
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two pairs of enantiomeric α-pyrone dimers from the endophytic fungus Phoma sp. YN02-P-3†
Xia-Nan Sangab,
Shao-Fei Chenab,
Gang Chenab,
Xiao Anab,
Sheng-Ge Liab,
Xiao-Jie Luab,
Dan Zhaoab,
Jiao Baiab,
Hai-Feng Wang*ab and
Yue-Hu Pei*ab
aKey Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China. E-mail: wanghaifeng0310@163.com; peiyueh@vip.163.com
bSchool of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, China
First published on 12th January 2017
Abstract
(±) Phomones A (1) and B (2), two pairs of novel enantiomeric α-pyrone dimers from the endophytic fungus Phoma sp. YN02-P-3 are reported. Compounds 1 and 2 are the first examples of 6-α,β-unsaturated ester-2-pyrone dimers, and compound 1 possesses a novel 6/4/5/6 tetracyclic ring system. Their structures and stereochemistry were determined by the analysis of extensive spectroscopic data, ECD calculations and single-crystal X-ray diffraction data.
[2 + 2] Cycloaddition reactions that construct two new C–C bonds and establish up to four new stereogenic centers in a single step1–3 are widely used in the synthesis of natural4,5 and bioactive products and have been utilized to synthesize many kinds of important compounds.6 The course of the addition reaction and the resulting regioselectivities have remained a topic of great interest in this area.7,8 α-Pyrone is a simple heterocyclic dienone system,9 which is frequently used as the substrate of intramolecular photochemical reactions, in order to investigate the intermolecular cycloaddition reactivity.10–12
During our continuing search for novel bioactive secondary metabolites from endophytic fungi, Phoma sp. was obtained from the sample collected in the plant Sumbaviopsis J. J. Smith from Yunnan Province, China. Previous study of this fungus resulted in the isolation of six novel compounds phomeketale A–F,13 one novel 3,4-dihydronaphthalen-1(2H)-one with spiro-butyrolactone and a new isocoumarin.14 Further investigation led to the discovery of two pairs of novel enantiomeric α-pyrone dimers, (±) phomones A (1) and B (2), and a known compound rosellsin (3).15 Phomones A and B are the first examples of 6-α,β-unsaturated ester-2-pyrone dimers via intermolecular