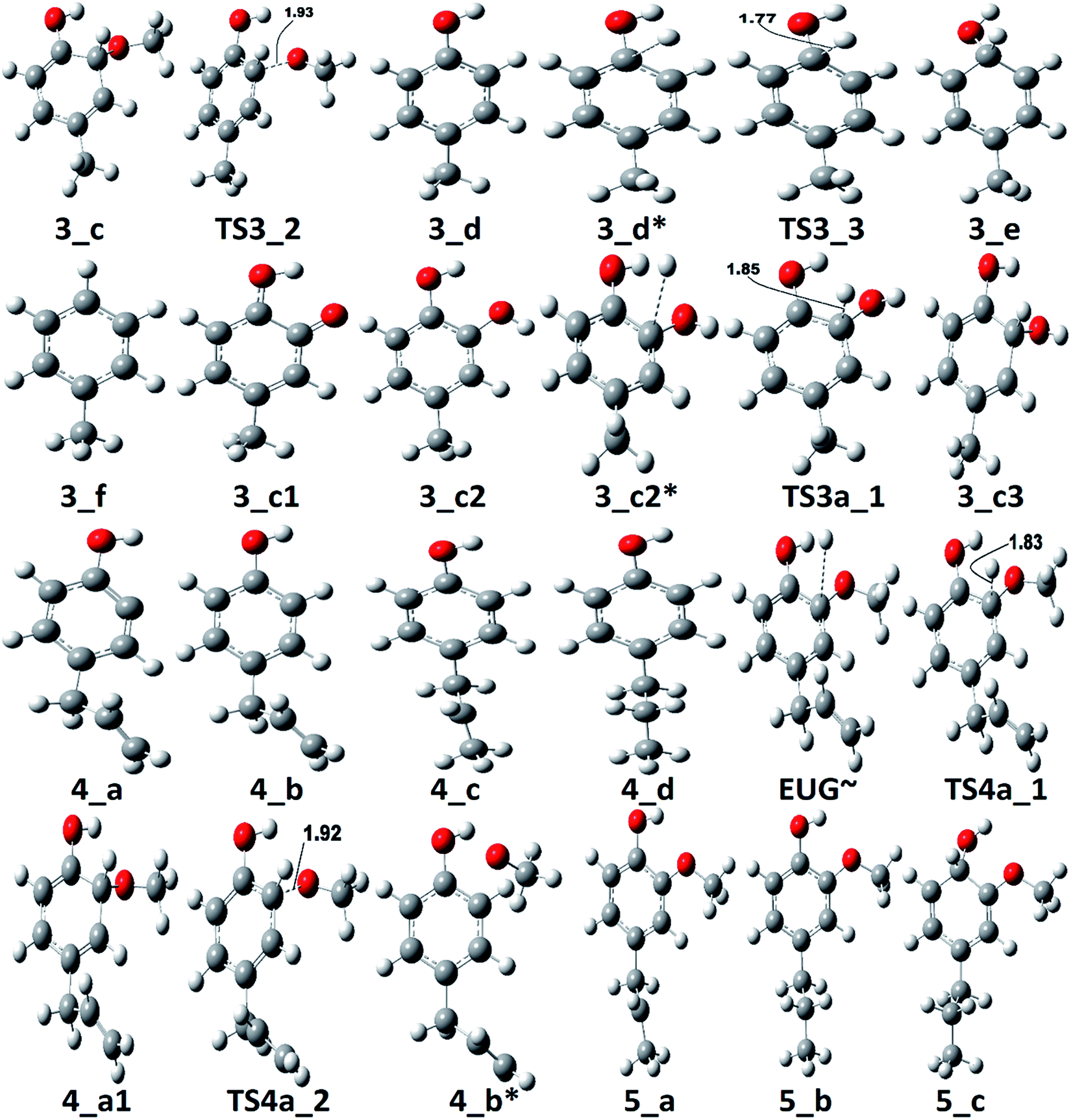
| Fig. 5 Potential energy surfaces of reaction pathways 4 and 4a. | ||
The reaction pathway 4 presents the methoxy group cleavage followed by a single step hydrogenation reaction to produce 4-allylphenol. BDE of the direct cleavage of methoxy group is given in Table 2, however, that is high energetics, 92.39 kcal mol−1, and cannot be achieved without appropriate catalyst. Nevertheless, the BDE of atomic hydrogen addition requires an even higher BDE, i.e., 111.76 kcal mol−1. Therefore, it is observed earlier and here too that the direct cleavages are not favourable. On the other hand, the reaction pathway 4a carries out an atomic hydrogenation at the aromatic carbon of Caromatic–OCH3 sigma bond prior to the methoxy group removal; and the barrier heights of these two steps are calculated to be 4.48 kcal mol−1 and 9.40 kcal mol−1, respectively. It can be seen that the direct cleavage of methoxy group from eugenol is not favourable; but, a single step hydrogenation reaction followed by methoxy group removal requires considerably less amount of barrier heights. The imaginary frequencies corresponding to TS4_1 and TS4_2 are calculated to be 767.66i and 410.44i cm−1, respectively.
It should be noted that the reaction pathway 1 also produces 4-allylphenol (1_d) from eugenol and requires an activation energy of 75.22 kcal mol−1 considering the 4-allylphenol as product and eugenol as reactant. This activation energy, i.e., 75.22 kcal mol−1, is less compared to the activation energy demanded by reaction pathway 4 considering the 4-allylphenol as product, but is considerably higher than the activation energy required by reaction pathway 4a for the same reactant and product. Furthermore, the saturation of double bond which is present in the allyl group is saturated via two atomic hydrogenation reactions which requires BDEs of 35.97 kcal mol−1 and 99.86 kcal mol−1, respectively. The reaction pathway 4a merges to the structure of 4-allylphenol (see structure 4_b in RS 4) and follows the similar pathway as of reaction pathway 4, therefore, the saturation of double bond of allyl group will follow the same energetics as of reaction pathway 4.
 | ||
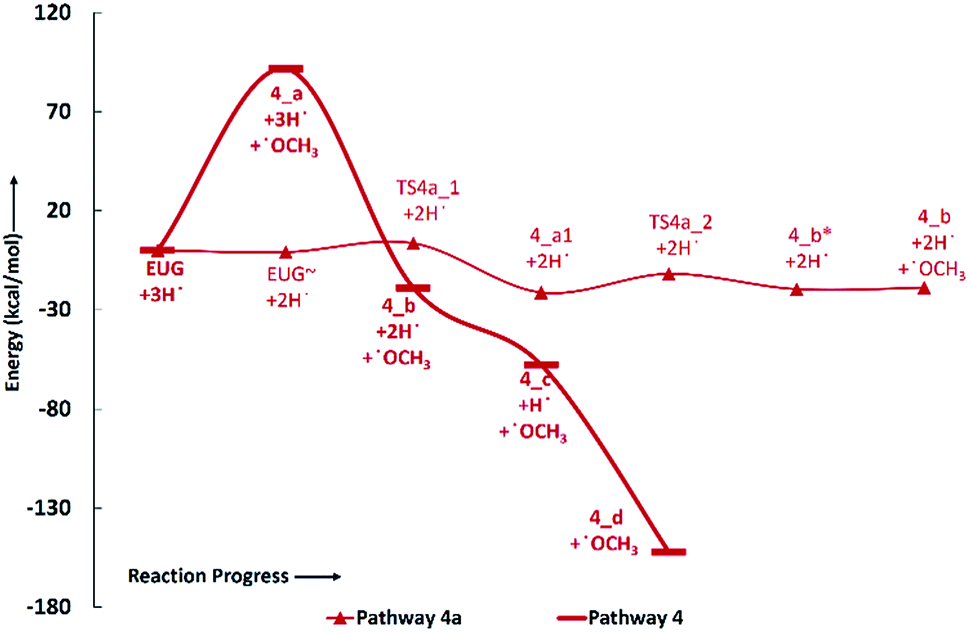
| Fig. 6 Potential energy surfaces of reaction pathways 5 and 6. | ||
Zhang et al.14 suggested the formation of 2-methoxy-4-propylcyclohexanol from eugenol in the presence of Pd/C catalyst with an intermediate called 2-methoxy-4-propylphenol. Similar phenomena have also been observed by Deepa and Dhepe24 in the presence of Pd catalyst and Chen et al.8 over Ru catalyst. Therefore, in this study, the reaction pathway 5 starts with the saturation of allyl group of eugenol which produces 2-methoxy-4-propylphenol. The first single step hydrogenation reaction is carried out at the terminal carbon of allyl according to the reaction mechanism suggested by Nimmanwudipong et al.;13 followed by the second atomic hydrogen addition to saturate the radical produced due to the first atomic hydrogen addition. The first hydrogen addition reaction requires an energy of 39.37 kcal mol−1 according to the BDE, whereas, the BDE of second step is calculated to be 96.11 kcal mol−1. This leads to the saturation of aromatic ring which requires six single step hydrogenation reactions to produce 2-methoxy-4-propylcyclohexanol. The formation of 2-methoxy-4-propylcyclohexanol from 4-propylguaiacol processes through the production of two intermediates, i.e., 6-methoxy-4-propylcyclohexa-2,4-dien-1-ol and 6-methoxy-4-propylcyclohex-2-en-1-ol. The bond dissociation energies of six single step hydrogenation reactions for ring saturation are calculated to be 22.75 kcal mol−1, 67.99 kcal mol−1, 52.51 kcal mol−1, 76.67 kcal mol−1, 36.79 kcal mol−1, and 97.49 kcal mol−1, respectively. The bond dissociation energies involving in reaction pathways are also enlisted in Table 3. The produced component after the ring saturation is identified as 2-methoxy-4-propylcyclohexanol which further undergoes the demethylation and a single step hydrogenation reactions to produce 4-propylcyclohexane-1,2-diol. The production of 4-propylcyclohexane-1,2-diol from 2-methoxy-4-propylcyclohexanol is carried out by Zhang et al.14 using hydrolysis over Pd/C in the aqueous phase environment, however, the present study represents the gas phase milieu. Nevertheless, a demethylation reaction is carried out at 2-methoxy-4-propylcyclohexanol followed by an atomic hydrogen addition reaction to produce 4-propylcyclohexane-1,2-diol. The BDEs of these two steps are calculated to be 73.28 kcal mol−1 and 100.31 kcal mol−1, respectively. Further, the production of propylcyclohexane involves the production of an important intermediate called 4-propylcyclohexanol which is formed during the dehydroxylation, the hydroxyl group which is meta positioned to propyl group, of 4-propylcyclohexane-1,2-diol followed by an atomic hydrogen addition reaction. The BDEs of hydroxyl cleavage and atomic hydrogenation reactions are calculated to be 89.76 kcal mol−1 and 97.72 kcal mol−1, respectively. The 4-propylcyclohexanol component can be a very good candidate to produce 4-propylcyclohexanone, an important component used as an intermediate of liquid crystals, by two single step dehydrogenation reactions. However, in the present study, the production of propylcyclohexane is also carried out. The production of propylcyclohexane requires the hydroxyl cleavage and a single step hydrogenation reactions similar to the production of 4-propylcyclohexanol from 4-propylcyclohexane-1,2-diol. The bond dissociation energies of these two steps to produce propylcyclohexane are calculated to be 87.32 kcal mol−1 and 96.44 kcal mol−1, respectively.