unsymmetrical [2 + 2] cycloaddition.16,17 Their structures and stereochemistry were elucidated on the basis of the spectral data, single-crystal X-ray diffraction, and ECD analysis. Interestingly, it was found that phomone B (2) slowly transformed to phomone A (1) in MeOH over one month. The effect of H2O-, pH- and temperature-dependent transformation between compounds 1 and 2, as well as the structural elucidation, postulated biogenetic origin and biological evaluation of these metabolites are reported herein.
(±)-Phomone A (1a/1b) was initially obtained as colorless block crystals. Its molecular formula was established to be C24H28O14 (eleven degrees of unsaturation) on the basis of HRESIMS at m/z 563.1352 [M + Na]+ (calcd 563.1371). Inspection of 1H and 13C NMR spectra (Table 1) indicated one 4-oxy-α-pyrone ring (δC 163.3, 110.0, 168.1, 116.3 and 156.3), one 4-oxy-α-dihydropyrone ring (δC 163.2, 107.4, 164.9, 53.1 and 82.6), four methoxyl groups (δH 3.54, δC 57.9; δH 3.65, δC 51.6; δH 4.03, δC 62.1 and δH 3.78, δC 51.7), five methylene groups (δH 4.28, δC 52.5; δH 4.12/4.29, δC 74.4; δH 4.35, δC 52.8; δH 2.53/2.85, δC 33.9 and δH 4.43/4.33, δC 52.2), three methine groups (δH 4.62, δC 79.8; δH 4.26, δC 40.8 and δH 3.92, δC 47.2) and two ester carbonyls (δC 170.5 and δC 169.2). The HMBC spectrum (Fig. 2) corroborated the presence of the 4-oxy-α-pyrone ring moiety based on correlations from methylene H2-10′ (δH 4.35) to C-2′ (δC 163.3), C-3′ (δC 110.0) and C-4′ (δC 168.1) and from H2-12′ (δH 4.43/4.33) to C-4′, C-5′ (δC 116.3) and C-6′ (δC 156.3), and due to the other HMBC correlations, the gross structure of 1 could not be established unambiguously. Fortunately, a crystal suitable for X-ray crystallographic study (CCDC 1504985) was obtained upon slow evaporation of MeOH by keeping the sample at room temperature for one month. The final refinement on the Cu Kα data resulted the crystal of 1 had a p21/c space group, indicating a racemic nature, which was in accordance with the lack of optical activity. Furthermore, the X-ray diffraction analysis (Fig. 3) allowed to unambiguously assign the absolute configurations of the two enantiomers of 1 to be (5R, 6R, 7R, 7′S, 8′S) and (5S*, 6S*, 7S*, 7′R*, 8′R*) as Fig. 1, respectively. Separation by using chiral-phase HPLC yielded 1a ([α]20D + 30 (c 0.20 MeOH)) and 1b ([α]20D − 39 (c 0.20 MeOH)) in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, whose absolute configurations were established by comparing the calculated ECD spectra with the experimental spectra (Fig. 4). From the above evidence, the absolute stereochemistry for 1a (5R, 6R, 7R, 7′S, 8′S) and 1b (5S*, 6S*, 7S*, 7′R*, 8′R*) were unambiguously determined as shown in Fig. 1.
:1, whose absolute configurations were established by comparing the calculated ECD spectra with the experimental spectra (Fig. 4). From the above evidence, the absolute stereochemistry for 1a (5R, 6R, 7R, 7′S, 8′S) and 1b (5S*, 6S*, 7S*, 7′R*, 8′R*) were unambiguously determined as shown in Fig. 1.
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH (m, J in Hz) | δC | δH (m, J in Hz) | |
| a Measured in DMSO-d6 at 400 MHz for 1H and 100 MHz for 13C. | ||||
| 2 | 163.2 | 166.5 | ||
| 3 | 107.4 | 112.4 | ||
| 4 | 164.9 | 169.0 | ||
| 5 | 53.1 | 58.1 | ||
| 6 | 82.6 | 80.1 | ||
| 7 | 79.8 | 4.62 (dd, 9.6, 3.2) | 141.4 | 7.31 (d, 15.6) |
| 8 | 33.9 | 2.53 (dd, 9.6, 16.4), 2.85 (dd, 3.2, 16.4) | 124.3 | 6.39 (d, 15.6) |
| 9 | 170.5 | 167.7 | ||
| 10 | 52.5 | 4.28 (s) | 55.1 | 4.56 (s) |
| 11 | 57.9 | 3.54 (s) | 63.0 | 4.03 (s) |
| 12 | 74.4 | 4.29(d, 9.6), 4.12 (d, 9.6) | 62.6 | 3.90 (d, 11.4), 3.75 (d, 11.4) |
| 13 | 51.6 | 3.65 (s) | 52.4 | 3.78 (s) |
| 2′ | 163.3 | 166.5 | ||
| 3′ | 110.0 | 111.3 | ||
| 4′ | 168.1 | 170.4 | ||
| 5′ | 116.3 | 117.9 | ||
| 6′ | 156.3 | 158.2 | ||
| 7′ | 40.8 | 4.26 (d, 10.0) | 37.2 | 4.20 (d, 11.4) |
| 8′ | 47.2 | 3.92 (d, 10.0) | 50.9 | 4.39 (d, 11.4) |
| 9′ | 169.2 | 170.4 | ||
| 10′ | 52.8 | 4,35(s) | 55.4 | 4.56 (s) |
| 11′ | 62.1 | 4.08 (s) | 63.3 | 4.20 (s) |
| 12′ | 52.2 | 4.43 (d, 12.4), 4.33 (d, 12.4) | 54.6 | 4.55 (d, 12.4), 4.46 (d, 12.4) |
| 13′ | 51.7 | 3.67 (s) | 52.8 | 3.64 (s) |
 | ||
| Fig. 1 Structures of compounds 1–3. | ||
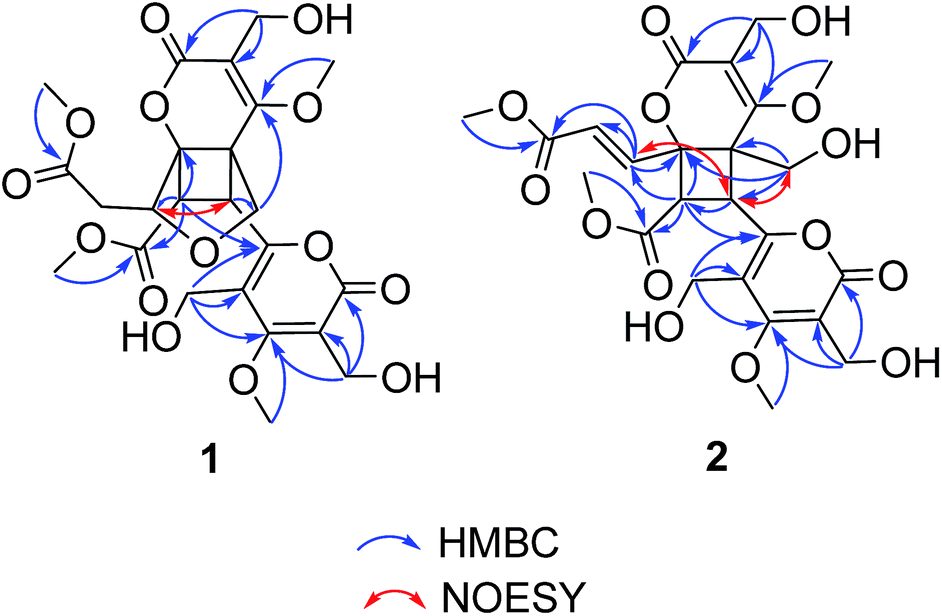 | ||
| Fig. 2 Key HMBC and Noesy correlations of compounds 1 and 2. | ||
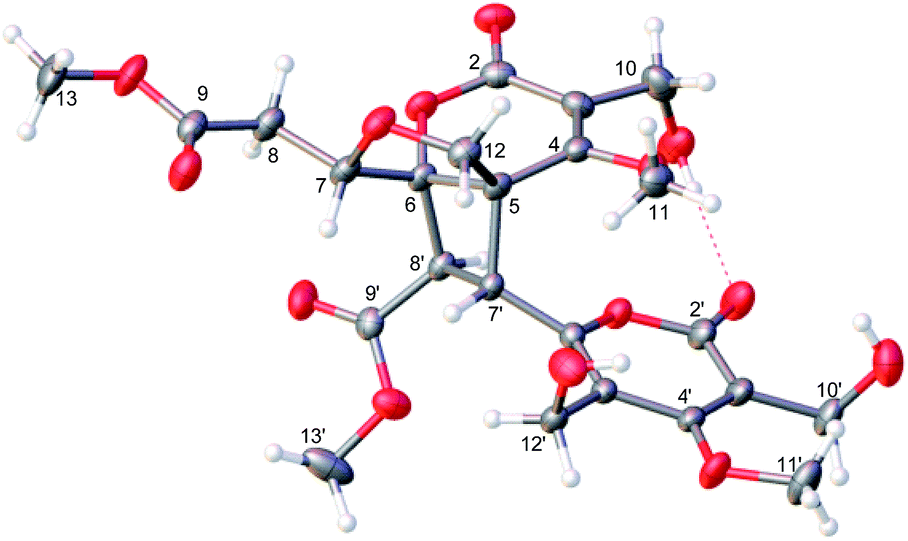 | ||
| Fig. 3 X-ray crystallographic data for 2. | ||
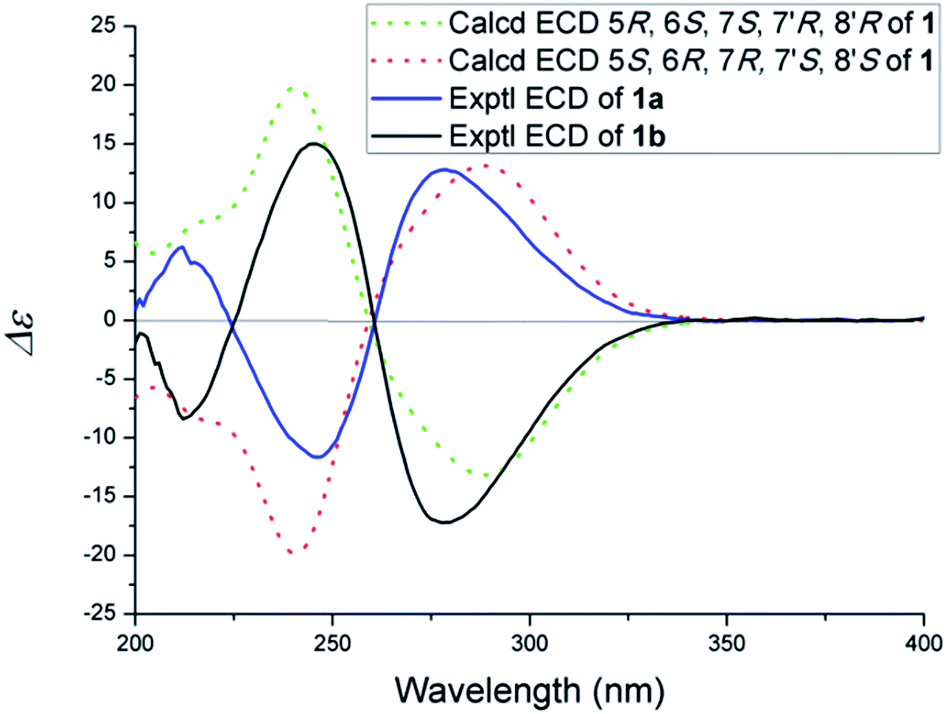 | ||