As mentioned in section of ‘Reaction schemes’, the production of 4-methylguaiacol from eugenol in pathways 3 and 3a is carried out using the vinyl group removal followed by an atomic hydrogen addition reaction. However, the literature reviews13,14,24 suggest the formation of 4-methylguaiacol through ethyl group removal of 4-propylguaiacol which is produced via the allyl group saturation of eugenol followed by an atomic hydrogen addition reaction. Thus, the reaction steps for the production of 4-methylguaiacol according to the literature13,14,24 can be laid as EUG → 5_a → 5_b → 3_a → 3_b. The bond dissociation energies of reaction steps EUG → 5_a → 5_b are calculated to be 39.37 kcal mol−1 and 96.11 kcal mol−1, respectively. The BDE of reaction step 5_b → 3_a is calculated to be 65.14 kcal mol−1 and the BDE of single step hydrogen reaction to structure 3_a to produce 3_b is calculated to be 86.55 kcal mol−1. Therefore, the activation energies for the production of 4-methylguaiacol according to the literature experiments13,14,24 and the reaction pathway 3 are calculated to be 96.11 kcal mol−1 and 86.55 kcal mol−1. Thus, it can be concluded that the production of 4-methylguaiacol under reaction pathway 3 is more favourable than the mechanisms given in literature experiments.13,14,24 However, it entirely depends on catalyst, support, temperature–pressure conditions, etc. which promote the reaction in specific way, nevertheless, the study for production of 4-methylguaiacol from eugenol in the presence of various catalysis conditions is still limited and needs to be unravelled to finally understand the true mechanism.
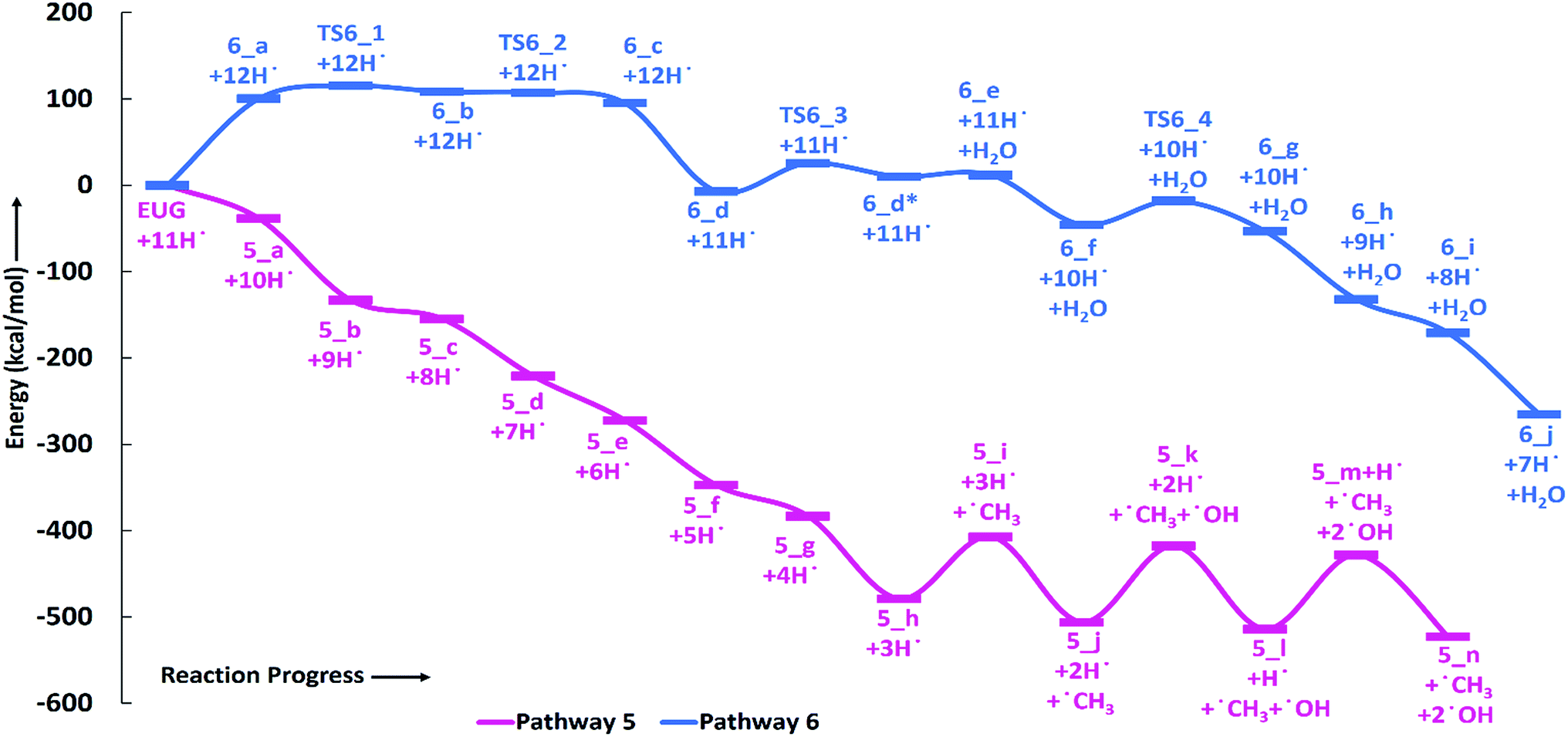
The reaction pathway 6 is about the production of 2-methyl-4-propylphenol from eugenol. The reaction starts from the dehydrogenation reaction of methyl group for which the BDE is calculated to be 96.23 kcal mol−1. The dehydrogenation of methyl group leads the methylene group to attach with one of the aromatic carbon (see Fig. 7 for corresponding molecular structures) which completes via a transition state structure, TS6_1. Further, the detachment of oxygen is carried out using TS6_2 in order to remove the C–O bond from the aromatic ring. The barrier heights corresponding to both transition state structures are calculated to be 14.71 kcal mol−1 and 0.03 kcal mol−1, respectively. However, the detachment of oxygen atom from phenyl ring generates a radical which is saturated using an atomic hydrogen addition to the oxygen atom. The BDE of the reaction step 6_c → 6_d is calculated to be 102.76 kcal mol−1. Further, the water compound removal from 6_d structure causes a barrier height of 32.26 kcal mol−1 with transition structure identified as TS6_3. The produced component after water removal is recognized as 4-allyl-6-methylenecyclohexa-2,4-dien-1-one. An atomic hydrogenation reaction to the oxygen atom is carried out, with calculated BDE of 58.54 kcal mol−1, to convert the keto group into an enol group; however, this reaction generates a radical on the carbon group (meta positioned to allyl group). The hydrogen atom of hydroxyl group is migrated to convert the methylene group into methyl group; and the barrier height for the same is calculated to be 27.74 kcal mol−1. In addition, another single step hydrogenation to the oxygen atom is carried out to produce 2-methyl-4-allylphenol with calculated BDE of 80.30 kcal mol−1. The produced component 2-methyl-4-allylphenol further undergoes two single step hydrogenation reactions to convert the double bond of allyl to single bond thus producing 2-methyl-4-propylphenol. The BDEs of allyl saturation are calculated to be 39.46 kcal mol−1 and 96.32 kcal mol−1, respectively. The imaginary frequencies corresponding to TS6_1, TS6_2, TS6_3, and TS6_4 are calculated to be 660.0i, 328.11i, 558.04i, and 2178.67i cm−1, respectively.