| Fig. 4 Experimental and calculated ECD spectra of 1. | ||
(±)-Phomone B (2a/2b) shared the same molecular formula of C24H28O14 with compound 1 based on HRESIMS and 13C NMR data. Detailed comparison of its NMR data (Table 1) with those of compound 1 indicated that the main differentiation between compounds 2 and 1 was the absence of the methene [δH 2.53 (1H, dd, J = 16.4, 9.6 Hz), δH 2.85 (1H, dd, J = 16.4, 3.2 Hz)] and the methine [δH 4.62 (1H, dd, J = 9.6, 3.2 Hz)] and the presence of one trans-double bond signals [δH 6.39 (1H, d, J = 15.6 Hz), δH 7.31 (1H, d, J = 15.6 Hz)] in compound 2, which suggested that the furan ring might be open loop to the double bond additive and the C-12 primary alcohol. The HMBC spectrum (Fig. 2) corroborated the presence of a dihydropyrone ring from H2-10 (δH 4.56) to C-2 (δC 166.5), C-3 (δC 112.4) and C-4 (δC 169.0) and from H2-12 (δH 3.75/3.90) to C-4, C-5 (δC 58.1) and C-6 (δC 80.1) and an α-pyrone moiety based on correlations from H2-10′ (δH 4.56) to C-2′ (δC 166.5), C-3′ (δC 111.3) and C-4′ (δC 171.4) and from H2-12′ (δH 4.55/4.46) to C-4′, C-5′ (δC 117.9) and C-6′ (δC 158.2). Further HMBC correlations observed from H-7′ (δH 4.20) to C-5 and C-8′ (δC 50.9) and from H-8′ (δH 4.39) to C-7′ (δC 37.2), C-9′ (δC 170.4), C-6 and C-7 (δC 141.4) demonstrated two α-pyrone rings should be conjugated through a cyclobutane ring and the additional α,β-unsaturated ester moiety was obviously attached to C-6 due to the confirmation of the HMBC correlations from H-7 (δH 7.31) to C-6 and C-8 (δC 124.3), and C-9 (δC 167.7) established the planar structure of compound 2 as shown in Fig. 1.
The NOESY spectrum gave diagnostic correlations of H-7′ with H2-12 and H-7′ with H-7, which illustrated H-7′, H2-12, H-7 oriented in the same direction, and analyses of the coupling constants placed H-8′ on the opposite side of the cyclobutane ring. Subsequent chiral resolution of compound 2 was performed on a chiral column to yield 2a and 2b in a ratio of 1:1, which were virtually opposite in terms of their CD curves (Fig. 5). The final assignment of 2a (5S, 6S, 7′R, 8′R) and 2b (5R, 6R, 7′S, 8′S) was made by the comparison of the calculated electronic circular dichroisms (ECD) via a quantum method with the experimental data (Fig. 5).
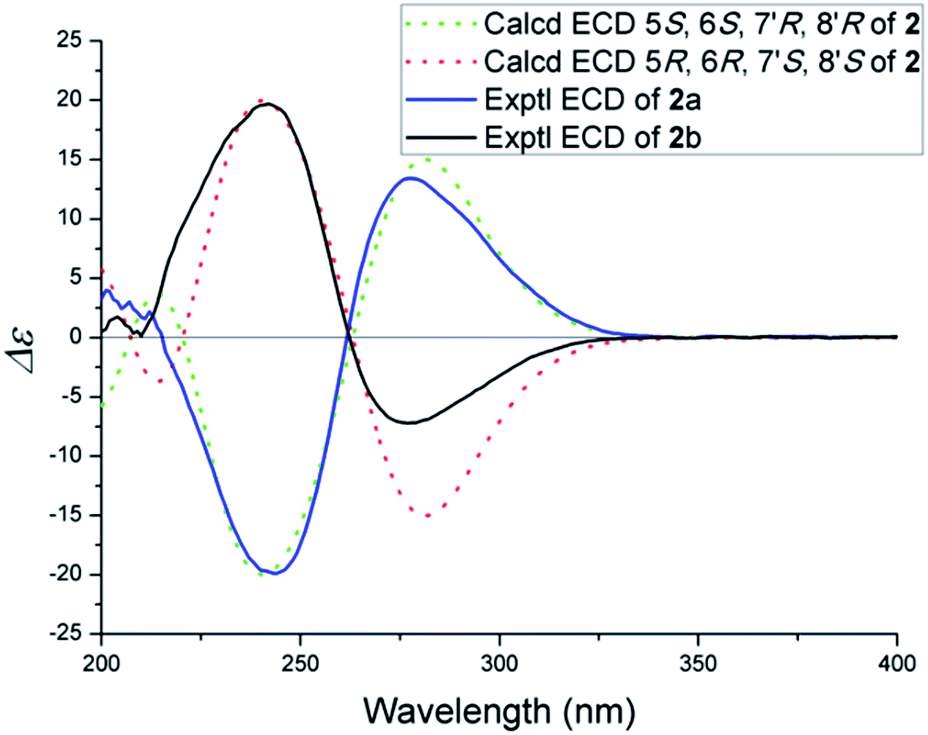 | ||
| Fig. 5 Experimental and calculated ECD spectra of 2. | ||
It was found that compound 2 slowly transformed to compound 1 in MeOH over one month, which indicated that a cyclization reaction was occurring. The effect of H2O-, pH - and temperature-dependent transformation between phomones A (1) and B (2) were further studied. As shown in Fig. S21,† the H2O-temperature heating experiment suggested that H2O could promote transformation and the epimerization was quite sensitive to H2O. The pH-dependent experiment revealed that the transformation was smothered by acid (Fig. S24†) and promoted by alkali (Fig. S22†). Meanwhile, it was not going to make the alkali promote retransformation successful (Fig. S23†). The variable-temperature heating experiment (Fig. S25†) revealed that the single compound 2 was stable in anhydrous ethanol solution below 50 °C for 12 hours. Based on the above results, epimerization of 2 and 1 could be induced by H2O and alkali. The main effect of these transformed observations was H2O, since H2O might come into being along with the fermentation. In our study, three pairs of enantiomers, (±) phomones A (1), B (2) and the acetylated products 4a/4b (Scheme 2) were successfully separated by HPLC employing a CHIRALPAK AD-H chiral column (250 × 4.6 mm, 5 μm) and using anhydrous ethanol as the mobile phase at a flow rate of 0.3 mL min−1.