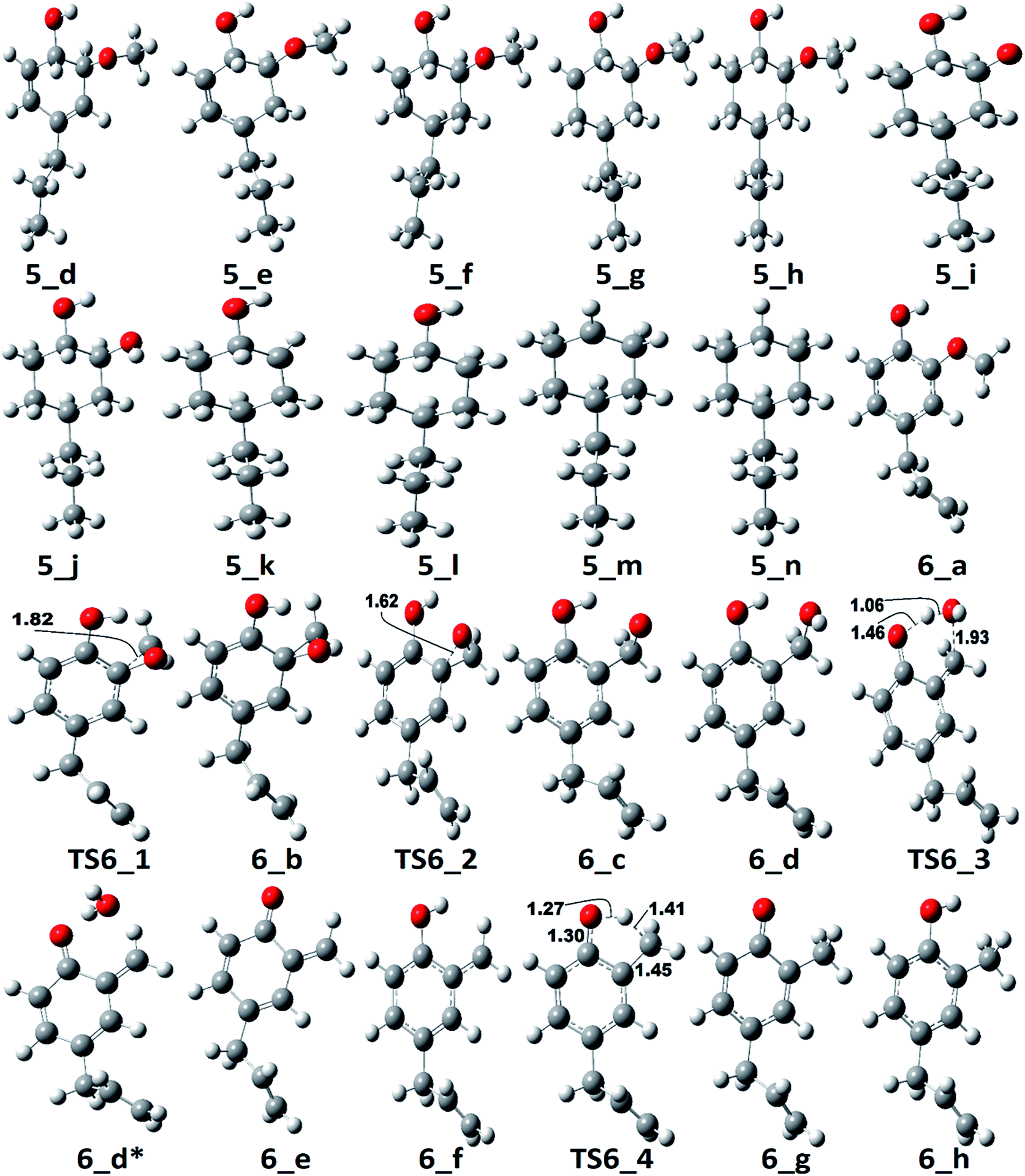 | ||
| Fig. 7 The optimized molecular structures corresponding to PESs of pathways 5 and 6. | ||
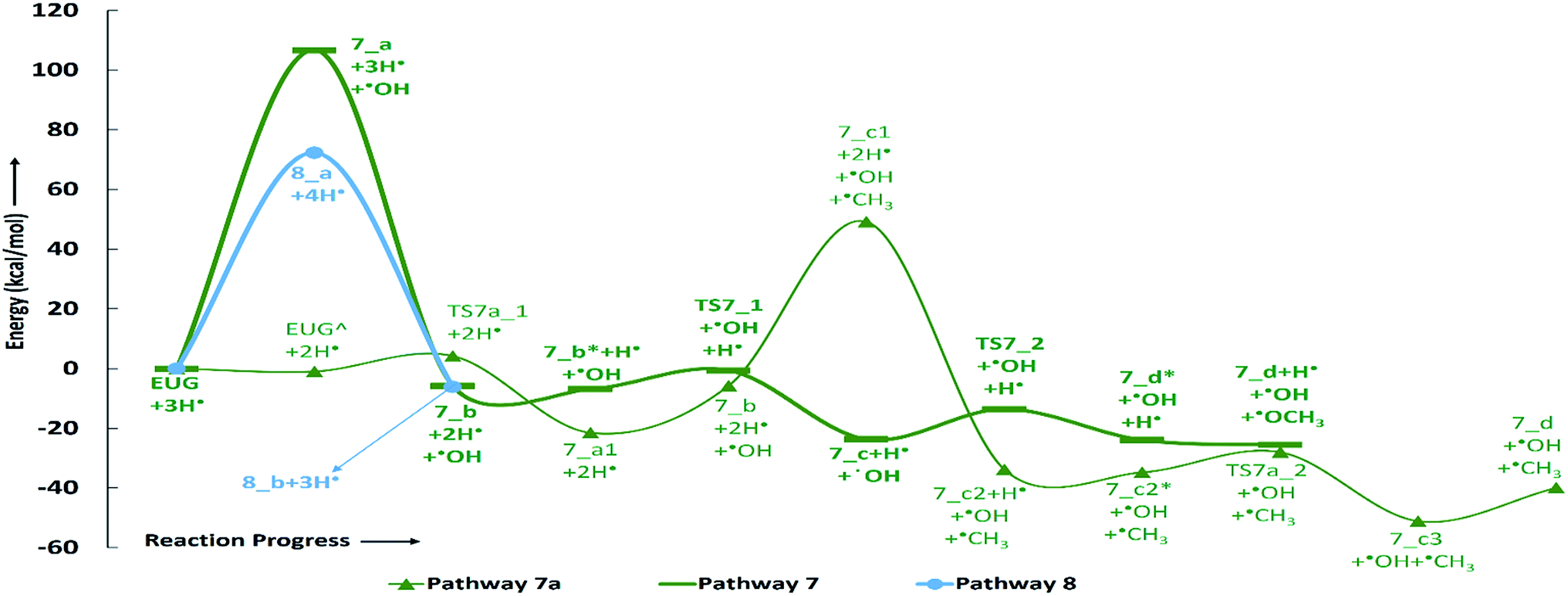 | ||
| Fig. 8 Potential energy surfaces of reaction pathways 7, 7a, and 8. | ||
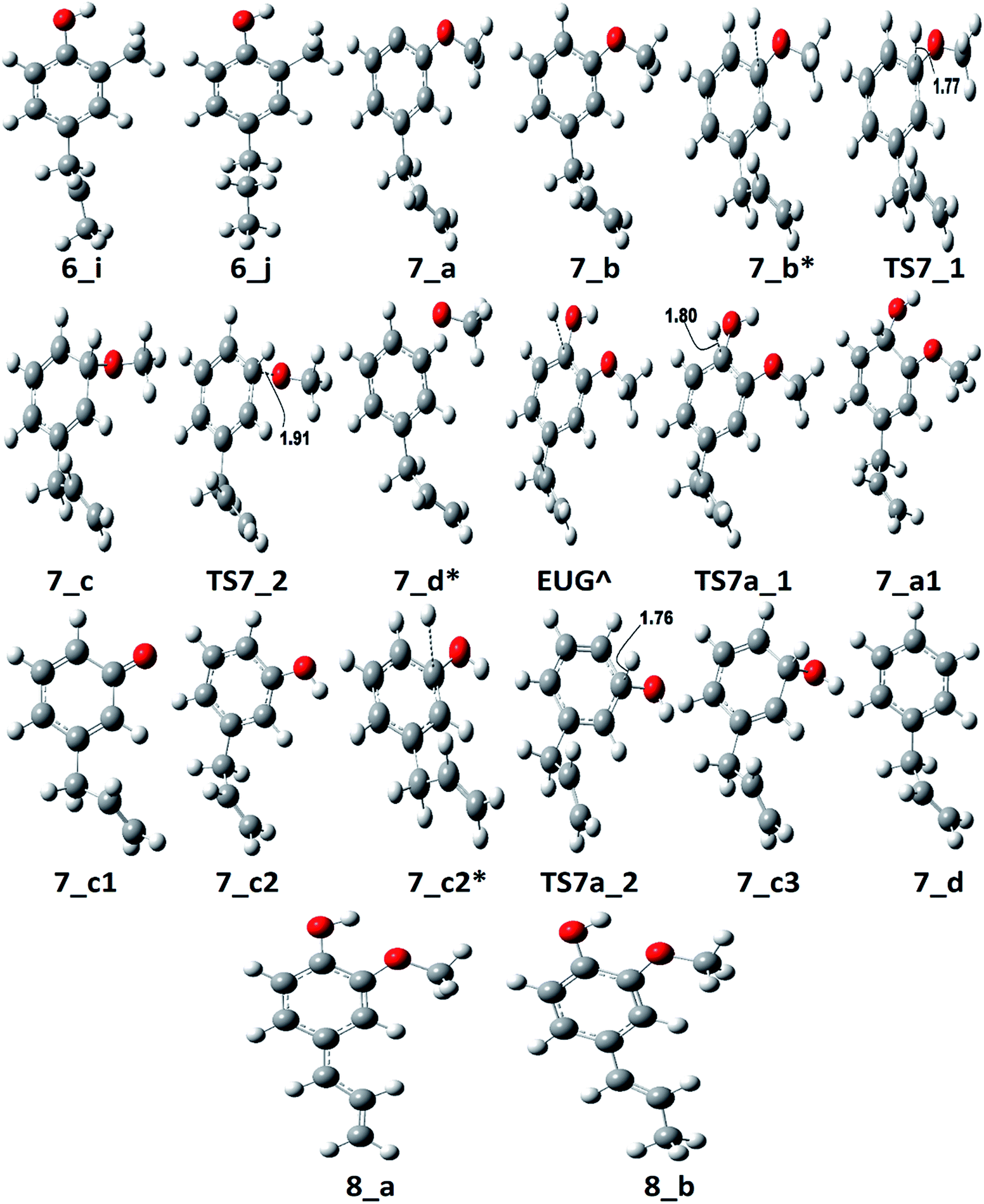 | ||
| Fig. 9 The optimized molecular structures corresponding to PESs of pathways 6, 7, 7a, 8, and 7a. | ||
The reaction pathway 7 starts from the cleavage of hydroxyl group followed by an atomic hydrogenation reaction to produce 3-methoxy-allylbenzene. The BDEs of the first two steps of pathway 7 are calculated to be 108.02 kcal mol−1 and 113.91 kcal mol−1, respectively. These energetics are quite high and are not possible without the use of any appropriate catalyst; however, the reaction pathway 7a finds a way to reduce the energetics. The reaction pathway 7a first carries out an atomic hydrogenation reaction to the aromatic carbon of Caromatic–OH sigma bond by a transition state structure TS7a_1 followed by hydroxyl group removal to produce 3-methoxy-allylbenzene. The barrier height and BDE of first and second reaction steps of reaction pathway 7a are calculated to be 5.17 kcal mol−1 and 16.70 kcal mol−1, respectively. Therefore, pathway 7a becomes favourable if one considers the product as 3-methoxy-allylbenzene and eugenol as reactant. Since, the direct cleavage of oxy-functionals is not favourable, referring to reaction pathways 2, 4 and 7, the adsorption of hydrogen atom is carried out on the aromatic carbon of Caromatic–OCH3 sigma bond under primary reaction pathway 7 followed by the removal of methoxy group to produce allylbenzene. The barrier heights corresponding to TS7_1 and TS7_2 are calculated to be 6.23 kcal mol−1 and 10.07 kcal mol−1, respectively. On the other hand, the secondary reaction pathway 7a carries out demethylation reaction of structure 7_b followed by an atomic hydrogen addition to produce 3-hydroxy-4-allylbenzene. The BDEs of these two steps are calculated to be 56.41 kcal mol−1 and 84.29 kcal mol−1, respectively. Further, 3-hydroxy-4-allylbenzene undergoes an atomic hydrogen addition to the aromatic carbon of Caromatic–OH sigma bond followed by the removal of hydroxyl functional to produce allylbenzene. The barrier height corresponding to TS7a_2 and BDE of the reaction step 7_c3 → 7_d are calculated to be 6.73 kcal mol−1 and 11.98 kcal mol−1, respectively. The imaginary frequencies corresponding to TS7_1, TS7_2, TS7a_1, and TS7a_2 are calculated to be 908.09i, 487.08i, 855.78i, and 957.67i cm−1, respectively. It can be seen that the optimum reaction pathway for the production of allylbenzene from both reaction pathways 7 and 7a follows as EUG → 7_a1 → 7_b → 7_c → 7_d. Therefore, the rate determining step of the optimum reaction steps can be identified as the reaction step 7_a1 → 7_b which provides an activation energy of 16.70 kcal mol−1. On the other hand, the activation energy for the production of allylbenzene from eugenol under RS 1 is calculated to be 75.22 kcal mol−1 which is much higher than the activation energy given by optimum reaction pathway of allylbenzene. Thus, the production of allylbenzene should occur according to the optimum reaction pathway of allylbenzene, i.e., EUG → 7_a1 → 7_b → 7_c → 7_d. However, the production of propylbenzene from allylbenzene follows the same pathway as of the reaction steps given in RS 1.