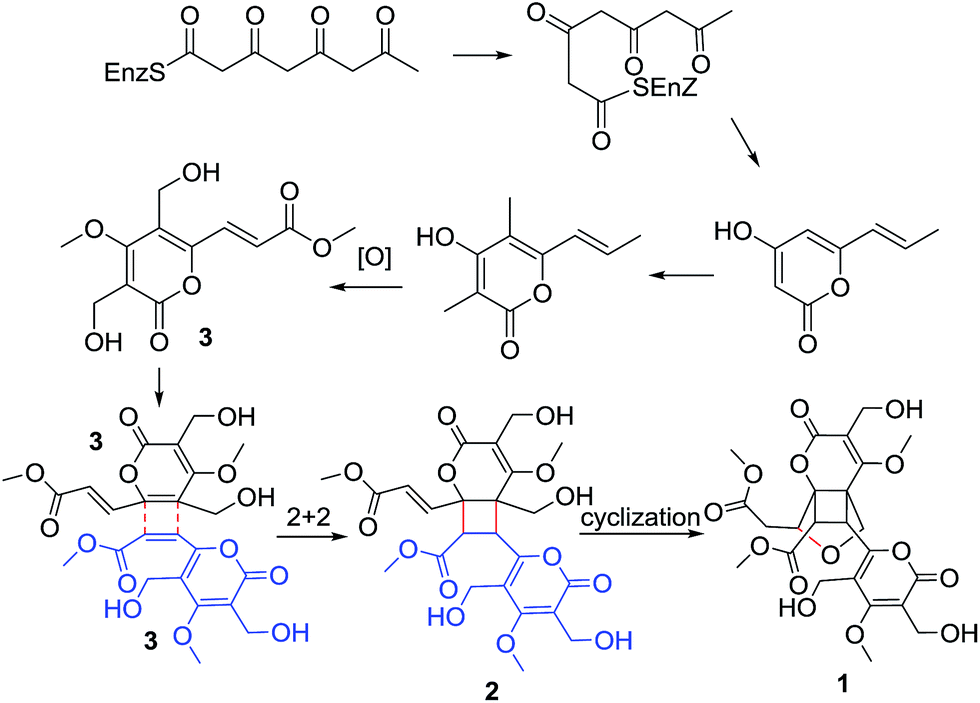 | ||
| Scheme 1 Suggested biosynthetic pathway of 1 and 2. | ||
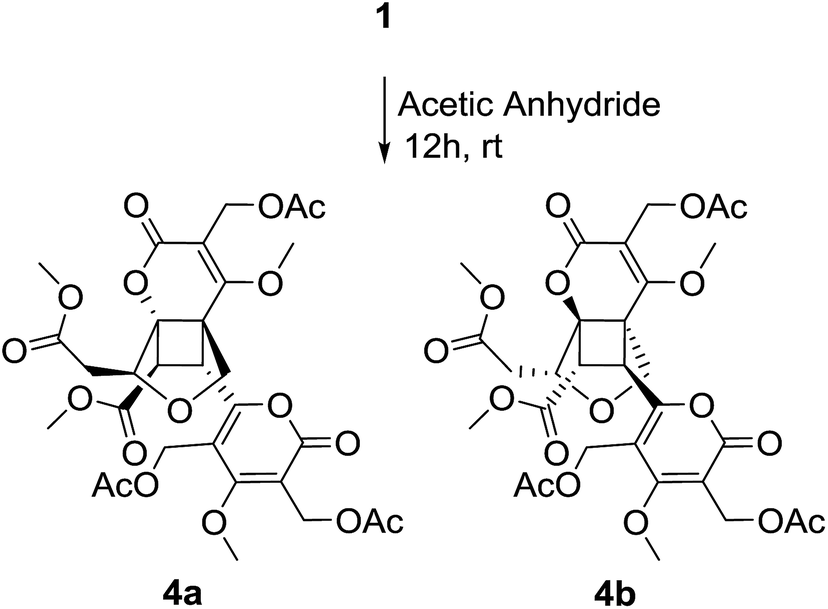 | ||
| Scheme 2 Acetylated of phomone A (1). | ||
As phomone A (1) and B (2) are 6-α,β-unsaturated ester-2-pyrone dimers, a close biosynthetic relationship could be speculated. Phomone B (2) may be derived through a intermolecular unsymmetrical [2 + 2] cycloaddition reaction of two ethylenic bonds between two rosellsin (3) molecules phomone A (1) could be formed through an intramolecular cyclization reaction of phomone B (2) (Scheme 1).
Compounds 1 and 2 showed no activity (IC50 > 50 μM) when evaluated for their cytotoxic activity against three human cancer cell lines, including human leukemia HL-60, human prostatic carcinoma PC-3 and human colon cancer HCT-116, using 5-fluorouracil as positive control. The acetylated products (4a/4b) of compound 1 showed moderate cytotoxic activity against HL-60 cell line with IC50 value of 11.05 μM and 14.18 μM, respectively.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81502951).Notes and references
- H. I. Omar, T. Shimo and K. Somekawa, J. Mol. Struct.: THEOCHEM, 2006, 763, 115–121 CrossRef CAS.
- Y. Yanagisawa, Y. Nishiyama, H. Tanimoto, T. Morimoto and K. Kakiuchi, Tetrahedron Lett., 2014, 55, 2123–2126 CrossRef CAS.
- A. Furutani, K. Tsutsumi, H. Nakano, T. Morimoto and K. Kakiuchi, Tetrahedron Lett., 2004, 45, 7621–7624 CrossRef CAS.
- M. Zhou, X. R. Li, J. W. Tang, Y. Liu, X. N. Li, B. Wu, H. B. Qin, X. Du, L. M. Li, W. G. Wang, J. X. Pu and H. D. Sun, Org. Lett., 2015, 17, 6062–6065 CrossRef CAS PubMed.
- C. S. Yang, X. B. Wang, J. S. Wang, J. G. Luo, J. Luo and L. Y. Kong, Org. Lett., 2011, 13, 3380–3383 CrossRef CAS PubMed.
- T. Sunazuka and S. Omura, Chem. Rev., 2005, 105, 4559–4580 CrossRef CAS PubMed.
- H. I. Omar, Y. Odo, Y. Shigemitsu, T. Shimo and K. Somekawa, Tetrahedron, 2003, 59, 8099–8105 CrossRef CAS.
- T. Obata, T. Shimo, M. Yasutake, T. Shinmyozu, M. Kawaminami, R. Yoshida and K. Somekawa, Tetrahedron, 2001, 57, 1531–1541 CrossRef CAS.
- Y. Li, D. Z. Ye, X. L. Chen, X. H. Lu, Z. Z. Shao, H. Zhang and Y. S. Che, J. Nat. Prod., 2009, 72, 912–916 CrossRef CAS PubMed.
- T. Suishu, T. Shimo and K. Somekawa, Tetrahedron, 1997, 53, 3545–3556 CrossRef CAS.
- T. Shimo, M. Matsushita, H. I. Omar and K. Somekawa, Tetrahedron, 2005, 61, 8059–8064 CrossRef CAS.
- T. Shimo, S. Ueda, T. Suishu and K. Somekawa, J. Heterocycl. Chem., 1995, 32, 727–730 CrossRef CAS.
- X. N. Sang, S. F. Chen, G. Chen, X. An, S. G. Li, X. N. Li, B. Lin, J. Bai, H. F. Wang and Y. H. Pei, RSC Adv., 2016, 6, 64890–64894 RSC.
- X. N. Sang, S. F. Chen, X. An, G. Chen, H. F. Wang and Y. H. Pei, J. Asian Nat. Prod. Res., 2016, 7, 1–8 CrossRef PubMed.
- M. S. R. Nair and S. T. Carey, Tetrahedron Lett., 1975, 41, 3517–3518 CrossRef.
- T. Sagawa, Y. Takaishi, Y. Fujimoto, C. Duque, C. Osorio, F. Ramos, C. Garzon, M. Sato, M. Okamoto, T. Oshikawa and S. U. Ahmed, J. Nat. Prod., 2005, 68, 502–505 CrossRef CAS PubMed.
- I. Ortmann, S. Werner, C. Kruger, S. Mohr and K. Schaffner, J. Am. Chem. Soc., 1992, 114, 5048–5054 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1D and 2D NMR, HRESIMS, UV, IR, and ECD spectra of phomones and detailed experimental procedures. CCDC 1504985. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26319d |
| This journal is © The Royal Society of Chemistry 2017 |