The reaction pathway 8 is about the production of trans-isoeugenol by dissociation of benzylic hydrogen followed by the atomic hydrogen addition to the terminal carbon (see 8_b in Fig. 9). As it is established in the subsection of ‘Bond dissociation energies’ that the dissociation of benzylic hydrogen (see D8 in Table 2) is the second most favourable bond cleavage, therefore, the reaction based on the dissociation of benzylic hydrogen as the first reaction step is considered in reaction pathway 8. Deepa and Dhepe24 reported the formation of trans-isoeugenol as an intermediate product in their experiment of eugenol conversion over supported Pd catalyst. Furthermore, the production of isoeugenol is also studied by Nimmanwudipong et al.13 in their experimental study on conversion of eugenol in the presence of HY zeolite catalyst; they reported this component as major product. Therefore, the reaction pathway 8 incorporates the possibility of formation isoeugenol. The BDE of dissociation of the benzylic hydrogen is calculated to be 73.68 kcal mol−1. The BDE due to the atomic hydrogen addition to produce trans-isoeugenol is calculated to be 79.64 kcal mol−1. Further dissociation of trans-isoeugenol is not considered in the present study because experimental evidences due to Nimmanwudipong et al.13 do not recommend. However, trans-isoeugenol can be hydrogenated at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond present in the chain to produce 4-propylguaiacol which can follow the reaction forward as schemed in reaction scheme 5.
C double bond present in the chain to produce 4-propylguaiacol which can follow the reaction forward as schemed in reaction scheme 5.
Finally, summarizing all reaction schemes, the activation energy for the production of guaiacol from eugenol under reaction scheme 2 is reported to be 10.53 kcal mol−1 only which is the least activation energy required amongst all activation energies corresponding to each reaction scheme. This verifies the experiment of eugenol conversion carried out by Nimmanwudipong et al.13 who reported guaiacol as one of the major product in the presence of HY zeolite catalyst. However, they witnessed that deallylation of eugenol, i.e., formation of guaiacol from eugenol over zeolite catalyst, is relatively slower than the isomerization reaction of eugenol. It can be observed that some of the reaction schemes require a high amount of activation energies especially atomic hydrogen addition reactions after direct cleavages of functional groups. The activation energies of reactions can be reduced using various ways such as application of appropriate catalysts, solution phase reactions, etc. For instance, Verma and Kishore37 carried out a numerical study for the conversion reaction of acetaldehyde into ethanol in gas phase as well as in aqueous phase. They reported that aqueous phase reaction required 14.45 kcal mol−1 less activation energy compared to the gas phase reaction at CCSD/6-311+g(3df,2p)//B3LYP/6-31g(d) level of theory. However, the component used by them is different than the one is in the present study but the aqueous phase milieu can significantly reduce the activation energy. Similarly, the variation of temperature and pressure conditions can also affect the rate of reactions, therefore, the thermochemistry calculation is also carried out. Further thermochemical details are presented in next subsection:
Thermochemistry
The thermochemistry study for conversion of eugenol into various products using all reaction schemes is presented here at wide range of temperature of 298–898 K with an interval temperature of 100 K. The pressure is constant as 1 atm for each temperature variation. The reactions whose reactants and products are same, the thermodynamic parameters (ΔG and ΔH) are also identical, e.g., reaction pathways 2, 2a, 4, and 4a. On the other hand, the reactions whose reactants and products are different, the thermodynamic parameters (ΔG and ΔH) are different, e.g., reaction pathways 3, 3a, 7, and 7a. The thermodynamic parameters, ΔG and ΔH, are enlisted in Table 4.| Pathways | Parameters | Temperature (K) | ||||||
|---|---|---|---|---|---|---|---|---|
| 298 | 398 | 498 | 598 | 698 | 798 | 898 | ||
| 1 | ΔG | −170.10 | −167.89 | −165.33 | −162.48 | −159.39 | −156.10 | −152.64 |
| ΔH | −176.06 | −177.44 | −178.86 | −180.28 | −181.69 | −183.07 | −184.41 | |
| 2 & 2a | ΔG | −38.63 | −40.02 | −41.31 | −42.52 | −43.66 | −44.75 | −45.79 |
| ΔH | −34.31 | −34.70 | −35.09 | −35.48 | −35.87 | −36.28 | −36.69 | |
| 3 | ΔG | −46.54 | −51.41 | −56.16 | −60.79 | −65.33 | −69.78 | −74.16 |
| ΔH | −31.81 | −32.28 | −32.81 | −33.37 | −33.96 | −34.56 | −35.15 | |
| 3a | ΔG | −62.31 | −67.83 | −73.25 | −78.54 | −83.72 | −88.79 | −93.76 |
| ΔH | −45.67 | −46.04 | −46.57 | −47.21 | −47.95 | −48.75 | −49.58 | |
| 4 & 4a | ΔG | −144.54 | −140.78 | −136.74 | −132.48 | −128.06 | −123.52 | −118.88 |
| ΔH | −155.20 | −156.37 | −157.47 | −158.47 | −159.38 | −160.21 | −160.97 | |
| 5 | ΔG | −449.80 | −432.41 | −414.36 | −395.88 | −377.15 | −358.26 | −339.29 |
| ΔH | −500.34 | −503.16 | −505.46 | −507.25 | −508.57 | −509.46 | −509.96 | |
| 6 | ΔG | −219.20 | −213.61 | −207.96 | −202.32 | −196.74 | −191.25 | −185.86 |
| ΔH | −235.65 | −236.05 | −236.13 | −235.91 | −235.42 | −234.69 | −233.74 | |
| 7 | ΔG | −3.96 | −7.71 | −11.53 | −15.45 | −19.48 | −23.65 | −27.94 |
| ΔH | 7.18 | 7.33 | 7.72 | 8.33 | 9.13 | 10.09 | 11.20 | |
| 7a | ΔG | −19.72 | −24.13 | −28.62 | −33.20 | −37.87 | −42.65 | −47.53 |
| ΔH | −6.68 | −6.43 | −6.04 | −5.51 | −4.86 | −4.10 | −3.23 | |
| 8 | ΔG | 23.59 | 22.15 | 20.45 | 18.49 | 16.30 | 13.87 | 11.21 |
| ΔH | 27.49 | 28.40 | 29.55 | 30.92 | 32.47 | 34.18 | 36.03 | |
It can be seen from Table 4 that all reaction schemes are spontaneous at each temperature condition except reaction pathway 8. The reaction scheme 1, i.e., the production of propylbenzene from eugenol, shows high spontaneity and exothermicity at all temperature conditions; however, it shows decrement in spontaneity as the temperature is increasing but it shows incremental behaviour in the case of exothermicity with increasing temperature. On the contrary, the reaction schemes 2 and 2a, i.e., the production of guaiacol from eugenol, show increment in spontaneity and exothermicity with increasing temperature. The reaction schemes 3 and 3a, i.e., the production of methylbenzene from eugenol, also show similar behaviour as of reaction schemes 2 and 2a, i.e., they become more favourable with increasing temperature, though, both reactions are reported to be favourable at each temperature. It should be noted that both thermodynamic parameters are different for reaction schemes 3 and 3a unlike the reaction schemes 2 and 2a (see Table 4). The reaction schemes 4 and 4a, i.e., the production of 4-propylphenol from eugenol, show similar behaviour as of reaction scheme 1, i.e., reaction schemes 4 and 4a are spontaneous at each temperature but become less spontaneous with increasing temperature. On the other hand, increase in exothermicity is reported for both reaction schemes 4 and 4a with increasing temperature. The reaction scheme 5, i.e., the production of propylcyclohexane from eugenol, is reported to be highest exothermic and spontaneous amongst all reaction schemes but it becomes significantly less spontaneous with increasing temperature because the difference between ΔG898 and ΔG298 is calculated to be 110.51 kcal mol−1. However, reaction scheme 5 becomes slightly more exothermic with increasing temperature as the difference between ΔH898 and ΔH298 is calculated to be −9.62 kcal mol−1. Thus, it can be observed that reaction scheme 5 is most favourable at 298 K. Hence, the highest exothermicity and spontaneity of reaction scheme 5 validate the work done by Deepa and Dhepe24 for one of the major production of propylcyclohexane from eugenol in the presence of supported Pd catalysts. The reaction scheme 6, i.e., the production of 2-methyl-4-propylphenol from eugenol, is second most favourable reaction with similar observation of reaction free energy as of reaction scheme 5, however, the difference between ΔG898 and ΔG298 is only 33.34 kcal mol−1. But, unlike reaction scheme 5, reaction scheme 6 becomes slightly less exothermic with increasing temperature. The reaction schemes 7 and 7a, i.e., the production of allylbenzene from eugenol, show opposite behaviour compared to reaction scheme 1. The ΔG of reaction schemes 7 and 7a decreases with increasing temperature or in other words it becomes more spontaneous with increasing temperature but the ΔH of reaction schemes 7 and 7a increases with increasing temperature. However, it should be noted that the ΔH values are positive for reaction scheme 7 and negative for reaction scheme 7a at all temperatures. Moreover, ΔG values of reaction scheme 7a are lower compared to reaction scheme 7. Therefore, reaction scheme 7a is more favourable compared to reaction scheme 7 at all temperature variations. The reaction pathway 8, i.e., the formation of trans-isoeugenol is neither spontaneous nor exothermic at any given temperature condition. The ΔG values for the formation of trans-isoeugenol decrease as the temperature increases but do not reach to negative values so that the reaction could become spontaneous. On the other hand, ΔH values of reaction pathway 8 increase as the temperature increases.
The reaction pathways 1, 4, 4a, 5, and 6 show decrement in spontaneity with increasing temperature, therefore, these reactions are most spontaneous at 298 K only. Out of all the reaction schemes, only reaction schemes 6 and 7a are the ones which report a reduction in exothermicity; however, reaction pathways 7 and 8 are the ones which are endothermic at 298 K and become further endothermic with increasing temperature. On the other hand, reaction schemes 2, 2a, 3 and 3a are the only reactions which show an increment in both spontaneity and exothermicity with increasing temperatures. The overall conversion of eugenol over zeolite catalyst by Nimmanwudipong et al.13 is represented as first-order kinetics.
Conclusions
The conversion of eugenol, a bio-oil model component, using eight primary and four secondary reaction schemes is studied numerically under gas phase environment at B3LYP/6-311+g(d,p) level of theory. The present study concludes that direct cleavages of functional groups of eugenol followed by an atomic hydrogenation reaction, to produce lower fraction product, are not favourable. Instead, an atomic hydrogenation reaction prior to the cleavage of functional groups of eugenol makes reactions much more favourable. The activation energy for the production of guaiacol from eugenol under reaction scheme 2 is reported to be 10.53 kcal mol−1 only which is the least activation energy required amongst all activation energies corresponding to each reaction scheme. The activation energies of some of the reactions are high which cannot be carried out in the gas phase milieu, can be tried in the solution phase using various solvents to decrease the activation energy. The reaction scheme 5, i.e., the production of propylcyclohexane from eugenol, is reported to be most exothermic and spontaneous reaction at all temperature conditions, however, ΔG values increase with increasing temperature and ΔH values decrease with increasing temperature.Acknowledgements
Authors gratefully acknowledge the financial support (sanction no. 34/20/17/2016-BRNS) received from Board of Research in Nuclear Sciences (India) for this work.References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. R. K. Gollakota, M. Reddy, M. D. Subramanyam and N. Kishore, Renewable Sustainable Energy Rev., 2016, 58, 1543–1568 CrossRef CAS.
- A. R. K. Gollakota, M. D. Subramanyam, N. Kishore and S. Gu, RSC Adv., 2015, 5, 41855–41866 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- Y. Wang, T. He, K. Liu, J. Wu and Y. Fang, Bioresour. Technol., 2012, 108, 280–284 CrossRef CAS PubMed.
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075 RSC.
- M.-Y. Chen, Y.-B. Huang, H. Pang, X.-X. Liu and Y. Fu, Green Chem., 2015, 17, 1710–1717 RSC.
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef PubMed.
- D. L. Klass, Encyclopedia of Energy, 2004, 1, 193–212 Search PubMed.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 CAS.
- J. Horáček, G. Šťávová, V. Kelbichová and D. Kubička, Catal. Today, 2013, 204, 38–45 CrossRef.
- T. Nimmanwudipong, R. C. Runnebaum, S. E. Ebeler, D. E. Block and B. C. Gates, Catal. Lett., 2012, 142, 151–160 CrossRef CAS.
- C. Zhang, J. Xing, L. Song, H. Xin, S. Lin, L. Xing and X. Li, Catal. Today, 2014, 234, 145–152 CrossRef CAS.
- W. Mu, H. Ben, A. Ragauskas and Y. Deng, BioEnergy Res., 2013, 6, 1183–1204 CrossRef CAS.
- D. J. Nowakowski, A. V. Bridgwater, D. C. Elliott, D. Meier and P. de Wild, J. Anal. Appl. Pyrolysis, 2010, 88, 53–72 CrossRef CAS.
- A. M. Verma and N. Kishore, Mol. Simul., 2017, 43, 141–153 CrossRef CAS.
- J. Lu, S. Behtash, O. Mamun and A. Heyden, J. Catal., 2015, 321, 39–50 CrossRef CAS.
- A. M. Verma and N. Kishore, ChemistrySelect, 2016, 1, 6196–6205 CrossRef CAS.
- J. Lu, S. Behtash, O. Mamun and A. Heyden, ACS Catal., 2015, 5, 2423–2435 CrossRef CAS.
- M. V. Bykova, O. Bulavchenko, D. Y. Ermakov, M. Y. Lebedev, V. Yakovlev and V. N. Parmon, Catal. Ind., 2011, 3, 15–22 CrossRef.
- M. Wang, C. Liu, X. Xu and Q. Li, Chem. Phys. Lett., 2016, 654, 41–45 CrossRef CAS.
- C. Liu, Y. Deng, S. Wu, H. Mou, J. Liang and M. Lei, J. Anal. Appl. Pyrolysis, 2016, 118, 123–129 CrossRef CAS.
- A. K. Deepa and P. L. Dhepe, ChemPlusChem, 2014, 79, 1573–1583 CrossRef CAS.
- E. B. Ledesma, J. N. Hoang, Q. Nguyen, V. Hernandez, M. P. Nguyen, S. Batamo and C. K. Fortune, Energy Fuels, 2013, 27, 6839–6846 CrossRef CAS.
- L. Simón and J. M. Goodman, Org. Biomol. Chem., 2011, 9, 689–700 Search PubMed.
- R. G. Parr, Density-functional theory of atoms and molecules, Oxford Univ. Press, 1994 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev. [Sect.] A, 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. Sham, Phys. Rev. [Sect.] A, 1965, 140, 1133–1138 CrossRef.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed.
- C. Liu, Y. Zhang and X. Huang, Fuel Process. Technol., 2014, 123, 159–165 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revis. B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, Gauss View 5, Semichem Inc., Shawnee Mission, KS, 2009.
- A. Olbert-majkut and M. Wierzejewska, J. Phys. Chem. A, 2008, 112, 5691–5699 CrossRef CAS PubMed.
- A. M. Verma and N. Kishore, International Journal of Research in Engineering and Technology, 2016, 5, 53–57 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26357g |
| This journal is © The Royal Society of Chemistry 2017 |